DOI:
10.1039/D4SC02536A
(Edge Article)
Chem. Sci., 2024,
15, 9192-9200
Palladium-catalyzed selective C–C bond cleavage of keto-vinylidenecyclopropanes: construction of structurally rich dihydrofurans and tetrahydrofurans†
Received
17th April 2024
, Accepted 14th May 2024
First published on 14th May 2024
Abstract
Palladium-catalyzed selective cleavage of the distal C–C bond and proximal C–C bond of keto-vinylidenecyclopropanes by altering the sterically bulky phosphine ligands has been realized. The proximal C–C bond cleavage can be achieved by using dtbpf as a phosphine ligand, affording bicyclic products containing dihydrofuran skeletons in good yields along with broad substrate scope. In proximal C–C bond cleavage reactions, the eight-membered cyclic palladium intermediate plays a key role in the reaction. The [3 + 2] cycloaddition of keto-vinylidenecyclopropanes through the distal C–C bond cleavage can be effectively accomplished with tBuXPhos as a phosphine ligand and ZnCl2 as an additive, delivering bicyclic products containing tetrahydrofuran skeletons in good yields. The further transformation of these bicyclic products has been demonstrated, and the reaction mechanisms of two different C–C bond cleavage reactions have been investigated by control experiments and DFT calculations.
Introduction
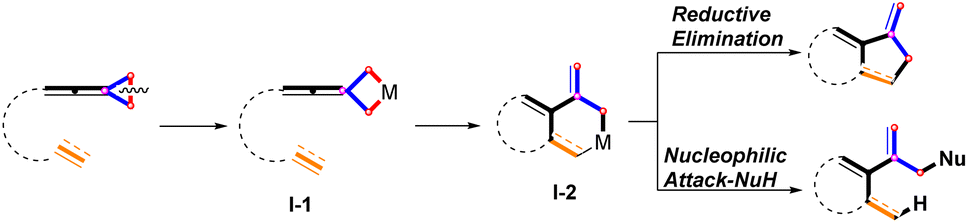
Organic compounds with a bicyclic skeleton are widely found in natural products and pharmaceutical molecules.1 In the past few decades, significant progress has been made in the construction of bicyclic compounds through transition metal (TM)-catalyzed C–C bond cleavage of alkylidenecyclopropanes (ACPs) and vinylidenecyclopropanes (VDCPs).2,3 In the synthesis of bicyclic compounds,4 these interesting functionalized strained small rings played key roles as three-carbon components. Particularly, several novel examples of cycloaddition reactions with regard to ACPs and VDCPs have been recently disclosed using Pd,5 Rh,6 Co,7 and Ni8 as the catalysts. For unactivated VDCPs, it has been established that oxidative addition of TM to the distal C–C bond of VDCP produces a metallacyclobutane species I-1, accompanied by inter- or intramolecular migratory insertion into the unsaturated C–C bond to afford metallacyclohexane species I-2. The metallacyclohexane species I-2 undergoes further transformations via two pathways: (a) direct reductive elimination;6e,f,i (b) nucleophilic reagent attack,6g,6h furnishing the cyclized products (Scheme 1).
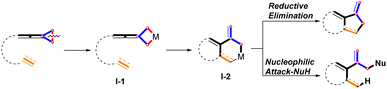 |
| | Scheme 1 TM catalyzed cycloaddition reactions of VDCPs through distal C–C bond cleavage (well-established). | |
In the past few decades, the reaction of proximal C–C bond cleavage of alkylidenecyclopropanes has made significantly effective progress.9 As for activated VDCPs, our group's previous work has disclosed the palladium-catalyzed and Lewis acid-assisted proximal C–C bond cleavage of the vinylidenecyclopropane-diester as shown in Scheme 2.10 In this proximal C–C bond cleavage, a η1-(allenyl)palladium species II-1 is produced, which can be transformed into η3-(propargyl)palladium species II-2 and η1-(propargyl)palladium species II-3 to undergo the downstream transformations.11 Moreover, Chen's group recently also revealed that the vinylidenecyclopropane-diester12 reacted with nucleophilic copper species to form a vinyl copper intermediate III-1,13 which underwent a β-carbo elimination to give a copper species III-2 and a protonation to deliver products III-3 (Scheme 2).
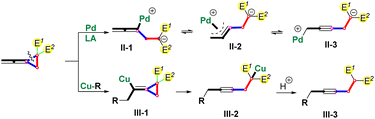 |
| | Scheme 2 Ring-opening reactions of proximal C–C bond cleavage of the VDCP-diester. | |
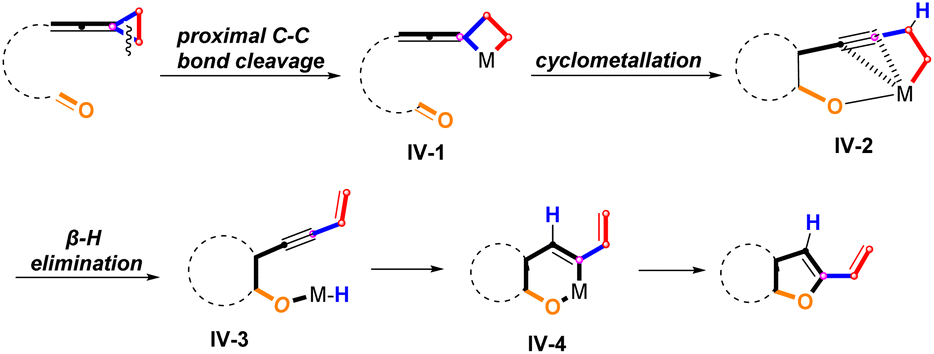
Thus far, the proximal C–C bond cleavage of unactivated vinylidenecyclopropane has never been reported before. As a consequence, we envisaged that the activation of the proximal C–C bond of VDCPs tethered with a carbonyl group would produce a metallacyclobutane species IV-1, which can lead to the metallacyclic species IV-2 through a cyclometallation process. IV-2 is energetically and structurally stabilized by coordination of the alkyne to palladium.14 Then, the intermediate IV-2 undergoes a β-hydrogen elimination to generate an intermediate IV-3. Subsequently, intramolecular migratory insertion affords intermediate IV-4, which undergoes a reductive elimination to give the bicyclic product bearing a dihydrofuran skeleton (Scheme 3).15
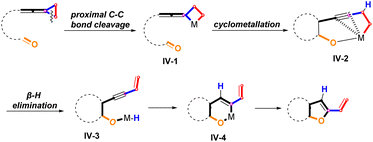 |
| | Scheme 3 Proposed ring opening reaction of proximal C–C bond cleavage of unactivated VDCP (unexplored). | |
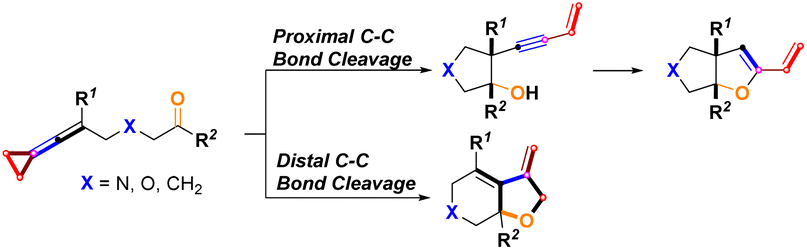
In this work, we wish to report the combined results of palladium-catalyzed and sterically bulky tertiary phosphine ligand-controlled selective proximal and distal C–C bond cleavage of keto-VDCPs for the rapid construction of a series of bicyclic products containing dihydrofuran and tetrahydrofuran skeletons (Scheme 4).
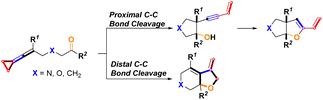 |
| | Scheme 4 This work. | |
Results and discussion
Experimental investigations
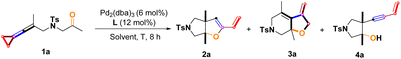
At the start of our investigations, keto-VDCP 1a was used as the model substrate to examine the reaction outcomes. Interestingly, the [3 + 2] cycloaddition reactions successfully took place in the presence of 6 mol% of Pd2(dba)3 and 12 mol% of tBuXPhos in anhydrous toluene at 100 °C after 8 hours, giving 2a in 8% isolated yield and 3a in 46% isolated yield along with the intramolecular hydroalkoxylation product 4a in 12% isolated yield (Table 1, entry 1). These [3 + 2] cycloaddition reactions can take place with another bulky biaryl phosphine-RuPhos, while affording the corresponding products 2a, 3a and 4a in lower yields (Table 1, entry 2). Some other bisphosphine ligands including (rac)-BINAP were also screened, but no reaction occurred (Table 1, entries 3–5). On increasing the reaction temperature to 120 °C and increasing the amount of Pd catalyst and tBuXPhos, we found that the yield of 3a was improved to 60% yield along with 2a in 12% yield and 4a in 10% yield (Table 1, entry 6). However, even under such rigorous reaction conditions, substrate 1a still did not react completely. Inspired by Yu and Dong's work,4i,p,q we next considered that using ZnCl2 as an additive in the catalytic system may improve the reaction outcome, and found that the yield of 3a was considerably increased to 86% with the addition of 20 mol% ZnCl2 without the formation of 2a and 4a (Table 1, entry 7). Furthermore, the product 3a was exclusively obtained in 92% yield with the addition of 100 mol% ZnCl2 (Table 1, entry 8). Accordingly, a series of Lewis acids were screened, such as InCl3, GaCl3, Zn(OTf)2, Yb(OTf)3 and ZnI2, and none of them showed a better yield of the desired products than ZnCl2, probably due to the good solubility of ZnCl2 in toluene (Table 1, entries 9–13). The examination of solvent revealed that toluene should be used for the production of 3a (Table 1, entries 14 and 15). Next, we tried to explore the role of K2CO3 in this reaction. The addition of 6 mol% Pd2(dba)3, 12 mol% tBuXPhos and 20 mol% K2CO3 produced 3a in 50% yield along with 4a in 20% yield and the result is similar to entry 1 (Table 1, entry 16). On the other hand, we also identified that the cyclized product 2a was obtained in 80% yield along with 4a in 12% yield using 6 mol% of Pd2(dba)3 as the catalyst and 12 mol% HPtBu3BF4 as the ligand in the presence of 20 mol% K2CO3 (Table 1, entry 17). As the loading of K2CO3 increased to 50 mol%, the yield of product 2a reduced to 20% and the yield of product 4a increased to 60% in the presence of 6 mol% of Pd2(dba)3 and 12 mol% of HPtBu3BF4 (Table 1, entry 18). Upon increasing the amount of K2CO3 to 100 mol%, 4a was formed in 82% yield (Table 1, entry 19). Fortunately, the cyclized product 2a with proximal C–C bond cleavage of 1a was achieved in 96% yield as a sole product using dtpbf as a ligand in the absence of K2CO3 (Table 1, entry 20). When the reaction temperature was lowered to 90 °C, the yield of 2a decreased to 72% (Table 1, entry 21). On adding 100 mol% K2CO3 into the catalytic system in the presence of 6 mol% of Pd2(dba)3 and 12 mol% of dtbpf, product 4a becomes the major product (Table 1, entry 22). We hypothesize that a large amount of K2CO3 in the reaction system promotes the reductive elimination of intermediate 2-Int4 (shown in Scheme 10) more quickly than the alternative migratory insertion, affording 4a as the major product.16 On adding 100 mol% ZnCl2 into this catalytic system, no reaction occurred (Table 1, entry 23). Solvent screening was performed and toluene was found to be the optimal solvent for this reaction as well (Table 1, entries 24–25) (for more information, see page S7 in the ESI†).
Table 1 Optimization of the reaction conditionsa
|
|
| Entrya |
Catalyst |
L |
Additive |
Solvent |
T (°C) |
Yieldb/% |
|
2a
|
3a
|
4a
|
|
Reaction conditions: substrate 1a (0.10 mmol), Pd2(dba)3 (6 mol%) and L (12 mol%) in 1.0 mL anhydrous toluene under an argon atmosphere for 8 h.
Isolated yield.
Pd2(dba)3 (10 mol%), L (20 mol%) was added.
K2CO3 (20 mol%) was added.
K2CO3 (50 mol%) was added.
K2CO3 (100 mol%) was added.
|
| 1 |
Pd2(dba)3 |
t
BuXPhos |
None |
Toluene |
100 |
8 |
46 |
12 |
| 2 |
Pd2(dba)3 |
RuPhos |
None |
Toluene |
100 |
6 |
26 |
6 |
| 3 |
Pd2(dba)3 |
dppf |
None |
Toluene |
100 |
— |
— |
— |
| 4 |
Pd2(dba)3 |
dppb |
None |
Toluene |
100 |
— |
— |
— |
| 5 |
Pd2(dba)3 |
(rac)-BINAP |
None |
Toluene |
100 |
— |
— |
— |
| 6c |
Pd2(dba)3 |
t
BuXPhos |
Nonejlv |
Toluene |
100 |
12 |
60 |
12 |
| 7 |
Pd2(dba)3 |
t
BuXPhos |
20 mol% ZnCl2 |
Toluene |
100 |
— |
86 |
Trace |
|
8
|
Pd
2
(dba)
3
|
t
BuXPhos
|
100 mol% ZnCl
2
|
Toluene
|
100
|
—
|
96
|
Trace
|
| 9 |
Pd2(dba)3 |
t
BuXPhos |
100 mol% InCI3 |
Toluene |
100 |
— |
60 |
Trace |
| 10 |
Pd2(dba)3 |
t
BuXPhos |
100 mol% GaCI3 |
Toluene |
100 |
— |
72 |
Trace |
| 11 |
Pd2(dba)3 |
t
BuXPhos |
100 mol% Zn(OTf)2 |
Toluene |
100 |
— |
62 |
Trace |
| 12 |
Pd2(dba)3 |
t
BuXPhos |
100 mol% Yb(OTf)3 |
Toluene |
100 |
— |
48 |
Trace |
| 13 |
Pd2(dba)3 |
t
BuXPhos |
100 mol% ZnI2 |
Toluene |
100 |
— |
82 |
Trace |
| 14 |
Pd2(dba)3 |
t
BuXPhos |
100 mol% ZnCl2 |
Toluene |
100 |
— |
80 |
Trace |
| 15 |
Pd2(dba)3 |
t
BuXPhos |
100 mol% ZnCl2 |
DCE |
100 |
— |
80 |
12 |
| 16d |
Pd2(dba)3 |
t
BuXPhos |
K2CO3 |
Toluene |
100 |
— |
50 |
20 |
| 17d |
Pd2(dba)3 |
HPtBu3BF4 |
K2CO3 |
Toluene |
100 |
80 |
— |
12 |
| 18e |
Pd2(dba)3 |
HPtBu3BF4 |
K2CO3 |
Toluene |
100 |
20 |
— |
60 |
| 19f |
Pd2(dba)3 |
HPtBu3BF4 |
K2CO3 |
Toluene |
100 |
6 |
— |
82 |
|
20
|
Pd
2
(dba)
3
|
dtbpf
|
None
|
Toluene
|
100
|
96
|
—
|
—
|
| 21 |
Pd2(dba)3 |
dtbpf |
None |
Toluene |
90 |
72 |
— |
6 |
| 22f |
Pd2(dba)3 |
dtbpf |
K2CO3 |
Toluene |
100 |
12 |
— |
82 |
| 23 |
Pd2(dba)3 |
dtbpf |
100 mol% ZnCl2 |
Toluene |
100 |
— |
— |
— |
| 24 |
Pd2(dba)3 |
dtbpf |
None |
Dioxane |
100 |
80 |
— |
10 |
| 25 |
Pd2(dba)3 |
dtbpf |
None |
DCE |
100 |
82 |
— |
12 |
|
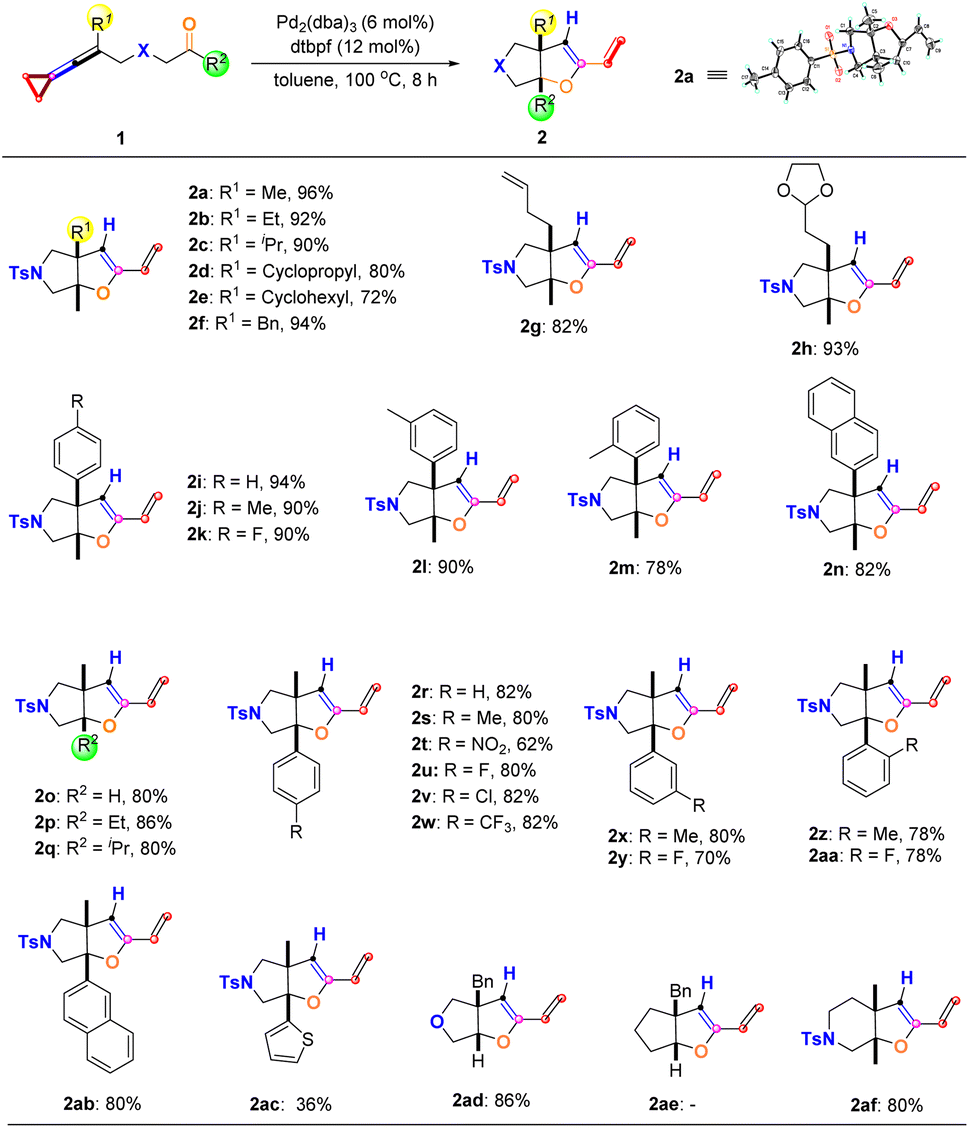
With the optimal reaction conditions established, we next explored the substrate scope of the production of 2 derived from the proximal C–C bond cleavage, and the results are summarized in Scheme 5. It can be found that R1 can be an alkyl, cycloalkyl, or benzyl group, affording the desired products 2a–2f in good yields ranging from 72% to 96%. The structure of 2a was identified by X-ray diffraction, and its CIF data have been deposited in CCDC with the number 2206931. Introducing functionalized alkyl substituents such as an olefinic moiety and acetal moiety in keto-VDCPs 1 gave the desired products 2g and 2h in 82% yield and 93% yield, respectively. We also investigated aryl substituted 1 in this reaction and found that whether an electron-donating group, an electron-withdrawing group, or methyl substitution was introduced at different positions of the benzene ring, the reactions proceeded smoothly, giving the target products 2i–2m in good yields ranging from 78% to 94%. 2-Naphthyl-substituted keto-VDCP 1n was also tolerated, giving the desired product 2n in 82% yield. We then explored the R2 substituent in this reaction and found that R2 can be a hydrogen atom, ethyl, and isopropyl substituents, furnishing the desired products 2o–2q in 80% to 86% yields. In addition, R2 can be an aromatic group as well, in which regardless of whether an electron-donating group or electron-withdrawing group was introduced at the different positions of the benzene ring, the reactions were compatible, affording the desired products 2r–2aa in moderate to good yields ranging from 62% to 82%. Furthermore, the use of 2-naphthyl substituted and 2-thiophene substituted keto-VDCPs 1ab and 1ac as substrates delivered the desired products 2ab and 2ac in 80% yield and 36% yield, respectively. Among them oxygen atom-linked 1ad could also be used in the reaction, giving 2ad in 86% yield although the carbon atom-linked substrate 1ae did not provide the corresponding product, presumably due to this substrate without the Thorpe-Ingold effect. On extending the carbon chain in the designed substrate 1af, we found that the reaction also occurred efficiently, affording 2af in 80% yield.
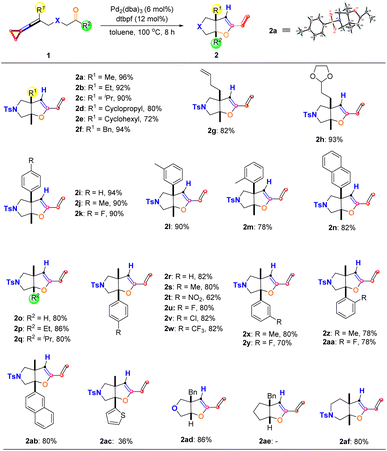 |
| | Scheme 5 Substrate scope of 2 derived from the proximal C–C bond cleavage. Reaction conditions: substrate 1 (0.10 mmol), Pd2(dba)3 (6 mol%) and dtbpf (12 mol%) in 1.0 mL anhydrous toluene under an argon atmosphere for 8 h. | |
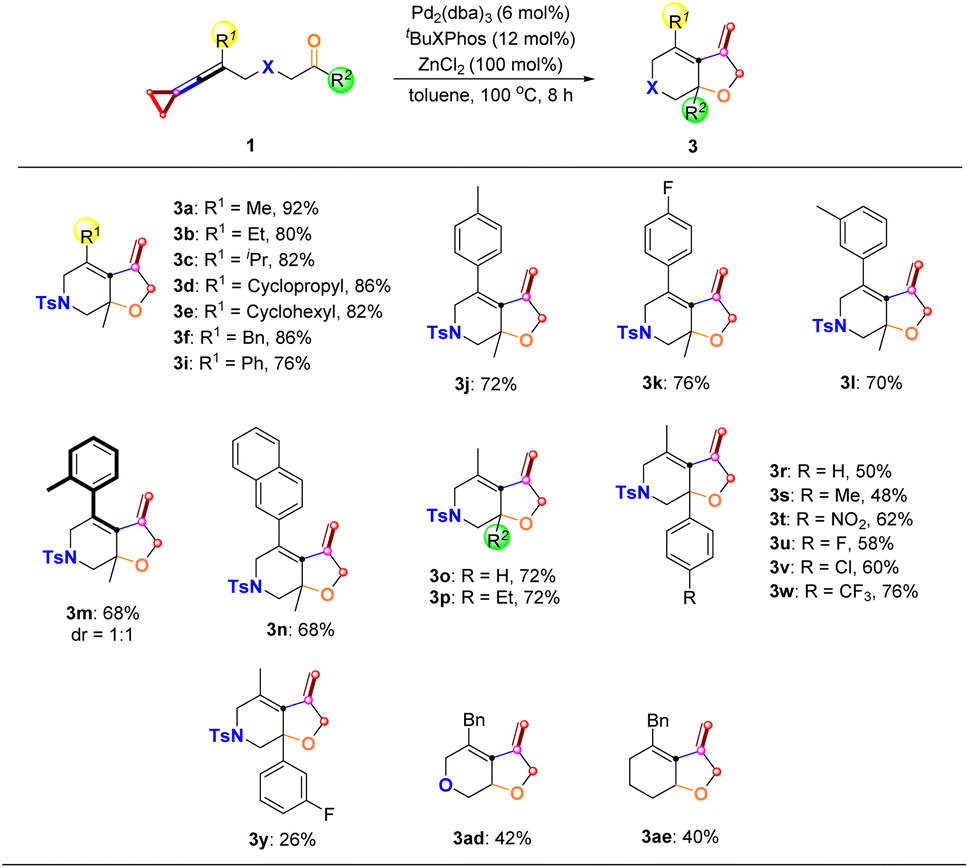
We next explored the substrate scope for the [3 + 2] cycloaddition products 3 derived from the distal C–C bond cleavage (Scheme 6). The R1 substituent of keto-VDCPs 1 can be an alkyl, cycloalkyl, or benzyl substituent or a phenyl group, affording the desired products 3a–3i in 76–92% yields. A variety of electron-rich or electron-poor aryl-substituted keto-VDCPs 1 as well as naphthyl-substituted VDCP 1n underwent this cyclization reaction smoothly, affording the corresponding products 3j–3n in 68–76% yields. For ortho-methyl group substituted keto-VDCP 1m, the desired product 3m was a pair of atropisomers. Besides, we tried to probe the R2 substituents of keto-VDCPs 1 and found that for the aldehyde substituted substrate 1o and the ethyl carbonyl substituted substrate 1p, the reactions proceeded smoothly, affording the desired products 3o and 3p in 72% yields, respectively. A series of electron-rich or electron-poor aryl-substituted keto-VDCPs 1r–1y could be converted to the bicyclic products 3r–3y in 26% to 76% yields along with a small amount of cyclized 2-chlorocyclobutene derivatives (see Scheme 8f, side effect of ZnCl2). When the bridge linkage was replaced with an oxygen atom and a CH2 moiety, the reactions were also tolerated, giving the desired products 3ad and 3ae in 42% yield and 40% yield, respectively.
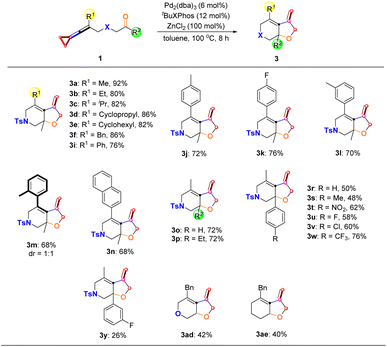 |
| | Scheme 6 Substrate scope of 3 derived from the distal C–C bond cleavage. Reaction conditions: substrate 1 (0.10 mmol), Pd2(dba)3 (6 mol%), tBuXPhos (12 mol%) and ZnCl2 (100 mol%) in 1.0 mL anhydrous toluene under an argon atmosphere for 8 h. | |
Synthetic applications
In order to explore the synthetic applicability of these protocols, gram-scale syntheses were first performed as shown in Scheme 7a & c by employing 1.0 g (3.2 mmol) of 1a, producing 0.9 g of 2a in 90% yield and 0.6 g of 3a in 60% yield, respectively under the standard conditions. Furthermore, the synthetic utilities of products 2a and 3a were investigated as that Pd-catalyzed hydrogenation of 2a and 3a furnished the corresponding reduced products 5 and 8 as diastereomeric mixtures in 96% yield and 80% yield, respectively (Scheme 7b & d). Product 2a can be used as a dienophile and undergo a D–A cycloaddition reaction with electron deficient olefins. After screening a series of electron-deficient olefins (e.g., N-phenylmaleimide, benzoquinone, diethyl maleate, diethyl acetylenedicarboxylate and ethene-1,1,2,2-tetracarbonitril), the product 2a only reacted with ethene-1,1,2,2-tetracarbonitril to give the desired tricyclic product 6 in 90% yield. This reaction probably required strongly electron-deficient olefins to take place.17 The N-tosyl group of 2f was removed upon treating with sodium naphthalenide in THF at −78 °C, affording the product 7 in 90% yield (Scheme 7b). The product 3a could be transformed into the functionalized cyclic ketone 9 in 90% yield as well as the lactone product 10 in 36% yield upon using two different oxidation methods as shown in Scheme 7d.18,19 Removal of the N-tosyl group of 3f with sodium naphthalenide afforded the isomerization product 11 in 62% yield (Scheme 7d).
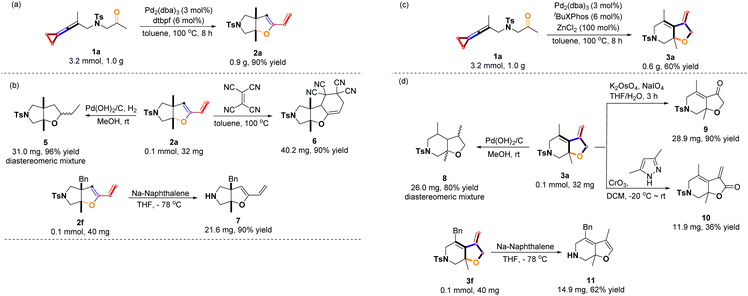 |
| | Scheme 7 Scale-up experiments and synthetic transformations: (a) Gram-scale experiment of 2a; (b) synthetic applications of 2; (c) Gram-scale experiment of 3a; (d) synthetic applications of 3. | |
Mechanistic studies
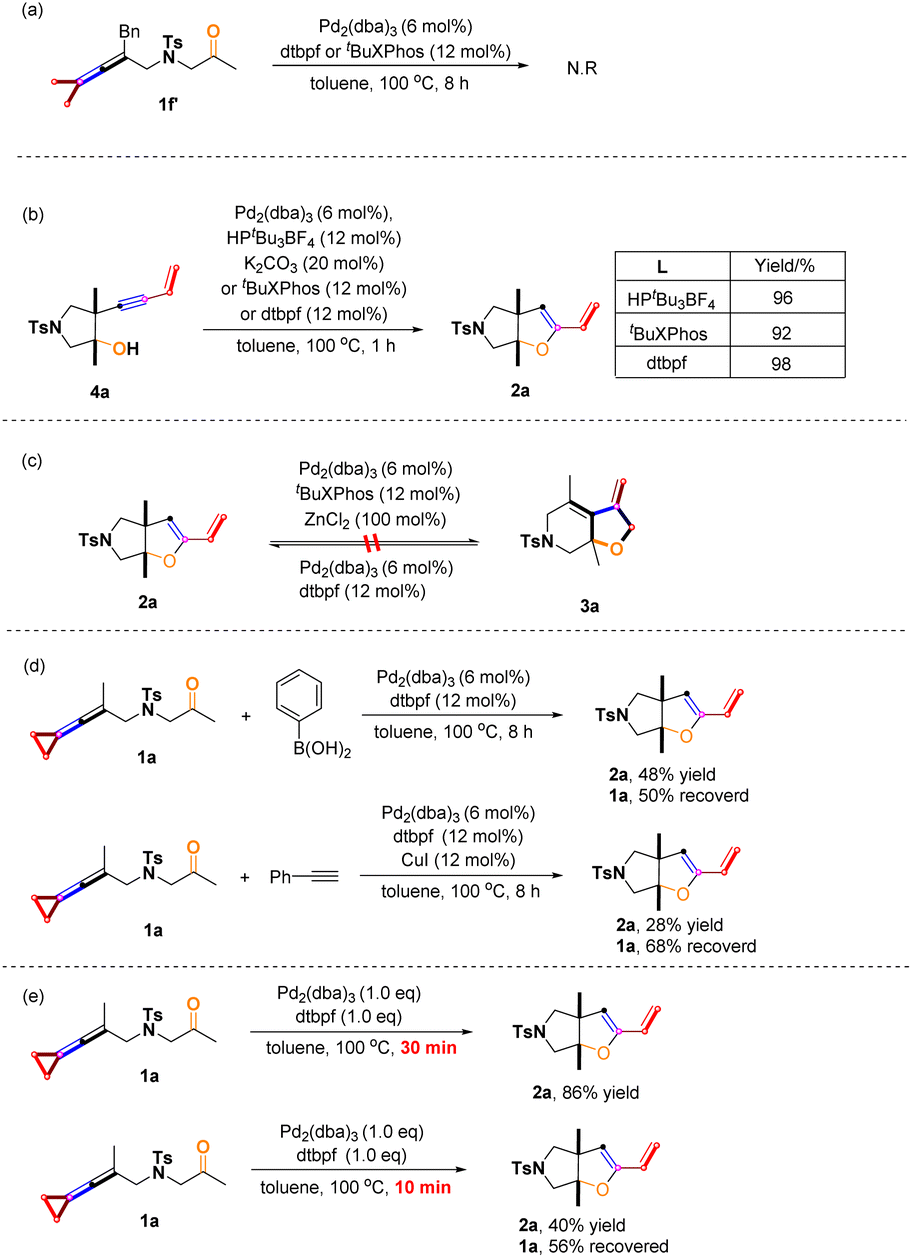
We designed and synthesized the substrate 1f′ without a cyclopropane moiety; however, the reaction of 1f′ did not occur under the standard reaction conditions. Apparently, the cyclopropane moiety plays a critical role in initiating the reaction (Scheme 8a). The control experiments indicated that 4a could be rapidly converted to product 2a in the presence of Pd2(dba)3 with the participation of HPtBu3BF4 combined with K2CO3 (20 mol%) or tBuXPhos or dtpbf in high yields, showing that product 4a derived from proximal C–C bond cleavage could undergo hydroalkoxylation to give the product 2a (Scheme 8b). In addition, product 2a cannot be converted to 3a under the standard conditions, suggesting that products 2 and 3 are produced by their own selective C–C bond cleavages of the cyclopropane unit at the beginning of the reaction (Scheme 8c). Based on previous work, we added nucleophile like arylboronic acid and phenylacetylene in the reaction system, and attempted to obtain cross-coupled products;5d,6e but we only isolated the product 2a and the unreacted substrate 1a (for more information, see page S14 in the ESI†), probably due to intramolecular reactions taking place faster than intermolecular reactions at such a high temperature (Scheme 8d). We added 1.0 equiv of Pd2(dba)3 and 1.0 equiv of dtpbf to participate in the reaction, and product 2a was obtained in 86% yield in 30 min. Product 2a was also obtained in 40% yield in 10 min, and 56% of the substrate 1a was recovered. Obviously, employing 1.0 equiv of Pd2(dba)3 and 1.0 equiv of dtpbf makes the reaction very rapid; therefore, it is difficult to obtain the reaction intermediates at such a high temperature (for more information, see page S13 in the ESI†) (Scheme 8e).
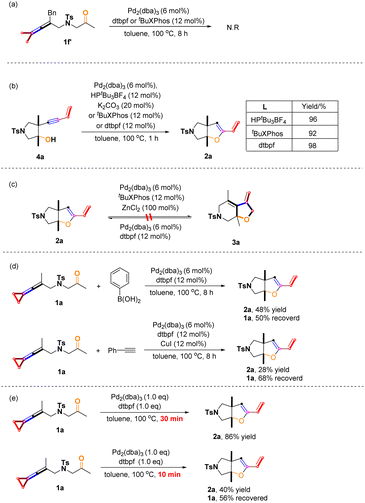 |
| | Scheme 8 Control experiments: (a) reations of 1f′ under standard conditions; (b) hydroalkoxylation of 4a to 2a; (c) experiments on the interconversion between 2a and 3a; (d) reactions of 1a with arylboronic acids and phenylacetylene; (e) reactions under 1.0 eq. catalyst and ligand condition. | |
As described above, adding ZnCl2 facilitated the completion of the [3 + 2] cycloaddition reaction; however, we also found that the deliquescence of ZnCl2 by ambient moisture caused one side reaction, giving another cyclization product as the byproduct.20 We attempted to explore the reaction results by adding H2O (2.0 equiv.) under the standard conditions. The results showed that the desired [3 + 2] cycloaddition reaction and the side reaction proceeded simultaneously, affording [3 + 2] cycloaddition products 3s and 3u in 48% and 58% yields and the minor products 12 and 13 in 20% and 26% yields (Scheme 9a). As shown in Scheme 9b, treating 1u, 1s and 1ac with ZnCl2 (1.0 equiv) and water (2.0 equiv) in toluene at 100 °C provided the corresponding chlorocyclobutene derivatives 12–14 in 38–46% yields (Scheme 9b). The structure of 14 has been confirmed by X-ray diffraction and the CIF data are deposited in CCDC with the number 2246469. This finding can explain why in some cases, the [3 + 2] cycloaddition products 3 were obtained in low yields such as 3y, 3ad, and 3ae.
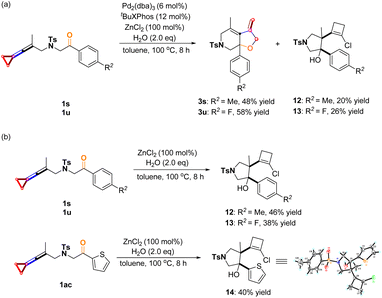 |
| | Scheme 9 (a) Reaction of adding 2.0 eq. H2O under standard condition; side effects of aqueous ZnCl2. | |
To further understand the mechanistic paradigm, density functional theory (DFT) calculations were performed. The calculations on the tandem cyclization reaction derived from proximal C–C bond cleavage have been performed at the SMD(toluene)/B3LYP/6-311+G(d,p)/Lanl2dz//B3LYP/6-31G(d)/Lanl2dz level with the Gaussian 16 program. We investigated the reaction pathway starting from a stable palladium complex 2-Int1 (shown in Scheme 10a), in which the allene units of 1a are coordinated to a palladium catalyst. The insertion of the palladium catalyst into the proximal C–C bond of VDCP produces a palladacyclic 2-Int2 through 2-Ts1 with an energy barrier of 22.4 kcal mol−1. We also investigate the oxidative cyclometallation of 2-Int1 through 2-Ts1′ to generate a palladacyclic intermediate 2-Int2′; however, this process has an extremely high energy barrier of 50.4 kcal mol−1, and thus, we exclude this pathway (for more details, see Scheme S1 in page S15 of the ESI†). The intermediate 2-Int2 subsequently undergoes cyclometallation to generate an intermediate 2-Int3via the transition state 2-Ts2. The energy barrier for cyclometallation is 13.8 kcal mol−1, which is lower than that of the competitive pathway involving the so-called a β-H elimination step (41.7 kcal mol−1via2-Ts2′′) (for more details, see Scheme S2 in page S16 of the ESI†). The palladium intermediates 2-Int3 presents the alkyne moiety coordinated to palladium (Pd–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, 2.49 Å and 2.35 Å). The intermediate 2-Int3 is a stable intermediate, probably due to the alkyne moiety coordinated to palladium.13 We also attempted to locate the intermediate in which the alkyne moiety is not coordinated·to palladium; however, we failed to obtain such a kind of intermediate and it always reverses to the previous intermediates 2-Int1 or 2-Int2 after geometry optimization. The intermediate 2-Int3 subsequently undergoes β-hydrogen elimination to generate an intermediate 2-Int4via2-Ts3 with a low energy barrier of 4.4 kcal mol−1. The intermediate 2-Int4 can undergo reductive elimination to afford product 4a, which was accessed in the experiment. Alternatively, 2-Int4 undergoes migratory insertion via2-Ts4 to generate 2-Int5 with a free energy of 10.9 kcal mol−1. Subsequently, reductive elimination of 2-Int5 is performed to afford 2-Int6via2-Ts5 with an energy barrier of 14.7 kcal mol−1 and then releasing the palladium catalyst to form product 2a. This is in line with the result of the control experiment shown in Scheme 8b, in which 4a could be rapidly converted to product 2a under standard conditions. We also investigated another possible reaction pathway that involves 2-Int2 undergoing β-C elimination followed by β-H elimination (for details, see Scheme S2 in the page S16 of ESI†).
C, 2.49 Å and 2.35 Å). The intermediate 2-Int3 is a stable intermediate, probably due to the alkyne moiety coordinated to palladium.13 We also attempted to locate the intermediate in which the alkyne moiety is not coordinated·to palladium; however, we failed to obtain such a kind of intermediate and it always reverses to the previous intermediates 2-Int1 or 2-Int2 after geometry optimization. The intermediate 2-Int3 subsequently undergoes β-hydrogen elimination to generate an intermediate 2-Int4via2-Ts3 with a low energy barrier of 4.4 kcal mol−1. The intermediate 2-Int4 can undergo reductive elimination to afford product 4a, which was accessed in the experiment. Alternatively, 2-Int4 undergoes migratory insertion via2-Ts4 to generate 2-Int5 with a free energy of 10.9 kcal mol−1. Subsequently, reductive elimination of 2-Int5 is performed to afford 2-Int6via2-Ts5 with an energy barrier of 14.7 kcal mol−1 and then releasing the palladium catalyst to form product 2a. This is in line with the result of the control experiment shown in Scheme 8b, in which 4a could be rapidly converted to product 2a under standard conditions. We also investigated another possible reaction pathway that involves 2-Int2 undergoing β-C elimination followed by β-H elimination (for details, see Scheme S2 in the page S16 of ESI†).
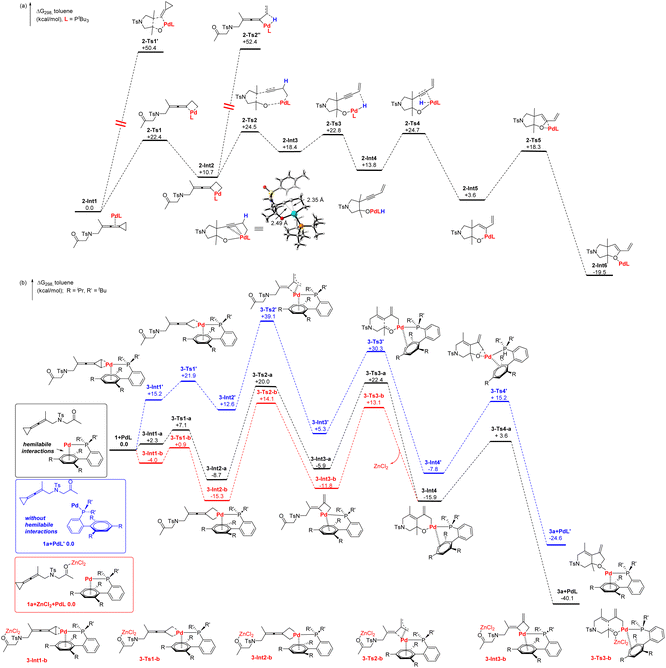 |
| | Scheme 10 DFT calculations on the possible reaction pathways: (a) DFT calculations of 1a to 2a; (b) DFT calculations of 1a to 3a. | |
In conjunction with the previously proposed concept of hemilabile interactions between the palladium, and bulky biaryl phosphine ligands,5c,d we also compared the difference between the presence and absence of hemilabile interactions by DFT calculations to show the role of the bulky biaryl phosphine ligand in the [3 + 2] cycloaddition reaction (shown in Scheme 10b). The DFT calculations for this reaction were performed at the SMD(toluene)/ωB97X-D/6-31G(d)/Lanl2dz//ωB97X-D/6-31G(d)/Lanl2dz level with the Gaussian 16 program.21 We used the hemilabile palladium complex and substrate 1a as the first step. The palladium complex coordinated to the distal C–C bond of VDCP to produce 3-Int1-a with an activation free energy of 2.3 kcal mol−1. The hemilabile interactions between the palladium and bulky phosphine ligands allows the energy of 3-Int1-a to be 12.9 kcal mol−1 less than that of 3-Int1′. The insertion of a palladium catalyst into the distal C–C bond of VDCP gives a palladacyclic intermediate 3-Int2-a through 3-Ts1-a with an energy barrier of 4.8 kcal mol−1. 3-Int2-a then isomerizes to 3-Int3-avia3-Ts2-a with an energy barrier of 28.7 kcal mol−1, which is lower than that of 3-Ts2′ due to the presence of hemilabile interactions. Subsequently, carbonyl group insertion occurs via3-Ts3-a with an activation free energy of 28.3 kcal mol−1 to give a palladacyclohexane intermediate 3-Int4. Then, the reductive elimination reaction (via3-Ts4) converts 3-Int4 to 3a+PdL with an activation free energy of 19.5 kcal mol−1. For comparison, the reaction pathway without hemilabile interactions is also investigated systematically (Scheme 10b, blue letters). Generally, the energies of all intermediates and transition states without hemilabile interactions are higher in the range of 7.9–21.3 kcal mol−1, indicating that hemilabile interactions play important roles in this reaction. Moreover, DFT calculations on the reaction pathway involving ZnCl2-promoted the [3+2] cycloaddition were also performed (Scheme 10b, red letters). It is noteworthy that the intermediates and transition states are more energetically stable (5.9–9.3 kcal mol−1) due to binding between ZnCl2 and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond in 1a. The addition of ZnCl2 decreases the energy barrier of carbonyl group insertion (3-Int3-b to 3-Ts3-b) which is lower than that without ZnCl2 (3-Int3-a to 3-Ts3-a) by 3.4 kcal mol−1, and indicates that the addition of ZnCl2 facilitates the migratory insertion of the CO bond. For comparison, the insertion of a palladium catalyst with the bulky biaryl phosphine ligand into the proximal C–C bond of VDCP which produces a palladacyclic intermediate 3-Int2-b′ was also investigated. The energy of 3-Int2-b′ is higher than that of 3-Int2-b by 8 kcal mol−1, probably due to steric hindrance. This may account for the distal C–C bond cleavage being preferred, utilizing the bulky biaryl phosphine ligand. In general, the phosphine ligand can tune the modes of the initial C–C bond cleavage, which leads to different reaction patterns and generates different products.
O bond in 1a. The addition of ZnCl2 decreases the energy barrier of carbonyl group insertion (3-Int3-b to 3-Ts3-b) which is lower than that without ZnCl2 (3-Int3-a to 3-Ts3-a) by 3.4 kcal mol−1, and indicates that the addition of ZnCl2 facilitates the migratory insertion of the CO bond. For comparison, the insertion of a palladium catalyst with the bulky biaryl phosphine ligand into the proximal C–C bond of VDCP which produces a palladacyclic intermediate 3-Int2-b′ was also investigated. The energy of 3-Int2-b′ is higher than that of 3-Int2-b by 8 kcal mol−1, probably due to steric hindrance. This may account for the distal C–C bond cleavage being preferred, utilizing the bulky biaryl phosphine ligand. In general, the phosphine ligand can tune the modes of the initial C–C bond cleavage, which leads to different reaction patterns and generates different products.
Asymmetric studies
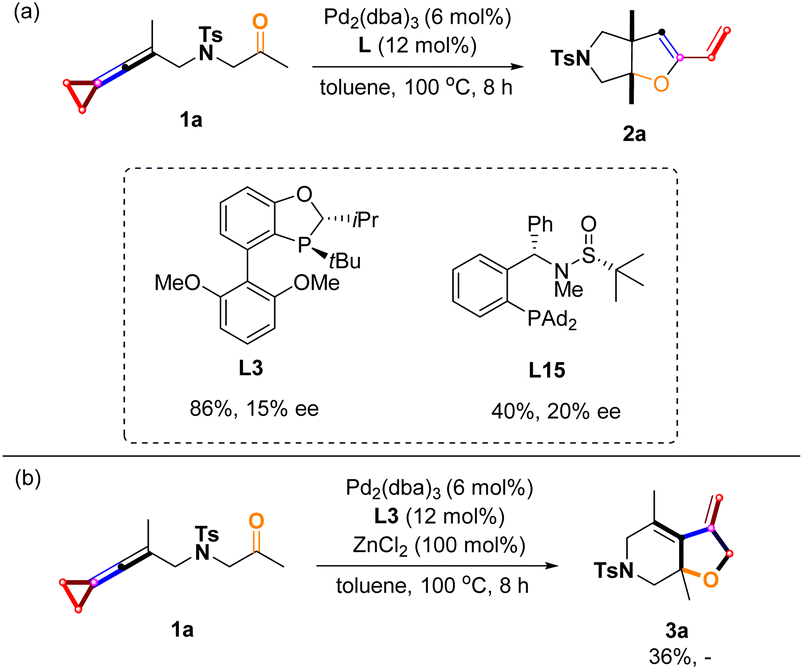
The asymmetric variants of these reactions were investigated with a series of sterically bulky chiral phosphine ligands,22,23 and we found that the use of ligands L3 and L15 gave 2a in 86% yield along with 15% ee and 40% yield along with 20% ee, respectively (Scheme 11a). However, for the product 3a, no chiral induction was observed, presumably due to the high reaction temperature (Scheme 11b) (for more information, see pages S10–S12 in the ESI†). In asymmetric catalysis, high ee values were difficult to be obtained at such a high temperature (100 °C), probably because the chiral ligands dissociate on the central palladium.
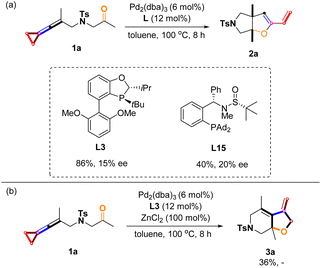 |
| | Scheme 11 (a) Asymmetric studies of 2a; (b) asymmetric studies of 3a. | |
Conclusions
In summary, we have developed a divergent protocol for the palladium-catalyzed and ligand-controlled selective cleavage of the distal C–C bond and proximal C–C bond of cyclopropane units in keto-vinylidenecyclopropanes 1, affording a series of bicyclic products containing dihydrofuran skeletons and tetrahydrofuran skeletons in moderate to good yields with a broad substrate scope and good functional group tolerance. The further transformations of the obtained bicyclic products have been also conducted to obtain a variety of their derivatives. Control experiments and DFT calculations have been used to elucidate the reaction mechanisms of the selective C–C bond cleavage. We propose a palladium intermediate in which the alkyne moiety coordinated to palladium is the key intermediate for the reaction involving the proximal C–C bond of unactivated vinylidenecyclopropane. To the best of our knowledge, this is the first example of transition metal catalyzed cleavage of the proximal C–C bond of unactivated vinylidenecyclopropane, opening up numerous avenues for further development. The utilization of these synthetic methodologies for the synthesis of biologically active molecules is currently under investigation.
Data availability
Experimental and computational data have been made available as the ESI.†
Author contributions
C. N. contributed to the experimental work; Z. Q. Y and Y. W. contributed to the computational work. C. N., Y. W. and M. S. contributed to ideation and writing of the paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for the financial support from the National Key R & D Program of China (2023YFA1506700), the National Natural Science Foundation of China (21372250, 21121062, 21302203, 20732008, 21772037, 21772226, 21861132014, 91956115 and 22171078), Project supported by the Shanghai Municipal Science and Technology Major Project (Grant No. 2018SHZDZX03) and the Fundamental Research Funds for the Central Universities 222201717003.
References
-
(a) R. R. A. Kitson, A. Millemaggi and R. J. K. Taylor, Angew. Chem. Int. Ed., 2009, 48, 9426–9451 CrossRef CAS PubMed;
(b) K.-D. Umland, A. Palisse, T. T. Haug and S. F. Kirsch, Angew. Chem., Int. Ed., 2011, 50, 9965–9968 CrossRef CAS PubMed;
(c) X. Cai, W. Liang, M. Liu, X. Li and M. Dai, J. Am. Chem. Soc., 2020, 142, 13677–13682 CrossRef CAS PubMed;
(d) C. Chen, W. Cheng, J. Wang, T. Chao, M. Cheng and R. Liu, Angew. Chem., Int. Ed., 2021, 60, 4479–4484 CrossRef CAS PubMed;
(e) W. Zhang, L. Li and C.-C. Li, Chem. Soc. Rev., 2021, 50, 9430–9442 RSC.
-
(a) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2003, 103, 1213–1270 CrossRef CAS PubMed;
(b) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117–3179 CrossRef CAS PubMed;
(c) M. Shi, J.-M. Lu, Y. Wei and L.-X. Shao, Acc. Chem. Res., 2012, 45, 641–652 CrossRef CAS PubMed;
(d) D.-H. Zhang, X.-Y. Tang and M. Shi, Acc. Chem. Res., 2014, 47, 913–924 CrossRef CAS PubMed.
-
(a) M. Shi, L.-X. Shao, J.-M. Lu, Y. Wei, K. Mizuno and H. Maeda, Chem. Rev., 2010, 110, 5883–5913 CrossRef CAS PubMed;
(b) S. Yang and M. Shi, Acc. Chem. Res., 2018, 51, 1667–1680 CrossRef CAS PubMed.
-
(a) M. Shanmugasundaram, M.-S. Wu and C.-H. Cheng, Org. Lett., 2001, 3, 4233–4236 CrossRef CAS PubMed;
(b) E. S. Johnson, G. J. Balaich and I. P. Rothwell, J. Am. Chem. Soc., 1997, 119, 7685–7693 CrossRef CAS;
(c) T. Kawasaki, S. Saito and Y. Yamamoto, J. Org. Chem., 2002, 67, 4911–4915 CrossRef CAS PubMed;
(d) M. Shanmugasundaram, M.-S. Wu, M. Jeganmohan, C.-W. Huang and C.-H. Cheng, J. Org. Chem., 2002, 67, 7724–7729 CrossRef CAS PubMed;
(e) J. A. Varela, S. G. Rubín, C. González-Rodríguez, L. Castedo and C. Saá, J. Am. Chem. Soc., 2006, 128, 9262–9263 CrossRef CAS PubMed;
(f) T. N. Tekavec and J. Louie, J. Org. Chem., 2008, 73, 2641–2648 CrossRef CAS PubMed;
(g) A. Geny, S. Gaudrel, F. Slowinski, M. Amatore, G. Chouraqui, M. Malacria, C. Aubert and V. Gandon, Adv. Synth. Catal., 2009, 351, 271–275 CrossRef CAS;
(h) M. Gulías, A. Collado, B. Trillo, F. López, E. Oñate, M. A. Esteruelas and J. L. Mascareñas, J. Am. Chem. Soc., 2011, 133, 7660–7663 CrossRef PubMed;
(i) P. Chen, T. Xu and G. Dong, Angew. Chem., Int. Ed., 2014, 53, 1674–1678 CrossRef CAS PubMed;
(j) K. Masutomi, H. Sugiyama, H. Uekusa, Y. Shibata and K. Tanaka, Angew. Chem., Int. Ed., 2016, 55, 15373–15376 CrossRef CAS PubMed;
(k) H. Ueda, K. Masutomi, Y. Shibata and K. Tanaka, Org. Lett., 2017, 19, 2913–2916 CrossRef CAS PubMed;
(l) S. Yoshizaki, Y. Shibata and K. Tanaka, Angew. Chem., Int. Ed., 2017, 56, 3590–3593 CrossRef CAS PubMed;
(m) Z. Tian, Q. Cui, C. Liu and Z. Yu, Angew. Chem., Int. Ed., 2018, 57, 15544–15548 CrossRef CAS PubMed;
(n) L. Deng, Y. Fu, S. Y. Lee, C. Wang, P. Liu and G. Dong, J. Am. Chem. Soc., 2019, 141, 16260–16265 CrossRef CAS PubMed;
(o) S.-H. Hou, X. Yu, R. Zhang, L. Deng, M. Zhang, A. Y. Prichina and G. Dong, J. Am. Chem. Soc., 2020, 142, 13180–13189 CrossRef CAS PubMed;
(p) Y. Xue and G. Dong, J. Am. Chem. Soc., 2021, 143, 8272–8277 CrossRef CAS PubMed;
(q) G.-Y. Zhang, P. Zhang, B.-W. Li, K. Liu, J. Li and Z.-X. Yu, J. Am. Chem. Soc., 2022, 144, 21457–21469 CrossRef CAS PubMed;
(r) S.-H. Hou, X. Yu, R. Zhang, C. Wagner and G. Dong, J. Am. Chem. Soc., 2022, 144, 22159–22169 CrossRef CAS PubMed.
-
(a) I. Nakamura, B. H. Oh, S. Saito and Y. Yamamoto, Angew. Chem., Int. Ed., 2001, 40, 1298–1300 CrossRef CAS;
(b) F. Verdugo, L. Villarino, J. Durán, M. Gulías, J. L. Mascareñas and F. López, ACS Catal., 2018, 8, 6100–6105 CrossRef CAS;
(c) F. Verdugo, E. Da Concepción, R. Rodiño, M. Calvelo, J. L. Mascareñas and F. López, ACS Catal., 2020, 10, 7710–7718 CrossRef CAS;
(d) F. Verdugo, R. Rodiño, M. Calvelo, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2022, 61, e20220229 CrossRef PubMed;
(e) J. Zhou, L. Meng, S. Lin, B. Cai and J. Joelle Wang, Angew. Chem., Int. Ed., 2023, 62, e202303727 CrossRef CAS PubMed.
-
(a) P. A. Evans and P. A. Inglesby, J. Am. Chem. Soc., 2008, 130, 12838–12839 CrossRef CAS PubMed;
(b) S. Mazumder, D. Shang, D. E. Negru, M.-H. Baik and P. A. Evans, J. Am. Chem. Soc., 2012, 134, 20569–20572 CrossRef CAS PubMed;
(c) P. A. Inglesby, J. Bacsa, D. E. Negru and P. A. Evans, Angew. Chem., Int. Ed., 2014, 53, 3952–3956 CrossRef CAS PubMed;
(d) P. A. Evans, D. E. Negru and D. Shang, Angew. Chem., Int. Ed., 2015, 54, 4768–4772 CrossRef CAS PubMed;
(e) S. Yang, K.-H. Rui, X.-Y. Tang, Q. Xu and M. Shi, J. Am. Chem. Soc., 2017, 139, 5957–5964 CrossRef CAS PubMed;
(f) S. Yang, Q.-Z. Li, C. Xu, Q. Xu and M. Shi, Chem. Sci., 2018, 9, 5074–5081 RSC;
(g) K.-H. Rui, S. Yang, Y. Wei and M. Shi, Org. Chem. Front., 2019, 6, 2506–2513 RSC;
(h) K.-H. Rui and M. Shi, Org. Chem. Front., 2019, 6, 1816–1820 RSC;
(i) X. Chen, C. Ning, S. Yang, Y. Wei and M. Shi, Adv. Synth. Catal., 2021, 363, 1727–1732 CrossRef.
-
(a) E. Da Concepción, I. Fernández, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2021, 60, 8182–8188 CrossRef PubMed;
(b) X. Xiao and Z. Yu, Chem.–Eur. J., 2021, 27, 7176–7182 CrossRef CAS PubMed.
-
(a) S. Saito, M. Masuda and S. Komagawa, J. Am. Chem. Soc., 2004, 126, 10540–10541 CrossRef CAS PubMed;
(b) S. Komagawa and S. Saito, Angew. Chem., Int. Ed., 2006, 45, 2446–2449 CrossRef CAS PubMed;
(c) L. Saya, G. Bhargava, M. A. Navarro, M. Gulías, F. López, I. Fernández, L. Castedo and J. L. Mascareñas, Angew. Chem., Int. Ed., 2010, 49, 9886–9890 CrossRef CAS PubMed;
(d) S. Saito, K. Maeda, R. Yamasaki, T. Kitamura, M. Nakagawa, K. Kato, I. Azumaya and H. Masu, Angew. Chem., Int. Ed., 2010, 49, 1830–1833 CrossRef CAS PubMed;
(e) L. Saya, I. Fernández, F. López and J. L. Mascareñas, Org. Lett., 2014, 16, 5008–5011 CrossRef CAS PubMed.
-
(a) L.-Z. Yu, K. Chen, Z.-Z. Zhu and M. Shi, Chem. Commun., 2017, 53, 5935–5945 RSC;
(b) L. Yu and M. Shi, Chem.–Eur. J., 2019, 25, 7591–7606 CrossRef CAS PubMed;
(c) H.-S. Li, S.-C. Lu, Z.-X. Chang, L. Hao, F.-R. Li and C. Xia, Org. Lett., 2020, 22, 5145–5150 CrossRef CAS PubMed;
(d) Z. Shen, I. Maksso, R. Kuniyil, T. Rogge and L. Ackermann, Chem. Commun., 2021, 57, 3668–3671 RSC;
(e) X.-L. Liu, Y.-Y. Zhang, L. Li, L.-Q. Tan, Y.-A. Huang, A.-J. Ma and J.-B. Peng, Org. Lett., 2022, 24, 6692–6696 CrossRef CAS PubMed;
(f) M.-M. Ji, P.-R. Liu, J.-D. Yan, Y.-Y. He, H. Li, A.-J. Ma and J.-B. Peng, Org. Lett., 2024, 26, 231–235 CrossRef CAS PubMed.
-
(a) B. Niu, Y. Wei and M. Shi, Chem. Commun., 2021, 57, 4783–4786 RSC;
(b) Z. Yang, B. Zhang, Y. Long and M. Shi, Chem. Commun., 2022, 58, 9926–9929 RSC;
(c) B. Wu, Q.-J. Ding, Z.-L. Wang and R. Zhu, J. Am. Chem. Soc., 2023, 145, 2045–2051 CrossRef CAS PubMed.
-
(a) K. Tsutsumi, S. Ogoshi, S. Nishiguchi and H. Kurosawa, J. Am. Chem. Soc., 1998, 120, 1938–1939 CrossRef CAS;
(b) T. Murahashi, S. Ogoshi and H. Kurosawa, Chem. Rec., 2003, 3, 101–111 CrossRef CAS PubMed.
-
(a) J. Chen, S. Gao and M. Chen, Chem. Sci., 2019, 10, 10601–10606 RSC;
(b) J. Chen, S. Gao and M. Chen, Org. Lett., 2019, 21, 8800–8804 CrossRef CAS PubMed;
(c) M. Chen, Chem. Commun., 2021, 57, 9212–9215 RSC.
- W. Yuan, L. Song and S. Ma, Angew. Chem., Int. Ed., 2016, 55, 3140–3143 CrossRef CAS PubMed.
-
(a) Y. Yamamoto, A. Nagata, Y. Arikawa, K. Tatsumi and K. Itoh, Organometallics, 2000, 19, 2403–2405 CrossRef CAS;
(b) Y. Yamamoto, A. Nagata, H. Nagata, Y. Ando, Y. Arikawa, K. Tatsumi and K. Itoh, Chem.–Eur. J., 2003, 9, 2469–2483 CrossRef CAS PubMed;
(c) Y. Yamamoto, S. Kuwabara, Y. Ando, H. Nagata, H. Nishiyama and K. Itoh, J. Org. Chem., 2004, 69, 6697–6705 CrossRef CAS PubMed;
(d) X. Lian and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 8255–8258 CrossRef CAS PubMed;
(e) Y. Feng, N. Tian, Y. Li, C. Jia, X. Li, L. Wang and X. Cui, Org. Lett., 2017, 19, 1658–1661 CrossRef CAS PubMed;
(f) H. Miura, Y. Tanaka, K. Nakahara, Y. Hachiya, K. Endo and T. Shishido, Angew. Chem., Int. Ed., 2018, 57, 6136–6140 CrossRef CAS PubMed.
-
(a) R. M. P. Veenboer, S. Dupuy and S. P. Nolan, ACS Catal., 2015, 5, 1330–1334 CrossRef CAS PubMed;
(b) Q. Li, X. Fang, R. Pan, H. Yao and A. Lin, J. Am. Chem. Soc., 2022, 144, 11364–11376 CrossRef CAS PubMed;
(c) Q. He, L. Zhu, Z.-H. Yang, B. Zhu, Q. Ouyang, W. Du and Y.-C. Chen, J. Am. Chem. Soc., 2021, 143, 17989–17994 CrossRef CAS PubMed.
-
(a) S.-C. Sha, J. Zhang and P. J. Walsh, Org. Lett., 2015, 17, 410–413 CrossRef CAS PubMed;
(b) H. Zhang, P. Ruiz-Castillo and S. L. Buchwald, Org. Lett., 2018, 20, 1580–1583 CrossRef CAS PubMed;
(c) P.-X. Zhou, X. Yang, X. Du, S. Zhao, H. Wang, X. Li, N. Liu, X. Tan, F. Ren and Y.-M. Liang, Org. Chem. Front., 2022, 9, 4097–4103 RSC;
(d) S. Yu, Y. Ai, L. Hu, G. Lu, C. Duan and Y. Ma, Angew. Chem., Int. Ed., 2022, 61, e202200052 CrossRef CAS PubMed.
-
(a) D. L. Boger and C. E. Brotherton, J. Am. Chem. Soc., 1986, 108, 6695–6713 CrossRef CAS;
(b) M. H. Cheng, J. Chem. Soc., Chem. Commun., 1992, 934–936 RSC.
- W. G. Salmond, M. A. Barta and J. L. Havens, J. Org. Chem., 1978, 43, 2057–2059 CrossRef CAS.
- R. Pappo, D. Allen Jr, R. Lemieux and W. Johnson, J. Org. Chem., 1956, 21, 478–479 CrossRef CAS.
-
(a) T. Xu, Z. Yu and L. Wang, Org. Lett., 2009, 11, 2113–2116 CrossRef CAS PubMed;
(b) T. Xu, Q. Yang, D. Li, J. Dong, Z. Yu and Y. Li, Chem.–Eur. J., 2010, 16, 9264–9272 CrossRef CAS PubMed;
(c) S. Yang, W. Yuan, Q. Xu and M. Shi, Chem.–Eur. J., 2015, 21, 15964–15969 CrossRef CAS PubMed.
- For ωB97X-D functional, see: J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
-
(a) G. Xu, C. H. Senanayake and W. Tang, Acc. Chem. Res., 2019, 52, 1101–1112 CrossRef CAS PubMed;
(b) H. Yang and W. Tang, Chem. Rec., 2020, 20, 23–40 CrossRef CAS PubMed;
(c) H. Yang and W. Tang, Nat. Commun., 2022, 13, 4577 CrossRef CAS PubMed.
-
(a) Y. Tu, B. Xu, Q. Wang, H. Dong, Z.-M. Zhang and J. Zhang, J. Am. Chem. Soc., 2023, 145, 4378–4383 CrossRef CAS PubMed;
(b) S. Zhang, S. Wu, Q. Wang, S. Xu, Y. Han, C. Yan, J. Zhang and L. Wang, Angew. Chem., Int. Ed., 2023, 62, e202300309 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of new compounds. CCDC 2206931 and 2246469. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02536a |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Min
Shi
a,
Min
Shi