
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical semi-hydrogenation of adiponitrile over copper nanowires as a key step for the green synthesis of nylon-6†
Shutao
Wu
,
Jia
Cheng
,
Yang
Xiang
,
Yunchuan
Tu
 *,
Xun
Huang
* and
Zidong
Wei
*
*,
Xun
Huang
* and
Zidong
Wei
*
Center of Advanced Electrochemical Energy, State Key Laboratory of Advanced Chemical Power Sources, School of Chemistry and Chemical Engineering, Chongqing University, Chongqing, 400044, China. E-mail: huangxun@cqu.edu.cn; zdwei@cqu.edu.cn; yctu@cqu.edu.cn
First published on 22nd June 2024
Abstract
The industrial production of nylon 6 usually includes synthesizing caprolactam through the cyclohexanone–hydroxylamine route. This approach requires complex protocols, elevated temperatures, noble metal catalysts and the use of hazardous strong acids or hydroxylamine. Additionally, a significant quantity of ammonium sulphate is generated during the synthesis procedure. This study aims to develop an electrochemical reduction system for the conversion of ADN generated from the electrolytic dimerization of acrylonitrile (AN) to 6-aminocapronitrile (ACN), a precursor of nylon 6. This system utilizes a cost-effective Cu nanomaterial under eco-friendly conditions, avoiding lengthy and harsh processes, eliminating NH2OH use, and reducing low-value ammonium sulfate generation. This electrosynthesis method maintains approximately 85% ACN selectivity at 40–100 mA cm−2 when passing the charge required for 37% theoretical conversion. When extending the reaction time to achieve an 80% conversion, ACN selectivity still reached 81.6%, exceeding the theoretical value of non-selective hydrogenation by 20%. The pseudo-first-order reaction kinetic modeling proves that the reaction rate constant for ADN hydrogenation is significantly greater than that for ACN hydrogenation, highlighting the selectivity advantage of the system for ACN. This study establishes the foundation for developing a continuous electrolysis process to produce the nylon 6 precursor from AN feedstock.
Introduction
Caprolactam (CPL) is a key monomer for nylon-6 production. With global production of nylon-6 expected to reach 8.9 million tons a year in 2024, the demand for CPL will increase accordingly.1 Currently, the cyclohexanone–hydroxylamine route is crucial in the industrial production of CPL.2 This strategy involves the preparation of cyclohexanone oxime and the synthesis of CPL via the Beckmann rearrangement of cyclohexanone oxime.3 As illustrated in Fig. 1a, the traditional method for synthesizing cyclohexanone oxime includes the sequential processes of hydrogenation and oxidation of benzene to cyclohexanone, followed by its reaction with hydroxylamine (NH2OH).4 This process generates a substantial amount of ammonium sulfate, a by-product of low-value. Moreover, the production of NH2OH directly involves high-pressure H2 and corrosive SO2 and NO2 at high temperature, and the direct utilization of an unstable high-concentration NH2OH solution poses a significant risk of explosion, thereby raising apprehension regarding safety, cost, and sustainability.5–8 The Beckmann rearrangement step is hindered by the requirement to maintain harsh and strongly acidic reaction conditions.3,9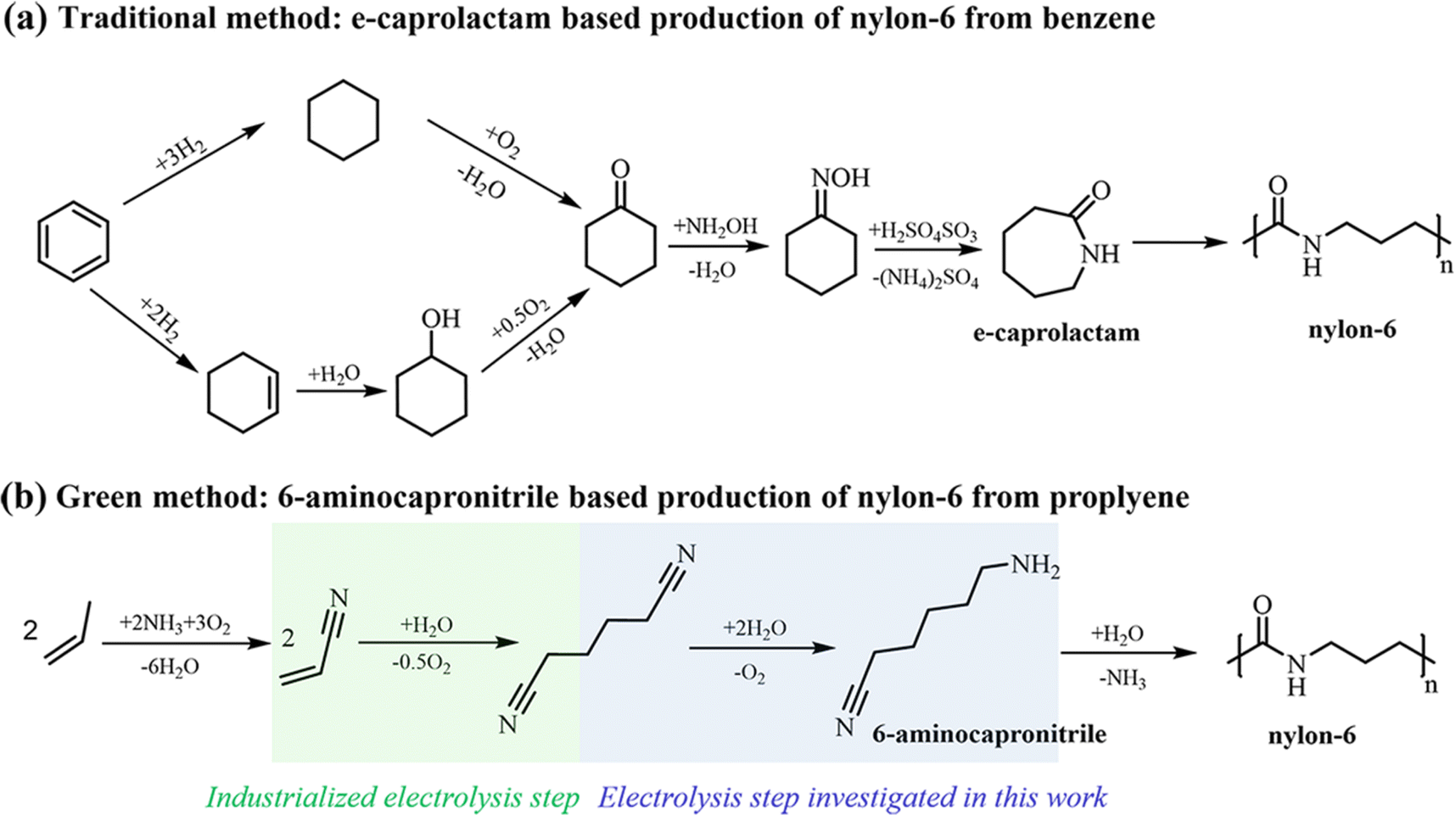 | ||
| Fig. 1 Process steps for the formation of nylon-6 from (a) benzene and (b) propylene. | ||
Accordingly, it is significant to explore alternative methods for the synthesis of nylon 6. As shown in Fig. 1b, one intriguing approach involves the production of nylon 6 from adiponitrile (ADN) through selective hydrogenation to produce intermediate 6-aminocapronitrile (ACN), which is subsequently subjected to hydrolysis polymerization.10 Some progress has been made in traditional hydrogenation of ADN, but current studies rely on Raney-type catalysts or noble-metal complexes with H2 as the hydrogen source, operating at high-temperature and pressure, posing concerns about resource cost, safety, and feedstock/product thermal degradation.11 Since the photo-/electrocatalytic technology offers a mild and sustainable alternative to organic synthesis,12,13 in particular, the electrocatalyst utilizing water as a hydrogen/oxygen source can avoid the above disadvantages existing in ADN reduction. In addition, ADN can also be produced from the electrochemical hydrogenation of acrylonitrile (AN). As a potent and sustainable method in synthetic industries,14–17 the above electrosynthesis route offers significantly enhanced atom efficiency and reduced fossil energy consumption. According to the electrochemical hydrogenation pathway of ADN depicted in Scheme 1 (see the ESI),† the primary competitive by-products involving hexamethylenediamine (HMDA) and H2 would hinder the efficient synthesis of ACN. Modestino18 has comprehensively studied the hydrogenation of ADN on the Raney nickel electrode and emphasized the consequence of higher ADN concentration and alkaline electrolyte on promoting conversion. Subsequently, HMDA was obtained with a Faraday efficiency (FE) of 92%, yet the ACN was regarded as a by-product. However, Song19 has reported the electrocatalytic hydrogenation of ADN and nonanenitrile, resulting in the production of corresponding aminonitrile compounds with good selectivity and FE when utilizing a Raney Ni cathode, demonstrating the feasibility for ACN production through selectivity regulation. Nevertheless, achieving the selective synthesis of ACN or other aminonitriles at higher conversion and current density poses a challenge due to the limitations of reactant concentration and current density, which impact the ability to extend the reaction time. The primary challenge remains to find an optimal electrocatalyst and reaction system capable of high-yield synthesis over a wide range of current densities.
In this study, we investigate a selective semi-hydrogenation system for the conversion of ADN to ACN employing a cost-effective free-standing copper nanowire array as the cathode. By integrating electrochemical testing with density functional theory (DFT) calculations, we elucidate the influence of cosolvents in the reaction system on improving the FE and altering the distribution of products. Furthermore, the system exhibits outstanding adaptability to a wide range of current densities. Within the current density range of 40–100 mA cm−2, an ACN selectivity of approximately 85% is maintained without notable reduction in FE when passing the charge required for 37% theoretical conversion. Additionally, the selectivity toward ACN reaches approximately 81.6% at a constant current density of 80 mA cm−2, when the reaction duration is extended to achieve an 80% conversion of ADN. This selectivity is approximately 20% higher than the theoretical value expected for the non-selective hydrogenation of two cyano-groups. The pseudo-first-order chemical kinetics modelling provides additional support for the selectivity advantage of ACN within this reaction system. Our research highlights the feasibility of employing an inexpensive copper nanowire array cathode for the highly selective semi-hydrogenation of ADN. This approach shows promise in the nylon 6 industry by enhancing safety, cost-effectiveness, and sustainability.
Results and discussion
Recent studies have indicated that adjusting the hydrogen binding energy is essential for suppressing competitive reactions, such as hydrogen production.20–23 Initially, we conducted a screening of various metal foils with diverse hydrogen affinity properties as cathode materials for ADN hydrogenation in a 1 M KOH solution. According to the LSV curves depicted in Fig. 2a, the hydrogen evolution reaction (HER) activity aligns with the trend Pt > Ni > Cu > Ag > Pb, consistent with their respective hydrogen binding energies. After the addition of ADN, there was a reduction in the current density on the Pt cathode. This decrease can be attributed to the competitive adsorption of organic molecules, which hindered the HER process. In contrast, the current densities exhibit an increase on Ag and Pb, metals known for their poor HER activity, but these values are significantly lower than other metals.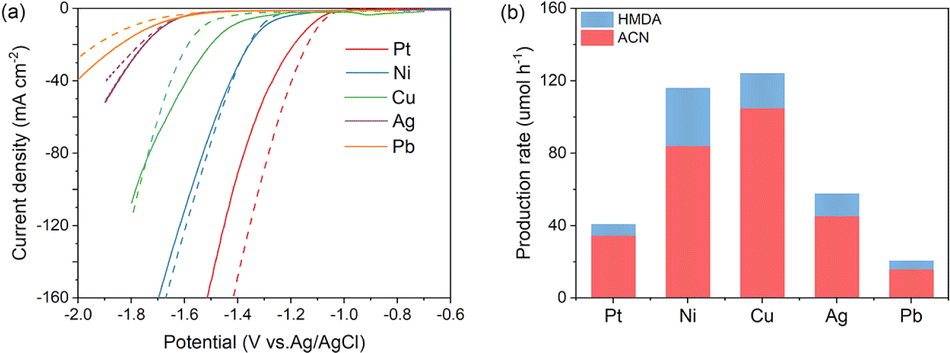 | ||
| Fig. 2 Electrochemical performance of different metals in ADN hydrogenation. (a) LSV curves (dashed line: in a 1 M KOH solution; solid lines: in 1 M KOH solution with 0.1 M ADN); (b) production rates of HMDA and ACN on different metals. | ||
On Cu and Ni electrodes with moderate HER activity, the current density response demonstrates an increase upon the addition of ADN at lower potentials, yet decreases at higher potentials. Combining with the product analysis results shown in Fig. 2b, Ni and Cu demonstrate the highest hydrogenation rates, but the FE and selectivity of ACN are notably smaller. Consequently, Cu is considered as the most suitable cathode for ACN production.
Though high ACN selectivity is obtained, the current density on the 2-dimensional Cu foil is below the industrial requirement. This low current density can be improved by constructing nanostructured electrodes that increase the density of electrochemically active sites. Herein, this work reports the preparation of copper nanowire arrays (Cu NWAs) supported on Cu foam (CF). As shown in Fig. 3a, Cu(OH)2 NWAs were first synthesized by oxidizing the CF substrate with (NH4)2S2O8. Then, CuO NWAs were prepared by thermal treatment of Cu(OH)2 NWAs at 300 °C under an air atmosphere. Finally, Cu NWAs were obtained through electrochemical reduction of CuO NWAs at −0.55–−1.5 V vs. Ag/AgCl in 1 M KOH. After the electrolysis procedure, the cathodic reduction peak disappeared, and the current density became stable, indicating that CuO is successfully transformed into Cu (Fig. 3b). After electrochemical reduction, all the diffraction peaks in XRD patterns match the standard characteristic peaks of Cu (JCPDS No. 04-0836) (Fig. 3c). The “Shoulder” satellite peak located at 962.1 eV and 942.3 eV in the XPS spectrum also disappeared (Fig. 3d), confirming the transformation of CuO into Cu. Moreover, SEM images show the morphology of nanowire arrays in Cu(OH)2 NWAs, and this appearance did not obviously change overall after electrochemical reduction (Fig. 3e, S1 and S2†). The TEM images of the selected regions and the corresponding electron diffraction patterns further indicated the lattice spacing of 0.209 nm, which coincides with the Cu (111) plane, also indicating the successful preparation of Cu NWAs (Fig. 3f and g).
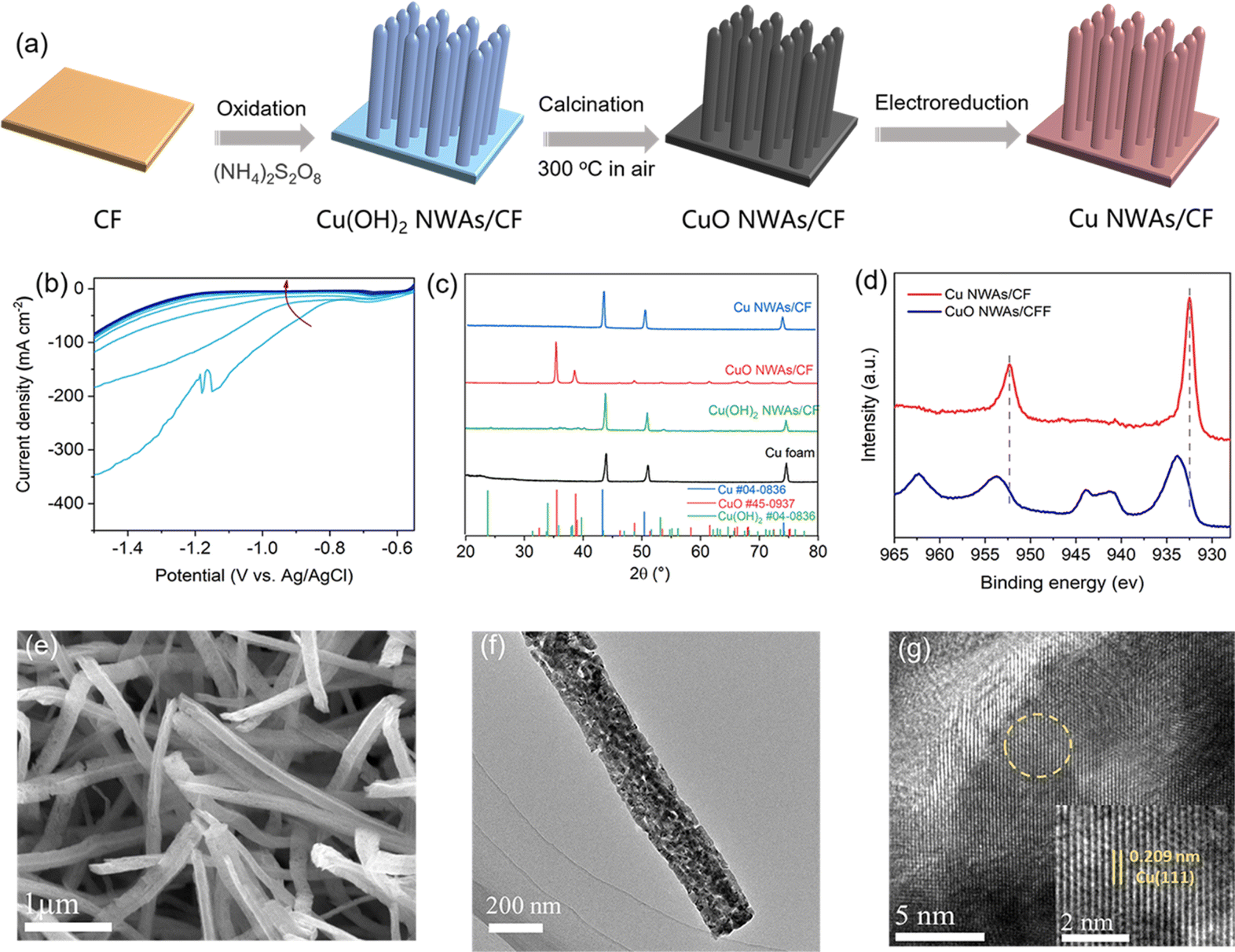 | ||
| Fig. 3 Preparation and characterization of the Cu NWAs catalyst. (a) Synthesis procedure of Cu NWAs; (b) LSV curves during electroreduction; (c) XRD, (d) XPS, (e) SEM, and (f and g) TEM characterization. | ||
Electrochemical performance of the prepared Cu NWAs was first determined in a divided electrolytic cell in a 1 M KOH solution with 0.1 M ADN. The LSV curves exhibited obviously increased current density compared to electrolysis without ADN addition (Fig. 4a), and the current density is about ten fold higher than that on Cu foil at 1.6 V vs. RHE. Constant current measurement was carried out, and qualitative analysis was performed by Gas Chromatography (GC). After continuous electrolysis at 80 mA cm−2 for 2 h, the selectivity of ACN is still as high as 88%, with the total FE of ACN and HMDA maintaining at 60.8% (Fig. 4b). To increase the solubility of ADN in electrolyte, we attempted to add co-solvent such as methanol, ethanol, and tert-butanol into the system. Due to the decrease in the proton concentration of the system,24 the total FE increased to over 80% after the addition of various alcohols (Fig. 4b). Nevertheless, the co-solvent was observed to affect product distribution as well. As illustrated in Fig. 4c, the selectivity in solution with methanol as the co-solvent is similar to that in water alone. However, with the increase of the carbon number in alcohols, the selectivity of ACN decreases slightly. Therefore, in order to balance the FE and ACN selectivity, we chose methanol as the co-solvent in subsequent experiments.
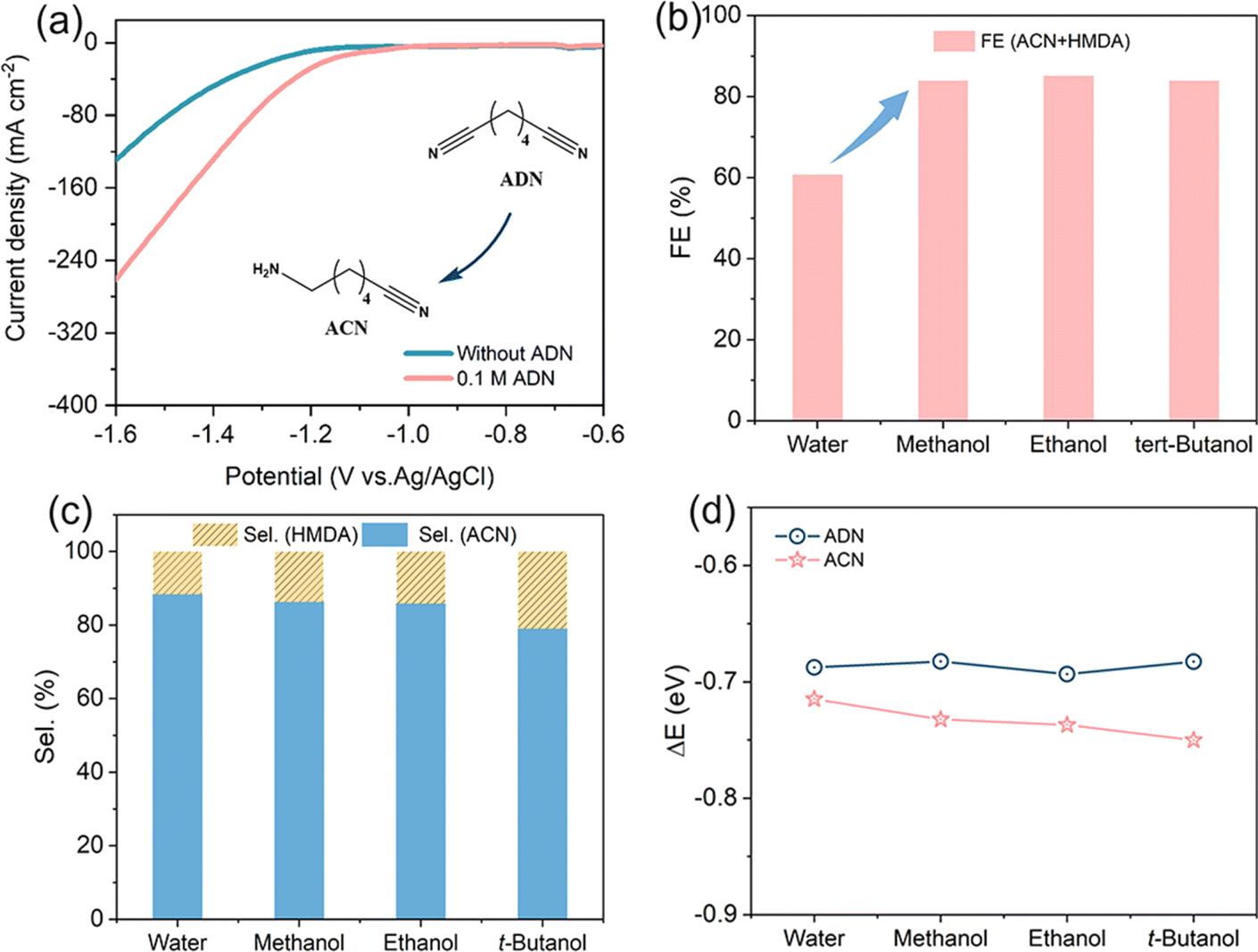 | ||
| Fig. 4 Effect of cosolvents on hydrogenation of ADN. (a) LSV curves over the Cu NWAs electrode in 1 M KOH with and without ADN, scan rate: 10 mV s−1. (b) Total FE and (c) selectivity for ADN reduction in 1 M KOH with different co-solvents at 80 mA cm−2, charge passed = 290 C. (d) Adsorption energy of ADN and ACN on the Cu (111) crystal plane under different relative permittivity. | ||
To further investigate the impact of co-solvents on product distribution, the adsorption of ADN and ACN on Cu (111) was computationally evaluated by the DFT method. The influence of the solvent was included in the model through an implicit solvent model, wherein dielectric constants were used to characterize the polarities of electrolytes. As illustrated in Fig. 4d, the adsorption energies (ΔE) of ADN and ACN in aqueous solution are extremely similar; this phenomenon arises from the presence of a six-carbon alkyl chain in both molecules, facilitating their interaction with the Cu (111) surface via the cyanide group. The different groups on the opposite end exhibit minimal impact on the adsorption strength. However, the change in ΔE of ACN shows a gradual increase, whereas that of ADN exhibits only slight variation, in response to the decrease in the dielectric constant of the co-solvent. This is due to the higher polarity of the amine group in ACN compared to the cyanide group in ADN, resulting in a stronger solvation effect of ACN. Thus, the inclusion of a less polar alcohol leads to a reduction of the solvation effect of ACN.25
Electrocatalytic semi-hydrogenation was further conducted over a wide range of current densities. As shown in the galvanostatic findings (Fig. 5a and S3†), the selectivity of ACN can reach 85.2% at a lower current density of 40 mA cm−2, with an actual ADN conversion of about 25.6% when passing 290 C (charge required for 37% theoretical conversion). The selectivity of ACN was observed to be largely preserved as the applied current density was increased from 40 to 100 mA cm−2. This current density is documented as the maximum achievable for the highly selective electrosynthesis of ACN at a comparable level of ADN conversion. A higher applied current density would lead to a slight decrease in ACN selectivity. Simultaneously, there could be a notable decrease in FE of ACN, which might be attributed to the competitive HER and the extensive hydrogenation of ACN induced by the elevated concentration of surface-absorbed H on the cathode.
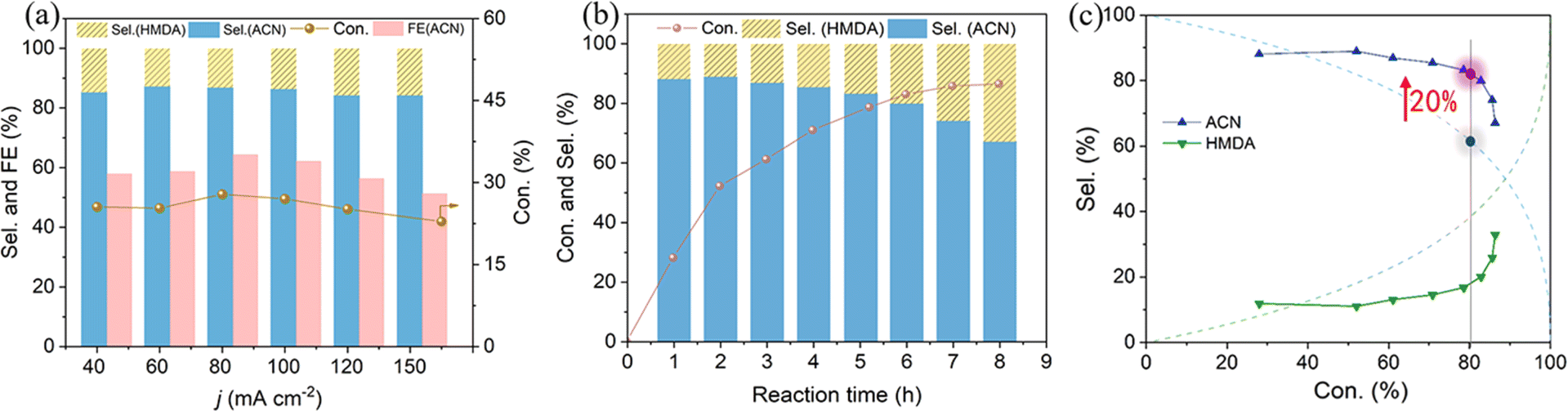 | ||
Fig. 5 (a) Current density-dependent reaction performance over the Cu NWAs cathode in a mixture solution of 1 M KOH/methanol = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v), charge passed = 290 C. (b) Time-dependent conversion and selectivity for ADN electrochemical reduction at −80 mA cm−2. (c) Conversion-dependent reduction product selectivity obtained theoretically (dashed line) via non-selective hydrogenation and experimentally over the Cu NWAs cathode (solid line). :1 (v/v), charge passed = 290 C. (b) Time-dependent conversion and selectivity for ADN electrochemical reduction at −80 mA cm−2. (c) Conversion-dependent reduction product selectivity obtained theoretically (dashed line) via non-selective hydrogenation and experimentally over the Cu NWAs cathode (solid line). | ||
To ascertain the efficacy of our system in preventing the continued hydrogenation of ACN, an extended galvanostatic electrolysis was conducted and changes in selectivity throughout the reaction were recorded. Time-dependent conversion of ADN and selectivity of products are illustrated in Fig. 5b. Due to the structural resemblance of the cyanogen group (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) in ADN and ACN, the intermediate ACN typically undergoes further hydrogenation to produce HMDA when the raw material ADN is consumed. The diagram illustrating the selectivity of ACN and HMDA in relation to ADN conversion, derived from the kinetic reaction rate equation of this continuous reaction by assuming the same kinetic constant for the two steps demonstrates that the ACN selectivity is only 60% at 80% conversion (Fig. 5c). This finding highlights the challenge in sustaining high ACN selectivity in the semi-hydrogenation process. Therefore, prior studies demonstrating excellent ACN selectivity were performed at elevated ADN concentration, passing only about 10% theoretically necessary charge.16 In contrast, although the deep hydrogenation process on Cu NWAs is inevitably facilitated after ADN conversion exceeds 50%, the ACN selectivity decreases slowly and remains at about 81.6% when the conversion reaches 80%, which is about 20% higher than the theoretically expected value for the non-selective hydrogenation of the two cyano-groups in the ADN molecule (Fig. 5c). This observation highlights the superior performance of the system in semi-hydrogenation of ADN.
N) in ADN and ACN, the intermediate ACN typically undergoes further hydrogenation to produce HMDA when the raw material ADN is consumed. The diagram illustrating the selectivity of ACN and HMDA in relation to ADN conversion, derived from the kinetic reaction rate equation of this continuous reaction by assuming the same kinetic constant for the two steps demonstrates that the ACN selectivity is only 60% at 80% conversion (Fig. 5c). This finding highlights the challenge in sustaining high ACN selectivity in the semi-hydrogenation process. Therefore, prior studies demonstrating excellent ACN selectivity were performed at elevated ADN concentration, passing only about 10% theoretically necessary charge.16 In contrast, although the deep hydrogenation process on Cu NWAs is inevitably facilitated after ADN conversion exceeds 50%, the ACN selectivity decreases slowly and remains at about 81.6% when the conversion reaches 80%, which is about 20% higher than the theoretically expected value for the non-selective hydrogenation of the two cyano-groups in the ADN molecule (Fig. 5c). This observation highlights the superior performance of the system in semi-hydrogenation of ADN.
To determine the reaction rate constants k1 and k2 of the two steps at various temperatures or potentials, we assumed the reaction to be first order, and their values were obtained by applying the least squares method to fit the experiment data at different temperatures (Fig. S4(a and b)†) or applied potentials (Fig. S4(c and d)†). The temperature dependence of k1 and k2 were modelled using the Arrhenius equation (Fig. 6a), whereas the potential dependence were determined by fitting the data to the Tafel equation (Fig. 6b). The good linearity indicates that the adopted first-order chemical kinetic model exhibits a high level of accuracy. Furthermore, the experimental and calculated concentrations of ACN and HMDA agree well with each other, thereby confirming the reliability of the model (Fig. 6c). The parameter values in k1 and k2 expressions of ADN electroreduction are summarized in Table 1. As shown in Fig. 6d, the calculated k1: k2 = 4.24 at −1.4 V vs. Ag/AgCl and 25 °C, indicating that the kinetic of ADN hydrogenation is much larger than that of the further hydrogenation of ACN in this system, and the electrochemical reduction system has a selectivity advantage for ACN.
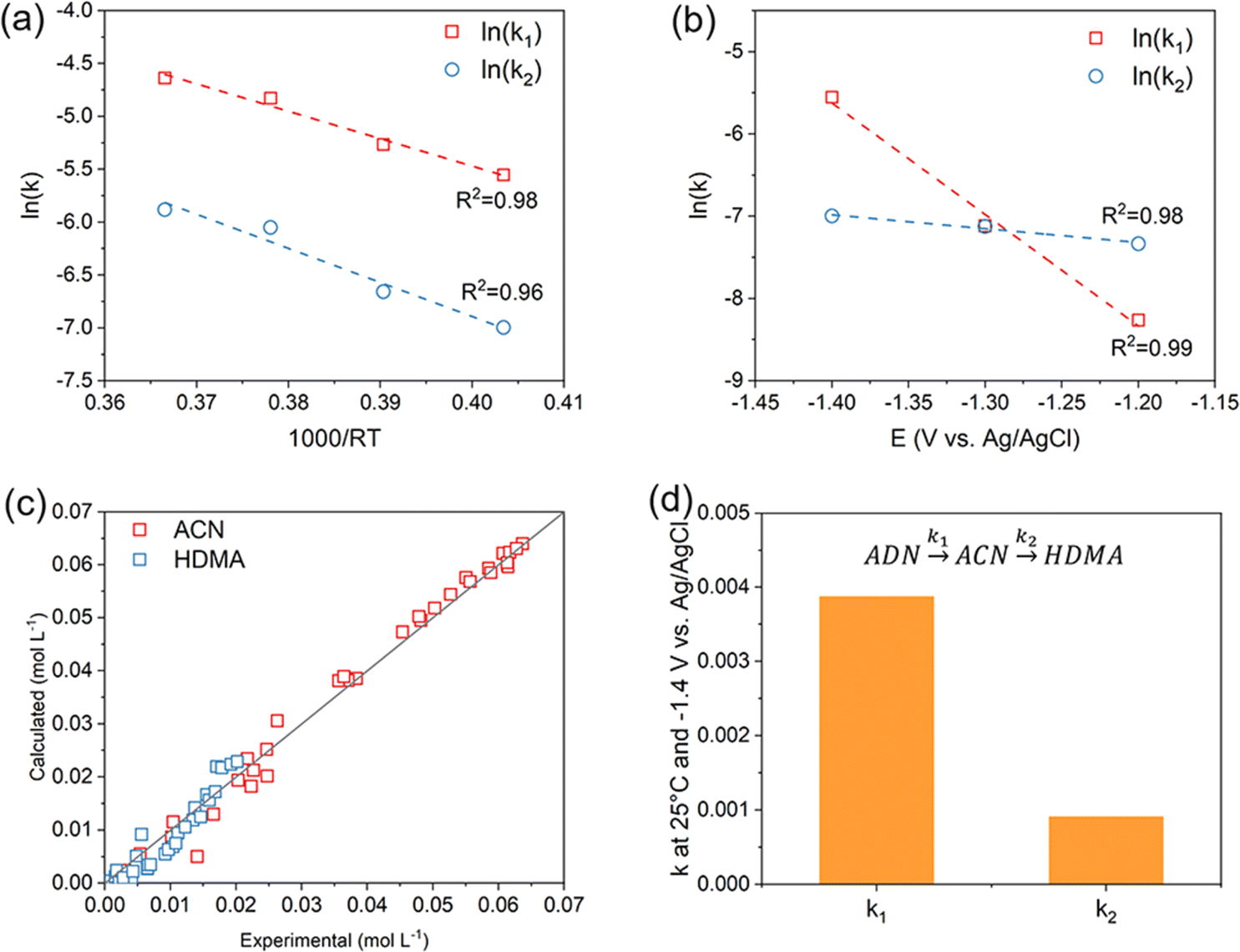 | ||
| Fig. 6 Results of the kinetic study. (a) Temperature dependence of the rate constant at −1.4 V vs. Ag/AgCl; (b) potential dependence of the rate constant at 25 °C; (c) comparison between experimental and calculated concentrations; (d) comparison between k1 and k2 at 25 °C and −1.4 V vs. Ag/AgCl. | ||
| Parameter | Value | Unit |
|---|---|---|
| k ref1 | 3.88 × 10−3 | mol cm−2 min−1 |
| k ref2 | 9.15 × 10−4 | mol cm−2 min−1 |
| T ref | 298.15 | K |
| E ref | −1.4 | V vs. Ag/AgCl |
| Ea1 | 25.92 | kJ mol−1 |
| Ea2 | 32.21 | kJ mol−1 |
| α 1 | −13.55 | V−1 |
| α 2 | −1.68 | V−1 |
Conclusion
In conclusion, a Cu NWAs nanomaterial was prepared for the electroreduction of ADN to ACN. The initial investigation demonstrates that Cu is the most suitable cathode for high ACN selectivity and FE owing to its moderate hydrogen adsorption strength. Then, a cathode composed of Cu NWAs self-supported on CF was fabricated to increase the specific surface area and the current density. Methanol was employed as a cosolvent to further enhance the FE to about 80% while maintaining a high ACN selectivity. At a current density of −80 mA cm−2, the selectivity of ACN remains approximately at 81.6% when ADN conversion reaches nearly 80%, which exceeds the theoretical value of 60% for non-selective hydrogenation. The pseudo-first-order chemical kinetic modelling provides additional confirmation that the rate constant of the ADN electroreduction reaction is about four times higher than that of the deep hydrogenation of ACN. The study results of this work provide a practical approach for the green synthesis of ACN, the nylon 6 precursor, from AN feedstock.Data availability
The datasets supporting this article have been uploaded as part of the ESI.† Also, other relevant data for this article are available from the corresponding authors, upon reasonable request.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (No. 2022YFA1504200) and National Natural Science Foundation of China (No. 22178033 and 22090030).Notes and references
- R. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. Davies, D. Morgan, L. Chen, J. Qi, J. Singleton and J. Edwardset, Science, 2022, 376, 615–620 CrossRef CAS PubMed.
- R. Mokaya and M. Poliakoff, Nature, 2005, 437, 1243–1244 CrossRef CAS PubMed.
- B. Zong, B. Sun, S. Cheng, X. Mu, K. Yang, J. Zhao, X. Zhang and W. Wu, Engineering, 2017, 3, 379–384 CrossRef.
- J. M. Thomas and R. Raja, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13732–13736 CrossRef CAS PubMed.
- L. O. Cisneros, W. J. Rogers and M. Sam Mannan, Thermochim. Acta, 2004, 414, 177–183 CrossRef CAS.
- J. Zheng, Z. Li, D. Zhang, Q. Yang, X. Zhao, B. Zhang and Y. Wang, J. Mol. Liq., 2022, 348, 118407 CrossRef CAS.
- L. A. Long, Process Saf. Prog., 2004, 23, 114–120 CrossRef CAS.
- G. Dahlhoff, J. P. M. Niederer and W. F. Hoelderich, Catal. Rev.: Sci. Eng., 2001, 43, 381–441 CrossRef CAS.
- Y. Wu, W. Chen, Y. Jiang, Y. Xu, B. Zhou, L. Xu, C. Xie, M. Yang, M. Qiu and D. Wang, Angew. Chem., Int. Ed., 2023, 62, e202305491 CrossRef CAS PubMed.
- J. Meuldijk, A. J. M. van Dijk, R. Duchateau and C. E. Koning, Macromol. Symp., 2007, 259, 164–173 CrossRef CAS.
- Y. Lv, J. Li, S. Feng, P. Liu, F. Hao and W. Xiong, Chem. Eng. J., 2018, 346, 203–216 CrossRef CAS.
- D. D. Wang, Z. W. Chen, Y. J. Wu, Y. C. Huang, L. Tao, J. Chen, C. L. Dong, C. V. Singh and S. Y. Wang, SmartMat, 2023, 4, e1117 CrossRef CAS.
- J. C. Xiang, J. li, X. Yang, S. Y. Gao and R. Cao, Acta Phys. Chim. Sin., 2023, 39, 2205039 Search PubMed.
- T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484–2496 CAS.
- Q. Gu, Z. Cheng, X. Qiu and X. Zeng, Chem. Rec., 2023, 23, e202200177 CrossRef CAS PubMed.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- R. Shi, Z. Wang, Y. Zhao, G. I. N. Waterhouse, Z. Li, B. Zhang, Z. Sun, C. Xia, H. Wang and T. Zhang, Nat. Catal., 2021, 4, 565–574 CrossRef CAS.
- D. E. Blanco, A. Z. Dookhith and M. A. Modestino, ACS Sustain. Chem. Eng., 2020, 8, 9027–9034 CrossRef CAS.
- Y. Song and P. N. Pintauro, J. Appl. Electrochem., 1991, 21, 21–27 CrossRef CAS.
- J. Bu, S. Y. Chang, J. J. Li, S. Yang, W. Ma, Z. Liu, S. An, Y. Wang, Z. Li and J. Zhang, Nat. Commun., 2023, 14, 1533 CrossRef CAS PubMed.
- S. T. Wu, H. L. Zhang, X. Huang, Q. Liao and Z. D. Wei, Ind. Eng. Chem. Res., 2021, 60, 8324–8330 CrossRef CAS.
- X. Huang, L. Tan, L. Zhang, C. P. Li and Z. D. Wei, Chem. Eng. J., 2020, 382, 123006 CrossRef CAS.
- Y. M. Wu, C. B. Liu, C. H. Wang, Y. F. Yu, Y. M. Shi and B. Zhang, Nat. Commun., 2021, 12, 3881 CrossRef CAS PubMed.
- E. B. Daniela and Z. D. Aaliyah, ACS Sustain. Chem. Eng., 2020, 8(24), 9027–9034 CrossRef.
- C. Sievers, Y. Noda, L. Qi, E. M. Albuquerque, R. M. Rioux and S. L. Scott, ACS Catal., 2016, 6, 8286–8307 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02280g |
| This journal is © The Royal Society of Chemistry 2024 |