
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-induced direct C–H alkylation of polycyclic aromatic hydrocarbons with alkylsulfones†
Motoo
Ohtsuka‡
a,
Koushik
Ghosh‡
a,
Jacky C.-H.
Yim‡
a,
Hikaru
Sotome
 *b,
Tsubasa
Okamoto
cd,
Kayo
Suda
e,
Yasuhiro
Kobori
*cd,
Daisuke
Yokogawa
*e,
Hiroshi
Miyasaka
*b,
Cathleen M.
Crudden
*afg and
Masakazu
Nambo
*ah
*b,
Tsubasa
Okamoto
cd,
Kayo
Suda
e,
Yasuhiro
Kobori
*cd,
Daisuke
Yokogawa
*e,
Hiroshi
Miyasaka
*b,
Cathleen M.
Crudden
*afg and
Masakazu
Nambo
*ah
aInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Chikusa, Nagoya, Aichi 464-8601, Japan. E-mail: cruddenc@chem.queensu.ca; nambo.masakazu.p3@f.mail.nagoya-u.ac.jp
bDivision of Frontier Materials Science and Centre for Advanced Interdisciplinary Research, Graduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan. E-mail: sotome@laser.chem.es.osaka-u.ac.jp
cMolecular Photoscience Research Center, Kobe University, 1-1 Rokkodai-cho, Nada-ku, Kobe 657-8501, Japan. E-mail: ykobori@kitty.kobe-u.ac.jp
dDepartment of Chemistry, Graduate School of Science, Kobe University, 1-1, Rokkodai-cho, Nada-ku, Kobe 657-8501, Japan
eGraduate School of Arts and Sciences, The University of Tokyo, Komaba, Meguro-ku, Tokyo, 153-8902, Japan. E-mail: c-d.yokogawa@g.ecc.u-tokyo.ac.jp
fDepartment of Chemistry, Queen's University, Chernoff Hall, Kingston, Ontario K7L 3N6, Canada
gCarbon to Metal Coating Institute, Queen's University, Kingston, Ontario K7L 3N6, Canada
hDepartment of Chemistry, Graduate School of Science, Nagoya University, Furo, Chikusa, Nagoya, Aichi 464-8601, Japan
First published on 4th June 2024
Abstract
Polycyclic aromatic hydrocarbons (PAHs) are fragments of graphene that have attracted considerable attention as a new class of carbon-based materials. The functionalization of edge positions in PAHs is important to enable the modulation of physical and chemical properties essential for various applications. However, straightforward methods that combine functional group tolerance and regioselectivity remain sought after. Here we report a photochemical approach for the direct alkylation of carbon–hydrogen bonds in PAHs that takes place in a regiospecific manner, an outcome that has never been achieved in related thermal reactions. A reaction mechanism involving a single electron transfer process from photo-excited PAHs to sulfones, and a rationale for the origin of regioselectivity are proposed on the basis of spectroscopic analyses and theoretical calculations.
Introduction
The synthesis of well-defined aromatic compounds has been of central importance in organic chemistry since the very beginning of the field.1–3 In modern times, aromatic substructures are found in virtually all pharmaceutical compounds and are key components of many polymers and materials.4–6 Among aromatic compounds, polycyclic aromatic hydrocarbons (PAHs) have attracted much attention both as fragments of graphene and also as interesting materials in their own right (Fig. 1a). Functionalized PAHs have already shown promise in organic electronic devices, bioimaging, sensing, and gene delivery.7,8 In recent years, several bottom-up approaches have been developed that enable the preparation of new atomically precise PAHs, including systems with embedded heteroatoms and rings of varying sizes.9,10 To explore the functions of these new PAHs, methods to modify edge sites are crucial because they enhance solubility and processability, and enable the tuning of intermolecular interactions and electronic properties – all critical factors in materials applications.11,12 However, the introduction of functionality into PAHs often requires harsh reaction conditions, which limit applicable functional groups or require long synthetic sequences to introduce desired substituents regioselectively. Therefore, the development of straightforward methods for site-selective functionalization of carbon–hydrogen (C–H) bonds in PAHs is essential for the field to progress.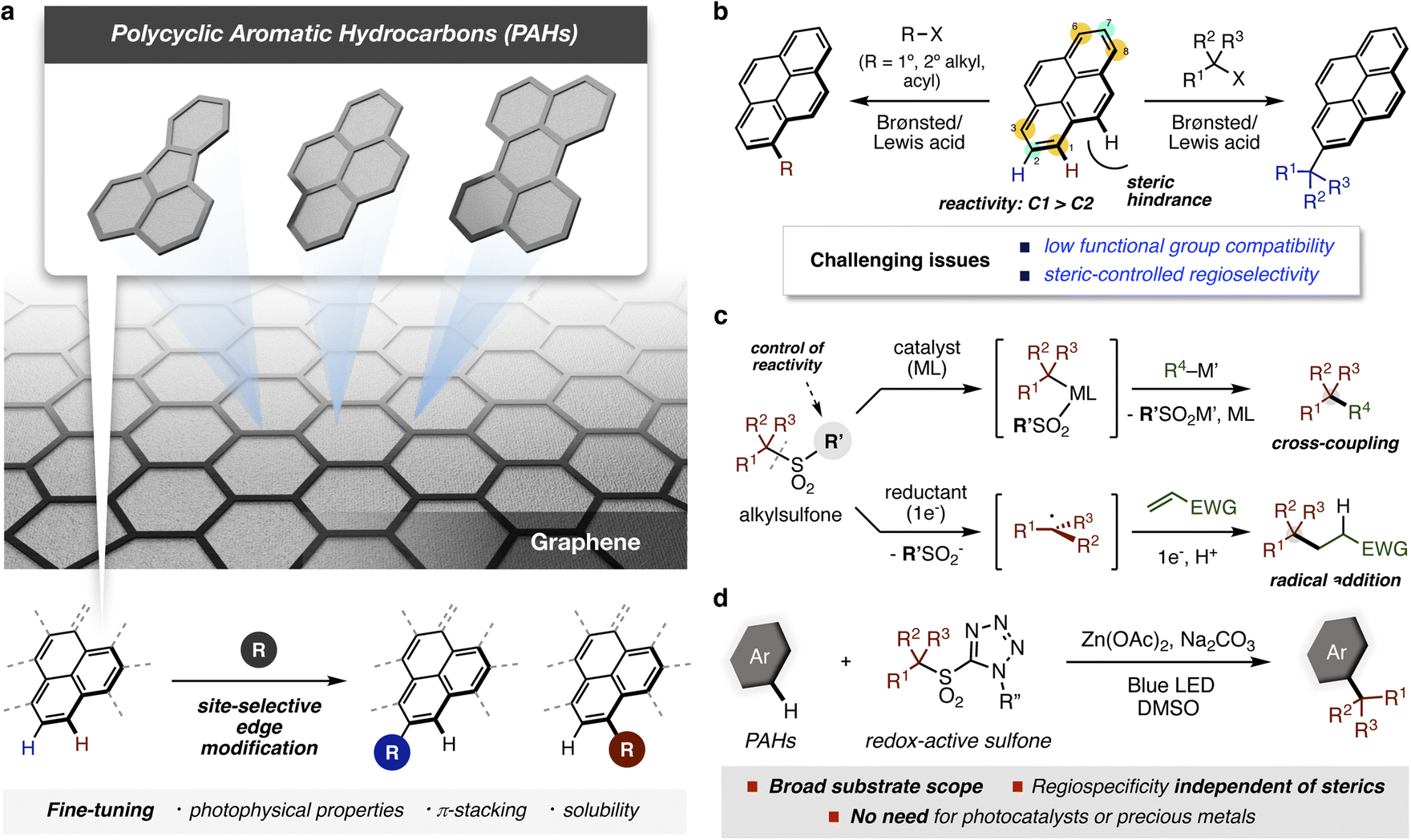 | ||
| Fig. 1 Edge modification of polycyclic aromatic hydrocarbons (PAHs): (a) site-selective functionalizations of PAHs for tuning molecular functions. (b) Regioselectivity in Friedel–Crafts reaction of pyrene (X = halides or OH). (c) Desulfonylative transformations of sulfones. (d) This work: visible-light-induced regiospecific C–H alkylation of PAHs with redox-active alkyl sulfones. | ||
Among direct methods to introduce functional groups onto PAHs,13–15 Friedel–Crafts reactions have been the most widely employed (Fig. 1b).16 As expected from this classical reaction, strong acids are needed,17,18 resulting in low functional group compatibility. Moreover, groups that can be introduced are limited to very simple alkyl substituents. Accurate control of the regioselectivity is also a significant challenge for Friedel–Crafts reactions on PAHs. For pyrene, electrophilic aromatic substitution generally occurs at the C1, C3, C6, and C8 positions, whereas bulky tertiary alkyl groups are installed at the C2 and C7 positions. Steric effects also control the regioselectivity of Ir-catalyzed C–H borylations, which have been reported by the Marder group to introduce synthetically useful boryl groups.19 To date, there is only one example of tert-butylation of pyrene at the C1 position under electrochemical conditions, which includes hydroalkylated pyrenes as side products.20 The regioselectivity in classical thermal functionalizations of PAHs has therefore depended on the bulkiness of substituents for control, illustrating the need for new synthetic approaches to prepare structurally diverse PAH derivatives.
Over the past few decades, sulfones have emerged as versatile, readily available building blocks owing to their bench stability and functional group compatibility (Fig. 1c).21 Compared with (pseudo)halides, the sulfonyl group is less reactive, and thus has received less attention as a leaving group in organic synthesis. However, recent results have shown that subtle changes to the non-reactive portion can result in versatile reactivity in transition-metal catalyzed reactions.22,23 Furthermore, reductive activation of alkyl sulfones through single electron transfer (SET) has enabled radical functionalization reactions under mild conditions.24–28
During our investigations into radical generation from sulfones using PAHs as photocatalysts, we observed the formation of alkylated PAHs. Building on this observation, we envisioned that a photo-induced process might provide a powerful solution to the issue of regioselective functionalization of PAHs without resorting to photocatalysts or precious metals. Herein, we report a simple photochemical method for the direct C–H alkylation of PAHs with redox-active alkylsulfones in a regiospecific manner never before achieved under thermal conditions (Fig. 1d). This method provides a variety of alkylated PAHs bearing useful functional groups under mild reaction conditions, increasing the potential utility for PAH-based organic materials. A reaction mechanism involving a SET and a regiospecific carbon–carbon bond forming processes is proposed based on spectroscopic analyses and theoretical calculations.
Results and discussion
We began our investigation of the direct C–H alkylation of 2-tert-butylpyrene 1a as a model PAH. A blue light-emitting diode (LED, 456 nm) was employed for irradiation. In early experiments using 1,1-dimethylpropyl N-phenyltetrazolyl sulfone 2a as the alkylating agent and DMSO as solvent, Na2CO3 was found to be the optimal base (see ESI, Table S1†), affording mono-alkylated- (8a) and di-alkylated products (8aa) in 30% and 5% yields, respectively. Surprisingly, the alkylation occurred exclusively at the sterically-hindered C6 and C8 positions, in contrast with Friedel–Crafts alkylation conditions, which yield the C7-alkylated product.The wavelength dependence of LEDs in C–H alkylation was investigated next (Fig. 2a). The yields of both products depended on the wavelength of light, with lower yields obtained at shorter wavelengths. When the progress of the reaction was monitored by HPLC, rapid decomposition of sulfone 2a was observed using a UV LED (370 nm), suggesting that sulfone stability suffers under UV light irradiation and that visible light in the blue region of the spectrum is optimal (see ESI, Fig. S4†).
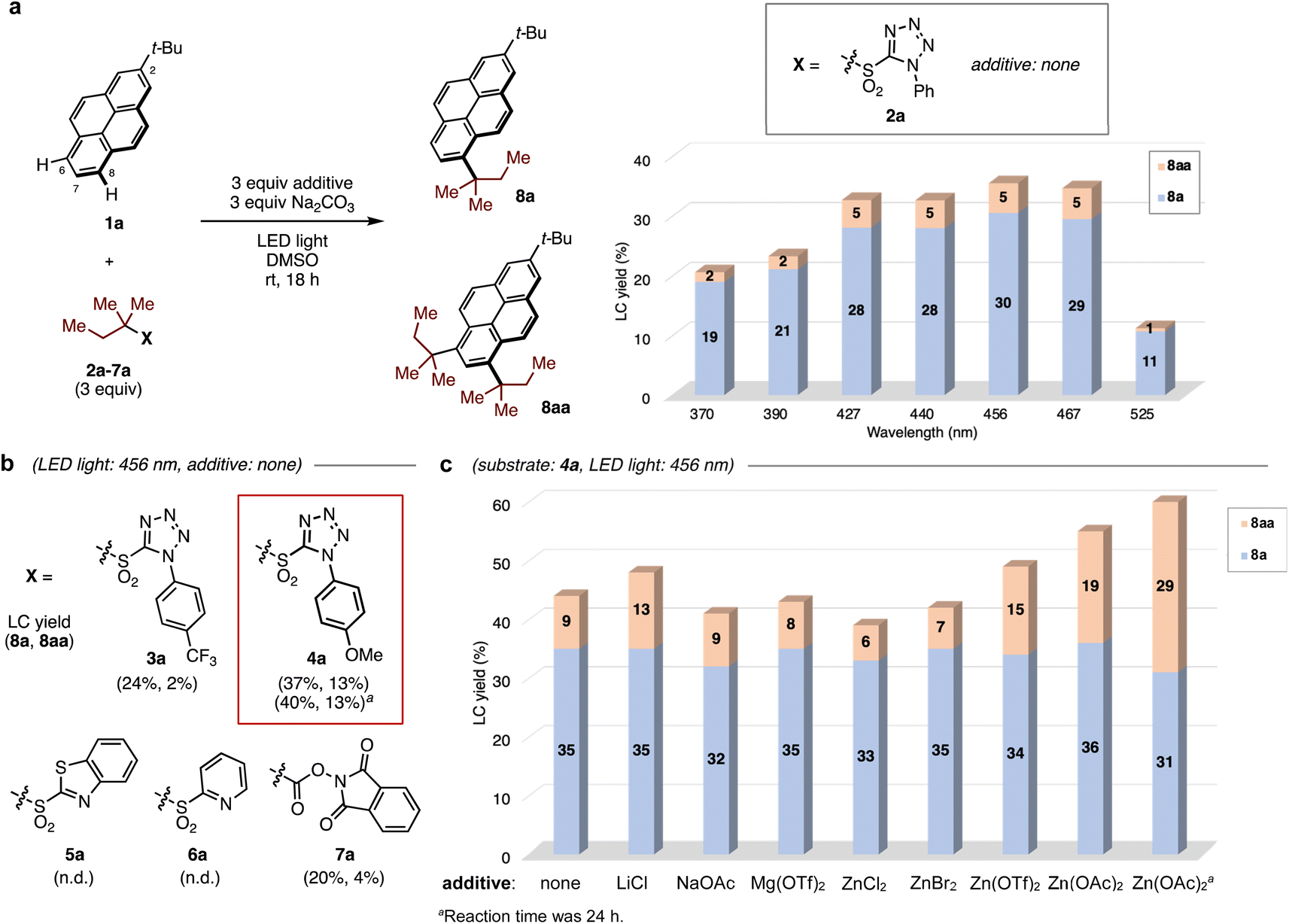 | ||
| Fig. 2 Optimization of reaction conditions in visible-light-induced C–H alkylation: (a) LED wavelength dependence in the reaction of 2-tert-butylpyrene 1a with alkylating agents. Yields were determined by high performance liquid chromatography (LC) of crude mixture using biphenyl as an internal standard. (b) Effect of substituents (X) in redox-active substrates. (c) Effect of additives when 4a was used. | ||
To improve the reaction efficiency, the aryl substituent on the tetrazolyl group was modified (Fig. 2b). The electron-deficient 4-trifluoromethylphenyl group (3a) showed similar reactivity, and the electron-rich 4-methoxyphenyl group (4a) slightly increased the yield of 8aa. Notably, the replacement of tetrazolyl group with other substituents such as 2-benzothiazolyl (5a) and 2-pyridyl (6a) groups shut down the reaction. A redox-active N-hydroxyphthalimide ester 7a, which is widely employed for radical transformations,29 gave low yields of both products. Further optimization was attempted by screening of various additives (Fig. 2c, see ESI, Table S3†). While metal salts including Li and Mg salts were not effective, the addition of Zn(OAc)2 led to increased yields of 8aa. Prolonging the reaction time increased the combined yield of 8a/8aa to 60%. However, if 8a alone is the desired product, the reaction without additive provides similar yields of this product, with only 9% double alkylation. The role of Zn salt is not clear, but the degradation of alkyl sulfone appeared to be inhibited during the reaction (see ESI, Fig. S6†), presumably leading to the improved total yield of products.
With optimized conditions in hand, the generality of the method was assessed (Scheme 1). 2-Substituted pyrenes 1 reacted with tertiary alkyl sulfones to give the corresponding mono- (8a–11a), and dialkylated products (8aa–11aa). In addition to alkylated PAHs, aryl- (1b), boryl- (1c) and silyl- (1d) substituted pyrene substrates were compatible with the reaction, leading to products incorporating these useful handles for further transformations. The exact structure of dialkylated product 8bb was successfully confirmed by single-crystal X-ray diffraction analysis. Pyrenes bearing substituents at the 6-position yielded single products alkylated at the 8-position; the scope of alkylsulfones was thus investigated using pyrene derivative.12 Tertiary alkyl sulfones that bear phenyl (4c) and silyloxy (4d) groups, which are easily prepared by α-alkylation, were converted into the desired products.
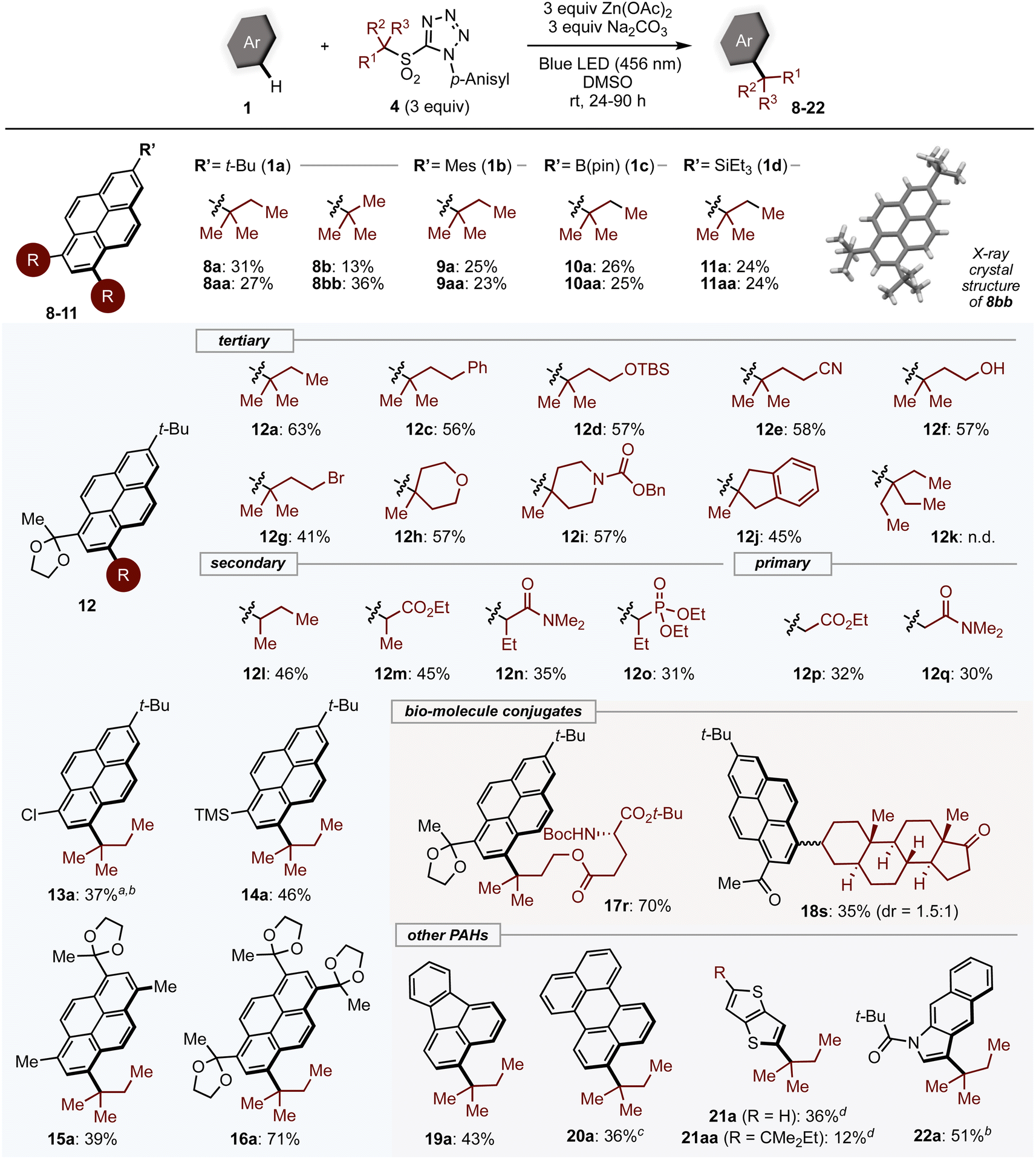 | ||
| Scheme 1 Substrate scope. Isolated yields. Reaction conditions: PAH 1 (0.1–0.2 mmol), sulfone 4 (3 equiv.), Zn(OAc)2 (3 equiv.), Na2CO3 (3 equiv.), DMSO (0.2 M), rt, 24–90 h. a5 equiv. sulfone was used. bwithout Zn(OAc)2. cGreen LED (525 nm) irradiation and N,N-dimethylformamide was used as a solvent. dBlue LED (427 nm) irradiation. | ||
The mild basic conditions provide broad functional group compatibility. Sensitive functional groups such as cyano, alcohol, and bromo groups (12e–12g) were all tolerated. Cyclic tertiary alkyl groups with heteroatoms could be installed in good yields (12h–12j). Interestingly, the most bulky group examined, 3-ethyl-3-pentyl group (12k), did not give the desired product. Secondary alkyl substrates (4l–4o) also reacted smoothly under standard conditions, while primary alkylsulfones (4p, 4q) showed lower reactivity. Benzyl, allyl, and methyl sulfones were not suitable (see ESI, Table S6†). Other 2,6-disubstituted pyrene derivatives (13–16) underwent alkylation with 4a in moderate to good yields. Pyrenes conjugated with complex structures such as amino acids (17r) or steroids (18s) could be produced, which may be useful for biological applications.
Next, we subjected a variety of PAHs to alkylation with tertiary alkyl sulfones. The C3 position of fluoranthene (19) and perylene (20) were alkylated exclusively, affording mono-alkylated products. In the case of 20, product yield was improved under green LED irradiation. It is worth noting that, as with pyrene derivatives, the regioselectivity was different from that observed under Friedel–Crafts conditions.30 PAHs containing heteroatoms such as thieno[3,2-b]thiophene (21) and benzo[f]indole (22) were also competent substrates. Unfortunately, small aromatics and highly π-extended PAHs were not applicable presumably due to weak absorption of visible light or a diminished reducing ability in the excited state (Table S6†).
The multiple C–H alkylation of pyrene (23) was also examined. To accomplish this, the low inherent solubility of pyrene was counteracted with excess sulfone 4d and Na2CO3, and the reaction was cycled three times. Under these conditions, symmetric tetrasubstituted pyrene 23d was obtained as a single regioisomer in 34% yield (Scheme 2).
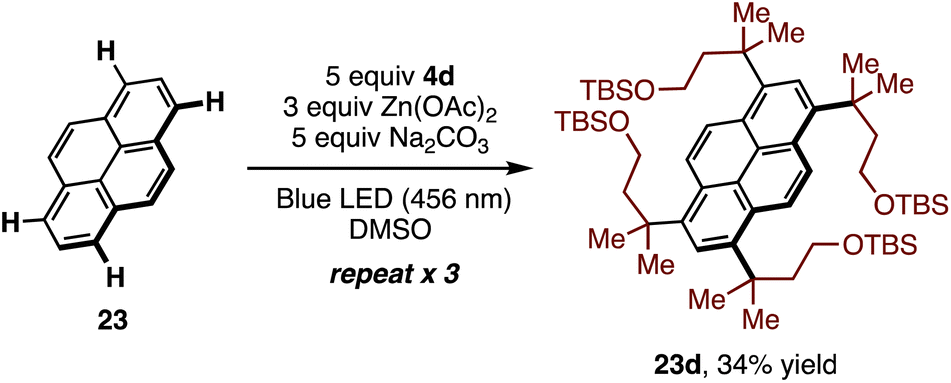 | ||
| Scheme 2 Multiple C–H alkylation of pyrene with 4d. | ||
The difference in regiospecificity compared with Friedel–Crafts alkylation suggests that the present photo-induced alkylation proceeds through a distinct reaction mechanism. To gain insight into the mechanism of this reaction, several control experiments were performed. Initially, we investigated the photoexcitation process. The UV-Vis absorption spectrum shows that pyrene 23 has almost no absorption in blue light region (>400 nm) at low concentration in DMSO solution (Fig. 3a). However, as the concentration is increased from 2 mM to 0.2 M, an absorption edge appears in the blue light region. Similar phenomena were observed with other PAHs in concentrated solution (see ESI, Fig. S3†).
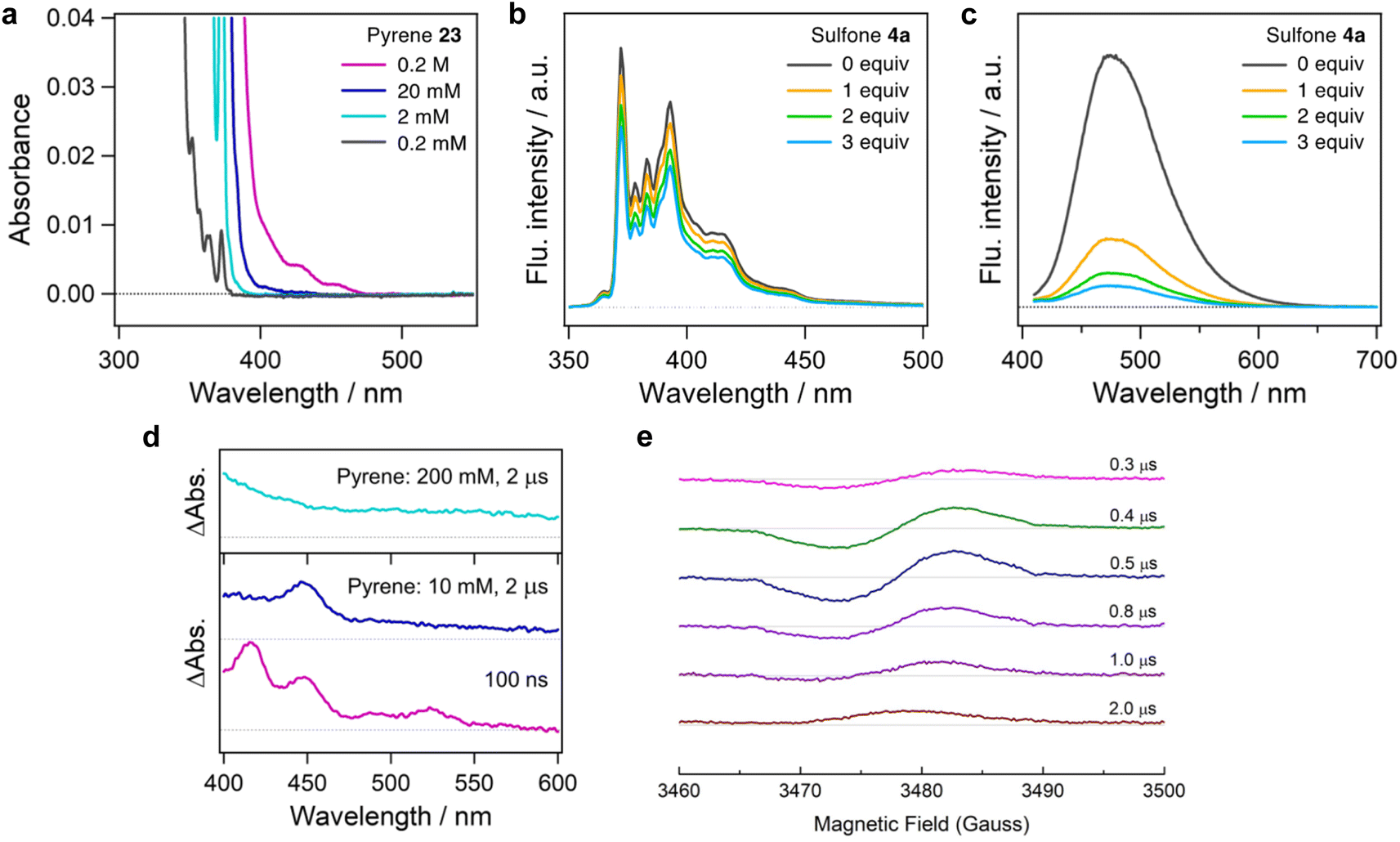 | ||
| Fig. 3 Mechanistic investigations by spectroscopic analysis: (a) concentration-dependent UV-Vis absorption spectra of pyrene 23 in DMSO. (b) and (c) fluorescence quenching of 23 in DMSO (0.2 mM in DMSO, λex = 300 nm, and 0.2 M in DMSO, λex = 390 nm) by addition of sulfone 4a. (d) Transient absorption spectra of 23 and 4a in DMSO excited with a nanosecond laser pulse at 355 nm. Concentrations of 23 were respectively 0.2 M and 10 mM in the top and bottom panels. 4a was dissolved at a concentration of 0.2 M in both the cases. (e) Delay time dependence of the time-resolved EPR spectrum. Concentrations of 23 and 4a were 10 mM and 200 mM, respectively. The corresponding delay time is shown after a pulsed excitation light (wavelength: 355 nm, power: 2 mJ per pulse). EPR signal above or below the baseline corresponds to the microwave-absorption or microwave-emission, respectively. | ||
The red-shifting of the absorption wavelength can result from a PAH monomer at high concentration or aggregates resulting from stacking of PAHs,31–33 which is essential for the photoexcitation of PAH substrates by blue LEDs. No absorption shift was observed in the solution of 23 with sulfone 4a, thus an electron donor–acceptor complex between pyrene and sulfone is unlikely (see ESI, Fig. S4†). Stern–Volmer quenching studies conducted with 4a reveal concentration-dependent quenching (Fig. 3b and c). The concentrated solution of pyrene exhibited a fluorescence maximum at 475 nm when excited by 390 nm light, which is derived from the excimer of pyrene. The quenching constants (kq) at 2 mM and 0.2 M solution of 23 were determined to be 3.4 × 109 M−1 s−1 and 1.8 × 108 M−1 s−1, respectively from the Stern–Volmer plot (see ESI, Fig. S9†) and the fluorescence lifetimes of the monomer (τ0 = 230 ns)34 and excimer (τ0 = 90 ns).35 These results suggest the SET process occurs from the lowest single excited state (S1) of the pyrene monomer or dimer to the singlet ground state (S0) of the alkylsulfone, resulting in the formation of pyrene monomer or dimer cation radicals and the sulfone anion radical.
To confirm the formation of cationic pyrene, we measured transient absorption spectra of pyrene in the presence of sulfone 4a (Fig. 3d). The transient spectrum of pyrene (10 mM) and sulfone 4a (200 mM) in DMSO at 100 ns is characterized by positive bands at 415, 445, 490 and 525 nm. The absorption peak at 445 nm is attributable to the radical cation of the pyrene monomer,36 while the other peaks due to the triplet state vanished in the microsecond region. It should be noted that no absorption bands due to other species were observed, indicating that the decomposition of sulfone is negligible under these conditions, although the excitation wavelength of 355 nm is shorter than that used in Fig. 2a. In addition, the spectrum of a solution containing 200 mM of pyrene showed an increasing trend toward 400 nm together with disappearance of the 445 nm peak. This spectral signature is in line with an absorption band of the dimer cation of pyrene.36 These results support the SET process as taking place between a pyrene monomer/dimer and sulfone.
A time-resolved electron spin resonance (TREPR) analysis also suggests the generation of radical species (Fig. 3e). The signal corresponding to the pyrene cation radical was confirmed, as reported for the intermolecular photoinduced charge-separation systems involving 23 (1 mM) and dicyanobenzene (10 mM).37 The spin polarization pattern of emission/absorption in Fig. 3d denotes that the radical ion pair (23+˙ and 4a−˙) generated by the charge-separation causes nuclear spin-state dependent electron spin polarization referred to as the radical pair mechanism (RPM)38 through diffusive separation between the radicals in the presence of weak positive exchange interaction.39
Combined with spectroscopic evidence for radical generation, the overall photo-induced alkylation process was evaluated by density-functional theory (DFT) and time-dependent DFT (TD-DFT). The energy profile for the reaction of pyrene 23 with tert-butyl sulfone 24 is shown in Fig. 4a. Initially, 23 reaches its S1 state (23′) from its S0 state by vertical excitation, and then interacts with 24 to form intermediate 26.
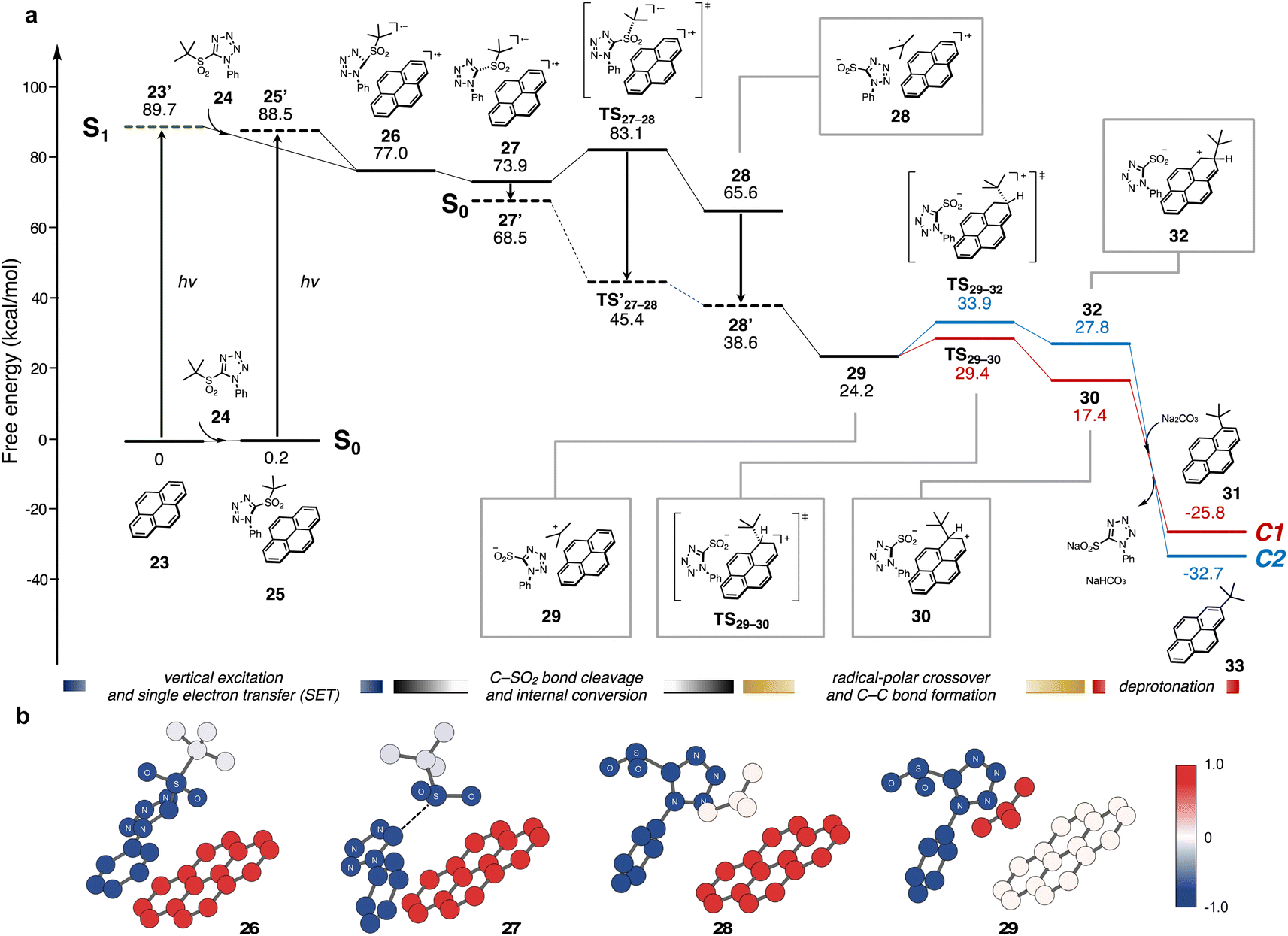 | ||
| Fig. 4 Theoretical calculations: (a) energy profile for visible-light-induced direct C–H alkylation of 23 with tert-butyl sulfone 24. Geometry optimizations of all structures in their ground state (S0) were performed at CAM-B3LYP/6-31+G(d) level with IEFPCM solvation modeling (DMSO). With the optimized geometries, we performed thermal correction at 298.15 K and single point calculations to obtain the corresponding Gibbs free energy. Structures in the S1 state were optimized at the TD-DFT level and the vertical S0–S1 excitation energy was also computed using the optimized S0-state geometry. (b) Change of natural charge from S0 equilibrium state (25) to each state. The whole charges of subgroups (N-phenyltetrazolyl sulfone, tert-butyl, and pyrene groups) were visualized. | ||
In 26, the pyramidalization of the tetrazole carbon center indicates that the carbon should have anionic character. Natural population analysis (NPA) of 26 revealed that charge separation occurs between pyrene (total charge: +0.94) and sulfone (total charge: −0.94) (Fig. 4b). This is consistent with a spontaneous SET from 23′ to 24, as supported by the results from spectroscopic analyses. The C(sp3)–SO2 bond of the metastable intermediate 27 was cleaved through a low activation energy (ΔG‡(TS27–28) = 9.2 kcal mol−1) path forming an excited-state complex between the tertiary carbon radical, the tetrazolyl sulfinate anion, and the pyrene cation (28).
Next, we assumed a radical coupling between the tert-butyl radical and pyrene radical cation in the S1 state, but this pathway required high activation energies (see ESI, Fig. S11†). Therefore, the internal conversion from an S1 state to an S0 state likely takes place prior to C–C bond formation (from 27 to 28). When the structure of 28 at the S0 state was re-optimized, natural charge revealed that the positive charge was transferred from pyrene to the tert-butyl group, which supports a radical-polar crossover occurring to generate neutral pyrene and the tert-butyl cation (29). In the C–C bond forming process, the activation energy for the C1 pathway (red line, ΔG‡(TS29–30) = 5.2 kcal mol−1) was lower than that for the C2 pathway (blue line, ΔG‡(TS29–32) = 9.7 kcal mol−1), and the Gibbs reaction energy of cation intermediate 30 was more exergonic than that of 32.40 This indicates that the regioselectivity should be determined by the electron density of the highest occupied molecular orbital (HOMO) of pyrene rather than by steric factors. Deprotonation by Na2CO3 was largely downhill in both pathways, providing C1-alkylated product (31) exclusively. A similar energy profile was obtained through the formation of pyrene dimer (see ESI, Fig. S8†).
The mechanism for Friedel–Crafts alkylation of pyrene 23 with tert-butyl chloride (34) promoted by AlCl3 as a Lewis acid was also computed to evaluate regiospecificity (Fig. 5a). AlCl3 exists as complicated Cl-bridged polymeric structures in the solid state, thus a simplified dimeric structure (Al2Cl6) was employed. The Friedel–Crafts alkylation should proceed through following steps: activation of 34 by coordination of AlCl3, electrophilic attack on 23, and then deprotonative rearomatization to form an alkylated pyrene with HCl loss and regeneration of Al2Cl6. As with the photo-induced reaction, the activation energy for the C1 pathway (red line, ΔG‡(TS35–36)) is lower than that for the C2 pathway (blue line, ΔG‡(TS35–38)) (12.1 kcal mol−1, 17.9 kcal mol−1, respectively). However, the deprotonation of intermediate 36 to afford 31 is energetically uphill. On the other hand, intermediate 38 can be converted by a 1,2-proton shift to tertiary carbocationic intermediate 39, which is even more stable than 33 formed by deprotonation.
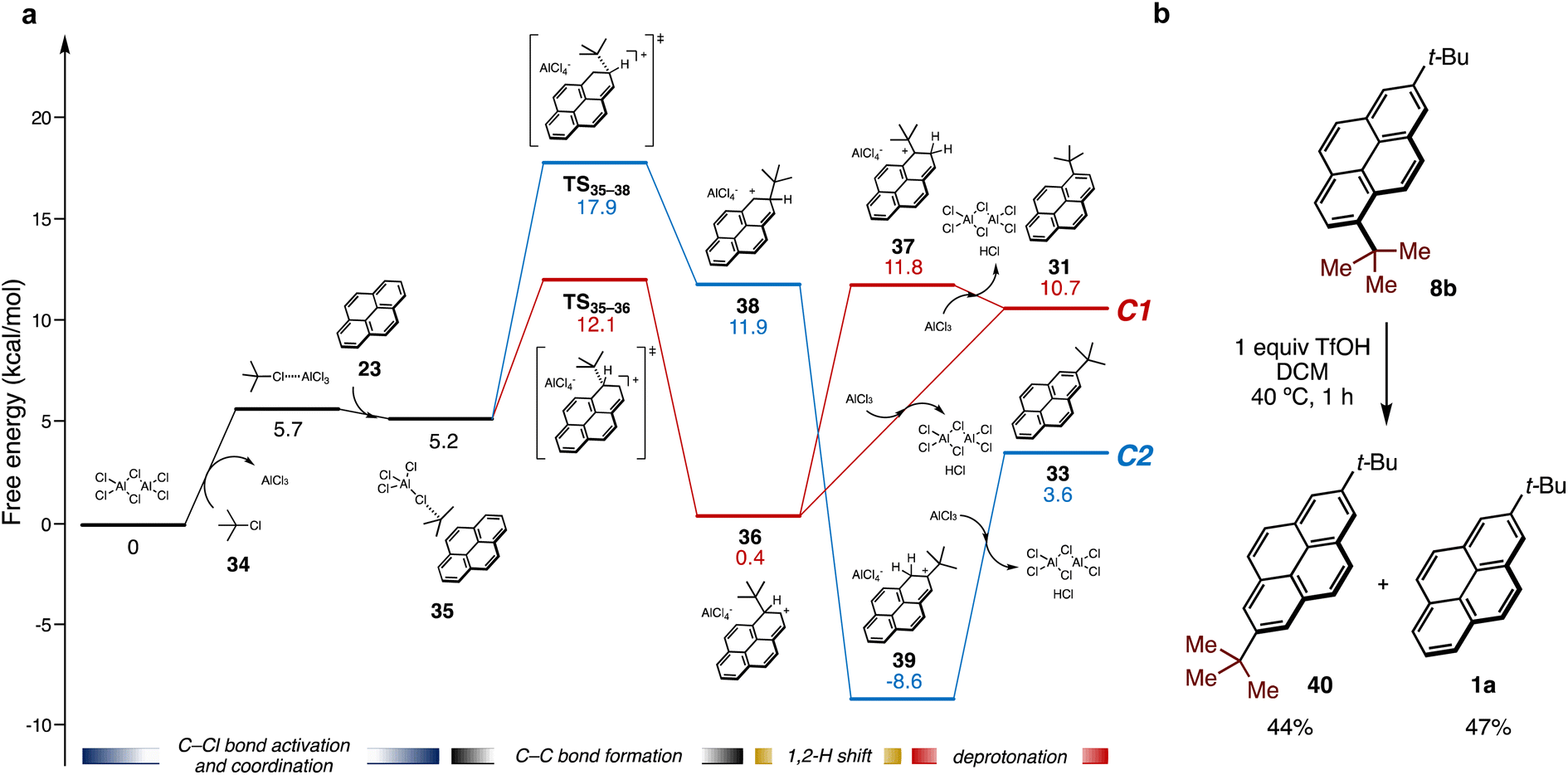 | ||
| Fig. 5 Investigation of Friedel–Crafts alkylation: (a) energy profile for AlCl3-mediated Friedel–Crafts alkylation of 23 with tert-butyl chloride 34 by DFT calculation. Gibbs free energies were calculated as in Fig. 4a with PCM solvation modeling (DCM). (b) The conversion of 8b in the presence of trifluoromethanesulfonic acid (TfOH) in DCM. Yields were determined by GC analysis of the crude mixture using 1,3-dimethoxybenzene as an internal standard. | ||
The formation of a similar carbocation intermediate (37) from 36 by a 1,2-proton shift was also unfavorable, probably due to the steric repulsion between the tert-butyl group and the hydrogen atom. Therefore, 36 preferentially reverts to 38 by reversible C–C bond formation under acidic conditions to form 39, resulting in exclusive formation of C2 product 33 after neutralization. Indeed, under experimental conditions, 1,7-di-tert-butylpyrene (8b) was rapidly converted into 2,7-di-tert-butylpyrene 40 and 1a in 44% and 47% GC yields, respectively, by treatment of stoichiometric amount of trifluoromethanesulfonic acid (TfOH) (Fig. 5b). This result clearly demonstrates formal rearrangement from the kinetically stable C1 product to the thermodynamically stable C2 product promoted by acid, which is consistent with the results from DFT calculations.
Conclusions
We have shown that the intrinsic photochemical properties of PAHs can promote a regiospecific C–H alkylation of PAHs without the need for separate photocatalysts or any type of catalysis. Based on all theoretical calculations, it is noteworthy that even though both reactions proceed through similar carbocationic intermediates, the regioselectivity changes depend completely on whether the reactions are under acidic or basic conditions rather than steric factors. The present photo-triggered process enables the generation of carbocations under basic conditions through a single electron shuttle between the PAH and the alkylating agent, providing kinetically favored products though thermodynamically disfavored intermediates by shunting acid-catalyzed isomerization pathways. This approach overcomes the substrate limitations of thermal reaction conditions, opening up a new avenue for the facile preparation of unexplored PAH-based functional materials, and highlighting the utility of organosulfones as redox-active substrates in organic synthesis.Data availability
All experimental data and computational data are available in the ESI.†Author contributions
C. M. C. and M. N. conceived the concept and supervised the project. M. O., K. G., and J. C.-H. Y. developed the reactions and performed and analyzed experiments. H. S. and H. M. performed transient absorption analysis. T. O. and Y. K. performed time-resolved EPR analysis. K. S., D. Y., and M. N. performed the DFT calculation. H. S., Y. K., D. Y., H. M., C. M. C., and M. N. wrote the manuscript with assistance from co-authors.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank Dr Yoshitaka Aramaki (Nagoya University) for assistance with X-ray crystal-structure analysis. This work is supported by Japan Society for the Promotion of Science (JSPS) (JP21H01888, JP21H05395, JP23H03956, JP23H04877 (H. S.), JP22H00344, JP20H05835 (Y. K.), JP21H01889, JP21K18934 (H. M.), JPMJPR21C9 (D. Y.), JP21K05068, JP21H05390 (M. N.)), Kansai Research Foundation for Technology Promotion (H. S.), Japan Science and Technology Agency (JST-CREST) (JPMJCR23I6 (Y. K.)). We acknowledge JSPS and Nagoya University through The World Premier International Research Centre Initiative (WPI) program. We thank the support from The Natural Sciences and Engineering Research Council of Canada (NSERC) (RGPIN/04667-2016) (C. M. C.), and the Canada Foundation for Innovation (CFI) (33355) (C. M. C.). We also thank Dr Jean Bouffard (Ewha Womans University) and Dr Issey Takahashi (Nagoya University) for helpful discussions and suggestions.Notes and references
- A. Kekulé, Bull. Soc. Chim. Fr., 1865, 3, 98 Search PubMed.
- L. T. Scott, Angew. Chem., Int. Ed., 2004, 43, 4994 CrossRef CAS PubMed.
- I. A. Stepek, M. Nagase, A. Yagi and K. Itami, Tetrahedron, 2022, 123, 132907 CrossRef CAS.
- H. Beck, M. Härter, B. Haß, C. Schmeck and L. Baerfacker, Drug Discovery Today, 2022, 27, 1560 CrossRef CAS PubMed.
- J. Hou, O. Inganas, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119 CrossRef CAS PubMed.
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, e2005630 CrossRef PubMed.
- N. Panwar, A. M. Soehartono, K. K. Chan, S. Zeng, G. Xu, J. Qu, P. Coquet, K. T. Yong and X. Chen, Chem. Rev., 2019, 119, 9559 CrossRef CAS PubMed.
- Y. Gu, Z. Qiu and K. Müllen, J. Am. Chem. Soc., 2022, 144, 11499 CrossRef CAS PubMed.
- H. Ito, Y. Segawa, K. Murakami and K. Itami, J. Am. Chem. Soc., 2019, 141, 3 CrossRef CAS PubMed.
- M. Grzybowski, B. Sadowski, H. Butenschon and D. T. Gryko, Angew. Chem., Int. Ed., 2020, 59, 2998 CrossRef CAS PubMed.
- A. Borissov, Y. K. Maurya, L. Moshniaha, W. S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565 CrossRef CAS PubMed.
- F. Würthner, C. R. Saha-Moller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962 CrossRef PubMed.
- K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739 CrossRef CAS PubMed.
- A. K. Pal, C. Li, G. S. Hanan and E. Zysman-Colman, Angew. Chem., Int. Ed., 2018, 57, 8027 CrossRef CAS PubMed.
- M. Fukazawa, F. Takahashi and H. Yorimitsu, Org. Lett., 2021, 23, 4613 CrossRef CAS PubMed.
- P. H. Gore, Chem. Rev., 1955, 55, 229 CrossRef CAS.
- L. Rodenburg, R. Brandsma, C. Tintel, J. Van Thujil, J. Lugtenburg and J. Cornelisse, Recl. Trav. Chim. Pays-Bas, 1986, 105, 156 CrossRef CAS.
- A. Wrona-Piotrowicz, A. Makal and J. Zakrzewski, J. Org. Chem., 2020, 85, 11134 CrossRef CAS PubMed.
- D. N. Coventry, A. S. Batsanov, A. E. Goeta, J. A. K. Howard, T. B. Marder and R. N. Perutz, Chem. Commun., 2005, 2172 RSC.
- P. E. Hansen, A. Berg and H. Lund, Acta Chem. Scand., Ser. B, 1976, 30, 267 CrossRef.
- B. M. Trost and C. A. Kalnmals, Chem.–Eur. J., 2019, 25, 11193 CrossRef CAS PubMed.
- M. Nambo, Y. Maekawa and C. M. Crudden, ACS Catal., 2022, 12, 3013 CrossRef CAS.
- J. Corpas, S. H. Kim-Lee, P. Mauleón, R. G. Arrayás and J. C. Carretero, Chem. Soc. Rev., 2022, 51, 6774 RSC.
- M. Nambo, Y. Tahara, J. C.-H. Yim, D. Yokogawa and C. M. Crudden, Chem. Sci., 2021, 12, 4866 RSC.
- T. Sengoku, D. Ogawa, H. Iwama, T. Inuzuka and H. Yoda, Chem. Commun., 2021, 57, 9858 RSC.
- Q. Wang, B. C. Lee, T. J. Tan, Y. Jiang, W. H. Ser and M. J. Koh, Nat. Synth., 2022, 1, 967 CrossRef.
- M. Nambo, K. Ghosh, J. C.-H. Yim, Y. Tahara, N. Inai, T. Yanai and C. M. Crudden, ACS Catal., 2022, 12, 9526 CrossRef CAS.
- S. Patel, B. Paul, H. Paul, R. Shankhdhar and I. Chatterjee, Chem. Commun., 2022, 58, 4857 RSC.
- S. Murarka, Adv. Synth. Catal., 2018, 360, 1735 CrossRef CAS.
- L. B. A. Johansson, J. G. Molotkovsky and L. D. Bergelson, J. Am. Chem. Soc., 1987, 109, 7374 CrossRef CAS.
- N. Mataga, Y. Torihashi and Y. Ota, Chem. Phys. Lett., 1967, 1, 385 CrossRef CAS.
- A. Itaya, T. Kawamura, H. Masuhara, Y. Taniguchi, M. Mitsuya, H. Uraki, K. Kano and S. Hashimoto, Chem. Lett., 1986, 15, 1541 CrossRef.
- F. Ito, S. Miyadera, H. Matsuda, Y. Ishibashi, S. Ito and H. Miyasaka, Photochem. Photobiol. Sci., 2018, 17, 910 CrossRef CAS PubMed.
- A. Nakajima, Bull. Chem. Soc. Jpn., 1973, 46, 2602 CrossRef CAS.
- J. B. Birks, D. J. Dyson and I. H. Munro, Proc. R. Soc. London, Ser. A, 1963, 275, 575 CAS.
- A. Kira, S. Arai and M. Imamura, J. Chem. Phys., 1971, 54, 4890 CrossRef CAS.
- S. N. Batchelor, H. Heikkila, C. W. M. Kay, K. A. Mclauchlan and I. A. Shkrob, Chem. Phys., 1992, 162, 29 CrossRef CAS.
- F. J. Adrian, Rev. Chem. Intermed., 1979, 3, 3 CrossRef CAS.
- Y. Kobori, S. Sekiguchi, K. Akiyama and S. Tero-Kubota, J. Phys. Chem. A, 1999, 103, 5416 CrossRef CAS.
- M. J. S. Dewar and R. D. Dennington, J. Am. Chem. Soc., 1989, 111, 3804 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2312745. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02577f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |