
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComplex electrochemiluminescence patterns shaped by hydrodynamics at a rotating bipolar electrode†
Leslie R.
Arias-Aranda
a,
Gerardo
Salinas
 a,
Alexander
Kuhn
a,
Guobao
Xu
bc,
Frédéric
Kanoufi
d,
Laurent
Bouffier
*a and
Neso
Sojic
*a
a,
Alexander
Kuhn
a,
Guobao
Xu
bc,
Frédéric
Kanoufi
d,
Laurent
Bouffier
*a and
Neso
Sojic
*a
aUniv. Bordeaux, CNRS UMR 5255, Bordeaux INP, Site ENSMAC, 33607 Pessac, France. E-mail: sojic@u-bordeaux.fr; laurent.bouffier@enscbp.fr
bState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, P. R. China
cSchool of Applied Chemistry and Engineering, University of Science and Technology of China, No. 96 Jinzhai Road, Hefei, Anhui 230026, P. R. China
dUniversité Paris Cité, CNRS, ITODYS, UMR 7086, 75013 Paris, France
First published on 21st May 2024
Abstract
Electrochemiluminescence (ECL) is a powerful analytical approach that enables the optical readout of electrochemical processes. Over the last few years, ECL has gained considerable attention due to its large number of applications, including chemical sensing, bioanalysis and microscopy. In these fields, the promotion of ECL at bipolar electrodes has offered unprecedented opportunities thanks to wireless electrochemical addressing. Herein, we take advantage of the synergy between ECL and bipolar electrochemistry (BE) for imaging light-emitting layers shaped by hydrodynamics, polarization effects and the nature of the electrochemical reactions taking place wirelessly on a rotating bipolar electrode. The proof-of-principle is established with the model ECL system [Ru(bpy)3]2+/tri-n-propylamine. Interestingly, the ECL-emitting region moves and expands progressively from the anodic bipolar pole to the cathodic one where ECL reactants should neither be generated nor ECL be observed. Therefore, it shows a completely unusual behavior in the ECL field since the region where ECL reagents are oxidized does not coincide with the zone where ECL light is emitted. In addition, the ECL patterns change progressively to an “ECL croissant” and then to a complete ring shape due to the hydrodynamic convection. Such an approach allows the visualization of complex light-emitting patterns, whose shape is directly controlled by the rotation speed, chemical reactivity and BE-induced polarization. Indeed, the bipolar electrochemical addressing of the electrode breaks the circular symmetry of the reported rotating system. This unexplored and a priori simple configuration yields unique ECL behavior and raises new curious questions from the theoretical and experimental points of view in analytical chemistry. Finally, this novel wireless approach will be useful for the development of original ECL systems for analytical chemistry, studies of electrochemical reactivity, coupling microfluidics with ECL and imaging.
Introduction
Optical analysis of electrochemical processes using light-emitting systems has gained considerable attention since it offers an easy and straightforward alternative to conventional methods.1–4 Commonly, light emission approaches are based on the coupling of chemical processes to microelectronic devices (such as light emitting diodes for example), fluorescent probes or electrochemiluminescence (ECL) reactions.5–14 In particular, ECL has become a powerful tool in optical imaging, due to its high sensitivity, broad dynamic range, fast response time and low cost of analysis.15–17 Briefly, by applying a potential step, a sequence of reactions is initiated by electrogenerated species in order to populate the excited state of a luminophore, which emits light upon relaxation. ECL has been known for decades but has evolved progressively from a purely academic experimental curiosity to a variety of well-established applications in multi-analysis detection, imaging, bioanalysis and sensing.18–23Although ECL emission can be triggered by classic electrochemical methods, a direct electric connection is indeed required in order to impose the potential at the working electrode, thus wireless approaches are highly desired. In this context, bipolar electrochemistry (BE) is an interesting alternative to generate localized ECL on the surface of conducting objects.15–17,24,25 BE is based on the induction of a polarization potential difference (ΔV) at the interface between a conducting device, acting as a bipolar electrode (BPE) and the surrounding electrolyte. This is achieved by applying an external electric field (ε) using two external feeder electrodes.15,16,26,27 When ΔV overcomes the thermodynamic threshold potential (ΔVmin), defined by a given set of electroactive species, redox reactions occur at each extremity of the BPE. The synergy between the wireless addressing capability of BE and ECL emission as a readout has already been employed for different applications ranging from sensing and electrocatalysis to the study of dynamic (i.e. mobile) systems.28–39
In particular, externally driven ECL-emitting systems have become a great alternative for imaging mass transport40–45 or exploring concentration gradients.34,35,37 In this context, the continuous motion of dynamic devices inside the electrolyte allows to circumvent and/or eventually overcome mass transfer limitations, which translates into an enhancement of the ECL intensity. Among the different types of motion, rotation is one of the most attractive due to the possibility of finely controlling forced convection. In theory, the controlled rotation of the electrode leads to a decrease of the diffusion layer thickness, allowing a constant renewal of electroactive species at the electrode|electrolyte interface, increasing the faradaic current.46–48 Several bipolar ECL rotors, involving mechanical, magnetic or chemical induction of motion have been developed.35,49–51 However, due to the anisotropy of the proposed BPE design together with the intrinsic principle of BE, light emission is only produced when the main dimension of the rotor is well-aligned with respect to the applied electric field lines.
In this work, we take advantage of a conventional rotating disk electrode (RDE), acting as a BPE, in order to shape the ECL reactive layer. This was achieved by monitoring the ECL emission of the model [Ru(bpy)3]2+/tri-n-propylamine (TPA) system, taking place normally at the anodic side of the rotating disk bipolar electrode (RDBPE) in the absence of rotation. The anodic reactions leading to ECL emission are intrinsically coupled with the water and dioxygen reduction occurring at the cathodic end (Scheme 1). The strength of the convection when increasing the rotation rate, leads to an unprecedented ring-type ECL emission governed by the rotation speed. To the best of our knowledge, such an experimental setup has never been investigated so far and we were able to generate complex ECL patterns. Indeed, the ECL emission is observed not only on the anodic bipolar pole where ECL is triggered but also on the cathodic pole as well as on non-polarized parts of the RDBPE where it should not be observed classically.
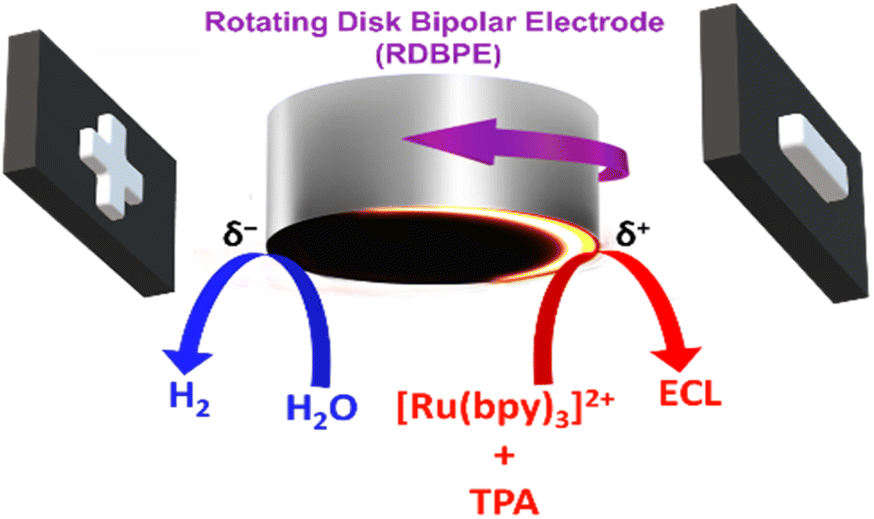 | ||
| Scheme 1 Illustration of the rotating disk bipolar electrode (RDBPE) polarized by the feeder electrodes schematized at both extremities in order to drive electrochemical reactions including electrochemiluminescence (ECL) emission triggered on the anodic pole. Simultaneous oxidation of ECL reagents (TPA stands for tri-n-propylamine) at the anodic pole (δ+) and reduction of dioxygen and water at the cathodic pole (δ−) induce light emission during the rotation of the RDBPE. The direction of the rotation is indicated with a purple arrow. | ||
Results and discussion
To evaluate the influence of the rotation speed (ω) on the ECL patterns, a conventional RDE was polarized in a wireless manner and used as a BPE. The ECL behavior of this device was studied by placing the RDBPE in a BE cell between two feeder electrodes (Scheme 1), containing first 0.1 mM [Ru(bpy)3]2+ and 0.25 mM TPA in 10 mM PBS solution (pH 7.4). [Ru(bpy)3]2+ and TPA were selected as they represent the most widely used luminophore/co-reactant ECL system for analytical and imaging applications.52–55 Several mechanistic pathways have already been discussed in the literature as a function of the applied potential, the electrode material, and the composition of the solution.14,56,57 In brief, the electrochemical oxidation of the ECL reagents leads to the typical red emission of the [Ru(bpy)3]2+ luminophore (620 nm) following the so-called general oxidative-reduction ECL mechanism.14,56,58–60 Thus, by applying a high enough external electric field, it is possible to trigger the oxidation of [Ru(bpy)3]2+/TPA and the reduction of water and dissolved dioxygen at the anodic (δ+) and cathodic (δ−) poles of the RDBPE, respectively. This generates a characteristic red ECL emission on the anodic side of the BPE (Scheme 1 and Fig. 1a). Indeed, BE breaks the circular symmetry of the polarization of the BPE. Under a constant electric field (8.3 V cm−1) and in the absence of rotation (i.e. quiescent solution), a well-defined semi-circular ECL emission is imaged on the anodic pole (Fig. 1a).35 A careful examination of the position of this ECL region revealed a slight displacement away from the edge of the anodic pole of the BPE. This is due to the relatively high electric field, which may trigger additional electrochemical reactions, i.e. the oxidation of water that can modify the local pH and affect the corresponding ECL efficiency, as reported previously.35,61–63 However, under controlled convection a substantial change of the ECL-emitting pattern was obtained for rotation speeds in the range from 10 to 2000 rpm (Fig. 1 and S1†). It is noteworthy that ECL is observed only at the level of the RDBPE (i.e. dotted white circle that materializes the electrode border on Fig. 1a) and not further in solution. Similarly, on a classic RDE (no bipolar addressing), the ECL was observed just at the level of the electrode even during the rotation (Fig. S2†). Please note that in this set of data recorded in a conventional RDE setup, the images were accumulated during the course of a cyclic voltammogram. Nevertheless, in this classic electrochemical configuration, the ECL shape on the RDE is observed as a disk corresponding to the entire electrode surface independently of the rotation speed (ω) and no ECL was observed further in solution. In the bipolar configuration, when ω increases, two main changes were observed: (i) a gradual shift of the ECL region in the direction of the rotation of the RDBPE (anticlockwise dragging), and (ii) a progressive decrease of the thickness of the emission area (Fig. 1b–e and S1†). Such combined effects can be observed at ω values ranging between 10 and 400 rpm, whereas, above this rotation speed interval, the ECL emission essentially decreases until it completely fades (Fig. 1e and S1†). For example, at ω = 25 and 100 rpm, one can see the ECL light, forming an “ECL croissant” shape (Fig. 1b and c), in the top part of the RDBPE, where the polarization tends to be null and is therefore not high enough to trigger the ECL reactions. Even more surprisingly, at ω = 100 and 400 rpm, the ECL emission is visible on the cathodic bipolar pole (i.e. δ−) where reduction reactions occur and where ECL cannot be produced with the [Ru(bpy)3]2+/TPA system (Fig. 1d).64,65 In fact, the ECL pattern forms progressively a quasi-complete ring (at ω = 400 rpm) over the entire periphery of the RDBPE, even if the ECL can be produced only in the anodically-polarized region (i.e. δ+). At ω = 2000 rpm, no ECL was observed under this experimental condition (Fig. S1†). Considering the isotropic disk shape of the RDBPE and the constant applied electric field, it can be assumed that the size of the RDBPE region that is sufficiently polarized to enable ECL remains uniform at all times during these dynamic experiments. Thus, the changes in the ECL-emitting zone are related mainly to the flow patterns of the electroactive species involved in the electrochemical reactions during the RDBPE rotation and to the bipolar polarization. Previously, a limited extension of the ECL-emitting region has been reported in a classic 3-electrode configuration in a flow cell. As ECL was dragged in the direction of the flowing solution,66 this work then demonstrated that the ECL provides a readout of the flow hydrodynamics developed in the cell. Indeed, we rationalized the results gathered in Fig. 1 based on this same assumption. | ||
| Fig. 1 ECL images of the RDBPE at different rotation speeds ω: (a) 0 rpm, (b) 25 rpm, (c) 100 rpm, (d) 400 rpm, and (e) 1000 rpm. A constant electric field of 8.3 V cm−1 was imposed by the two graphite feeder electrodes schematized at both extremities. Experiments were performed in a PBS (10 mM, pH 7.4) solution containing 0.1 mM [Ru(bpy)3]2+ and 0.25 mM TPA. The dotted white circle and the dotted arrow indicate the position of the RDBPE and the direction of rotation, respectively. Exposure time: 8 s. | ||
The first effect of hydrodynamics on ECL can be explained by the angular rotation of the ECL pattern with the electrode rotation rate. Indeed, the solution is dragged along the electrode surface with an angular velocity of vφ = (2πω/60)r. It increases with the radial position on the electrode disk, r, and with the electrode rotation speed ω in rpm, explaining that the electrogenerated species, and therefore the ECL patterns, are dragged along longer angular sectors when increasing the rotation rate, as can be seen for values from 25 to 400 rpm in Fig. 1b to d.
To provide a more quantitative understanding of these results, ECL profiles obtained during the BE experiments at different rotation speeds were analyzed following a two-step approach. At first, we carried out an azimuthal analysis of a defined region of interest (ROI) where ECL occurs, that corresponds to the arc area between two concentric circles (Fig. 2a, area between the internal yellow circle and the external white dotted circle). It is important to highlight that the dimensions of this ROI remained constant during the analysis of all the optical pictures. In this azimuthal description, the angle θ = 0° is defined as parallel to the electric field (i.e. orthogonal to the longitudinal axis of the feeder electrodes), and the ECL profile was extracted in a counterclockwise direction (Fig. 2a). As a result, the plots of the ECL intensity as a function of the angle were obtained for all the different ω values. For the sake of clarity, Fig. 2b shows a selection of the azimuthal profiles at different rotation speeds. Without rotation, the ECL intensity appears variable over the surface due to the surface heterogeneity of the electrode.41 The ECL signals are much smoother under rotation because the ECL-emitting layer is displaced by the convective flow. Based on such an azimuthal ECL plot, it is possible to identify the angle where the maximum light intensity is generated (θmax) at a given angular speed. For example, in the absence of rotation, the maximum ECL intensity is produced at θ ∼ 160° but another maximum is also visible at θ ∼ 200°. Without considering the rather large noise of the ECL signal in the absence of rotation, it suggests a symmetrical situation and the more appropriate angle to consider would be the average or the symmetry axis, which is ∼180°. For the ω values of 25 rpm and 300 rpm, the behavior is different with θmax found at 215° and 237°, respectively, meaning 35° to 57° anticlockwise compared to the electric field axis. It shows how the region where ECL is emitted moves progressively under the influence of the rotation. In other words, the location where the ECL reagents are oxidized (i.e. δ+ pole) does not coincide anymore with the region where ECL is emitted and observed. This is a totally unusual behaviour for an ECL system because ECL is a surface-confined technique due to the short lifetime of the electrogenerated TPA radicals involved in the coreactant mechanism.36,60,67
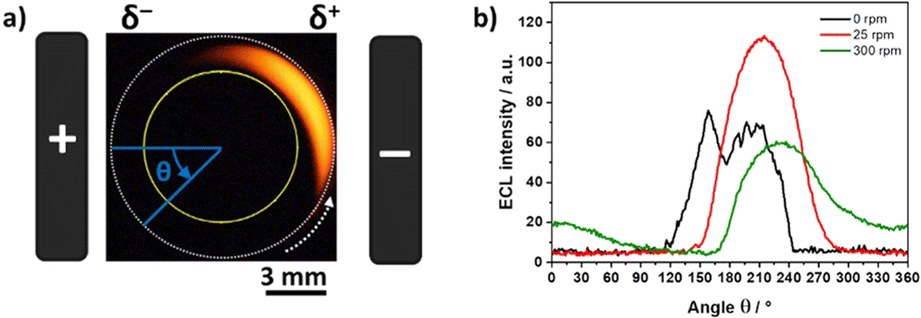 | ||
| Fig. 2 (a) Optical image of the ECL-emitting layer generated by a RDBPE at 25 rpm. The angle θ is used for the azimuthal description. θ = 0° is defined as parallel to the electric field and passing through the center of the RDBPE. The dashed white arrow indicates the direction of the rotation of the RDBPE. The yellow and dashed white circles define the region of interest where the azimuthal analysis was carried out. (b) Azimuthal ECL data in the absence of rotation (black line) and with rotation (25 rpm in red and 300 rpm in green). Same experimental conditions as in Fig. 1. | ||
Since the ECL intensity is an optical readout of the amount of luminophore relaxing from the excited to the ground state, it is possible to assume that θmax at a given ω value is the angular position on the RDBPE where the ECL pathway is the most favoured in the bipolar configuration, once convoluted with the hydrodynamics of the system. A possible explanation for the position of θmax, shifted in position compared to bipolar ECL in the absence of rotation, could be related to electrode polarization effects. Indeed, rotating the electrode in a fixed electric field would be analogous, from the electric point of view, to placing a static electrode in an alternating potential waveform. It is therefore comparable to performing ECL measurements under electrochemical impedance spectroscopy (EIS) conditions with electrode AC polarization described by a frequency 2πω/60, where ω is the rotation of the electrode in rpm. A combination of ECL and EIS was proposed by Itagaki et al.68–70 It was shown that the ECL response to AC polarization could be described by an ECL impedance of the form a/(1 + jω/k). This suggests that the ECL signal may appear with a phase shift from the electrode polarization. This could explain the observed angular shift θmax of the position of maximum ECL under electrode rotation. A quantitative understanding will require a more in-depth mechanistic study of the ECL co-reactant system by EIS. However, the observed angular shift of 30° to 50° could be qualitatively explained by mass transfer limitation, such as a control of the impedance of the system by a Warburg-type impedance.
In the next step, the ECL profiles were analysed as a function of the radial position on the RDBPE, defined as l = R(θ − π) with R the RDBPE disk radius and θ being expressed in radians in this equation. This was done by defining a new ROI from the center of the RDBPE to its edge, that was tilted at the corresponding θmax value (Fig. 3a, blue rectangle). The θmax values were obtained from the azimuthal ECL data. Notably, such a plot offers a quantitative analysis of the variations of the ECL-emitting patterns. At ω = 0, θmax is not obtained for θ = 0° where the polarization should be maximal, because of the side reactions of water oxidation and the heterogeneity of the electrode reactivity. The ECL patterns show a progressive shrinking of the emission area thickness as well as a slight increase of the ECL intensity with the angular speed (Fig. 3a and b). From this plot, one can evaluate the ECL intensity in two ways: (1) the maximum ECL intensity (ECLmax), which is given by the magnitude of the brightest pixel in the blue ROI, and (2) the integral of the ECL emission (ECLsum) that corresponds to the sum of the intensity of all the pixels. The plot of ECLsum as a function of the rotation speed exhibits a maximum at ω = 25 rpm, followed by a fast decrease, reaching a low plateau above 500 rpm (Fig. 3c, black dots). A similar tendency is obtained by evaluating the full width at half maximum (FWHM) of the ECL profile across the RDBPE (Fig. S3,† blue squares), illustrating the shrinking of the ECL layer. In contrast, ECLmax increases gradually with ω until reaching a maximum at 400 rpm (Fig. 3c, red squares). Such an increase in light intensity can be associated with an enhanced mass transport towards the surface of the electrode, as for the classic RDE configuration (Fig. S2†). However, the continuous decrease of the thickness of the emission area and the ECLmax at high ω values is related to the flux of mass from the bulk of the solution toward the electrode surface. This mass transport mostly occurs in the direction normal to the electrode surface. However, in theory, as the RDBPE rotation is isotropic, a radial laminar flow is also established in solution near the surface of the disk.45,46,71 This radial laminar flow (i.e. hydrodynamic pressure) is significant at short distances from the electrode surface, typically where the light-emitting species are generated (1–2 μm from the electrode).36,60 The expected flow velocity at radial position r at z = 1 μm from the electrode is typically:45,46
| vr = 0.51(2πω/60)3/2ν−1/2rz |
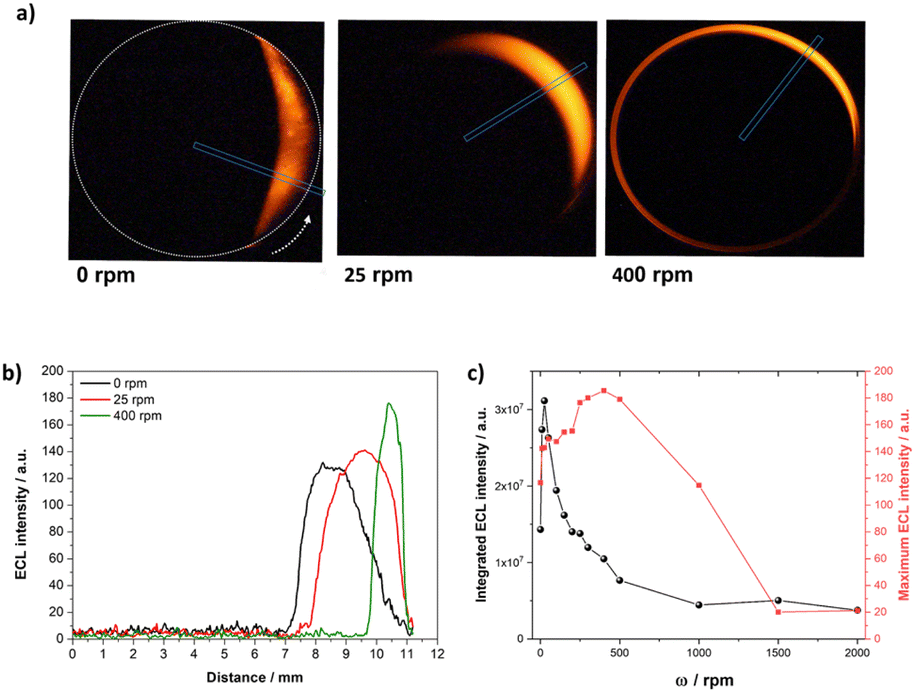 | ||
| Fig. 3 (a) ECL images of the RDBPE at different rotation speeds ω (indicated in the figure) with a representation of the ROI chosen at each ω. (b) Radial ECL intensity profiles at different rotation speeds extracted along the axes passing through the center of the RDBPE at the θmax angle (i.e. where maximum ECL intensity is obtained). (c) Comparison of the integrated ECL intensity (ECLsum, black dots) and maximum ECL intensity (ECLmax, red squares) as a function of the rotation speed ω. Same experimental conditions as in Fig. 1. | ||
For ω = 1000 rpm, the radial velocity vr = 0.55r ranges from zero at the center of the electrode up to 0.3 cm s−1 at the electrode edge (r = 0.55 cm), explaining that the light emitting species electrogenerated in this 1 μm region are pushed towards the edge.45,46,71 This is corroborated by the ECL intensity profiles at different ω values, for which the position where ECL appears is further apart from the center of the electrode (Fig. 3b). In addition, the radial velocity expands to the periphery of the electrode as a function of the rotation speed, limiting the ECL emission to an apparent ring-type region, which explains the gradual dragging of the light-emitting region in the direction of rotation. However, above a ω threshold value, the hydrodynamics overcomes the edge of the electrode, which translates into a loss of emission. These mass transport limitations, associated with the consumption of reactants, coupled with the characteristic polarization in BE, are characteristic of the reported RDBPE system.
To analyse further the ECL behaviors obtained in the reported wireless dynamic system, we changed the concentrations of the luminophore and of the co-reactant in the solution. Fig. S4† displays the sequence of ECL images recorded at increasing rotation speeds in a PBS solution containing 2 mM [Ru(bpy)3]2+ and 5 mM TPA. As expected, without rotation, strong ECL emission was observed on the anodic pole of the RDBPE. Upon increasing the ω values from 50 rpm to 2000 rpm, the ECL region is dragged by the rotation of the electrode and a complete ECL ring is observed over the RDBPE at 1500 rpm. As for lower concentrations (Fig. 1), the thickness of the ring tends to decrease with higher rotation rates.
Then, we increased the concentration of the TPA co-reactant up to 100 mM keeping constant the [Ru(bpy)3]2+ concentration (Fig. 4). Under these conditions, the optical images recorded at 50 and 100 rpm showed that the ECL reactive layer is dragged by the rotation but it kept the “croissant” shape that is mainly shifted in the direction of the rotation. At higher rotation speeds, the ECL “croissant” transformed into an ECL arc that became progressively thinner. Under these conditions, there was neither an important displacement nor the formation of a complete ring as for the lower TPA concentration (i.e. 5 mM, Fig. S4†). The comparison of the ECL images recorded at both TPA concentrations showed thus very different patterns, which reflects different chemical reactivities. These different patterns obtained at 5 mM and 100 mM TPA are related to the ECL mechanism operating at high and low concentrations of TPA. At concentrations of [Ru(bpy)3]2+ higher than >0.1 mM (as here), among the different competitive mechanistic pathways, the “catalytic route” (also called the EC' route described by reactions (1)–(5)) is the main path leading to the ECL emission.72,73 In this mechanism, the TPA co-reactant is catalytically oxidized by the electrogenerated [Ru(bpy)3]3+ and the predominant reaction pathway is as follows:
| [Ru(bpy)3]2+ → [Ru(bpy)3]3+ + e− | (1) |
| [Ru(bpy)3]3+ + TPA → [Ru(bpy)3]2+ + TPA˙+ | (2) |
| TPA˙+ → TPA˙ + H+ | (3) |
| [Ru(bpy)3]3+ + TPA˙ → [Ru(bpy)3]2+* + products | (4) |
| [Ru(bpy)3]2+* → [Ru(bpy)3]2+ + hν (620 nm) | (5) |
 | ||
| Fig. 4 Effects of high TPA co-reactant concentration on the ECL images of the RDBPE at different rotation speeds ω. Experiments were performed in a PBS (10 mM, pH 7.4) solution containing 2 mM [Ru(bpy)3]2+ and 100 mM TPA. The dotted white circle and the dotted arrow indicate the position of the RDBPE and the direction of the rotation, respectively. A constant electric field of 8.3 V cm−1 was imposed by the two graphite feeder electrodes schematized at both extremities. Exposure time: 8 s. | ||
This “catalytic route” implies that, at high concentrations of TPA, the rate of the global ECL reaction sequence is fast compared to the diffusional time scale. It means that the ECL-emitting region is confined to the immediate vicinity of the electrode surface by the catalytic reaction. In other words, in a quiescent solution, ECL is observed precisely where oxidation of the ECL reagents occurs. For instance, this characteristic has been exploited to image individual ECL-emitting layers developing at adjacent nanoelectrodes in an ordered array.44 The situation is different for the RDBPE where hydrodynamics plays a crucial role. At low TPA concentrations, the hydrodynamics determined essentially the ECL patterns and the corresponding light emission is observed far from the site where oxidation reactions of the ECL reagents occur (i.e. anodic pole, δ+). In contrast, at high TPA concentrations, ECL shapes are governed by the ECL mechanism. Indeed, the confinement of the ECL reaction by the “catalytic route” at high TPA concentrations limits the displacement of the ECL-emitting region far from the anodic pole. We performed additional control experiments without TPA in the solution and in this case, we were not able to detect any ECL emission (Fig. S5†). It demonstrates that the annihilation process between [Ru(bpy)3]3+ and [Ru(bpy)3]+ (reduced form potentially generated on the cathodic pole of the RDBPE) is negligible and that it cannot explain the complex ECL patterns. In brief, the ECL patterns result from the convolution of different processes involving the co-reactant ECL mechanisms, the asymmetric polarization of the RDBPE by BE and the hydrodynamics.
Conclusion
In summary, we developed a wireless approach, based on the ECL emission induced on a RDBPE in order to visualize the effects of hydrodynamics when the polarization is controlled by means of BE. Under a constant electric field and without rotation, a well-defined semi-circular anodic ECL emission was obtained. However, with increasing rotation speed, a gradual clockwise displacement of the ECL region and a progressive decrease of the thickness of the emission area were observed to afford an unexpected ring-like ECL region. Indeed, a complete ring is viewed on the periphery of the entire RDBPE. In other words, the ECL emission is observed on anodically-polarized, cathodically-polarized and non-polarized parts of the RDBPE whereas ECL reagents are oxidized only on the anodic pole. It is an unexpected behavior in ECL with the TPA co-reactant because usually the light emission is viewed only where the oxidation reactions take place. This essential feature constitutes the basis of the ECL microscopy. By changing the rotation speed and the concentration of the ECL reagents, we can conclude that the ECL patterns are determined mainly by the hydrodynamics and the electrode polarization effects at low co-reactant concentrations, whereas, at high co-reactant concentrations, the confinement of the ECL-emitting layer at the electrode surface by the electrochemical reactivity limits the displacement of the ECL-emitting region. Therefore, the different shapes of the ECL layer result from the convolution of the asymmetric bipolar polarization with the hydrodynamics and the ECL reactivity. This simple system opens new avenues for further investigation from theoretical and experimental points of view in analytical chemistry combining transient bipolar electrochemistry, electrochemical impedance spectroscopy and ECL. In addition, the reported wireless approach could be applied to design original dynamic bipolar ECL systems at different scales such as in microfluidics and should find promising applications in autonomous ECL-based sensing systems with improved sensitivity and increased complexity once coupled to enzymatic or immunorecognition reactions.74,75Experimental section
Tris(2,2′-bipyridine)ruthenium(II) dichloride hexahydrate, tri-n-propylamine, disodium hydrogen phosphate heptahydrate and sodium phosphate monobasic monohydrate were purchased from Sigma-Aldrich and used as received. All solutions were prepared with deionized water (Milli-Q Direct-Q, resistivity of 18.3 MΩ cm at 25 °C). After the TPA addition, the pH of the PBS solution was adjusted to 7.4 with concentrated H2SO4 solution. Potentiodynamic measurements were performed using a three-electrode cell composed of a graphite disk (diameter = 1.1 cm, thickness 1 mm) acting as the working electrode and a Pt cylinder and a Ag wire as the counter and reference electrodes, respectively. The ECL experiments were carried out with a PalmSens4 potentiostat connected to a personal computer, coupled to a commercial rotator holder connected to an external motor controller unit (RDE EDI-101 coupled to a speed control CTV 101-Radiometer Analytical). The bipolar electrochemical measurements were carried out by placing the graphite disk, acting as BPE, between two graphite feeder electrodes positioned at the extremities of the bipolar cell at a distance of 6 cm. The electric field along the cell was controlled by a commercial power supply (EA Elektro-Automatik, EA-PS 5080-10A) coupled to a voltage amplifier (A800DI FLC Electronics). Rotation was enabled by attaching the BPE to the commercial rotator holder and the angular speed was controlled by an external motor controller unit. ECL emission was monitored by using a CCD camera (Canon EOS 70D, Objective Canon Macro Lens, 100 mm, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.8) and the images were processed with open access ImageJ software.
:2.8) and the images were processed with open access ImageJ software.
Data availability
Additional data can be obtained from the corresponding author upon request.Author contributions
L. R. A.-A. and G. S. performed the ECL experiments and analysed the data. G. X., A. K., F. K., L. B. and N. S. directed the study and designed the research. All authors prepared the manuscript and approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. R. A.-A. acknowledges the University of Bordeaux for her PhD scholarship. This work was funded by the Agence Nationale de la Recherche (ELISE – ANR-21-CE42), the Sino-French International Research Network ELECTROSENS (CNRS) and the CAS President's International Fellowship Initiative (PIFI). G. S. and A. K. acknowledge the European Research Council for funding (grant agreement no 741251, ERC Advanced grant ELECTRA). In Memoriam: This contribution is dedicated to Prof. Allen J. Bard who passed away on the 11th of February 2024 at the age of 90. Allen was a pioneer in many fields of electrochemistry, including electrochemiluminescence.Notes and references
- N. Sojic and L. Bouffier, Curr. Opin. Electrochem., 2022, 34, 1–8 Search PubMed.
- Y. Wang, Z. Cao, Q. Yang, W. Guo and B. Su, Anal. Chim. Acta, 2019, 1074, 1–15 CrossRef CAS PubMed.
- J.-F. Lemineur, H. Wang, W. Wang and F. Kanoufi, Annu. Rev. Anal. Chem., 2022, 15, 57–82 CrossRef PubMed.
- H. Lee, J. Kim, M. Hwang and J. Kim, ACS Sens., 2023, 8(11), 4374–4383 CrossRef CAS PubMed.
- H. Xing, X. Zhang, Q. Zhai, J. Li and E. Wang, Anal. Chem., 2017, 89(7), 3867–3872 CrossRef CAS PubMed.
- T. J. Anderson, P. A. Defnet and B. Zhang, Anal. Chem., 2020, 92(9), 6748–6755 CrossRef CAS PubMed.
- T. Iwama, K. Y. Inoue and H. Shiku, Anal. Chem., 2022, 94(25), 8857–8866 CrossRef CAS PubMed.
- J. Zhai, L. Yang, X. Du and X. Xie, Anal. Chem., 2018, 90(21), 12791–12795 CrossRef CAS PubMed.
- X. Zhang, Y. Han, J. Li, L. Zhang, X. Jia and E. Wang, Anal. Chem., 2014, 86(3), 1380–1384 CrossRef CAS PubMed.
- X. Qin, J. Gao, H.-J. Jin, Z.-Q. Li and X.-H. Xia, Chem.–Eur. J., 2023, 29(8), e202202687 CrossRef CAS PubMed.
- M. Liu, G. Salinas, J. Yu, A. Cornet, H. Li, A. Kuhn and N. Sojic, Chem. Sci., 2023, 14(39), 10664–10670 RSC.
- J. S. Borchers, O. Riusech, E. Rasmussen and R. K. Anand, J. Anal. Test., 2019, 3(2), 150–159 CrossRef.
- G. Salinas, S. M. Beladi-Mousavi, L. Gerasimova, L. Bouffier and A. Kuhn, Anal. Chem., 2022, 94(41), 14317–14321 CrossRef CAS PubMed.
- S. Rebeccani, A. Zanut, C. I. Santo, G. Valenti and F. Paolucci, Anal. Chem., 2022, 94(1), 336–348 CrossRef CAS PubMed.
- S. E. Fosdick, K. N. Knust, K. Scida and R. M. Crooks, Angew. Chem., Int. Ed., 2013, 52(40), 10438–10456 CrossRef CAS PubMed.
- K. L. Rahn and R. K. Anand, Anal. Chem., 2021, 93(1), 103–123 CrossRef CAS PubMed.
- L. Bouffier, S. Arbault, A. Kuhn and N. Sojic, Anal. Bioanal. Chem., 2016, 408(25), 7003–7011 CrossRef CAS PubMed.
- C. Mwanza and S. N. Ding, Biosensors, 2023, 13(6), 666 CrossRef CAS PubMed.
- Z. Liu, W. Qi and G. Xu, Chem. Soc. Rev., 2015, 44(10), 3117–3142 RSC.
- C. Meng, S. Knežević, F. Du, Y. Guan, F. Kanoufi, N. Sojic and G. Xu, eScience, 2022, 2(6), 591–605 CrossRef.
- M. S. Alsalhi, J. Alam, L. A. Dass and M. Raja, Int. J. Mol. Sci., 2011, 12(3), 2036–2054 CrossRef CAS PubMed.
- H. Qi and C. Zhang, Anal. Chem., 2020, 92(1), 524–534 CrossRef CAS PubMed.
- Y. Zhao, L. Bouffier, G. Xu, G. Loget and N. Sojic, Chem. Sci., 2022, 13(9), 2528–2550 RSC.
- S. F. Douman, E. Brennan, E. I. Iwuoha and R. J. Forster, Anal. Chem., 2017, 89(21), 11614–11619 CrossRef CAS PubMed.
- S. F. Douman, D. Collins, L. R. Cumba, S. Beirne, G. G. Wallace, Z. Yue, E. I. Iwuoha, F. Melinato, Y. Pellegrin and R. J. Forster, Chem. Commun., 2021, 57(38), 4642–4645 RSC.
- L. Koefoed, S. U. Pedersen and K. Daasbjerg, Curr. Opin. Electrochem., 2017, 2(1), 13–17 CrossRef CAS.
- N. Shida, Y. Zhou and S. Inagi, Acc. Chem. Res., 2019, 52(9), 2598–2608 CrossRef CAS PubMed.
- Y. Liu, N. Zhang, J. B. Pan, J. Song, W. Zhao, H. Y. Chen and J. J. Xu, Anal. Chem., 2022, 94(6), 3005–3012 CrossRef CAS PubMed.
- W. Gao, K. Muzyka, X. Ma, B. Lou and G. Xu, Chem. Sci., 2018, 9(16), 3911–3916 RSC.
- A. J. Hsueh, N. A. A. Mutalib, Y. Shirato and H. Suzuki, ACS Omega, 2022, 7(23), 20298–20305 CrossRef CAS PubMed.
- W. Qi, J. Lai, W. Gao, S. Li, S. Hanif and G. Xu, Anal. Chem., 2014, 86(18), 8927–8931 CrossRef CAS PubMed.
- L. Qi, Y. Xia, W. Qi, W. Gao, F. Wu and G. Xu, Anal. Chem., 2016, 88(2), 1123–1127 CrossRef CAS PubMed.
- K. Hiramoto, K. Ino, K. Komatsu, Y. Nashimoto and H. Shiku, Biosens. Bioelectron., 2021, 181, 113123 CrossRef CAS PubMed.
- L. Bouffier, D. Zigah, C. Adam, M. Sentic, Z. Fattah, D. Manojlovic, A. Kuhn and N. Sojic, ChemElectroChem, 2014, 1(1), 95–98 CrossRef.
- M. Sentic, G. Loget, D. Manojlovic, A. Kuhn and N. Sojic, Angew. Chem., Int. Ed., 2012, 51(45), 11284–11288 CrossRef CAS PubMed.
- M. Sentic, M. Milutinovic, F. Kanoufi, D. Manojlovic, S. Arbault and N. Sojic, Chem. Sci., 2014, 5(6), 2568–2572 RSC.
- M. Sentic, S. Arbault, B. Goudeau, D. Manojlovic, A. Kuhn, L. Bouffier and N. Sojic, Chem. Commun., 2014, 50(71), 1020–10205 RSC.
- N. A. Ab Mutalib and H. Suzuki, Curr. Opin. Electrochem., 2024, 43, 101424 CrossRef CAS.
- L. Bouffier, D. Zigah, N. Sojic and A. Kuhn, Annu. Rev. Anal. Chem., 2021, 14(1), 65–86 CrossRef CAS PubMed.
- R. C. Engstrom, C. M. Pharr and M. D. Koppang, J. Electroanal. Chem. Interfacial Electrochem., 1987, 221, 251–255 CrossRef CAS.
- R. J. Bowling, R. L. McCreery, C. M. Pharr and R. C. Engstrom, Anal. Chem., 1989, 61(24), 2763–2766 CrossRef CAS.
- C. Amatore, B. Fosset, K. M. Maness and R. Mark Wightman, Anal. Chem., 1993, 65(17), 2311–2316 CrossRef CAS.
- C. Amatore, C. Pebay, L. Servant, N. Sojic, S. Szunerits and L. Thouin, ChemPhysChem, 2006, 7(6), 1322–1327 CrossRef CAS PubMed.
- A. Chovin, P. Garrigue, P. Vinatier and N. Sojic, Anal. Chem., 2004, 76(2), 357–364 CrossRef CAS PubMed.
- J. Ludvík and J. Volke, Electrochim. Acta, 1990, 35(11–12), 1983–1986 CrossRef.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, 2nd edn, 2001, pp. 335–347 Search PubMed.
- L. Qi, F. Yuan, F. Wu, X. Ma, C. Amatore and G. Xu, Anal. Chem., 2018, 90(22), 13217–13221 CrossRef CAS PubMed.
- J. T. Maloy and A. J. Bard, J. Am. Chem. Soc., 1971, 93(23), 5968–5981 CrossRef CAS.
- V. Eßmann, J. Clausmeyer and W. Schuhmann, Electrochem. Commun., 2017, 75, 82–85 CrossRef.
- A. L. Dauphin, A. Akchach, S. Voci, A. Kuhn, G. Xu, L. Bouffier and N. Sojic, J. Phys. Chem. Lett., 2019, 10(18), 5318–5324 CrossRef CAS PubMed.
- V. Eßmann, S. Voci, G. Loget, N. Sojic, W. Schuhmann and A. Kuhn, J. Phys. Chem. Lett., 2017, 8(19), 4930–4934 CrossRef PubMed.
- X. Gou, Z. Xing, C. Ma and J. J. Zhu, Chem. Biomed. Imaging, 2023, 1(5), 414–433 CrossRef CAS.
- L. Ding, P. Zhou, Y. Yan and B. Su, Chem. Biomed. Imaging, 2023, 1(6), 558–565 CrossRef CAS.
- W. Guo, Y. Liu, Z. Cao and B. Su, J. Anal. Test., 2017, 1(2), 1–17 Search PubMed.
- M. Sornambigai, L. Bouffier, N. Sojic and S. S. Kumar, Anal. Bioanal. Chem., 2023, 415(24), 5875–5898 CrossRef CAS PubMed.
- G. Valenti, A. Fiorani, H. Li, N. Sojic and F. Paolucci, ChemElectroChem, 2016, 3(12), 1990–1997 CrossRef CAS.
- W. Guo, P. Zhou, L. Sun, H. Ding and B. Su, Angew. Chem., Int. Ed., 2021, 60(4), 2089–2093 CrossRef CAS PubMed.
- E. T. Nurhan and A. J. Bard, J. Am. Chem. Soc., 1972, 94(8), 2862–2863 CrossRef.
- Y. Zu and A. Bard, J. Anal. Chem., 2000, 72(14), 3223–3232 CAS.
- W. Miao, J. P. Choi and A. J. Bard, J. Am. Chem. Soc., 2002, 124(48), 14478–14485 CrossRef CAS PubMed.
- J. F. L. Duval, M. Minor, J. Cecilia and H. P. Van Leeuwen, J. Phys. Chem. B, 2003, 107(17), 4143–4155 CrossRef CAS.
- J. F. L. Duval, J. Buffle and H. P. Van Leeuwen, J. Phys. Chem. B, 2006, 110(12), 6081–6094 CrossRef CAS PubMed.
- H. Li, L. Bouffier, S. Arbault, A. Kuhn, C. F. Hogan and N. Sojic, Electrochem. Commun., 2017, 77, 10–13 CrossRef CAS.
- A. G. Theakstone, E. H. Doeven, X. A. Conlan, L. Dennany and P. S. Francis, Chem. Commun., 2019, 55(49), 7081–7084 RSC.
- N. S. Adamson, A. G. Theakstone, L. C. Soulsby, E. H. Doeven, E. Kerr, C. F. Hogan, P. S. Francis and L. Dennany, Chem. Sci., 2021, 12(28), 9770–9777 RSC.
- L. L. Shultz, J. S. Stoyanoff and T. A. Nieman, Anal. Chem., 1996, 68(2), 349–354 CrossRef CAS.
- S. Voci, B. Goudeau, G. Valenti, A. Lesch, M. Jović, S. Rapino, F. Paolucci, S. Arbault and N. Sojic, J. Am. Chem. Soc., 2018, 140(44), 14753–14760 CrossRef CAS PubMed.
- M. Itagaki, N. Kobari and K. Watanabe, J. Electroanal. Chem., 2004, 572(2), 329–333 CrossRef CAS.
- M. Itagaki, T. Kikuchi and K. Watanabe, Anal. Sci., 1999, 15, 755–760 CrossRef CAS.
- M. Itagaki, E. Kasugai, N. Kobari and K. Watanabe, Electrochemistry, 2001, 69(2), 104–108 CrossRef CAS.
- M. Carpinella, M. I. Velasco, E. V. Silletta, J. M. Ovejero, S. A. Dassie and R. H. Acosta, J. Electroanal. Chem., 2015, 750, 100–106 CrossRef CAS.
- Y. Zu and A. Bard, J. Anal. Chem., 2000, 72(14), 3223–3232 CAS.
- F. Kanoufi, Y. Zu and A. J. Bard, J. Phys. Chem. B, 2001, 105(1), 210–216 CrossRef CAS.
- A. Barhoum, Z. Altintas, K. S. S. Devi and R. J. Forster, Nano Today, 2023, 50, 101874 CrossRef.
- I. Bist, B. Song, I. M. Mosa, T. E. Keyes, A. Martin, R. J. Forster and J. F. Rusling, ACS Sens., 2016, 1(3), 272–278 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02528h |
| This journal is © The Royal Society of Chemistry 2024 |