
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDissecting phospho-motif-dependent Shc1 interactome using long synthetic protein fragments†
Peizhong
Chen‡
ab,
Xiong
Chen‡
ac,
Xiaolei
Song
d,
An
He
a,
Yong
Zheng
*de,
Xuechen
Li
 *b and
Ruijun
Tian
*acd
*b and
Ruijun
Tian
*acd
aDepartment of Chemistry, College of Science, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: tianrj@sustech.edu.cn
bDepartment of Chemistry, State Key Lab of Synthetic Chemistry, University of Hong Kong, Pokfulam Road, Hong Kong SAR, P. R. China. E-mail: xuechenl@hku.hk
cShenzhen Key Laboratory of Functional Proteomics, Guangming Advanced Research Institute, Southern University of Science and Technology, Shenzhen, 518055, China. E-mail: yz@ncpsb.org.cn
dState Key Laboratory of Medical Proteomics, Beijing Proteome Research Center, National Center for Protein Sciences (Beijing), Beijing Institute of Lifeomics, Beijing 102206, China
eKey Laboratory of Prevention and Treatment of Cardiovascular and Cerebrovascular Diseases, Ministry of Education, School of Basic Medicine, School of Rehabilitation Medicine, Gannan Medical University, Ganzhou, 341000, China
First published on 12th August 2024
Abstract
Activated receptor tyrosine kinases (RTKs) rely on the assembly of signaling proteins into high-dimensional protein complexes for signal transduction. Shc1, a prototypical scaffold protein, plays a pivotal role in directing phosphotyrosine (pY)-dependent protein complex formation for numerous RTKs typically through its two pY-binding domains. The three conserved pY sites within its CH1 region (Shc1CH1) hold particular significance due to their substantial contribution to its functions. However, how Shc1 differentially utilizes these sites to precisely coordinate protein complex assembly remains unclear. Here, we employed multiple peptide ligation techniques to synthesize an array of long protein fragments (107 amino acids) covering a significant portion of the Shc1CH1 region with varying phosphorylation states at residues Y239, 240, 313, and S335. By combining these phospho-Shc1CH1 fragments with integrated proteomics sample preparation and quantitative proteomic analysis, we were able to comprehensively resolve the site-specific interactomes of Shc1 with single amino acid resolution. By applying this approach to different cancer cell lines, we demonstrated that these phospho-Shc1CH1 fragments can be effectively used as a diagnostic tool to assess cell type-specific RTK signaling networks. Collectively, these biochemical conclusions help to better understand the sophisticated organization of pY-dependent Shc1 adaptor protein complexes and their functional roles in cancer.
Introduction
In response to external cues, the activation of membrane receptors requires scaffold proteins to dynamically recruit high-dimensional protein complexes and precisely orient specific signaling output.1 Scaffold proteins often contain well-defined post-translational modification (PTM)-binding domains and specific PTM sites to sophisticatedly coordinate these dynamic signaling processes, which is essential for maintaining signaling fidelity and versatility. By genetically tagging these scaffold proteins and profiling their protein complexes typically by affinity purification-mass spectrometry (AP-MS), functional protein complexes and their coordination of multiple signaling pathways have been well studied under various physiological and pathological conditions of human cells.2 Despite these advancements, technical difficulties make structural resolving of protein complexes depending on specific PTM sites of scaffold proteins a long-term challenge.Shc1 is a prototypic scaffold protein that functions as a key regulator for protein complex assembly in a variety of receptor tyrosine kinases (RTKs) and immunoreceptors such as TCR and BCR.3 Both RTKs and immunoreceptors mainly use p52Shc1 (referred to as Shc1 hereafter), one of the three transcripts of the Shc1 gene, to transduce signals. This isoform possesses a linear collagen homology (CH1) domain flanked by an N-terminal phosphotyrosine-binding (PTB) domain and a C-terminal SH2 domain (Fig. 1A). Upon activation of RTKs, Shc1 is swiftly recruited to specific pY sites on the cytoplasmic tail of RTKs. This recruitment also leads to sequential phosphorylation of Shc1 on both pY and pS/T sites by the activities of receptor-associated tyrosine kinases and downstream kinases.4 We have systematically explored the temporal dynamics of the Shc1 interactome by immunoprecipitation-MS (IP-MS).5 But the site-specificity of the interactome remains vague. The CH1 domain of Shc1 contains two adjacent phosphotyrosine (pY) sites at position 239 and 240 (pY239/240) within the YYN motif, a pY site at position 313 (pY313) within the YVN motif, along with a phosphothreonine site at position 214 (pT214), and a phosphoserine site at position 335 (pS335). The phosphorylation sites pY239/240/313 and pT214 exhibit high conservation across species, whereas the pS335 site is found in mice and several other species but not in humans, suggesting its species-specific functions. These phosphorylation events create additional docking sites for numerous downstream effectors. As a result, the CH1 region mediates most of the functions of Shc1 by coupling the cytosolic signaling machinery to receptors. Since this signaling regulatory mechanism is adopted by several other RTK-scaffold systems,4 understanding how Shc1 differentially employs these phosphorylation sites to organize protein complex assembly can lead to a broader understanding of RTK signaling.
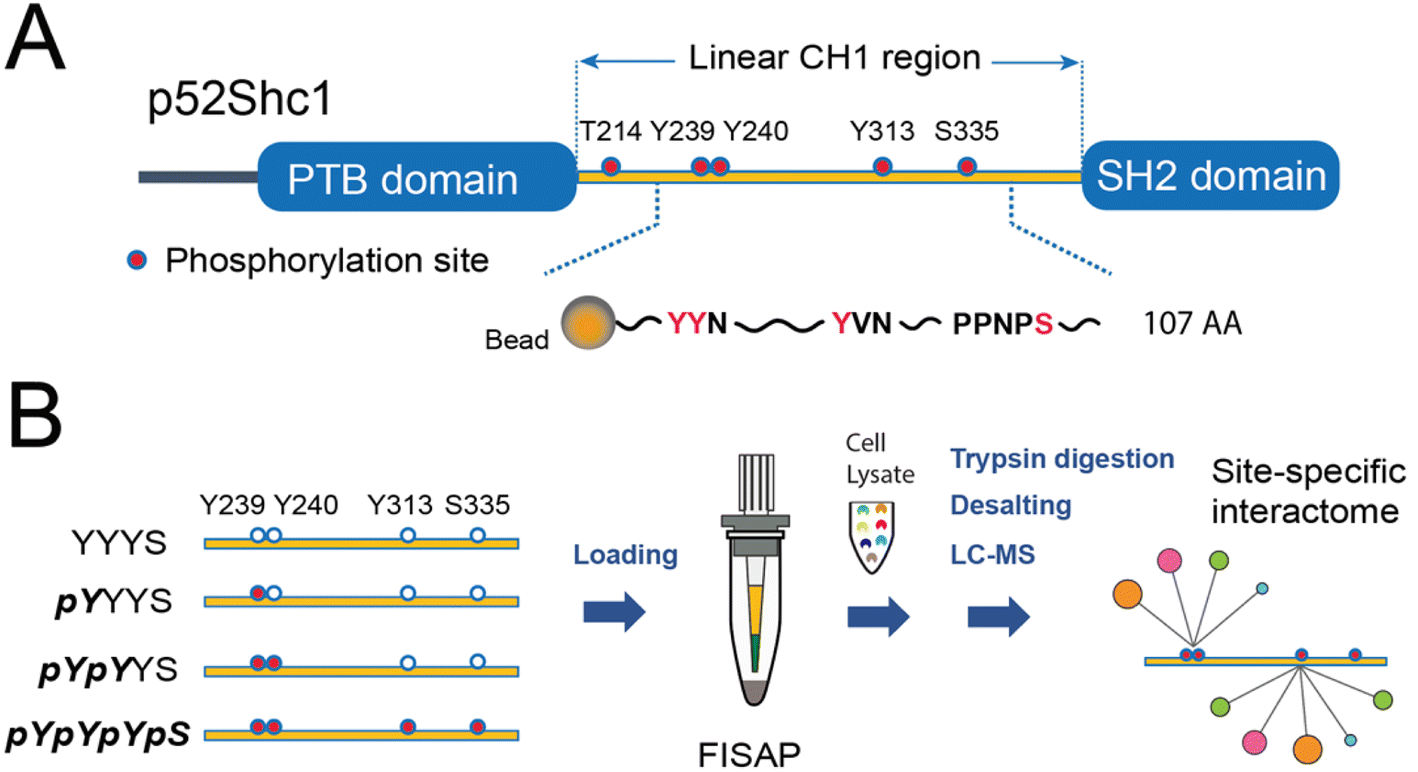 | ||
| Fig. 1 Schematic diagram of the synthetic peptide-based AP-MS for the phosphorylation site-specific interactome of Shc1CH1. (A) Designing 107 AA peptide probes for mouse Shc1CH1 with pY239, pY240, pY313, and pS335. (B) Schematic diagram of the 4 synthesized Shc1CH1 fragments with different phosphorylation patterns for integrated AP-MS analysis. | ||
The utilization of chemically synthesized protein fragments for AP-MS analysis has been demonstrated as an effective strategy for the analysis of interactomes specific to the PTM site in unstructured protein regions.6 For example, through integrating with MS-based proteomics and conventional solid-phase peptide synthesis (SPPS), we have successfully elucidated the site-specific interactome of the 41 amino acid (AA)-long uncoiled cytoplasmic tail of the immune coreceptor CD28.7 To extend the length of synthetic peptides over 100 AA for protein domains such as the Shc1 CH1 region, various chemical and enzymatic ligation strategies can be adopted.8 Among them the native chemical ligation (NCL) originally performed between a C-peptide terminal thioester and an N-terminal cysteine of another peptide remains the most widely applied approach.9 The expansions of NCL include convenient generation of peptide thioesters and the development of cysteine alternatives.8a Although the C-terminal peptide thioesters could be prepared by Boc-SPPS, it is not suitable for phosphorylated peptide because the employed hydrofluoric acid (HF) causes dephosphorylation of phospho-AAs.10 The developed peptide hydrazide chemistry not only offered peptide thioester surrogates for convenient Fmoc-SPPS, but also allowed consecutive assembly of multiple peptides in the N to C direction because it is inert during NCL without NaNO2 activation.11 Another constraint of NCL is the requirement of a cysteine at the ligation site, which is scarce in most proteins. The NCL-desulfurization of cysteine and other unnatural β, γ, or δ-mercapto amino acids greatly expands the ligation sites to cover almost all AAs at the ligating N-terminus.12 However, both metal-based and metal-free radical-based desulfurizations create drawbacks such as metal contamination and epimerization of secondary alcohols.12 The metal-free “add-and-done” (ADD) desulfurization we recently reported by employing TCEP and NaBEt4 is a mild and superfast method particularly suitable for the synthesis of peptides with methionine and phosphorylation.13
In this work, we adopted both the peptide hydrazine chemistry and the ADD desulfurization method for the construction of four 107 AA-length protein fragments containing all three pY sites along with the pS335 site in the Shc1 CH1 region (Shc1CH1, Fig. 1). By integrating these synthetic long phospho-protein fragments of Shc1CH1 with FISAP-based integrated proteomics sample preparation14 and label-free quantitative proteomic techniques, we have successfully revealed the sophisticated organization of pY-dependent Shc1 protein complexes and their utility for probing RTK signaling networks in various cancer cell lines.
Results and discussion
Synthesis of 4 Shc1CH1 protein fragments
As shown in Fig. 1A, Shc1CH1 contains five phosphorylation sites. Among them, Y239/240/313, and S335 play a crucial role in the assembly of Shc1-mediated protein complexes, while T214 likely influences the binding affinity of the PTB domain allosterically instead of directly participating in the complex formation.5 Therefore, to simplify the chemical synthesis, we chose a 107 amino acid long region that encompasses the four phosphorylation sites from the mouse Shc1CH1. Shc1CH1 contains a cysteine at position 302 (C302) (Fig. 2A), which can be ligated to proline by Pro–Cys ligation to overcome the low efficiency of NCL at this site.8a,15 However, C302 is not at the optimal location to allow convenient synthesis of NCL segments with moderate length. Therefore, we decided to utilize the available alanine sites, which can be regenerated through cysteine desulfurization. Particularly, we selected two ligation sites between two adjacent alanine residues as these sterically unhindered sites were reported to offer faster ligation speed.8b Meanwhile, the innate C302 was protected by an acetamidomethyl (ACM) group. We selected ligation sites located between phosphorylation sites to enable the generation of various phosphorylation patterns for the Shc1CH1 protein fragments by combining different short peptide segments. Specifically, we segmented the 107 AA section into four parts with lengths varying from 18 to 30 AAs for synthesis via Fmoc-based SPPS (Fmoc-SPPS). Among these segments, the four phosphorylation sites were distributed, with Y239/240 in Segment 1, Y313 in Segment 3, and S335 in Segment 4 (Fig. 2B).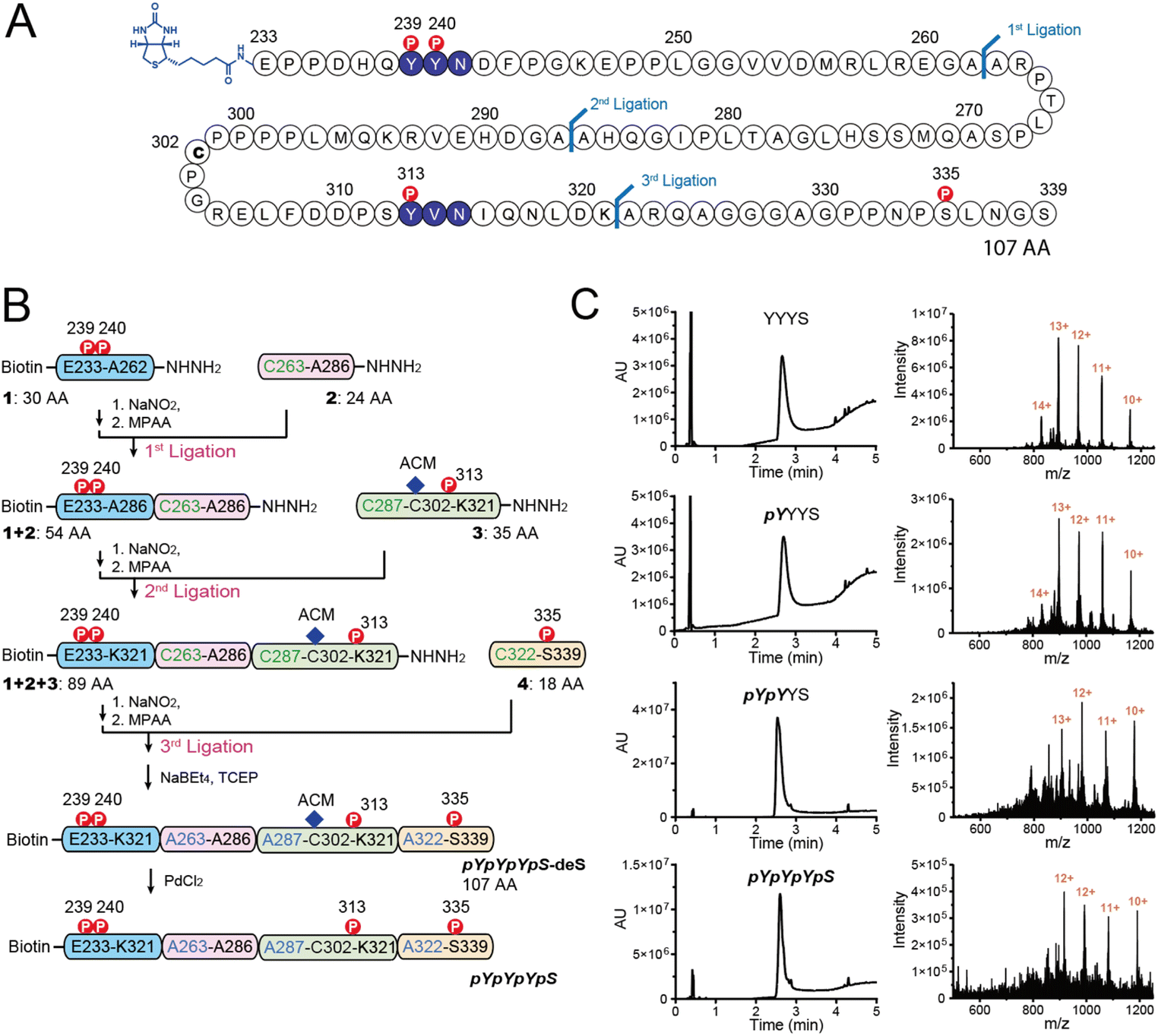 | ||
| Fig. 2 Schematic diagram of the assembly of Shc1CH1 fragments. (A) Chosen NCL sites on the Shc1CH1 peptides. (B) Assembly of the fragment pYpYpYpS by 3 NCLs with peptide hydrazides, followed by ADD desulfurization and ACM-deprotection. (C) LC-MS of the 4 purified Shc1CH1 fragments. | ||
For segments containing single phosphorylation site, only two either phosphorylated or unphosphorylated versions were required. For Y239/240-containing segment 1, we synthesized 3 versions, termed 1-YY, 1-pYY and 1-pYpY, to cover three possible phosphorylation patterns. The segment 2 that contains no phosphorylation sites was shared by all Shc1CH1 protein fragments, which reduced the synthetic workload. To allow sequential assembly of the Shc1CH1 protein fragments without self-circularization, we adopted peptide acyl hydrazide as the thioester surrogate, which was introduced on the C-terminal of the peptide segments via hydrazine-treated chlorotrityl chloride resin.11 One problem we encountered during our sequential NCL procedure is that the excessively introduced 4-mercaptophenylacetic acid (MPAA) during thiolysis often co-eluted with the ligation product during the purification by HPLC (Fig. S1†). This leftover MPAA in the HPLC-purified peptide could react with the newly added NaNO2 during the subsequent NCL, causing significant interference. We found that MPAA could be readily dissolved in either diethyl ether or ethyl acetate, while deprotected peptides were polar enough to remain in the aqueous phase. Thus, we optimized our multi-ligation approach by adding a washing step for all crude NCL products to prevent the carry-over of MPAA (Fig. S1†). After ADD desulfurization of the artificially introduced cysteines at the ligation site, the S-acetamidomethyl (S-ACM) protection of Cys302 was removed by PdCl2.16 Overall, we obtained 4 high-purity Shc1CH1 protein fragments termed YYYS, pYYYS, pYpYYS, and pYpYpYpS with the yield between 32% and 81% for each ligation, desulfurization, or ACM removal step (Fig. 2B, C and S2, Table S2†).
Site-specific interactome profiling by synthetic phospho-Shc1CH1-based proteomics
Due to the limited quantity of synthetic Shc1CH1 phospho-protein fragments, we adopted our fully integrated spintip-based AP (FISAP) technique14 for exploring phosphorylation site-specific interactomes of Shc1CH1 (Fig. 1B). The FISAP require only a few micrograms of the synthetic fragment during sample preparation with minimized sample loss in a simple setting.18 In brief, synthetic Shc1CH1 phospho-protein fragments were immobilized onto streptavidin beads within spintips and incubated with cell lysates. The enriched proteins underwent washing, tryptic digestion, and desalting in the same spintip.14 Eluted peptides were analyzed by LC-MS, and the relative protein amount was deduced by label-free quantification (LFQ, see 6. LC-MS analysis section in ESI† for details). Out of all the tryptic peptides derived from Shc1CH1 phospho-protein fragments, five peptide fragments were consistently identified across all experiments (colored in QC-1C, also underlined in Fig. S23†). The fragment with sequence “EPPLGGVVDMR” (highlighted in green in QC-1C) emerged as the most abundant tryptic peptide with the highest MS/MS count. It is also derived from the non-phosphorylated region of Shc1CH1 protein fragments, making it suitable for evaluating the loading of different fragments during AP-MS analysis. Thus, we used the intensity of this fragment to normalize our LFQ analysis across the YYYS, pYYYS, pYpYYS, and pYpYpYpS groups.Shc1 Y239/240/313 sites undergo rapid phosphorylation with indistinguishable kinetics during the initial stages of EGF signaling (<20 seconds after EGF treatment).5 Despite their crucial role in mediating Shc1 functions, earlier studies have only loosely linked these sites to Grb2-dependent interactions involved in the Shc1-mediated dynamic assembly of the EGFR receptor complex,17 which is conducted through the pTyr-binding SH2 domain on Grb2 that preferentially recruited to the “pYXN” motif.18 To identify the specific role of these sites in complex assembly, we analyzed the interactomes enriched by pYYYS, pYpYYS, and pYpYpYpS fragments in the mouse 4T1 cell line, a highly metastatic mouse cell line that very closely mimics human breast cancer. The interactome comparison between pYYYS and its non-phosphorylated counterpart YYYS indicated that Grb7 is the only high affinity interactor of the singly phosphorylated Y239 motif (Fig. 3A). Grb7 is also a strong in vivo binding partner of the EGF-induced endogenous Shc1 complex in human cancer cells.19 In contrast, the doubly phosphorylated Y239/240 fragment pYpYYS enriched a group of proteins known to participate in in vivo EGFR complex assembly during the early phase of EGFR activation (Fig. 3B and S20†), which is consistent with the dynamics of the phosphorylation of Y239/240.5 Alongside the direct interactors Grb2 and Grb7, there are also presumed indirect interactors, including Sos1/2, Arhgef5, Tns4, Pik3r1, Pik3cb, Src, and Yes1.20 Furthermore, the significantly increased binding of Grb7 to pYpYYS highlighted the requirement for double phosphorylation for optimal protein interactions within this motif. It is noteworthy that proteins bound to pY239/240 of Shc1CH1 predominantly consist of positive regulators of RTK signaling. Interestingly, Src has also been reported to initiate double-phosphorylation of the YYN motifs by priming phosphorylation of the 2nd tyrosine.21
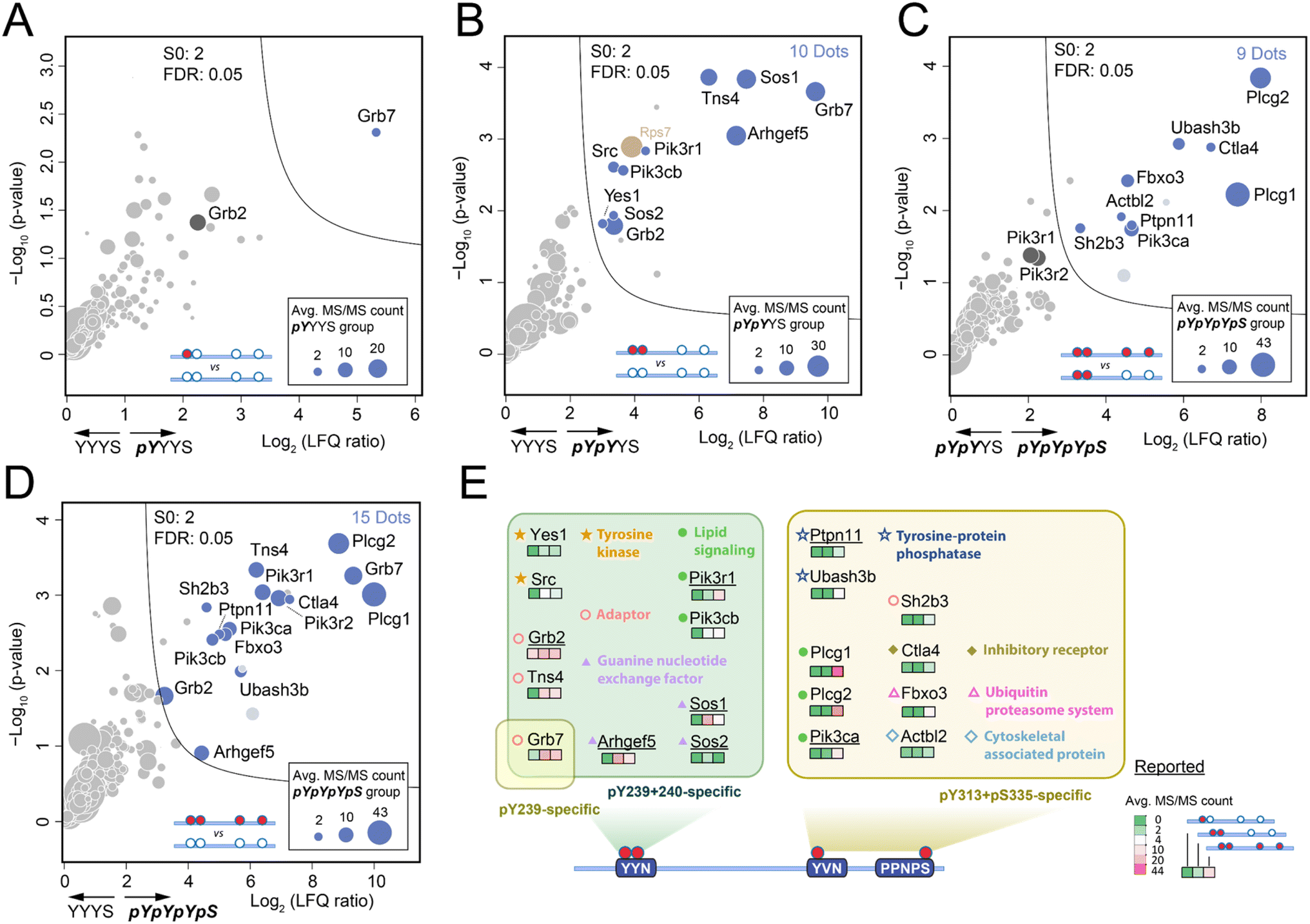 | ||
| Fig. 3 Site-specific interactome of Shc1 in mouse 4T1 cells (A–D). Volcano plot of the standard AP-MS comparing different Shc1CH1 fragments for the specific interactomes of pY239 (A), pY239+pY240 (B), pY313 (C), and the overall interactome of pYpYpYpS (D). The cut-off was set as S0 = 2, FDR = 0.05, average MS/MS count of the right-hand group >2, and at least 2 of the 3 replicates in the right-hand group have MS/MS count ≥ 1 for all volcano plots (A–D). Among the significant dots, the nucleotide-binding proteins were considered as noise, and were colored in tortilla brown. Other cytosolic proteins annotated by the database (https://www.proteinatlas.org) were counted and colored in blue. (E) Summary of the annotated Shc1CH1 interactome captured by AP-MS. | ||
The functions of pY313 and pS335 of Shc1 are particularly intriguing, as evidence from kinetic analysis and site-directed mutagenesis suggest that they are involved in mediating Grb2-independent interactions.17 Unfortunately, the synthesis of the pY313 singly-phosphorylated ShcCH1 segment was particularly challenging for unknown reasons. However, we could deduce the interactions of pY313 by comparing interactomes enriched by pY239/240/313/pS335 (pYpYpYpS) and pY239/240 (pYpYYS). Volcano plot analysis of the AP-MS results demonstrated that the phosphorylation of these two sites can attract a distinct set of proteins of RTK complex assembly. Remarkably, pY313/pS335 intensively recruits two lipid signaling proteins Plcg1 and Plcg2 (Fig. 3C). Previous studies have loosely attributed the recruitment of these two emerging cancer-driving genes to the tyrosine phosphorylation of RTKs.22 Therefore, our discovery hinted at a novel role of Shc1 in regulating lipid signaling through Plcg1/2. Additionally, the pY313/pS335 motif recruited a cluster of negative regulators of RTK signaling, including components of the ubiquitination machinery, such as Fxo3, tyrosine phosphatases Ubash3b and Ptpn11, the negative RTK adaptor Sh2b3, and the inhibitory receptor Ctla4. These observations were further supported by the comparison of interactomes enriched by pYpYpYpS and its non-phospho counterpart (YYYS, Fig. 3D), as well as various in vitro and in vivo studies.23 These pieces of evidence strongly hint at a negative role of these Shc1 phospho-sites in the RTK signaling pathway. However, further functional distinction of pY313 and pS335 relies on improved synthesis of corresponding phospho-protein fragments.
Collectively, the concurrent identification of both direct and indirect RTK interactors highlights the feasibility of our synthetic phospho-protein fragment-based proteomic approach in dissecting protein complex assembly. The analysis of the Shc1 interactome at site-specific resolution has elucidated surprisingly distinct roles for different phosphorylation sites. Specifically, the doubly phosphorylated pY239/240 sites primarily recruit interactors to positively regulate RTK signaling, while phosphorylation on Y313 and S335 sites is likely involved in lipid signaling and the negative regulation of signaling downregulation, respectively.
Verification of site-specific interactomes
First, we selected Plcg1, Pik3r1, Ubash3b, and Grb2 in our AP-MS result with specific affinity to either pY239-240 or pY313 for AP-western blot verification (Fig. 4A). For all 4 proteins, the western blot result matched the AP-MS quantification result (Fig. 4B). We then tested whether AlphaFold3 (AF3)24 can help provide molecular clues for these binding preferences. Specifically, we observed Grb2's enhanced association with the double-phosphorylated pY239/240 compared to the single-phosphorylated Y239, which has been reported and rationalized with the crystallography data.21,25 The AF3 prediction of the complex between mouse Grb2 and Shc1-pYpYpYS highly resembled the structure observed by crystallography (Fig. S21A†). Considering that the sequence of the GRB2-SH2 domain is identical between human and mouse, the predicted structure of the Grb2 complex by AF3 is convincing. Encouraged by the AF3 prediction of Grb2-pY239-240 interaction, we examined the complex between Pik3r1 and pYpYpYS. AF3 suggested a staple binding mode for Pik3r1, with its two SH2 domains for pY239-240 and pY313, respectively (Fig. 4C). The prediction matched our AP-MS and western blot results showing a weak association of Pik3r1 with pYpYYS and a much stronger binding to pYpYpYpS (Fig. 4A and B). Besides, AF3 also successfully predicted the association of Ubash3b (Sts1) with pYpYpYpS by its phosphatase domain, with the key residues specifically recognizing pY313 at its catalytic site (Fig. S21B†).26 This pY-dependent binding mechanism may also be adopted for the interaction between Ubash3b and the cytoplasmic tail of CD28 as we studied previously.7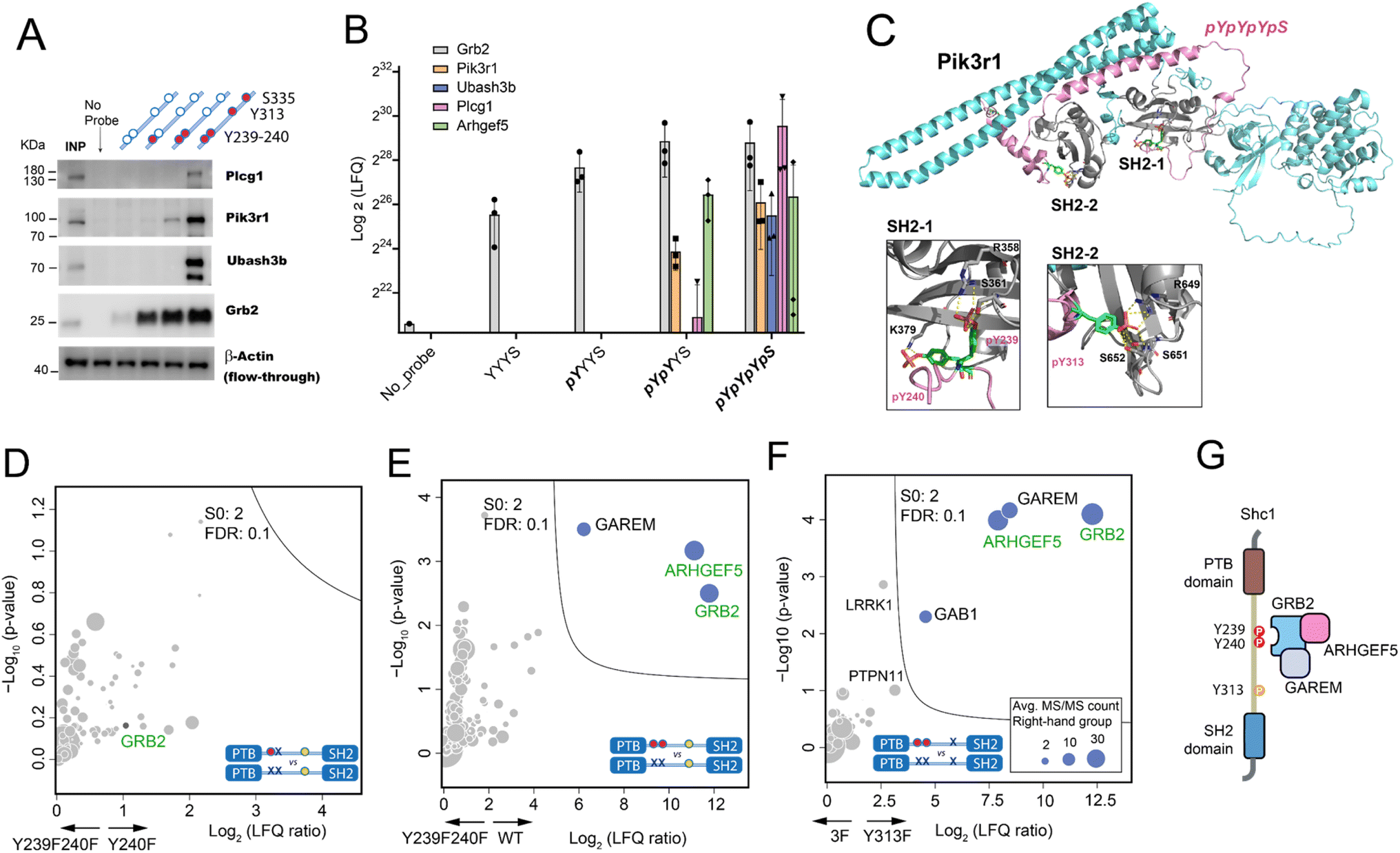 | ||
Fig. 4 Validation of the site-specific interactomes observed in the Shc1CH1 AP-MS analysis by western blot and mouse p52Shc1 IP-MS in HeLa cells. (A) AP-western blot experiment confirming the specific interactions of Plcg1, Pik3r1, Ubash3b, and Grb2 (INP: Input). (B) Log2 LFQ intensity of the selected proteins in the Shc1CH1 AP-MS result (Fig. 3). (C) AlphaFold3 prediction of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex between Shc1-pYpYpYpS and mouse Pik3r1 (D-F). Volcano plots of the IP-MS experiments to compare the interactome of stimulated Shc1 WT and various stimulated Shc1 mutants. Proteins found by our Shc1CH1 AP-MS in the pY239 + 240-specific category are shown in green. (G) Schematic diagram of the complex between Shc1, GRB2, ARHGEF5, and GAREM. :1 complex between Shc1-pYpYpYpS and mouse Pik3r1 (D-F). Volcano plots of the IP-MS experiments to compare the interactome of stimulated Shc1 WT and various stimulated Shc1 mutants. Proteins found by our Shc1CH1 AP-MS in the pY239 + 240-specific category are shown in green. (G) Schematic diagram of the complex between Shc1, GRB2, ARHGEF5, and GAREM. | ||
To further validate the findings from the AP analysis with our synthetic Shc1CH1 protein fragments, we conducted Shc1 IP-MS experiments in HeLa cells. We engineered HeLa cells to stably express a Flag-GFP-tagged wild type mouse p52Shc1 or its mutants with substituted tyrosine (Y) residues at positions Y239, 240, or 313 with structurally similar phenylalanine (F) to prevent phosphorylation. The simultaneous phosphorylation of wild type Shc1 at Y239/240 and Y313 upon EGF stimulation was confirmed by WB of the cell lysate (Fig. S22A†). For IP-MS analysis, the overexpressed Shc1 protein was immunoprecipitated by the anti-Flag antibody from cell lysates that were either treated with EGF to maximize protein complex assembly or left untreated. The resulting immunoprecipitates were then subjected to tryptic digestion and LC-MS analysis. The Flag-tagged GFP was included as a negative control (Fig. S22B†). The expression levels of Shc1 and its mutants were validated (QC-2C). For comparison between the cell lines with overexpressed Shc1, we normalized the LFQ intensity of all proteins in the IP samples based on the LFQ intensity of Shc1. Furthermore, we observed similar levels of EGFR activation upon EGF stimulation, which was indicated by the similar level of co-precipitated EGFR in Shc1 IP-MS analysis (Fig. S22C†).
We focused on comparing the interactomes mediated by different mutants of the Y239/240 sites with single AA difference. The results revealed that substitutions at either one of these two tyrosine residues would similarly impair protein binding, as no distinct interactors were found for either Shc1 Y239F (Fig. 4D) or Y240F (Fig. S22D†). The absence of phosphorylation on Y239/240 mostly compromised the binding of GRB2, ARHGEF5, and GAREM to Shc1, suggesting that Y313 alone cannot substitute for the function of Y239/240 in recruiting these interactors (Fig. 4E and F). The expanded interactome of double-phosphorylated Y239/240 obtained using synthetic Shc1CH1 protein fragments highlighted the advantage of our chemical proteomic approach (Fig. 3A and B). Specifically, our IP-MS analysis confirmed Grb2's reported binding to the double-phosphorylated Y239/240 with high affinity.21 Our IP-MS analysis also supported our AP-MS result for Arhgef5 (Figure 4B, E and F), which agrees with our previous study that Arhgef5 interacts with Shc1 indirectly through Grb2.5 It is worth noting that GAREM, an adaptor protein found to associate with Shc1 in a GRB2-dependent manner,27 consistently emerged as a significant interactor of the double-phosphorylated pY239/240 of the full-length Shc1.5 However, it was absent in the AP-MS analysis conducted with Shc1CH1 protein fragments, showing the necessity of combining different strategies to obtain detailed topological information of the Shc1-Grb2 complex (Fig. 4G).
Phospho-Shc1CH1 as diagnostic probes for RTK signaling networks in cancer cells
The PTB domain of Shc1 is responsible for recruiting Shc1 to RTKs, while the CH1 region helps implement most of its output functions in coupling receptors to downstream signaling machinery. Any compromise in the integrity of protein complex assembly can result in abnormal regulation of signaling pathways. Therefore, the capacity of phospho-Shc1CH1 to enrich both direct and indirect key signaling proteins has inspired us to investigate their potential in evaluating the activation status of RTK-dependent signaling networks in various human cancer cells. Since the sequence and function of Shc1CH1 show high homology between human and mouse (Fig. S23†), and especially has two identical pY-binding motifs, we used various mouse Shc1 phospho-protein fragments to enrich protein complexes in three human breast cancer cell lines with different ERBB expression patterns. MDA-231 is a triple-negative breast cancer (TNBC) cell line with low expressions of both EGFR and HER2 (EGFR-/HER2-).28 MDA-MB-468 is also a TNBC cell line but with significantly elevated EGFR expression (EGFR+/HER2-).29 The BT474 cell line is a frequently used cellular model for HER2-positive breast cancer.28 However, these cells express low levels of EGFR (EGFR-/HER2+). We conducted pilot experiments to further optimize our approach and confirmed the expressions of EGFR and HER2 in these cell lines by western blot and proteomic analysis (Fig. S24A and B†). Subsequently, cell lysates were collected and subjected to protein complex enrichment and sample processing using the FISAP-based workflow with phospho-Shc1CH1. We observed a similar site-specific Shc1CH1 interactome in MDA-MB-468 cells compared to that in mouse 4T1 cells, demonstrating the feasibility of our approach (Fig. S25†).Consistent with the low level of ERBB signaling in MDA-231 cells, fragment pYpYpYpS exhibited minimal recruitment of signaling proteins, as expected (Fig. 5A). In contrast, the phospho-Shc1CH1-enriched interactomes were substantially expanded in either MDA-468 or BT-474 cells (Fig. 5B and C), which validates the use of phospho-Shc1CH1 as a diagnostic probe for detecting signaling from RTKs such as ERBB receptors. Upon a more detailed comparison, it was found that components of the two interactomes were largely overlapped, which is consistent with the conservation of signaling between members of the ERBB family.30 Specifically, eight proteins were shared between MDA-468 and BT-474 cells, which included the adaptor GRB7, PIK3R1/2/3 of the lipid signaling, ARHGEF5, tyrosine kinase YES1, CTLA4 and PKD2L1 (Fig. 5B–D). Nevertheless, the proteins shared between the two interactomes exhibited quantitative differences, suggesting the potential to use phospho-Shc1CH1 as a diagnostic probe to assess quantitative difference in the activities of signaling pathway. A notable example is the significantly enhanced recruitment of GRB7 to Shc1CH1 in HER2-positive BT474 cells, which align with the frequent co-amplification of this adaptor protein with HER2 in breast cancer patients.31 By strongly binding to HER2, but weakly to other ERBB family members, GRB7 significantly enhances cellular survival and migration. Consequently, elevated levels of Grb7 have been correlated with decreased survival rates in breast cancer patients.32
 | ||
| Fig. 5 Comparison of the AP-MS results of Shc1CH1 between three human breast cancer cells with different EGFR and HER2 levels (A–C). Volcano plots of the AP-MS experiments for the interactome of pYpYpYpS in MDA-231 (EGFR-/HER2-), MDA-468 (EGFR+/HER2-), and BT-474 (EGFR-/HER2+) cells. Beside S0 and FDR, other cutoff criteria were the same as that in Fig. 3. (D) Comparison of the interactome of Shc1CH1pYpYpYpS in MDA-231, MDA-468, and BT-474. | ||
Despite the high similarity between phospho-Shc1CH1-enriched interactomes from MDA-MB-468 and BT474, there are still significant parts of the interactomes which are unique to each cell line, as summarized in Fig. 5D. Remarkably, the adaptor protein CRK, which is also a potent activator of tyrosine kinases such as Yes, plays a pivotal role in cell motility and metastasis in highly aggressive and motile cancer cells. The selective detection of CRK in BT474 but not the other two breast cancer cell lines by phospho-Shc1CH1 aligns with the highly invasive phenotype of BT-474.33 On the other hand, our phospho-Shc1CH1 selectively detect UBASH3B, a crucial regulator of the ubiquitination machinery, in the TNBC cell line MDA-MB-468, which overexpresses EGFR. UBASH3B is an oncogenic tyrosine phosphatase that has been found to be highly expressed in TNBC. It functions by deactivating the ubiquitin ligase CBL through tyrosine dephosphorylation, resulting in a significant increase in EGFR signaling.34 For diagnostic purposes, we also compared our AP-MS approach with the whole cell proteomic approach to evaluate the difference between the three cell lines (Fig. S24C and S24D†). Although certain differences between cell lines in our AP-MS analysis, such as that for Grb7, are also reflected in the proteomic analysis, most differences in our AP-MS are insignificant in the volcano plots of the proteomic comparison between cell lines. Also, the AP-MS approach offered significantly improved sensitivity over proteomic analysis for the key interacting proteins, such as PI3K family members PIK3R1, PIK3R2, and PIK3R3, thanks to the selective enrichment by AP.6b,c Collectively, these findings support the validity of employing our synthetic phospho-protein fragment-based method for both quantitative and qualitative evaluation of endogenous RTK signaling across various biological sample types, spanning from cell lines to freshly preserved clinical tissue specimens.
Conclusions
Similar to elucidating the 3D structures of individual proteins, characterizing the topology of high-dimensional protein complexes can yield valuable insights into the regulatory mechanisms of signaling networks. However, this has long been a challenging task from a technical perspective. To tackle this challenge, we developed a novel approach that combines long phospho-protein fragment synthesis using multiple peptide ligation techniques, integrated proteomics sample preparation, and quantitative proteomic analysis. We applied this approach to study a specific protein complex assembled by the CH1 region of Shc1 protein. Our approach successfully demonstrated the precise identification of interacting proteins at specific sites of Shc1CH1, enabling the proposal of novel molecular mechanisms of ERBB-regulated signaling. Additionally, we showcased the potential of utilizing long synthetic Shc1 phospho-protein fragments as diagnostic tools to assess the cell type-specific downstream signaling status of RTKs in cancer cells. This offers unique insights into the organization of protein networks of cancer. Significantly, our approach can be easily applied to elucidate the structural characteristics of diverse PTM-dependent endogenous protein complexes in different types of biological samples. The future progress in chemical and enzymological ligation techniques, including those facilitated by sequence-specific peptide ligases,35 holds great potential for expanding the scope of synthetic long peptides and even proteins. This opens up exciting possibilities for further advancements and broader applications in the field of proteomics.Data availability
All associated MS raw data files have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (https://www.ebi.ac.uk/pride/) with the dataset identifier PXD049113.Author contributions
R. T., X. L., and Y. Z. designed the project. P. C. synthesized the Shc1CH1 peptide fragments and performed AP-MS experiments. X. C. performd AP-western blot and whole cell proteomic experiments and analyzed all proteomic data. X. S. constructed all GFP/mShc1 cell lines and performed IP-MS experiments. X. C. analyzed AF3 results. X. C., Y. Z., X. L., and R. T. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (22125403, 92253304, 32201218, and 22104047), China State Key Basic Research Program Grants (2020YFE0202200, 2021YFA1301601, 2021YFA1301602, 2021YFA1302603, and 2022YFC3401104), the Shenzhen Innovation of Science and Technology Commission (ZDSYS20230626090803004, JSGGZD20220822095200001, JCYJ20200109141212325, JCYJ20200109140814408, JCYJ20210324120210029, and KJZD20230923114220041), and Hong Kong Research Grants Council-Senior Research Fellow Scheme (SPFS2324-7S01).References
- T. Pawson and J. D. Scott, Science, 1997, 278, 2075–2080 CrossRef CAS PubMed.
- (a) A. C. Gingras, M. Gstaiger, B. Raught and R. Aebersold, Nat. Rev. Mol. Cell Biol., 2007, 8, 645–654 CrossRef CAS PubMed; (b) J. Zheng, X. Chen, Y. Yang, C. S. H. Tan and R. Tian, Anal. Chem., 2021, 93, 598–619 CrossRef CAS PubMed.
- A. Mehlitz, S. Banhart, A. P. Maurer, A. Kaushansky, A. G. Gordus, J. Zielecki, G. Macbeath and T. F. Meyer, J. Cell Biol., 2010, 190, 143–157 CrossRef CAS PubMed.
- K. S. Ravichandran, Oncogene, 2001, 20, 6322–6330 CrossRef CAS PubMed.
- Y. Zheng, C. Zhang, D. R. Croucher, M. A. Soliman, N. St-Denis, A. Pasculescu, L. Taylor, S. A. Tate, W. R. Hardy, K. Colwill, A. Y. Dai, R. Bagshaw, J. W. Dennis, A. C. Gingras, R. J. Daly and T. Pawson, Nature, 2013, 499, 166–171 CrossRef CAS PubMed.
- (a) Z. Liu, X. Chen, S. Yang, R. Tian and F. Wang, Curr. Opin. Chem. Biol., 2023, 74, 102305 CrossRef CAS PubMed; (b) W. X. Schulze, L. Deng and M. Mann, Mol. Syst. Biol., 2005, 1, 2005 CrossRef PubMed; (c) A. Lundby, G. Franciosa, K. B. Emdal, J. C. Refsgaard, S. P. Gnosa, D. B. Bekker-Jensen, A. Secher, S. R. Maurya, I. Paul, B. L. Mendez, C. D. Kelstrup, C. Francavilla, M. Kveiborg, G. Montoya, L. J. Jensen and J. V. Olsen, Cell, 2019, 179, 543–560 CrossRef CAS PubMed.
- X. Chen, S. Ji, Z. Liu, X. Yuan, C. Xu, R. Qi, A. He, H. Zhao, H. Song, C. Xiao, W. Gao, P. R. Chen, R. Luo, P. Li, F. Wang, X. Yang and R. Tian, Cell Chem. Biol., 2022, 29, 1024–1036 CrossRef CAS PubMed.
- (a) Y. Tan, H. Wu, T. Wei and X. Li, J. Am. Chem. Soc., 2020, 142, 20288–20298 CrossRef CAS PubMed; (b) A. C. Conibear, E. E. Watson, R. J. Payne and C. F. W. Becker, Chem. Soc. Rev., 2018, 47, 9046–9068 RSC.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- J. W. Perich, E. Terzi, E. Carnazzi, R. Seyer and E. Trifilieff, Int. J. Pept. Protein Res., 1994, 44, 305–312 CrossRef CAS PubMed.
- J. S. Zheng, S. Tang, Y. K. Qi, Z. P. Wang and L. Liu, Nat. Protoc., 2013, 8, 2483–2495 CrossRef CAS PubMed.
- K. Jin and X. Li, Chemistry, 2018, 24, 17397–17404 CrossRef CAS PubMed.
- Z. Q. Sun, W. J. Ma, Y. H. Cao, T. Y. Wei, X. Y. Mo, H. Y. Chow, Y. Tan, C. H. P. Cheung, J. M. Liu, H. K. Lee, E. C. M. Tse, H. Liu and X. C. Li, Chem, 2022, 8, 2542–2557 CAS.
- Y. Mao, P. Chen, M. Ke, X. Chen, S. Ji, W. Chen and R. Tian, Anal. Chem., 2021, 93, 3026–3034 CrossRef CAS PubMed.
- Y. Tan, J. Li, K. Jin, J. Liu, Z. Chen, J. Yang and X. Li, Angew. Chem., Int. Ed., 2020, 59, 12741–12745 CrossRef CAS PubMed.
- (a) S. K. Maity, M. Jbara, S. Laps and A. Brik, Angew. Chem., Int. Ed., 2016, 55, 8108–8112 CrossRef CAS PubMed; (b) B. Zhang, Q. Deng, C. Zuo, B. Yan, C. Zuo, X. X. Cao, T. F. Zhu, J. S. Zheng and L. Liu, Angew Chem. Int. Ed. Engl., 2019, 58, 12231–12237 CrossRef CAS PubMed.
- W. R. Hardy, L. Li, Z. Wang, J. Sedy, J. Fawcett, E. Frank, J. Kucera and T. Pawson, Science, 2007, 317, 251–256 CrossRef CAS PubMed.
- P. van der Geer, S. Wiley, G. D. Gish and T. Pawson, Curr. Biol., 1996, 6, 1435–1444 CrossRef CAS PubMed.
- D. C. Han, T. L. Shen and J. L. Guan, Oncogene, 2001, 20, 6315–6321 CrossRef CAS PubMed.
- Y. Radhakrishnan, L. A. Maile, Y. Ling, L. M. Graves and D. R. Clemmons, J. Biol. Chem., 2008, 283, 16320–16331 CrossRef CAS PubMed.
- M. J. Begley, C. H. Yun, C. A. Gewinner, J. M. Asara, J. L. Johnson, A. J. Coyle, M. J. Eck, I. Apostolou and L. C. Cantley, Nat. Struct. Mol. Biol., 2015, 22, 983–990 CrossRef CAS PubMed.
- W. Shi, G. Zhang, Z. Ma, L. Li, M. Liu, L. Qin, Z. Yu, L. Zhao, Y. Liu, X. Zhang, J. Qin, H. Ye, X. Jiang, H. Zhou, H. Sun and Z. Jiao, Nat. Commun., 2021, 12, 2812 CrossRef CAS PubMed.
- (a) S. L. Harmer and A. L. DeFranco, Mol. Cell. Biol., 1997, 17, 4087–4095 CrossRef CAS PubMed; (b) A. E. Salcini, J. McGlade, G. Pelicci, I. Nicoletti, T. Pawson and P. G. Pelicci, Oncogene, 1994, 9, 2827–2836 CAS; (c) T. van der Meulen, S. Swarts, W. Fischer and P. van der Geer, Biochem. Biophys. Res. Commun., 2017, 490, 1334–1339 CrossRef CAS PubMed; (d) G. Koncz, G. K. Toth, G. Bokonyi, G. Keri, I. Pecht, D. Medgyesi, J. Gergely and G. Sarmay, Eur. J. Biochem., 2001, 268, 3898–3906 CrossRef CAS PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C. C. Hung, M. O'Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Zemgulyte, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Zidek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis and J. M. Jumper, Nature, 2024, 630, 493–500 CrossRef CAS PubMed.
- P. Nioche, W. Q. Liu, I. Broutin, F. Charbonnier, M. T. Latreille, M. Vidal, B. Roques, C. Garbay and A. Ducruix, J. Mol. Biol., 2002, 315, 1167–1177 CrossRef CAS PubMed.
- (a) A. Mikhailik, B. Ford, J. Keller, Y. Chen, N. Nassar and N. Carpino, Mol. Cell, 2007, 27, 486–497 CrossRef CAS PubMed; (b) B. San Luis, N. Nassar and N. Carpino, Biochem. J., 2013, 453, 27–35 CrossRef CAS PubMed.
- K. Tashiro, T. Tsunematsu, H. Okubo, T. Ohta, E. Sano, E. Yamauchi, H. Taniguchi and H. Konishi, J. Biol. Chem., 2009, 284, 20206–20214 CrossRef CAS PubMed.
- (a) X. Dai, H. Cheng, Z. Bai and J. Li, J. Cancer, 2017, 8, 3131–3141 CrossRef PubMed; (b) G. Gambardella, G. Viscido, B. Tumaini, A. Isacchi, R. Bosotti and D. di Bernardo, Nat. Commun., 2022, 13, 1714 CrossRef CAS PubMed.
- J. Filmus, M. N. Pollak, R. Cailleau and R. N. Buick, Biochem. Biophys. Res. Commun., 1985, 128, 898–905 CrossRef CAS PubMed.
- M. A. Olayioye, D. Graus-Porta, R. R. Beerli, J. Rohrer, B. Gay and N. E. Hynes, Mol. Cell. Biol., 1998, 18, 5042–5051 CrossRef CAS PubMed.
- S. Paik, S. Shak, G. Tang, C. Kim, J. Baker, M. Cronin, F. L. Baehner, M. G. Walker, D. Watson, T. Park, W. Hiller, E. R. Fisher, D. L. Wickerham, J. Bryant and N. Wolmark, N. Engl. J. Med., 2004, 351, 2817–2826 CrossRef CAS PubMed.
- Y. Nadler, A. M. Gonzalez, R. L. Camp, D. L. Rimm, H. M. Kluger and Y. Kluger, Ann. Oncol., 2010, 21, 466–473 CrossRef CAS PubMed.
- H. J. van Slooten, B. A. Bonsing, A. J. Hiller, G. T. Colbern, J. H. van Dierendonck, C. J. Cornelisse and H. S. Smith, Br. J. Cancer, 1995, 72, 22–30 CrossRef CAS PubMed.
- S. T. Lee, M. Feng, Y. Wei, Z. Li, Y. Qiao, P. Guan, X. Jiang, C. H. Wong, K. Huynh, J. Wang, J. Li, K. M. Karuturi, E. Y. Tan, D. S. Hoon, Y. Kang and Q. Yu, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11121–11126 CrossRef CAS PubMed.
- (a) K. B. Narayanan and S. S. Han, Enzyme Microb. Technol., 2022, 155, 109990 CrossRef CAS PubMed; (b) A. M. Weeks and J. A. Wells, Nat. Chem. Biol., 2018, 14, 50–57 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02350a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |