
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of constrained bicycloalkanes through bibase-promoted brook rearrangement/radical-polar crossover cyclization†
Xinke
Ouyang
a,
Bingyao
Shi
a,
Yuanyuan
Zhao
a,
Zhimin
Zhu
a,
Ziyang
Li
a,
Yuxin
Yang
a and
Chao
Shu
 *ab
*ab
aState Key Laboratory of Green Pesticide, Engineering Research Center of Photoenergy Utilization for Pollution Control and Carbon Reduction, CCNU-uOttawa Joint Research Centre, College of Chemistry, Central China Normal University (CCNU), 152 Luoyu Road, Wuhan, Hubei 430079, China. E-mail: chaoshu@ccnu.edu.cn
bWuhan Institute of Photochemistry and Technology, 7 North Bingang Road, Wuhan, Hubei 430083, China
First published on 13th June 2024
Abstract
Highly constrained bicyclic scaffolds are ubiquitous and attracting increasing interest in pharmaceutical and biotechnology discoveries owing to the enhanced activities. Herein, we report a protocol to access highly substituted constrained bicycloalkanes from readily accessible α-silyl alcohols and olefins through a bibase-promoted Brook rearrangement/radical-polar crossover cyclization (RPCC) process. Of note, the practical procedure features broad substrate scope and good group tolerance under mild and operationally simple conditions, using an inexpensive organic photocatalyst. Gram-scale preparation and diverse synthetic transformations demonstrate opportunities to rapidly construct molecular complexity. Mechanistic studies have indicated that the transformation involves a bibase-promoted radical transfer rearrangement addition/radical-polar crossover cyclization relay sequence, which differs from traditional solitary RPCC reactions.
Introduction
Constrained cycloalkanes, such as cyclopropanes, cyclobutanes, oxetanes and azetidines subunits, have received huge attention for more than 100 years not only due to their broad reactivity profile to improve drug's potency and biological activity, but also due to the opportunity to utilize these fragments as highly reactive building blocks in organic synthesis,1 and even cyclopropanes and cyclobutane units can easily undergo opening reactions because of their high ring strain energies (ca. 27.5 and 26.7 kcal.mol−1). Driven by the increased complexity and productivity demands of medicinal chemistry, these motifs have increasingly appeared in drugs and clinical candidates, for example, cyclopropanes and cyclobutanes are prevalent in medicinal chemistry, bioactive natural products, and pharmaceutical molecules, and used in the areas of agricultural chemistry and materials science as well.2 Specifically, compared to common single strained cycles in one molecule, it was found that constrained bicycloalkane frameworks have proven to have greater significance in numerous bioactive compounds and pharmaceuticals, resulting from the possible double enhanced biological activities, physicochemical properties, and metabolic profiles (Fig. 1).3 However, further investigation of bicycloalkanes for drug discovery has been hampered owing to their limited availability in nature and sophisticated preparation, even though substantial research efforts have been dedicated to the synthesis of single strained cyclic frameworks or bridged-rings.4,5 Therefore, the invention of new synthetic methods for efficiently achieving highly constrained bicycloalkanes is crucial and highly desired. These methods would not only enhance the capabilities of chemists, but also expand the range of compounds available for drug discovery efforts.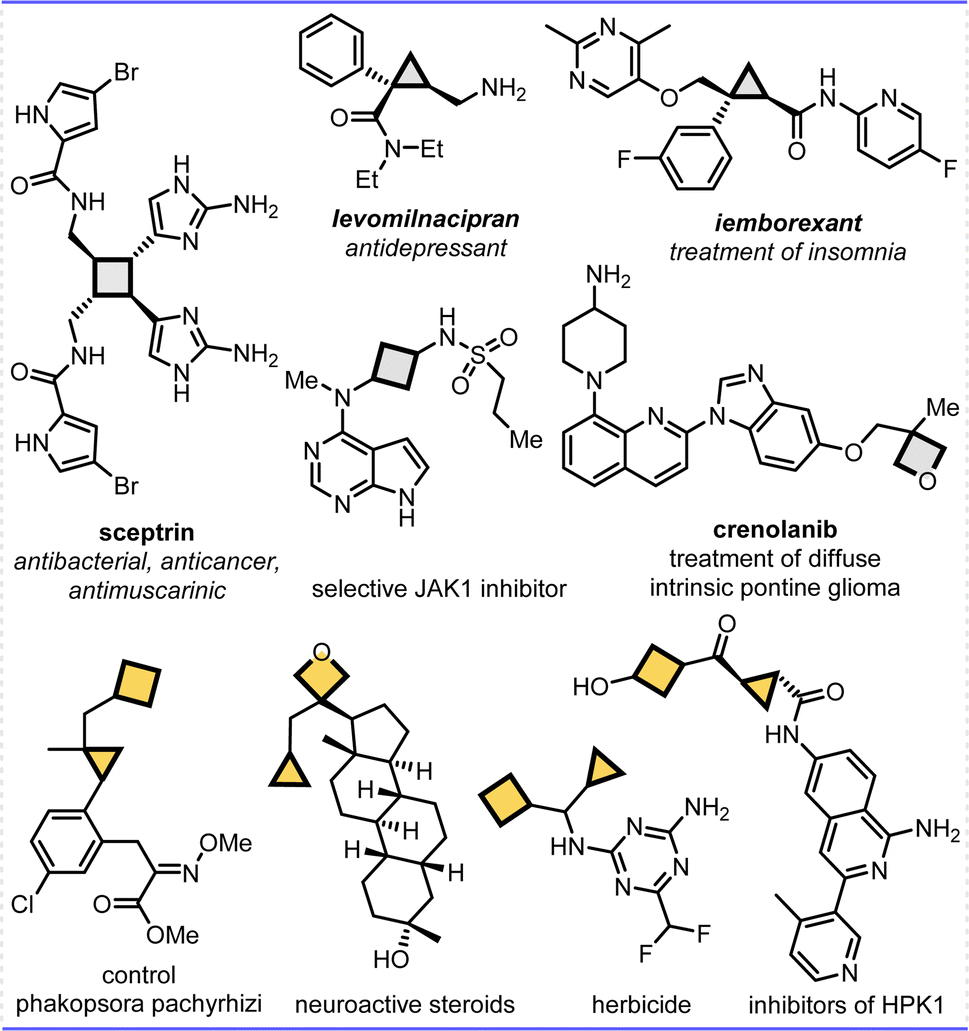 | ||
| Fig. 1 Selected application examples of bioactive cyclobutane and/or cyclopropane involved natural products and drug derivatives. | ||
Over the past decades, visible-light-mediated radical chemistry has become quite popular and attractive as a powerful tool to attain sustainability under mild reaction conditions, using cheap, abundant, and synthetically versatile starting materials. Wherein catalytic transformations involving photoinduced radical-polar crossover cyclization (RPCC) present a highly valuable strategy for converting easily accessible starting materials into structurally cyclic molecular complexity and are undergoing rapid development, particularly noteworthy is the net-neutral RPCC, in which both the single-electron oxidation and reduction steps occur through interaction with the photocatalyst, without the addition of exogenous oxidants or reductants (Fig. 2a).6,7
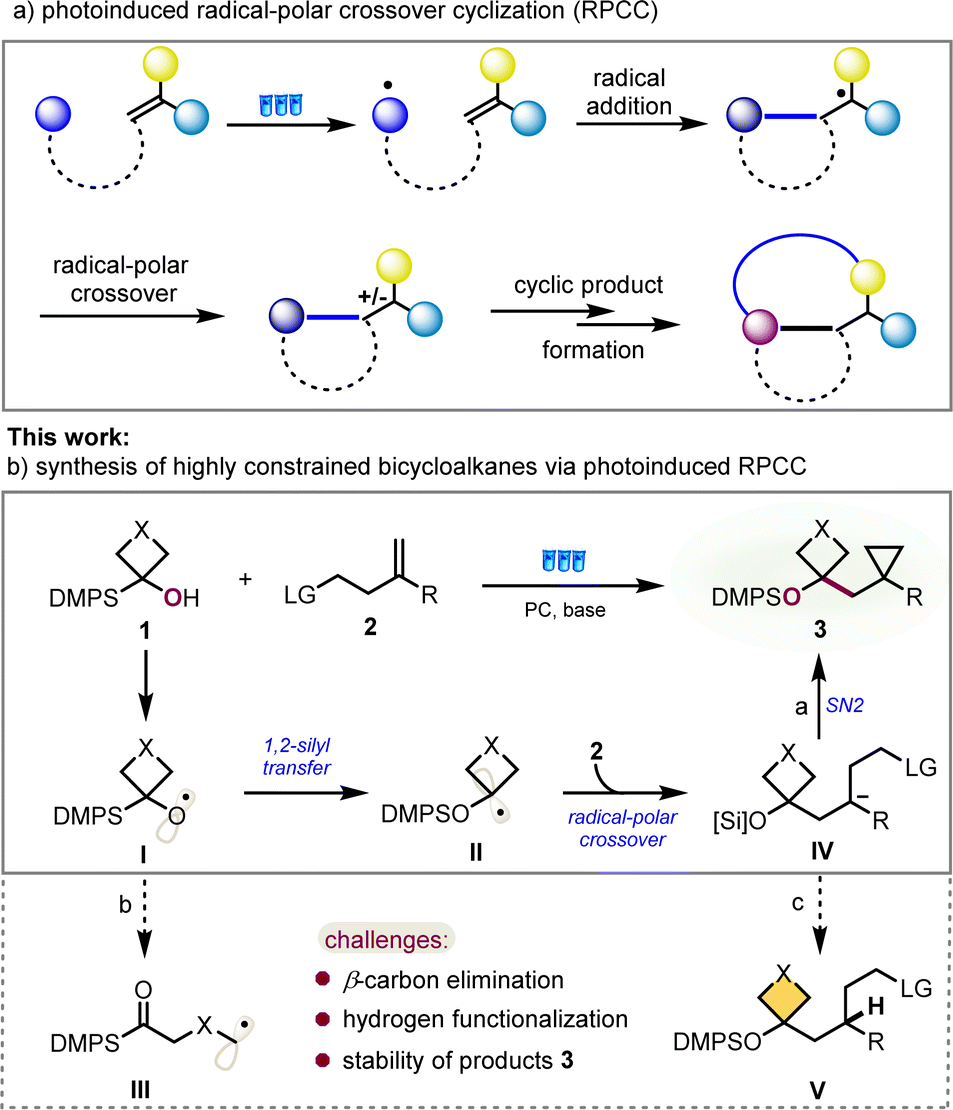 | ||
| Fig. 2 a) The development of net-neutral photoinduced radical-polar crossover cyclization (RPCC); (b) this work: synthesis of highly constrained bicycloalkanes via photoinduced Brook rearrangement/RPCC. | ||
With this in mind and our ongoing interests in radical cyclization reactions,8 we postulated that a bibase-promoted 1,2-silyl transfer of radical intermediate I, similar to 1,2-Brook rearrangement, from carbon to oxygen of the corresponding readily available α-silyl (hetero)cyclobutanol could produce an alkyl radical intermediate II under photoredox conditions.9 The highly nucleophilic character of radical II will enable it to readily react with the olefin to form a new radical species. This newly formed carbon radical intermediate will be converted to the carbanion through a radical-polar crossover process, which should provide a constrained bicycloalkane product after in situ intramolecular nucleophilic substitution (Fig. 2b, path a). However, the key challenges to the success of this idea at least are to suppress the competing pathway of β-carbon elimination to generate a carbon radical intermediate III (Fig. 2b, path b), to avoid hydrogen functionalization product IV of the carbanion intermediate in view of the high strain of formed cyclopropane (Fig. 2b, path c) and to keep enough stability of desired constrained bicycloalkane 3.
Results and discussion
Based on this design, we discovered a photoinduced radical transfer addition/radical-polar crossover/SN2 cyclization cascade for the synthesis of functionalized highly constrained bicycloalkanes with broad substrate scope and wide group compatibility, which are valuable moieties crucial for drug development but challenging to obtain, and now accessible from safe and easily available starting materials. Significantly, this would represent the inaugural synthesis of highly constrained bicycloalkanes through PRCC, showcasing an important advancement in the realm of small strained cycloalkane chemistry.At the outset of reaction, α-silyl alcohol 1a and homoallyl tosylate 2a were used as model substrates to optimize the cyclization conditions, and the selected results are displayed in Table 1. After a survey of reaction conditions, it was delight to find that when a solution of 1a and 2a with 2.0 equivalent of cesium carbonate (Cs2CO3) and 2.5 equivalent of s-collidine in the presence of 5 mol% of 4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile (4CzIPN) in MeCN was stirred for 24 h under a 30 W blue LED at ambient temperature, the desired product (cyclobutylmethyl)cyclopropane carboxylic ester 3a was obtained up to 90% yield (Table 1, entry 1). Reducing the catalyst loading to 1 mol% resulted in a decrease in the product yield, down to 70% (Table 1, entry 2). Interestingly, significantly lower efficiency was observed without base Cs2CO3 or s-collidine (Table 1, entries 3 and 4, please see more details in the ESI† for base optimization). Both two bases are conducive to the reaction outcome, implying that this is a new bibase-promoted radical reaction. 50% yield of 3a was observed when reducing the amount of 1a to 1.5 equivalent (Table 1, entry 6). The use of other metal- or metal-free photocatalysts led to decreased yields or even no product formation (Table 1, entries 7–9). The screening of solvents showed that MeCN was more suitable than others (Table 1, entry 10, please see more details in the ESI†). Control experiments revealed that the light source and photocatalyst (PC) were indispensable to this transformation. 75% yield of 3a was obtained under air.
|
|
|||
|---|---|---|---|
| Entry | Variation from standard conditions | Yield of 3ab (%) | Conversion of 2ab (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.1 mmol), 4CzIPN (5 mol%), Cs2CO3 (2.0 equiv.), s-collidine (2.5 equiv.), solvent (2.0 mL), 25 °C, 24 h, N2, 30 W blue LED, in vials; DMPS = SiMe2Ph. b Yields were determined after aqueous workup by 1H NMR analysis using 1,3,5-trimethoxybenzene as the internal standard. c Ir-dF = [Ir(dF(CF3)ppy)2(dtbbpy)](PF6). | |||
| 1 | None | 90 | 100 |
| 2 | 1 mol% 4CZIPN | 70 | 100 |
| 3 | Without CS2CO3 | 60 | 100 |
| 4 | Without s-collidine | 65 | 100 |
| 5 | Et3N instead of Cs2CO3 | 0 | 100 |
| 6 | 1.5 equiv. of 1a | 50 | 70 |
| 7 | Eosin Y instead of 4CZIPN | 44 | 100 |
| 8c | Ir-dF instead of 4CZIPN | 85 | 100 |
| 9 | Ru(bpy)2Cl2 | 0 | 0 |
| 10 | DCE instead of MeCN | 65 | 100 |
| 11 | Without light | 0 | 0 |
| 12 | Without 4CZIPN | 0 | 0 |
| 13 | Under air | 75 | 100 |

Having established the optimal reaction conditions, we then explored the scope of silyl alcohols 1 with homoallyl tosylates 2. As summarized in Scheme 1, a wide range of alkene substrates 2 bearing different functional groups were all compatible to afford the constrained (cyclobutylmethyl)cyclopropanes 3 smoothly (3a–3s) in generally moderate to good yields, since silyl ethers are common protecting groups in organic synthesis, especially in complex synthetic transformations, isolated silyl ether products were shown. Diverse carboxylic ester substituted alkenes proceeded well to give the corresponding bicycloalkane products 3a–3g in 63–90% yields. The reaction of alkenes with different substituents on the aryl ring (3i–3s), even with potentially reactive functional groups such as cyano (3p–3q), carbonyl (3r) and aldehyde (3s), could give the expected products in good yields. Delightfully, various heterocyclobutanol substrates such as oxetanol and azetidinol were compatible with this system as well, yielding the corresponding hetero atom containing constrained cycloalkanes 3t–3y in general moderate to good yields. Moreover, the current protocol could be employed in the late-stage functionalization of complex biological relevant molecules, for example the derivatives of Boc-DL-Phe-OH, piperonyl alcohol, lauryl alcohol, fenbufen and ibuprofen provide the desired highly substituted bicycloalkanes 4a–4e in 35–68% yields, which are hard-to-access with established methods, thus demonstrating the generality and adaptability of this method for the possible application in pharmaceutical and biotechnology industries.
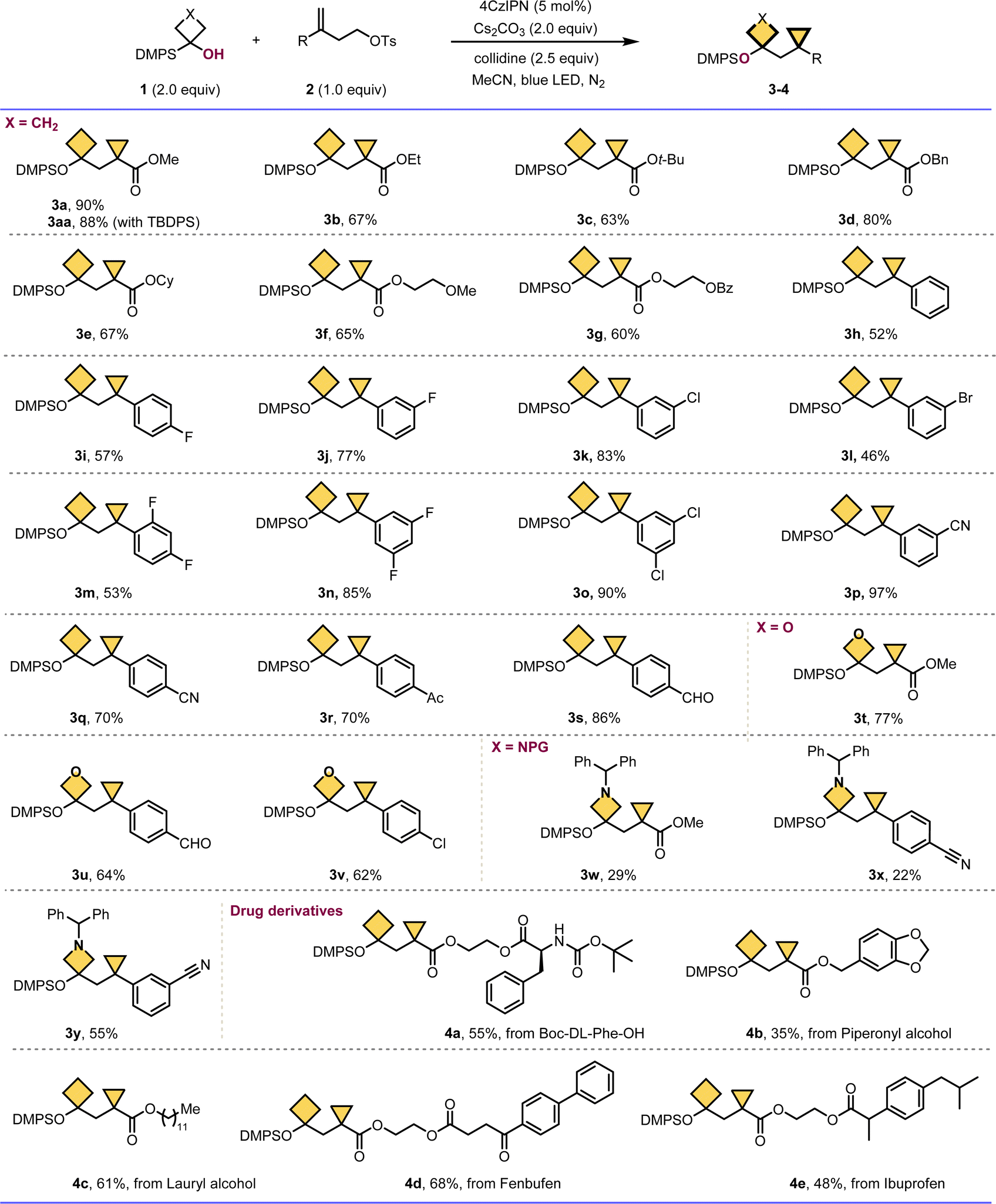 | ||
| Scheme 1 The scope of constrained bicycloalkanes containing cyclopropanes and four membered carbo- or hetero-cycles. Reaction conditions: 1 (2.0 equiv.), 2 (1.0 equiv.), 4CzIPN (5 mol%), Cs2CO3 (2.0 equiv.), s-collidine (2.5 equiv.), MeCN (0.05 M), 25 °C, 24 h, N2, 30 W blue LED, in vials; DMPS = SiMe2Ph; yields are of isolated products after chromatographic purification. | ||
After successfully exploring the generality of highly constrained bicycloalkanes containing cyclopropanes and four membered carbo- or hetero-cycles. We turned our attention to determining the scope of the reaction with respect to other ring size systems. Indeed, a diverse set of novel bicarbocycle products 5a–5p were obtained in moderate to excellent yields by varying both the radical acceptors and the silyl alcohol precursors (Scheme 2). The reaction was found to be not significantly affected by the substituents on carboxylic esters and the aryl ring of alkenes, and the ring sizes of silyl alcohols. For example, besides the substituted cyclopentylmethyl- and cyclohexyl methyl-cyclopropanes 5a–5l formed in 45–98% yields, the (cycloheptylmethyl)cyclopropane derivatives 5m and 5n, and (cyclooctylmethyl)cyclopropane derivatives 5o and 5p could also be delivered successfully in excellent yields, respectively. In addition, linear α-silyl alcohols were tolerated as well, affording corresponding β-protected hydroxyl cyclopropanes, such as 5q and 5r in 92% and 93% yields.
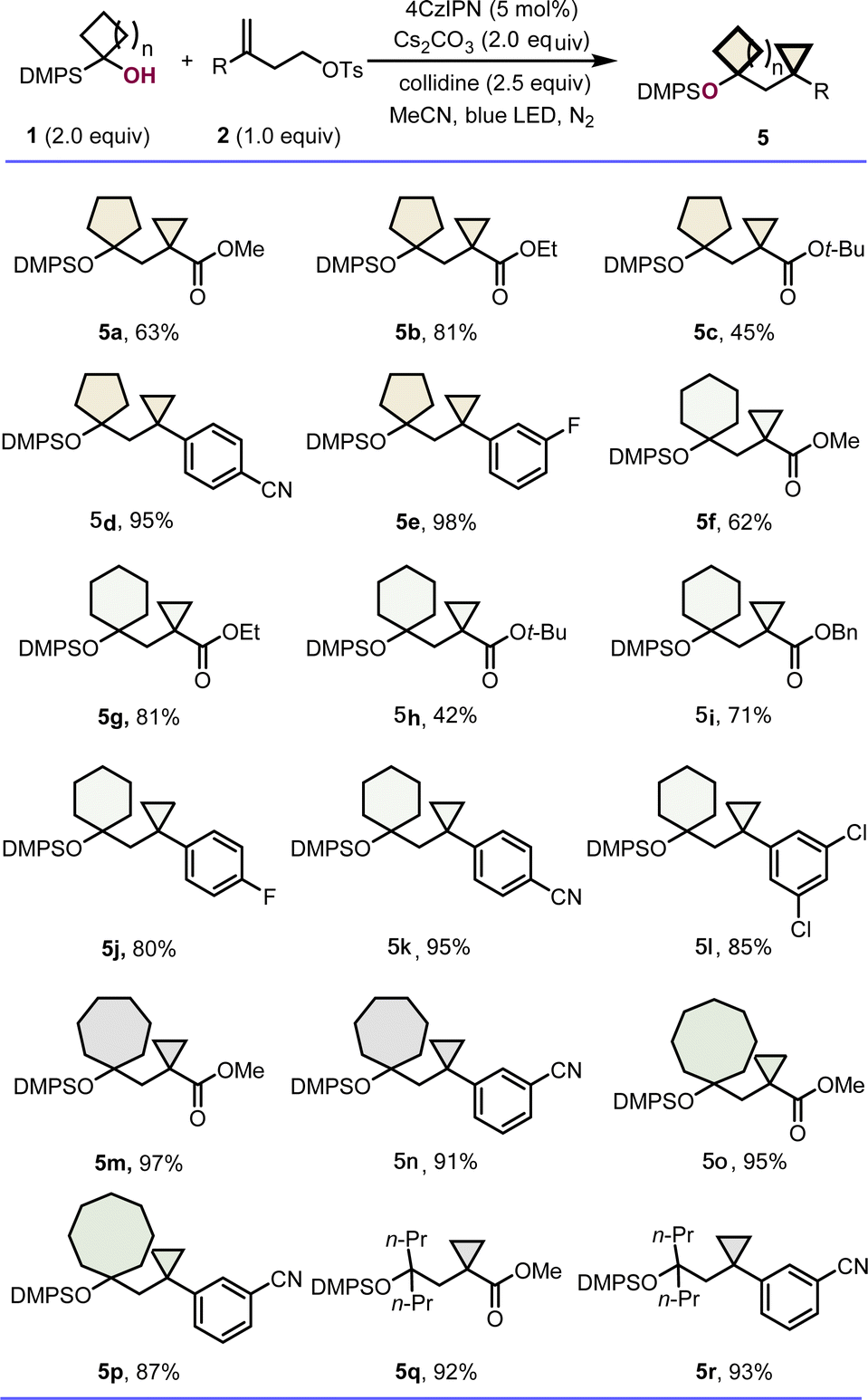 | ||
| Scheme 2 The scope of cyclopropanes tethered with other ring sizes system. Reaction conditions: 1 (2.0 equiv.), 2 (1.0 equiv.), 4CzIPN (5 mol%), Cs2CO3 (2.0 equiv.), s-collidine (2.5 equiv.), MeCN (0.05 M), 25 °C, 24 h, N2, 30 W blue LED, in vials; DMPS = SiMe2Ph; yields are of isolated products after chromatographic purification. | ||
To further demonstrate the potential synthetic utility of this protocol, representative product derivatizations were carried out (Scheme 3). First, the constrained bicarbocycle 3a was prepared on a gram scale in 84% yield, suggesting the practicability of this method for large-scale synthesis. Notably, 3a is also a versatile synthetic intermediate, such as free cyclobutanol 6a could be obtained in 85% yield via a base promoted desilylation, and 3a can be readily converted to a valuable tricycle γ-lactone 6b in 80% yield through an intramolecular tandem desilylation and esterification reaction.
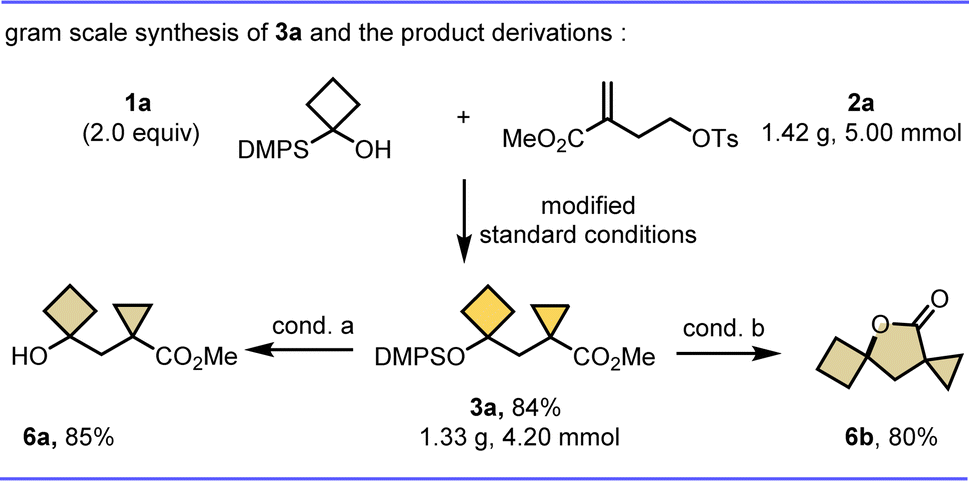 | ||
| Scheme 3 Scale-up reaction and follow-up chemistry. Isolated yields are shown; conditions for 6a: n-Bu4NF (2.5 equiv.), NaF (3.0 equiv.), THF/MeOH, rt, 6 h; conditions for 6b: n-Bu4NF (5.0 equiv.), dry DCM, rt, 10 min. See ESI† for details. | ||
To gain insight into the mechanism, a series of control experiments were performed (Scheme 4). First, the reaction was completely suppressed by adding radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or butylated hydroxytoluene (BHT) to the system, which suggested a possible stepwise radical process in this transformation (Scheme 4a). Hydrogen functionalization product 7b was isolated in 50% yield if using methyl acrylate as the radical acceptor (Scheme 4b). The free OH group is key for the success of the transformation given that no product 3a′ was formed from methoxycyclobutane substrate 1a′, implying that a radical 1,2-silyl transfer of an alkoxyl radical may be involved (Scheme 4c). The On/Off light-illumination influence experiments were performed10 indicating that the radical chain process was unlikely in this reaction (Scheme 4d). In addition, the Stern–Volmer luminescence quenching experiments were carried out with different reactants; silyl alcohol 1a showed more effective quenching in the presence of two bases (s-collidine and Cs2CO3) at the same time, which is pivotal to the high efficiency of this reaction and no obvious quenching was found with homoallyl tosylate 2a (Scheme 4e).
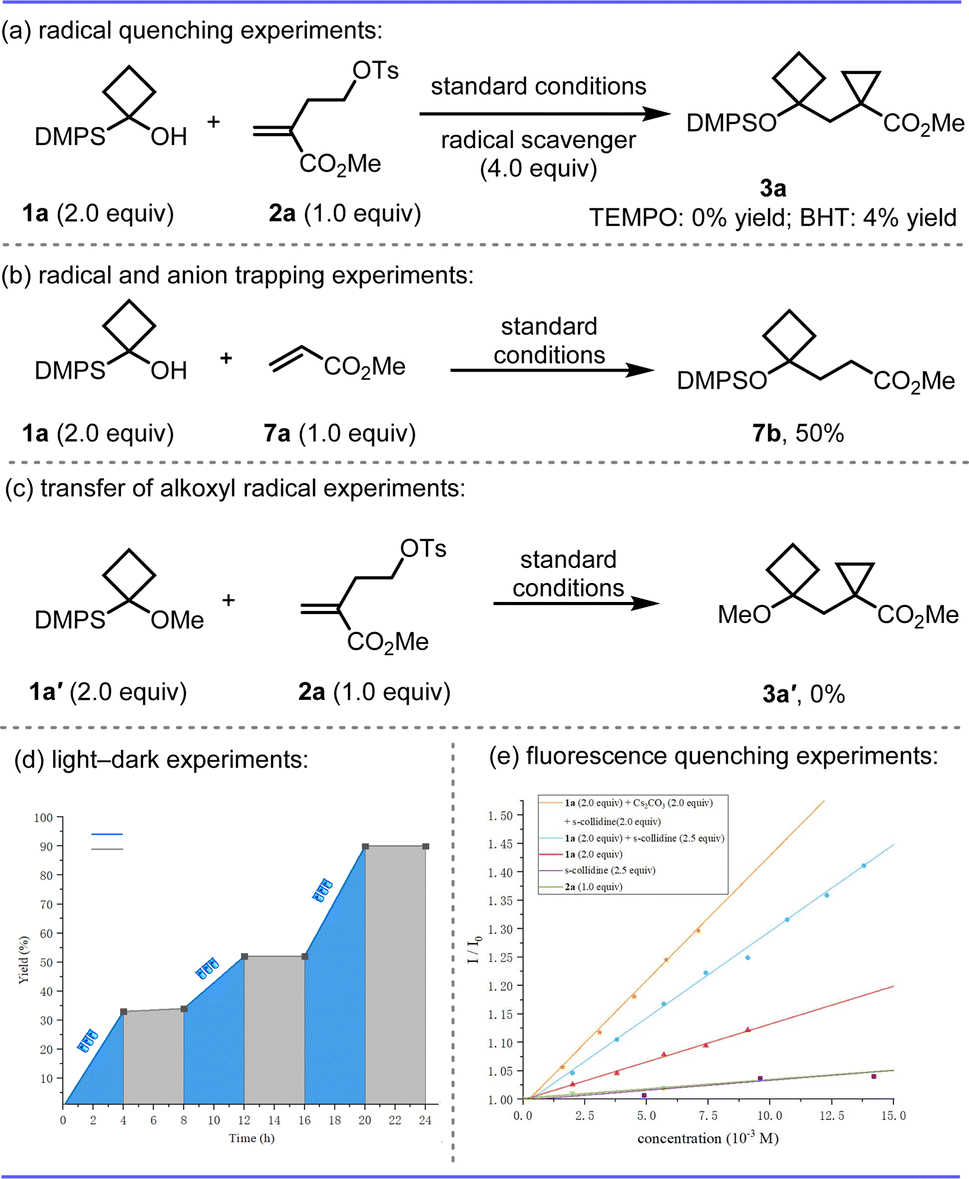 | ||
| Scheme 4 Mechanistic studies; see ESI† for details. | ||
On the basis of the above mechanistic studies and literature reports,11 a plausible mechanism is proposed in Scheme 5. First, light irradiation is performed on 4CzIPN to obtain the excited state catalyst 4CzIPN* (E1/2 [PC*/PC˙−] = 1.35 V vs. SCE in MeCN).12 Single-electron transfer (SET) with the silyl alcohol 1a in the presence of s-collidine and Cs2CO3 (Ep/2 = + 0.77 V vs. SCE for 1a)9c results in reduction of the photocatalyst to radical anion 4CzIPN˙−, and oxidation of the 1a to alkoxyl radical intermediate A, which will be transformed to an alkyl radical species B through a radical 1,2-silyl transfer. The facile addition of radical intermediate B to the double bond of 2a leads to the stabilized carbon center radical intermediate C. A second SET between radical species C and the 4CzIPN˙− state of the photocatalyst (E1/2 [PC/PC˙−] = −1.21 V vs. SCE in MeCN for 4CzIPN) finishes the photoredox catalytic cycle and brings about reductive termination of the radical process, namely the radical-polar crossover process, to deliver a carbanion intermediate D. Subsequently, the resulting carbanion D undergoes an intramolecular SN2 nucleophilic substitution to afford the expected constrained bicarbocycle 3a. The vigilant β-fluoride elimination of intermediate A and hydrogen functionalization of intermediate C were inhibited through this favorable radical transfer addition/radical-polar crossover/3-exo-tet ionic cyclisation relay process.
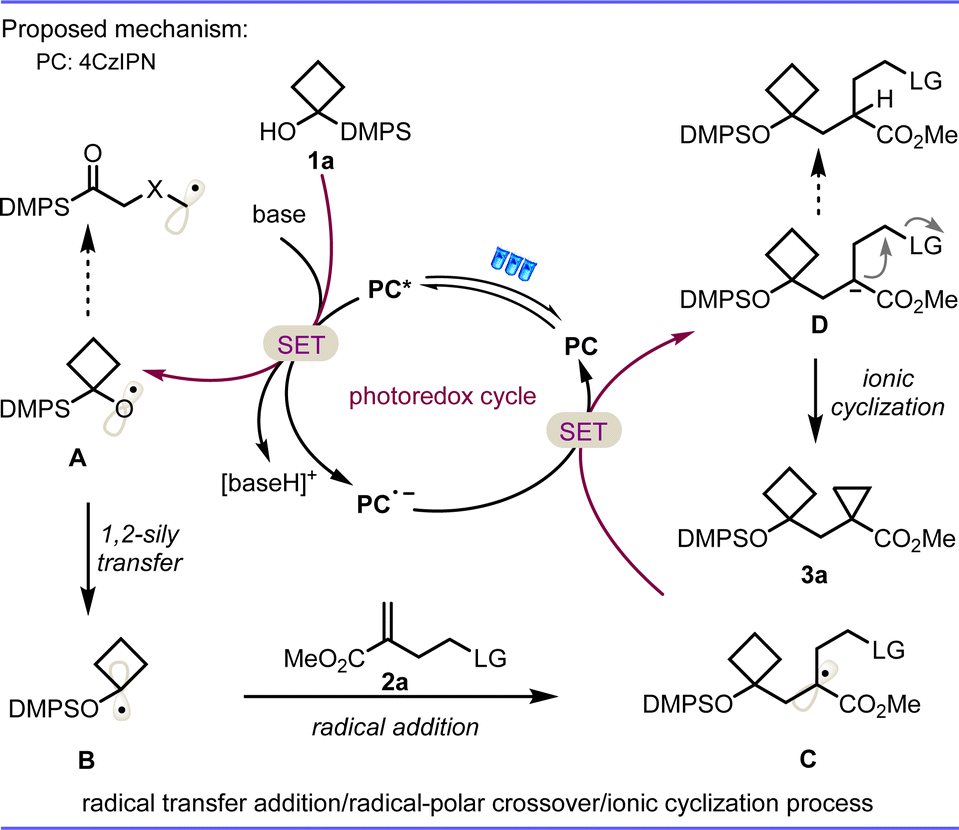 | ||
| Scheme 5 Mechanistic proposal of the photoinduced RPCC reaction. | ||
Conclusions
In summary, we have achieved a straightforward protocol for the use of readily available α-silyl alcohols as radical precursors which react with alkenes to form previously inaccessible highly constrained bicycloalkanes under mild photocatalytic conditions for the first time. The transformation is easy to set up and insensitive to common deviations from the reaction conditions, and features broad substrate scope and good functional group compatibilities. The synthetic utility of this protocol is further demonstrated by the gram-scale synthesis, late-stage functionalization of pharmaceuticals and the downstream derivatizations. Additionally, mechanism studies support a photosensitized bibase-promoted Brook rearrangement/RPCC process. Preliminary results revealed that the obtained highly constrained bicycloalkane in place of single strained cycloalkane resulted in an increase in biological activities in some agricultural chemicals. Given the novel reactivity and mechanism of this protocol, and the valuable constrained bicycloalkanes in drug discovery, we anticipate that the present methodology will be of interest for synthetic and medicinal chemists, and their toolbox for incorporation of constrained bicycles into drug-like molecules.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
X. O. and B. S. performed all the experiments and prepared the manuscript and ESI.† Y. Z., Z. Z., Z. L. and Y. Y. performed the preparation of raw materials. C. S. directed this project and revised the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Key R&D Program (2023YFD1700500), National Natural Science Foundation of China (22301093), the Fundamental Research Funds for the Central Universities, the Central China Normal University (CCNU) and Knowledge Innovation Program of Wuhan-Shuguang Project.References
- (a) O. G. Kulinkovich, Cyclopropanes in Organic Synthesis, Wiley, Hoboken, USA, 2015 CrossRef; (b) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740 CrossRef CAS PubMed; (c) J. Li, K. Gao, M. Bian and H. Ding, Org. Chem. Front., 2020, 7, 136 RSC; (d) M. R. van der Kolk, M. A. C. H. Janssen, F. P. J. T. Rutjes and D. Blanco-Ania, ChemMedChem, 2022, 17, e202200020 CrossRef CAS PubMed; (e) S. Ma, D. Mandalapu, S. Wang and Q. Zhang, Nat. Prod. Rep., 2022, 39, 926 RSC; (f) C. B. Kelly, L. Thai-Savard, J. Hu, T. B. Marder, G. A. Molander and A. B. Charatte, ChemCatChem, 2024, e202400110 CrossRef; (g) W. Wu, Z. Lin and H. Jiang, Org. Biomol. Chem., 2018, 16, 7315 RSC; (h) X. Tang, X. Li and Z. Qin, Adv. Agrochem, 2024, 3, 9 CrossRef CAS.
- Selected examples on single strained cyclopropanes and cyclobutanes in drug discovery, see: (a) M. R. Bauer, P. Di Fruscia, S. C. C. Lucas, I. N. Michaelides, J. E. Nelson, R. I. Storer and B. C. Whitehurst, RSC Med. Chem., 2021, 12, 448 RSC; (b) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed; (c) M. A. M. Subbaiah and N. A. Meanwell, J. Med. Chem., 2021, 64, 14046 CrossRef CAS PubMed; (d) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257 CrossRef CAS PubMed; (e) W. Ruzyllo, M. Tendera, I. Ford and K. M. Fox, Drugs, 2007, 67, 393 CrossRef CAS PubMed; (f) J. Singh, G. S. Bisacchi, S. Ahmad, J. D. Godfrey Jr, T. P. Kissick, T. Mitt, O. Kocy, T. Vu, C. G. Papaioannou, M. K. Wong, J. E. Heikes, R. Zahler and R. H. Mueller, Org. Process Res. Dev., 1998, 2, 393 CrossRef CAS; (g) L. R. Thomas, A. M. Foshage, A. M. Weissmiller and W. P. Tansey, Cancer Res., 2015, 75, 4012 CrossRef CAS PubMed; (h) C. V. Dang, Cell, 2012, 149, 22 CrossRef CAS PubMed.
- Selected examples on constrained bicycloalkanes in drug discovery, see: (a) G. A. Patani and E. J. LaVoie, Chem. Rev., 1996, 96, 3147 CrossRef CAS PubMed; (b) P. K. Mykhailiuk, Org. Biomol. Chem., 2019, 17, 2839 RSC; (c) M. R. Bauer, P. Di Fruscia, S. C. C. Lucas, I. N. Michaelides, J. E. Nelson, R. I. Storer and B. C. Whitehurst, RSC Med. Chem., 2021, 12, 448 RSC; (d) M. A. M. Subbaiah and N. A. Meanwell, J. Med. Chem., 2021, 64, 14046 CrossRef CAS PubMed; (e) M. Yuichi, N. Takaaki, T. Hidekatsu, T. Hiroto and A. Keisuke, World Intellectual Property Organization, WO2021153782A1, 2021; (f) L. Xu and G. Zhao, World Intellectual Property Organization, WO2021142477A1 2021.
- Selected reviews on single strained small carbo- or hetero-cycles, see: (a) D. Y.-K. Chen, R. H. Pouwer and J.-A. Richard, Chem. Soc. Rev., 2012, 41, 4631 RSC; (b) T. T. Talele, J. Med. Chem., 2016, 59, 8712 CrossRef CAS PubMed; (c) A. Fawcett, Pure Appl. Chem., 2020, 92, 751 CrossRef CAS; (d) Y.-Y. Fan, X.-H. Gao and J.-M. Yue, Sci. China Chem., 2016, 59, 1126 CrossRef CAS; (e) A. Reichelt and S. F. Martin, Acc. Chem. Res., 2006, 39, 433 CrossRef CAS PubMed; (f) B. P. Raiguru, S. Nayak, D. R. Mishra, T. Das, S. Mohapatra and N. P. Mishra, Asian J. Org. Chem., 2020, 9, 1088 CrossRef; (g) J. Pietruszka, Chem. Rev., 2003, 103, 1051 CrossRef CAS PubMed; (h) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748 CrossRef CAS PubMed; (i) N. K. Valego and J. C. Rose, Eur. J. Org Chem., 2022, e202201066 Search PubMed; (j) J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9052 CrossRef CAS PubMed; (k) J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. F. Morgan, Chem. Rev., 2016, 116, 12150 CrossRef CAS PubMed; (l) Z. Fu and J. Xu, Prog. Chem., 2021, 33, 895 CAS; (m) J. Anaya and R. M. Sánchez, Prog. Heterocycl. Chem., 2023, 34, 79 CAS.
- Selected examples on the synthesis of constrained bicycloalkanes, mainly on bridged-rings, see: (a) S. Cuadros, J. Paut, E. Anselmi, G. Dagousset, E. Magnier and L. Dell′Amico, Angew. Chem., Int. Ed., 2024, e202317333 CAS; (b) J. M. Anderson, N. D. Measom, J. A. Murphy and D. L. Poole, Angew. Chem., Int. Ed., 2021, 60, 24754–24769 CrossRef CAS PubMed; (c) M. M. D. Pramanik, H. Qian, W.-J. Xiao and J.-R. Chen, Org. Chem. Front., 2020, 7, 2531 RSC; (d) S. J. Sujansky and X. Ma, Asian J. Org. Chem., 2024, e202400045 CrossRef CAS; (e) P. Bellotti and F. Glorius, J. Am. Chem. Soc., 2023, 145, 20716 CrossRef CAS PubMed.
- Selected reviews, see: (a) M. Liu, X. Ouyang, C. Xuan and C. Shu, Org. Chem. Front., 2024, 11, 895 RSC; (b) Z. Zhu, Y. Zhang, Z. Li and C. Shu, Chem Catal., 2024, 4, 100945 CrossRef CAS; (c) S. K. Nanda, Adv. Synth. Catal., 2023, 365, 834 CrossRef; (d) S. Sharma, J. Singh and A. Sharma, Adv. Synth. Catal., 2021, 363, 3146 CrossRef CAS; (e) R.-J. Wiles and G. A. Molander, Isr. J. Chem., 2020, 60, 281 CrossRef CAS PubMed.
- Selected recent examples, see: (a) D. M. Fischer, H. Lindner, W. M. Amberg and E. M. Carreira, J. Am. Chem. Soc., 2023, 145, 774 CrossRef CAS PubMed; (b) J. Hu, M. Tang, J. Wang, Z. Wu, A. Friedrich and T. B. Marder, Angew. Chem., Int. Ed., 2023, 62, e202305175 CrossRef CAS PubMed; (c) W. Zuo, L. Zuo, X. Geng, Z. Li and L. Wang, Org. Lett., 2023, 25, 6062 CrossRef CAS PubMed; (d) P. R. D. Murray, I. N.-M. Leibler, S. M. Hell, E. Villalona, A. G. Doyle and R. R. Knowles, ACS Catal., 2022, 12, 13732 CrossRef CAS PubMed; (e) X. Shen, C. Huang, X.-A. Yuan and S. Yu, Angew. Chem., Int. Ed., 2021, 60, 9672 CrossRef CAS PubMed; (f) W. Li, X. Chen and L. Zhou, Org. Lett., 2022, 24, 5946 CrossRef CAS PubMed; (g) Y. Liu, W. Luo, T. Xia, Y. Fang, C. Du, X. Jin, Y. Li, L. Zhang, W. Lei and H. Wu, Org. Chem. Front., 2021, 8, 1732 RSC.
- (a) C. Shu, A. Noble and V. K. Aggarwal, Nature, 2020, 586, 714 CrossRef CAS PubMed; (b) C. Shu, R. S. Mega, B. J. Andreassen, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 15430 CrossRef CAS PubMed; (c) H. Li, Y. Zhang, X. Yang, Z. Deng, Z. Zhu, P. Zhou, X. Ouyang, Y. Yuan, X. Chen, L. Yang, M. Liu and C. Shu, Angew. Chem., Int. Ed., 2023, 62, e202300159 CrossRef CAS PubMed; (d) C. Shu, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 3870 CrossRef CAS PubMed; (e) H. Li, Y. Zhang, X. Zou, X. Yang, P. Zhou, X. Ma, S. Lu, Q. Sun and C. Shu, ACS Catal., 2024, 14, 3664 CrossRef CAS; (f) Y. Zhang, P. Zhou, X. Yang, H. Li, Z. Deng, M. Ren and C. Shu, Adv. Synth. Catal., 2023, 365, 3478 CrossRef CAS.
- Selected examples: (a) M. D. Paredes and R. Alonso, J. Org. Chem., 2000, 65, 2292 CrossRef CAS PubMed; (b) X. Chen, X. Gong, Z. Li, G. Zhou, Z. Zhu, W. Zhang, S. Liu and X. Shen, Nat. Commun., 2020, 11, 2756 CrossRef CAS PubMed; (c) Y. Zhang, Y. Zhang, Y. Guo, S. Liu and X. Shen, Chem Catal., 2022, 2, 1380 CrossRef CAS; (d) T. Qin, C. Xu, G. Zhang and Q. Zhang, Org. Chem. Front., 2023, 10, 1981 RSC; (e) Z. Yang, Y. Niu, X. He, S. Chen, S. Liu, Z. Li, X. Chen, Y. Zhang, Y. Lan and X. Shen, Nat. Commun., 2021, 12, 2131 CrossRef CAS PubMed; (f) Y. Zhang, Y. Zhang, C. Ye, X. Qi, L.-Z. Wu and X. Shen, Nat. Commun., 2022, 13, 6111 CrossRef CAS PubMed.
- See the ESI† for details..
- (a) H. G. Yayla, H. Wang, K. T. Tarantino, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2016, 138, 10794 CrossRef CAS PubMed; (b) R. Ren, Z. Wu and C. Zhu, Chem. Commun., 2016, 52, 8160 RSC.
- J. Luo and J. Zhang, ACS Catal., 2016, 6, 873 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02532f |
| This journal is © The Royal Society of Chemistry 2024 |