
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing the nature of covalently tethered distonic radical anions in the generation of heteroatom-centered radicals: evidence for the polarity-matching PCET pathway†
Kang
Fu‡
,
Xihui
Yang‡
,
Zhiyou
Yu
,
Lijuan
Song
 * and
Lei
Shi
*
* and
Lei
Shi
*
School of Chemistry and Chemical Engineering, School of Science(shenzhen), Harbin Institute of Technology, Harbin 150001, China. E-mail: songlijuan@hit.edu.cn; lshi@hit.edu.cn
First published on 28th June 2024
Abstract
Recognition of the intermediacy and regulation of reactivity patterns of radical intermediates in radical chemistry have profound impacts on harnessing and developing the full potential of open-shell species in synthetic settings. In this work, the possibility of in situ formation of O/N–X intermediates from Brønsted base covalently tethered carbonyl hypohalites (BCTCs) for the generation of heteroatom-centered radicals has certainly been excluded by NMR experiments and density functional theory calculations. Instead, the spectroscopic analyses reveal that the BCTCs serve as precursors of tether-tunable distonic radical anions (TDRAs) which have been unequivocally substantiated to be involved in the direct cleavage of O/N–H bonds to generate the corresponding heteroatom-centered radicals. Meanwhile, a deep insight into the properties and reactivities of the resulting TDRAs indicates that the introduction of a tethered Brønsted base on the parent open-shell species reinforces their stabilities and leads to a reversal of electrophilicity. Moreover, the dual descriptor values and electrophilicity indices are calculated based on eleven reported radical reactions involving various electrophilic/nucleophilic radical species, further confirming their validity in the prediction of the polar effect and the polarity-matching consistency between nucleophilic TDRAs and protic O/N–H bonds. The additional halogen-free experiments mediated by the combination of phthaloyl peroxide and TEMPO also prove the feasibility of the TDRA-assisted philicity-regulation approach. Lastly, detailed intrinsic bond orbital (IBO) and Hirschfeld spin population analyses are employed to elucidate that the H-atom abstraction processes are the polarity-matching proton-coupled electron transfer (PCET) pathways, with a degree of oxidative asynchronicity.
Introduction
Heteroatom-centered radicals (E˙, E = O, N) are crucial chemical entities extensively involved in organic synthesis,1,2 materials science3 and biology.4 The distinct characteristics of these open-shell species such as the existence of unpaired electrons, the high electronegativity of heteroatoms, and diverse hybridizations impart them with versatilities, reactivities and selectivities.5 Despite their power and influence, the synthetic utilities of heteroatom-centered radicals are still less explored than those of carbon radicals owing to the shortage of convenient access to their efficient generation and the lack of awareness of their reactivity patterns and properties.1,6Generally, such reactive radical species (Fig. S4 and S5†) can be accessed by the cleavage of relatively weak E–E, E–S, and E–X (X = F, Cl, Br, I) bonds from pre-activated substrates through mechanically different processes including: (i) homolytic cleavage of an in situ generated weak E–X bond,7 (ii) mesolytic bond cleavage via single electron transfer (SET),8,9 and (iii) homolytic bond cleavage via energy transfer (EnT)10,11 (Fig. 1a). Notably, direct cleavage of the E–H bonds represents another nontrivial activation mode with a higher step and atom economy. By virtue of ligand-to-metal charge transfer (LMCT),2,12,13 hydrogen atom transfer (HAT)14,15 or proton-coupled electron transfer (PCET),16,17 an array of heteroatom-centered radicals could be accessed from common closed-shell E–H containing compounds without any pre-functionalization (Fig. 1b left). However, the pinpoint controllable transfers of the proton and electron, which are closely correlated with direct generation of E˙ from E–H containing compounds, have not been well-addressed due to a series of intrinsic restrictions. For instance, an interdependent and inverse correlation of the acidity constant (pKa) and E° presents the challenge of structural modification for more potent HAT reagents.18 In contrast, a collaboration of an independent electron acceptor and a Brønsted base in PCET overcomes the extrinsic thermodynamic limitations, while termolecular systems (substrate, base, and oxidant) are intrinsically disfavored on entropic grounds.19 To circumvent these obstacles, many researches started to utilize tethering strategies enlightened by natural biological precedents such as the covalent integration of oxidation centers and intramolecular Brønsted base scaffolds (e.g., the carboxylate anion),20 typically represented by heme-propionate controlling the redox potential of heme upon interaction with ascorbate (Fig. 1b right and Fig. S3†).
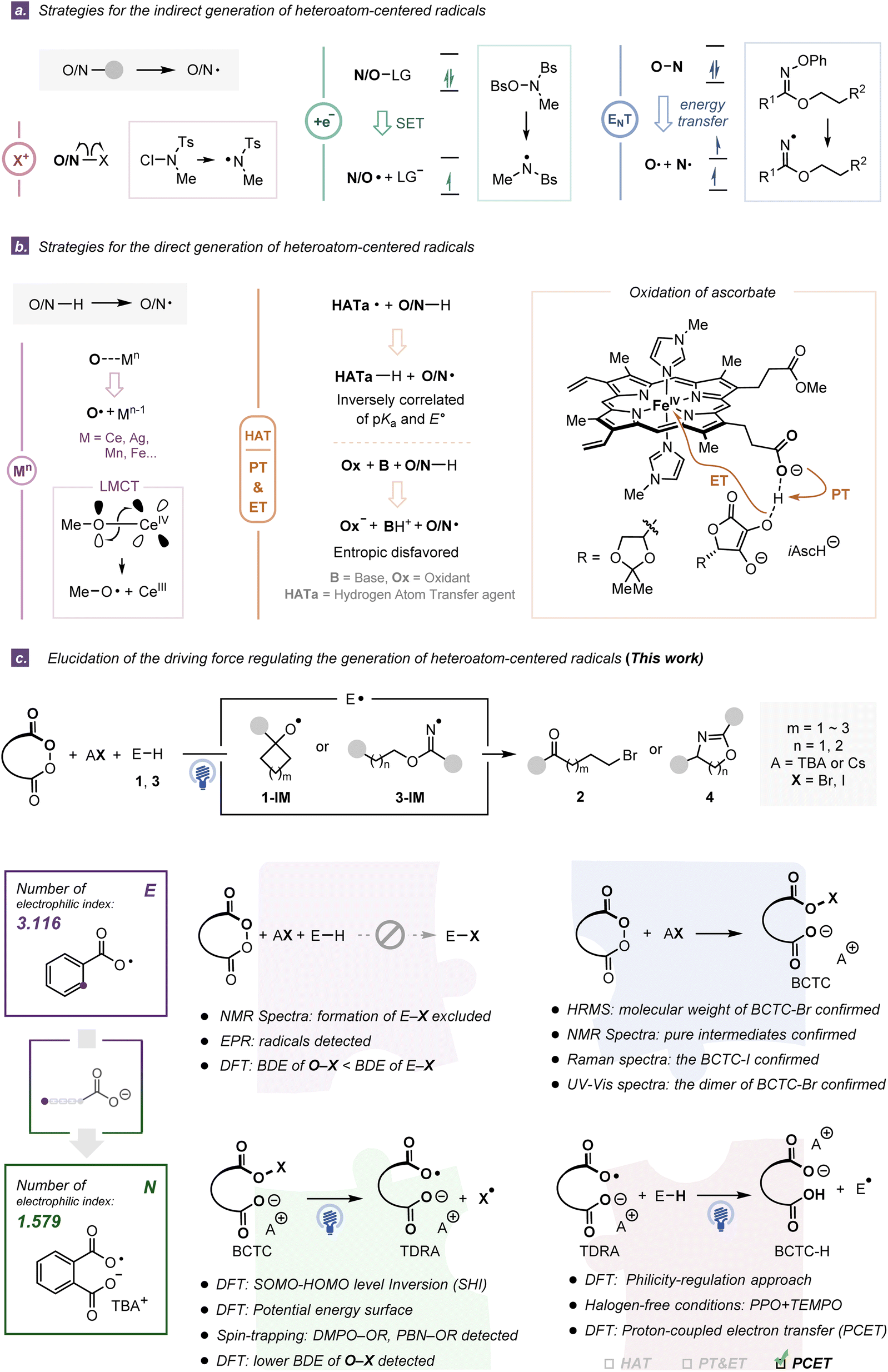 | ||
| Fig. 1 (a) Strategies for the indirect generation of heteroatom-centered radicals. (b) Strategies for the direct generation of heteroatom-centered radicals. (c) Critical experimental and theoretical studies for the TDRA-mediated generation of heteroatom-centered radicals through the philicity-regulation approach and PCET step. | ||
Different types of heteroatom-centered radicals have distinct configurations and substituents, which influences their electronic characters and radical philicities.2,21 For example, alkoxy radicals exhibit electrophilic behaviors and high reactivities, due to the unpaired electron at more electronegative oxygen atoms and the dearth of spin delocalization.2 Different substituents on oxygen-centered radicals could increase or reduce the electron deficiency at the corresponding radical center. Similarly, nitrogen-centered radicals, such as iminyl, aminyl, amidyl, and aminium species,21 offer diverse reactivities from nucleophilic to electrophilic. In particular, heteroatom-centered distonic radical anion species, which possess spatially separated charge and spin sites, have been documented to display orbital energy-level conversion and be more stable than their protonated or nonsubstituted counterparts.22
In our previously reported ring-opening halogenation reaction23 and C(sp3)–H amination reaction,24 the intermediacy of the tether-tunable distonic radical anion (TDRA) was proposed to facilitate the formation of heteroatom-centered radicals, primarily on the basis of radical trapping experiments and density functional theory (DFT) calculations. However, alternatively, Muñiz and Nagib suggested that the involved heteroatom-centered radicals were generated from the corresponding in situ formed E–X compounds.7,25 Indeed, the inevitable participation of the halogen source led to an uncertain mechanism, that is, the generation of heteroatom-centered radicals may occur through the following two pathways: (1) homolytic cleavage of weak E–X bonds of in situ generated stoichiometric 1-X or 3-X and (2) direct cleavage of strong E–H bonds. In this report, we present a series of experimental and computational pieces of evidence, including (i) NMR analyses and potential energy surface calculations to exclude the in situ formation of the E–X (X = Br, I) compounds; (ii) investigations on identifying the structures of Brønsted base covalently tethered carbonyl hypohalites (BCTCs) and the properties and reactivities of the resulting TDRAs; (iii) rigorous investigation of the radical philicity and polar effect to evaluate our two TDRA-mediated protocols together with eleven other reported radical reactions, and the philicity-regulation strategy in two halogen-free HAA processes, and (iv) intrinsic bond orbital (IBO) and the Hirschfeld spin population analyses for the revelation of a polarity-matching PCET process in an oxidatively asynchronous manner (Fig. 1c). These results not only reveal the activation mode of TDRA-induced heteroatom-centered radical generations but also provide a practical and efficient philicity-regulation approach for parent heteratom-centered radicals by introducing a suitable anionic fragment.
Results and discussion
Investigation on the possibility of in situ formation of O–Br or N–I compounds as heteroatom-centered radical precursors
While Singleton and co-workers recently proposed a hypobromite radical chain mechanism for BCTC-1-induced ring-opening bromination of 1-methylcyclobutanol,26 we deemed that evaluating the possibility of in situ formation of the E–X (X = Br, I) containing species would be a good starting point for the mechanistic investigation. E–X cleavage is one of the most common indirect methods to access heteroatom-centered radicals through thermolysis and UV photolysis or with radical initiators.27,28 In this regard, E–halogenated derivatives can be easily synthesized from native E–H containing compounds using common halogenating reagents, and they are also formed in situ owing to their innate instability.29,30 In general, structural elucidation by NMR spectroscopy can provide convincing evidence for the in situ formation of E–halogenated species.31 To distinguish the difference between the above two possible reaction pathways (homolytic cleavage of E–X bonds or direct cleavage of E–H bonds), the first critical question is whether the in situ formation of the E–halogenated species could be detected by NMR spectroscopy of the reaction mixture with appropriate standards.Initially, the corresponding hypobromites were prepared from 1-phenylcyclohexanol (1b) and 1-methylcyclohexanol (1d) in the presence of Br2 and HgO in the dark,32 and their identities were further confirmed by NMR spectroscopy. For convenient comparison, the strong carbon resonates in the 35–36 ppm region for O–Br derivatives were employed as decisive signals for the existence of corresponding hypobromites in the ring-opening halogenation (Fig. 2a). With the assessment criteria in hand, the in situ13C NMR experiments of 1b and 1d were performed using the combination of phthaloyl peroxide (PPO) and n-tetrabutylammonium bromide (TBAB) in 1,2-dichloroethane (DCE) at ambient temperature. After reacting in the dark for 70 min, a detectable trace amount of O–Br species could be observed in the in situ13C NMR spectroscopy, while most cycloalkanol substrates were left and only a trace amount of ring-opening products could be isolated. In contrast, after irradiation with blue LEDs for 40 min, no O–Br species were detected in the in situ NMR experiments, while the corresponding ring-opening products from 1b and 1d were isolated in 73% and 86% yield, respectively (Fig. S7 and S8†). Meanwhile, although we failed to prepare the hypobromite intermediate 1a-Br due to high instability, the obviously low synthetic efficiency was observed when 1a was subjected to the combination of PPO and TBAB in the dark at the same and even longer reaction time (Table S5†). Analogously, in situ1H NMR experiments were conducted to investigate the formation of N–I compounds in the C(sp3)–H amination of 3a and 3b by using the combination of malonoyl peroxides (MPO) and CsI in MeCN. As a result, no oxazoline product or N-iodo species was observed with only imidate substrates recovered under the dark conditions for·2 h, while no labile N-iodo species was detected by 1H NMR analysis under blue LED irradiation for 30 min (Fig. 2b and S12–S16†).31 Taken in concert, the above observations reveal that the E–X species are not indeed formed for the generation of heteroatom-centered radicals under our standard conditions.
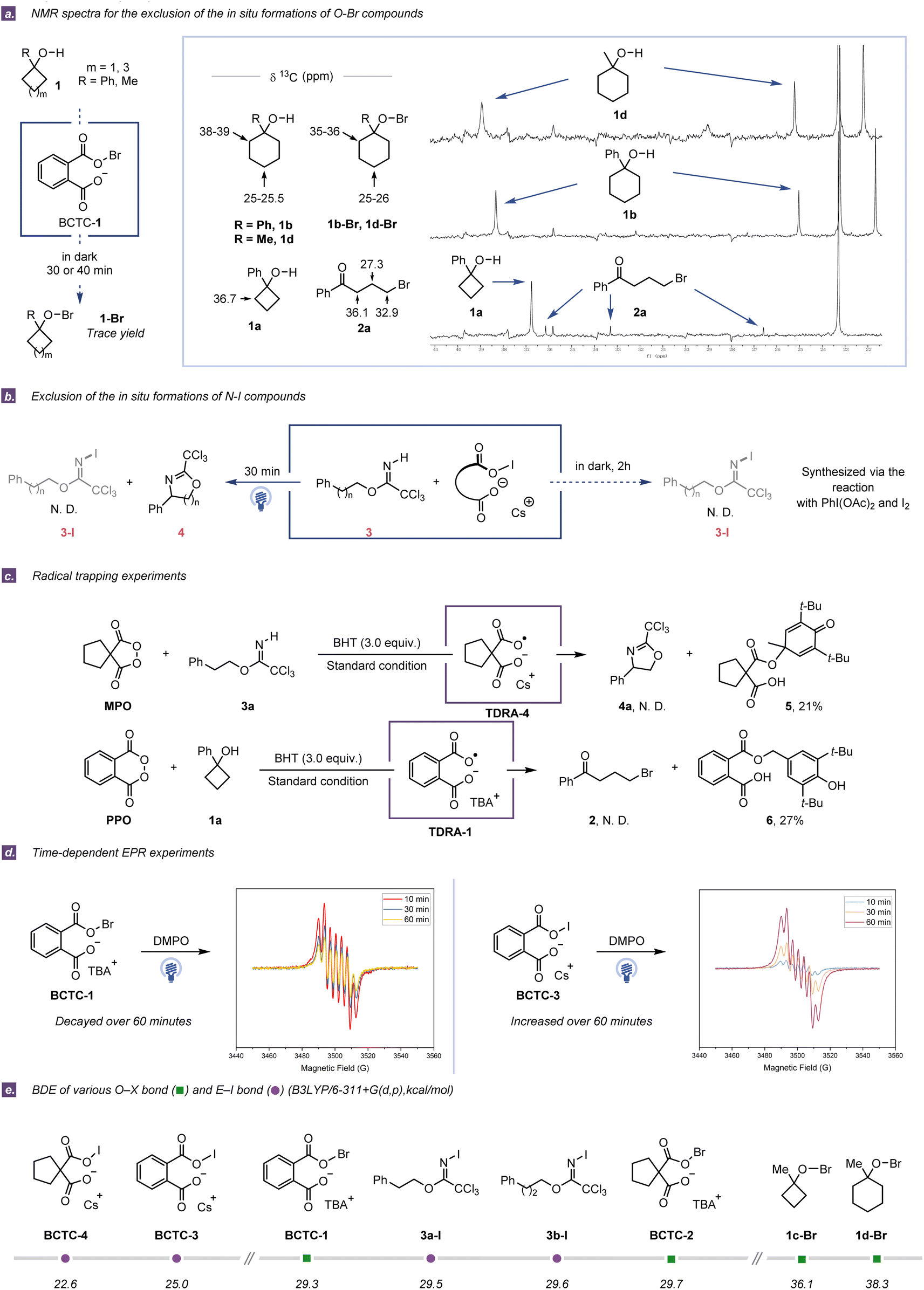 | ||
| Fig. 2 (a) NMR spectra for the exclusion of the in situ formation of O–Br compounds. (b) Exclusion of the in situ formation of N–I compounds. (c) Radical trapping experiments. (d) EPR spectra of the spin-trapped radical time-dependent on the pronounced irradiation time. (e) The E–X (E = O, N and X = Br, I) bond dissociation energies (BDEs) for BCTCs and the supposed O–Br/N–I intermediates formed from cycloalkanols and imidates. N.D. = not detected. | ||
Subsequently, a series of radical trapping experiments were carried out. The addition of 2,2,6,6-tetramethylpiperidinooxy (TEMPO) or butylated hydroxytoluene (BHT) completely inhibited either ring-opening halogenation or C(sp3)–H amination (Tables S1 and S2†). Interestingly, two different dicarboxylic radical anion adducts with BHT were successfully isolated in the presence or absence of starting materials, strongly implying that the in situ generation of TDRAs was reasonable (Fig. 2c and Tables S3 and S4†). To further assess the generation of TDRAs, electron paramagnetic resonance (EPR) experiments were conducted, wherein 5,5-dimethyl-1-pyrroline N-oxide (DMPO) or N-tert-butyl-α-phenylnitrone (PBN) was used as the spin-trapping agent. EPR spectra of PPO/MPO with or without TBAB/CsI showed distinct signals at g ∼ 2.0, which certified the generation of new radical species before the addition of E–H containing compounds (Section S4.5†).33,34 Additionally, the intensity of the DMPO-adduct signal was found to be irradiation-time-dependent (Fig. 2d), further suggesting that the new radical species was generated from the gradual photoinduced process of the combination of cyclic diacyl peroxides with halide salts. The signal intensity decreasing with time was observed in the measurement of the DMPO spin adduct of the PPO/TBAB system due to the fast generation and innate instability of the O–Br species. Furthermore, an increase in signal intensity was observed in the measurement of the DMPO spin adduct of the PPO/CsI system with the lapse of time, probably due to the poor solubility of CsI in MeCN. As a matter of fact, when the mixture of MPO and CsI was irradiated for 30 min before the addition of 3a, only a slight decrease in yield was observed (Table S12†). Also of note, if the mixture of PPO and TBAB was irradiated for 60 min prior to the addition of 1a, no ring-opening product was detected (Table S13†). These experimental results are consistent with the intensity changes in the EPR spectra.
On the other hand, the E–X (E = O, N and X = Br, I) bond dissociation energies (BDEs) were calculated for BCTCs and the supposed O–Br/N–I intermediates that formed from cycloalkanols and imidates. The much lower BDEs of O–halogen bonds in the BCTCs (<30.0 kcal mol−1 O–Br bonds and <25.0 kcal mol−1 O–I bonds) suggest that the homolytic cleavage of their O–halogen bonds should occur more facilely than that of proposed O–Br/N–I intermediates (>36.0 kcal mol−1 O–Br bonds and >29.0 kcal mol−1 N–I bonds) (Fig. 2e).35 Based on these experimental and computational results, it could be concluded that TDRAs could be easily generated in situ from the combined system of cyclic diacyl peroxides and halide salts with cation-dependent properties.
Identification and characterization of the Brønsted base covalently tethered carbonyl hypohalites (BCTCs)
A series of spectroscopic analyses such as NMR, UV-vis, Raman, and high-resolution mass spectroscopy (HRMS) were carried out to verify the exact structures of the BCTCs. The 1H and 13C NMR experiments by mixing PPO/MPO and halide salts indicated that two separate aromatic/cycloalkyl peaks were clearly labelled, due to the symmetrical structures of dimeric or even oligomeric BCTCs (Section S5.1†). In contrast, four separate aromatic peaks were observed and no such dimer was formed in the benzoyl peroxide (BPO)/TBAB system (Section S5.1†). The UV-vis spectra of the PPO/TBAB and MPO/TBAB systems in CCl4 exhibited absorption features at 266 nm (ε = 748.4 M−1 cm−1) and 273 nm (ε = 792.4 M−1 cm−1) respectively (Fig. 3a) and did not show the broad features between 400 and 500 nm, which completely excluded the initial formation of Br2 (λmax = 415 nm) (Section S5.3†).36 The clearly decreased absorption intensity and slight blue-shift were observed in the UV-vis spectrum when the PPO/TBAB system was illuminated for 60 min, thus revealing again that the key active species was unstable and photosensitive (Fig. S25†). Furthermore, the Raman measurements of several cyclic diacyl peroxide/iodized salt systems were conducted in MeCN (Fig. 3b and Section S5.4†). The band near 168 cm−1 was assigned to BCTCs and the band at 110 cm−1 was characteristic of I3−.37 By comparing the computational and experimental UV-vis and Raman spectra, the dimer of carboxylate-tethered acyl hypohalites was deemed to be unstable active species (Section S7.2 and S7.3†). Moreover, the consistency between the structure and the molecular weight obtained by HRMS analysis further proves the existence of BCTC-1 (Section S5.2†). The operative role of tribromide or triiodide as possible active species (Fig. S26 and S28–S30†) has also been ruled out,24 although it might be observed in solution.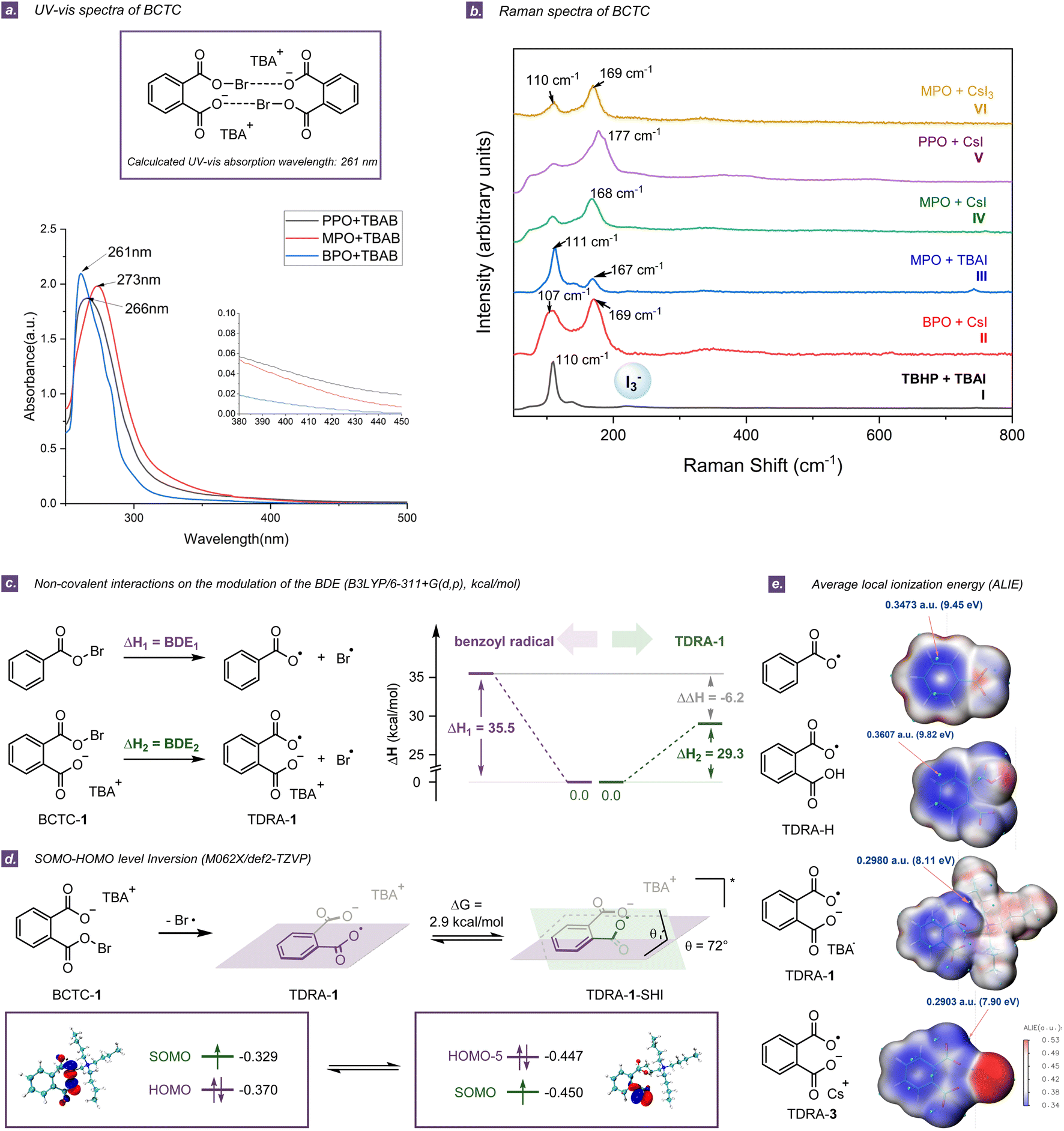 | ||
| Fig. 3 (a) UV-vis spectra of BCTCs. (b) Raman spectra of BCTCs and other species. (c) Non-covalent interactions on the modulation of the BDE. (d) SOMO–HOMO energy-level inversion (SHI) for TDRA-1. (e) ALIE to evaluate the intrinsic chemical reactivity of TDRAs. | ||
Properties of the tether-tunable distonic radical anions (TDRAs)
To explore the inherent chemical properties of structurally unique TDRAs, we conducted a comprehensive comparison with common acyloxy radicals. In contrast to the standard conditions for ring-opening halogenation and C(sp3)–H amination, rather low yields were obtained by utilizing acyclic benzoyl peroxides and other open-chain peroxides instead of cyclic diacyl peroxides (Tables S7 and S8†), indicating that the two respective carboxyl groups in TDRAs work together and the cooperation of intramolecular Brønsted base brought by the cyclic peroxides plays a key role in these transformations.For a better comprehension of differences in reactivity and properties between dicarboxylic radical anions and acyloxy radicals, it is important to understand the influence of the presence of COO−A+ (A = TBA or Cs) on the modulation of the BDEs of the acyloxy O–X (X = Br, I), which in turn affects the stability of the acyl hypohalites or the acyloxy radical (Section S7.5†).38 It is found that the BDE of the O–Br bond in BCTC-1 (BDEPc = 29.3 kcal mol−1) is much lower than that of acyl hypobromite substituted by COOH or without substitution (BDEPb = 36.4 kcal mol−1 and BDEPa = 35.5 kcal mol−1), implying that the presence of COO−A+ is critical for the stability of the resulting TDRAs (Fig. 3c and S37, S38†). This difference in BDE is even larger in the system of aliphatic acyl hypobromite (BDEMc = 29.7 kcal mol−1, BDEMb = 37.3 kcal mol−1, and BDEMa = 39.2 kcal mol−1). The same trend is found in the case of substituted acyl hypoiodite (Fig. S37 and S38†). Overall, the tethered carboxylate anion substituents contribute to the preferential stabilization of TDRAs. Considering that the photochemical homolysis of the O–X bond of benzoyl hypohalite (derived from BPO) might require higher energy light due to the higher BDE (>33.0 kcal mol−1), illumination with light of wavelength 300 nm was chosen to perform ring-opening halogenation and C(sp3)–H amination in the BPO/TBAB or BPO/CsI system, to compare the reactivity of the O-radicals generated by the processes. As a result, the quite high energy light did not bring significant improvement in yields (29% vs. 20%, and 40% vs. 31% respectively, Section S5.6†), suggesting that the efficiency of generating acyloxy radicals may not be a restrictive factor for smooth transformations.
Similar to other distonic radical anions,22 a SOMO (singly occupied molecular orbital)–HOMO (highest occupied molecular orbital) energy-level inversion (SHI) was observed for TDRA-1, showing that the SOMO in TDRA-1 is located on the oxygen atom of the COO moiety and the corresponding energy level is lower than HOMO−5 (Fig. 3d). This SHI is accompanied by tuning the tethered carboxylic radical and carboxylate anion moieties from a planar to a non-planar conformation with an energy difference of 2.9 kcal mol−1. In most reported distonic radical anions, the orbital conversion with lowered SOMO energy largely contributes to the stability of the radical anion intermediate.22 The stabilization is largest when a highly delocalized radical (e.g., TDRA-1) is combined with the deprotonation of an acid–base group (e.g., COO−).39 However, the TDRA-2 does not display SHI, because the stabilization is negligible for highly localized radicals (Section S7.6†).39
The calculations of the electrostatic potential (ESP)40 and average local ionization energy (ALIE)41 were carried out to evaluate the intrinsic chemical reactivities of the TDRAs. The ESP mapped van der Waals surface shows a negative region (minima) located at the carboxylic radical, indicating a preferable site for electrophilic attack.40 It is noticeable that the minimal value decreased significantly when a carboxylate anion was attached to the radical species (Table S16†). It is generally recognized that the ALIE could reflect the ionization energy of an electron in a local region.41 As the ALIE decreases, the weaker the electron binding, the higher the reactivity of the electrons. As shown in Fig. 3e and Table S17,† the electrophilic reaction site is located at the benzene moiety in the benzoyl radical or TDRA-H. However, once it is tailed with the carboxylate anion, the result differs to some extent. In TDRA-1/3 and TDRA-2/4, the electrophilic reaction site would move to the remote carboxylic radical moiety. Both the ESP and ALIE analyses suggest that the carboxylate-tethered distonic radical anions are nucleophilic (Fig. 3e).42
Based on the aforementioned findings, the potential energy profiles of the ring-opening halogenation of cycloalkanols and C(sp3)–H amination of imidates were thoroughly re-evaluated to examine the rationality and feasibility of TDRA-mediated generation of the heteroatom-centered radicals. PPO initially reacts with tetramethylammonium bromide (TMAB) (simplified model for TBAB) to generate BCTC-1, with a free energy barrier of 12.6 kcal mol−1. It is found that mono BCTC-1 is easily converted into a more stable dimer, with a free energy of −24.7 kcal mol−1. This is also supported by the computational analysis of UV-vis and Raman spectra, as well as the occurrence of two identical NMR shifts (Fig. S31, Table S14 and Section S5.1†). The O–Br bond of the dimer would then undergo homolytic cleavage under the blue LEDs, producing TDRA-1 and a bromine radical. Subsequently, the hydrogen atom of the O–H bond is abstracted from the six-membered cycloalkanol to TDRA-1, with an energy barrier of 19.3 kcal mol−1. Nevertheless, a higher energy barrier of 43.0 kcal mol−1 is required for the in situ generation of the E–X intermediate. Similar trends are found for the β- and γ-aminations of imidates. Hence, both computational studies and the above experimental results could rule out the possibility of the polar pathway for in situ generation of E–X intermediates (Section S7.4†).
Assessment of radical philicity and the polar effect as well as radical-regulation for hydrogen-atom abstraction
Following the initial findings (from ESP and ALIE) that TDRA species are nucleophilic, we thoroughly examined the electronic characteristics of TDRAs and some common radical species by utilizing the electrophilicity index (ω)43 and nucleophilicity index (NNu)44 (Fig. 4a). The computational results indicate that the benzoyloxy radical and acetoxy radical are typical electrophilic radicals with a high electrophilicity index (ω: 3.116 eV and 2.807 eV, respectively), while alkoxyl 1a-IM (ω: 2.056 eV) and iminyl radicals 3a-IM/3b-IM show weak to moderate electrophilicity (ω: 2.121 eV and 2.117 eV, respectively).45 According to the definition of the polar effect,46,47 the HAA of the protic O–H bond of 1a or N–H bond of 3a/3b with an electrophilic carboxylic radical is unfavorable due to the polarity mismatch. By covalently integrating a carboxylate anion, typical electrophilic carboxylic radicals are changed to weak nucleophilic dicarboxylic radical anions via a philicity-regulation strategy (ωω: 1.579 eV for TDRA-1 and 1.698 eV for TDRA-2, respectively).48–50 We also calculated more indices (e.g. condensed local electrophilicity/nucleophilicity index (ωre+/Nre+) and cubic electrophilicity index (ωcubic)) (Table S16†).51 All the indices suggest that the TDRAs are nucleophilic species and preferred attacking protic hydrogen atoms via polarity-matching HAA processes. Consequently, a philicity-regulation strategy is crucial to allow our two TDRA-mediated protocols to occur.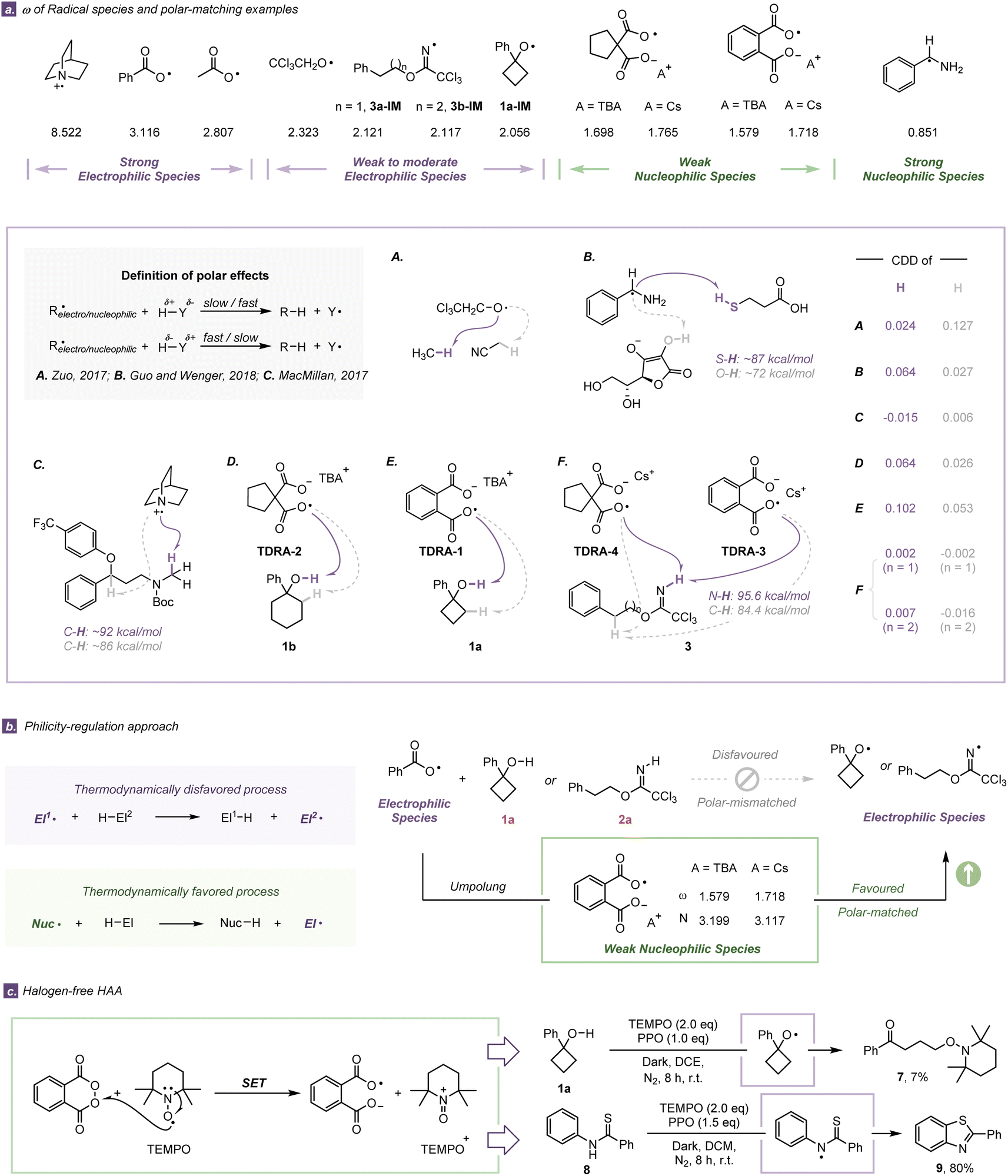 | ||
| Fig. 4 (a) ω of radical species and polarity-matching examples. (b) Typical electrophilic carboxylic radicals were changed to weak nucleophilic dicarboxylic radical anions via philicity-regulation. (c) Halogen-free HAA reactions via philicity-regulation approaches. | ||
In order to gain a better understanding of the polar effect, we investigated the selected HAA processes of another eleven related radical reactions (Fig. 4a and S41†). Herein, we employed dual descriptor values52 to evaluate the polarity of the hydrogen atom and the electrophilicity index to determine the electronic properties of open-shell radicals. This evaluation rule has proven successful in these eleven reported examples, in which electrophilic radicals prefer to attack the hydridic C–H (showing less positive Fukui function values) and vice versa.13,53,54 For example, the electrophilic radical Cl3CH2CO˙ prefers to attack the more hydridic hydrogen atom of methane (condensed dual descriptor (CDD) = 0.024) rather than that of acetonitrile.13 Analogically, the nucleophilic radical PhCH·NH2 tends to react with the more protic hydrogen atom of the S–H bond, even though its BDE is larger than that of the O–H bond of ascorbate.53 In our cases, the nucleophilic TDRAs would abstract the relative protic hydrogen atom of E–H bonds (CDD = 0.064, 0.102 for O–H bonds and 0.002, 0.007 for N–H bonds) rather than the hydridic hydrogen atom of the C–H bond (CDD = 0.027, 0.026 for cycloalkanols and −0.002, −0.016 for imidates). The consistency is astonishingly good between our calculation and experiment results, which demonstrates that the cooperation of dual descriptor values and the electrophilicity index is a useful tool for assessing the polar effect and explaining the chemo-selectivity during HAA processes.
To further verify the effectiveness of the philicity-regulation approach, halogen-free HAA reactions were designed and carried out. TEMPO is a well-known and thermodynamically stable nitroxyl radical, which easily undergoes a one-electron redox process.55 We hypothesized that one-electron oxidation of TEMPO with PPO might afford the active TDRA oxoammonium species even under dark conditions. It was found that the combination of 2 equivalents of TEMPO and 1 equivalent of PPO under dark conditions could dominate the HAA process and be followed by the ring-opening process. The ring-opening product 7 was obtained via C–O bond coupling with an albeit low yield (7%) (Fig. 4c). Analogously, we carried out the cyclization of N-phenylbenzothioamide 8 under the guidance of the philicity-regulation approach in which the cyclization product 9 was isolated in a good yield (80%) (Fig. 4c), using 2 equivalents of TEMPO and 1.5 equivalents of PPO under dark conditions. Control experiments prove that no desired product is isolated without the participation of PPO or TEMPO.56 These results under halogen-free conditions represent strong experimental evidence for demonstrating successful execution of the TDRA-assisted philicity-regulation approach, in which large polarity differences lead to lower barriers.
Intrinsic nature of proton and electron transfer processes
After identifying the E–H activation mediated by TDRAs, we employed the intrinsic bond orbital (IBO) analysis to elucidate the proton and electron transfer processes. IBO analyses allow us to analyze the electron transformation in the electronic structure by following changes in the localized IBO for the α and β spin manifolds separately in an open-shell system, providing an intuitive picture throughout a chemical transformation along an intrinsic reaction coordinate (IRC).57–60 Notably, this approach is particularly useful in dissecting the nuances between HAT and PCET in the context of HAA reactions. Herein, Fig. 5 shows illustrative examples of the evolutions of IBOs for α- and β-electrons from the σ O–H bond and lone pair of oxygen during the cleavage of O–H bonds in the six-membered cycloalkanol 1b along the IRC (calculations were carried out at the same level of theory as optimization, for up to 196 steps). The evolution of the α-IBO (green lobe) of the σ O–H bond is illustrated in Fig. 5d, and the corresponding β-IBO (orange lobe) follows almost the same evolution in Fig. 5e. Since both the α- and β-IBOs of the σ O–H bond remain on the alkoxy substrate throughout the hydrogen abstraction process, the σ O–H bond is only involved in the proton transfers and electron transfer should occur on another IBO to accomplish the overall HAA, reflecting the concerted nature of the reaction. As shown in Fig. 5g (purple lobe), in the transition state (TS), it is the β-IBO of the oxygen lone pair in the six-membered cycloalkanol that evolves into the oxygen p-orbital of the remote carboxylic radical moiety in TDRA-1, while the proton is transferred to the carboxylate moiety in TDRA-1. As the proton and electron transfers involve distinct donor/acceptor centers, this process could be most aptly described as a PCET pathway, as shown by the curly arrows in Fig. 5a. A similar scenario for the ring-opening halogenation of four- and five-membered cycloalkanol reactions is presented, i.e., both α- and β-IBOs of the σ O–H bond remain on the cycloalkanols and the β-IBO of the oxygen lone pair in the cycloalkanols transits into the oxygen p-orbital of the remote carboxylic radical moiety in TDRA-1 (Fig. S42 and S43†).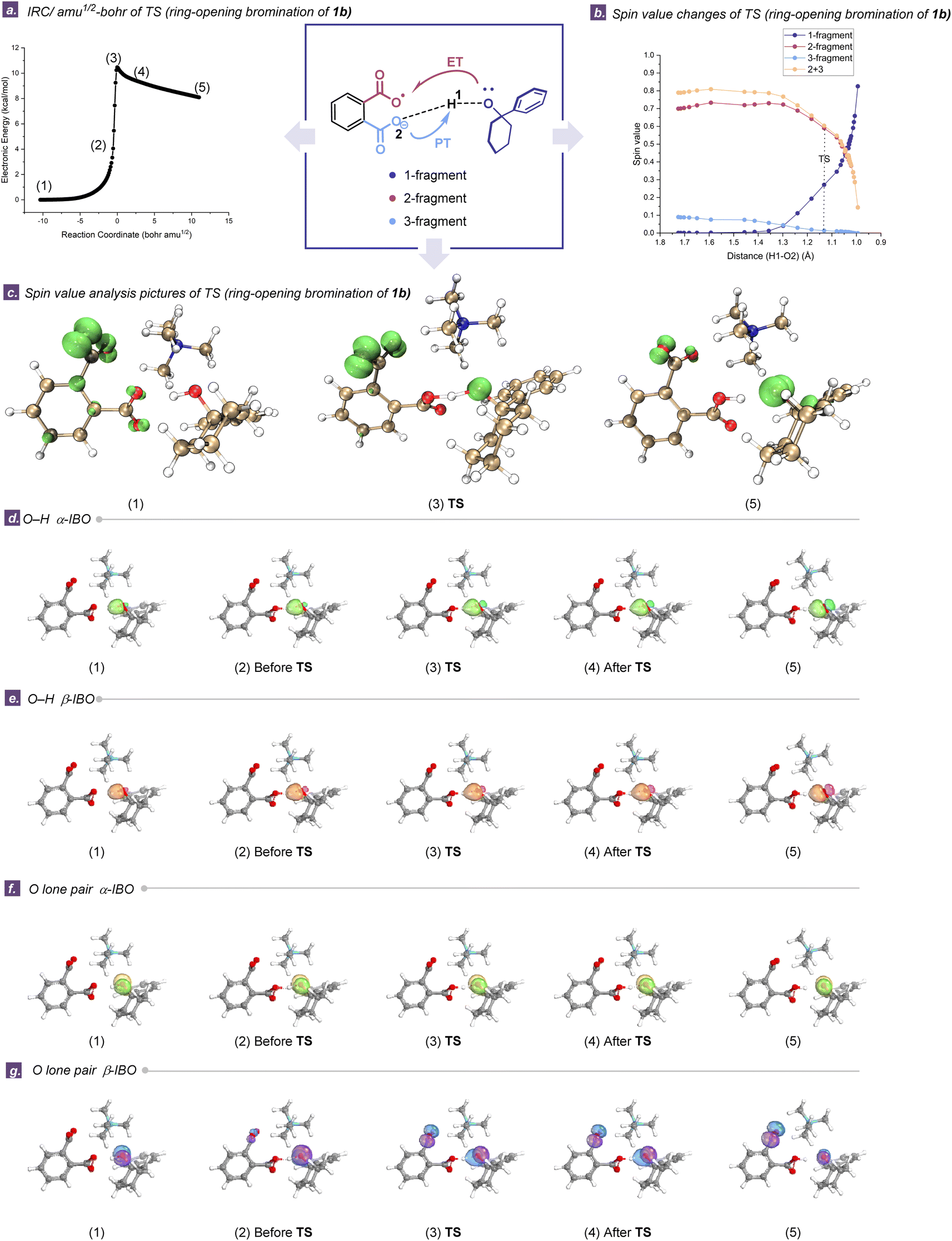 | ||
| Fig. 5 (a) Plots of the change in electronic energies (computed at UB3LYP-D3(BJ)/6-31+G(d)) along the intrinsic reaction coordinate (IRC amu−1/2 bohr−1) for the ring-opening halogenation of six-membered cycloalkanols. (b) Changes in the Hirschfeld spin-population for the ring-opening halogenation of the six-membered cycloalkanols. (c) Spin value analysis pictures of open-shell singlet transition states. (d) The evolution of the α-IBO (green lobe) of the σ O–H bond. (e) The evolution of the β-IBO (orange lobe) of the σ O–H bond. (f) The evolution of the α-IBO (yellow lobe) of the oxygen lone pair. (g) The evolution of the β-IBO (purple lobe) of the oxygen lone pair. | ||
In addition, the evolutions of the IBOs for the σ N–H bond of the imidate 3a, engaging in bonding interactions with TDRA-4 along the IRC path, were scrutinized. In this case, the β-IBO (Fig. S45,† blue lobe) undergoes a transformation from an N–H bond in imidate 3a to an oxygen p-orbital of the remote carboxylic radical moiety and the α-IBO (Fig. S45,† orange lobe) evolves from an N–H bond to an N-radical, while the proton is transferred to the carboxylate moiety in TDRA-4. The overall electron flow indicates that the reaction between TDRA-3 and imidate 3b can also be described as a PCET mechanism (Fig. S46†). These analyses lead to the conclusion that TDRAs abstract a hydrogen atom from cycloalkanols or imidates through a PCET pathway.
Comparative analyses of the PCET mechanisms in TDRAs/cycloalkanols and TDRAs/imidates are provided in Fig. 5b, c and S47–S56.† The plots illustrate the time evolutions of the Hirschfeld spin population along the TDRA-induced HAA processes, which is consistent with the bond length changes of the key transition states.61,62 In the TDRA mediated ring opening of the six-membered cycloalkanol, the spin-density on the 1-fragment begins to increase (and that of the remote carboxylic radical moiety, 2-fragment, starts to decrease) well before the TS, implying an early transfer of electrons from the substrate (Fig. 5b). Additionally, the spin orbitals of the transition states (3), initial state (1), and final state (5) are demonstrated in Fig. 5c.63 The single electron is predominantly distributed in the carboxylic radical (2-fragment) at the initial state and oxygen of the cycloalkanol at the final state. These calculated results indicate that the SET between the carboxylic radical of TDRAs and cycloalkanol occurs concomitantly with proton transfer at distinct sites, providing robust confirmation of the PCET pathway. In addition, the evaluation of the BDFEs offer further proofs that the PCET processes are actually involved in the two TDRA-mediated transformations (Section S9.5†).64
Within the framework of a PCET process, there is a variable degree of asynchronicity in the transfer of hydrogen ions (H+) and electrons (e−), which could exert a noteworthy influence on the overall reaction rate. Therefore, the determination of the asynchronicity factor (η) is an effective method for such quantification,47 delineating the relative impacts of two competing factors governing the HAA process: the pKa and the redox potential. In scenarios where acidobasic contributions hold greater significance, a negative value for the asynchronicity factor (η) is anticipated.47 Conversely, a positive value of η shows a prevailing influence of the redox potential playing a more dominant role. In this case, our two TDRA-mediated protocols display a more electron transfer (ET)-like character to be oxidatively asynchronous as we obtained a positive value for η (+123, +180 and +229 mV for the TDRA-induced ring-opening halogenation reaction and +64 and +381 mV for the C(sp3)–H amination reaction, respectively; Section S9.4†).
Conclusions
In summary, we have implemented comprehensive experimental and theoretical investigations to elucidate the mechanism of TDRA-mediated generation of heteroatom-centered radicals. The results from in situ NMR experiments and DFT calculation have excluded the possibility that the E–X species are formed for the generation of heteroatom-centered radicals. Instead, a series of radical trapping experiments and EPR experiments together with assessment of BDEs have clearly revealed the probability and rationality of in situ formation of TDRAs under our original conditions. Additional diverse spectroscopic analyses have verified the symmetrical dimeric or even oligomeric structures of the BCTCs, which suggests the intermediacy of TDRAs. Further investigations focusing on the chemical properties and reactivities of the TDRAs have revealed that the introduction of a tethered Brønsted base on the parent open-shell species reinforces their stabilities and leads to a reversal of electrophilicity, and this in turn predominantly induces philicity-regulation, thus leading to its distinguished reactivities in heteroatom-centered radical involved reactions. Furthermore, dual descriptor values and electrophilicity indices have been calculated based on eleven reported radical mediated selected HAA processes, indicating their validity in the prediction of the polar effect and in the polarity-matching consistency between nucleophilic TDRAs and protic O/N–H bonds. Additionally, two halogen-free HAA reactions have been also designed and carried out to illustrate the effectiveness of the TDRA-assisted philicity-regulation approach. Ultimately, the IBO analyses, Hirschfeld spin population analyses and asynchronicity factor analyses have enabled us to discern the proton and electron transfer modes during HAA processes, the results of which suggest the PCET pathway with a degree of oxidative asynchronicity. We hope that these exhaustive and substantial studies could not only help to directly resolve generation of heteroatom-centered radicals in TDRA-mediated transformations but also facilitate quantitative understanding and controlled modulation of structurally diversified radical species.Data availability
The data supporting the findings of this study are available within the article and its ESI.†Author contributions
K. F., X. Y. and L. S. conceived the project and designed the experiments. K. F. and X. Y. performed the experiments and analyzed the data. K. F. and L. J. S. performed computational chemistry. All the authors wrote the manuscript. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (22271069, 22203023, and 21871067), the Guangdong Basic and Applied Basic Research Foundation (2023A1515012457, 2022A1515011859, and 2021A1515010190), the Shenzhen Science and Technology Program (GXWD20231130100539001), the Fundamental Research Funds for the Central Universities (HIT.OCET.2021035), the Open Project Program of State Key Laboratory of Elemento-Organic Chemistry (202009), and the Shenzhen Government for a Talent Development Starting Fund. This work was also supported by the Shenzhen Bay Laboratory. We thank Prof. Min Wang (Hangzhou Normal University) for assistance in the measurement of HRMS spectra.Notes and references
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC.
- L. Chang, Q. An, L. Duan, K. Feng and Z. W. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed.
- C. Chatgilialoglu and A. Studer, Encyclopedia of Radicals in Chemistry, Biology and Materials, Wiley-Scrivener, 2012 Search PubMed.
- S. C. Von, Free-Radical-Induced DNA Damage and Its Repair, Springer Berlin Heidelberg, 2006 Search PubMed.
- A. F. Parsons, An introduction to free radical chemistry, Wiley-Blackwell, 2000 Search PubMed.
- D. P. Curran, N. A. Porter and B. Giese, Radical Reactions in Organic Synthesis, John Wiley & Sons, Ltd, 2007 Search PubMed.
- C. Martínez and K. Muñiz, Angew. Chem., Int. Ed., 2015, 54, 8287–8291 CrossRef PubMed.
- Q. Qin and S. Yu, Org. Lett., 2014, 16, 3504–3507 CrossRef CAS PubMed.
- J. Davies, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2017, 56, 13361–13365 CrossRef CAS PubMed.
- S. Dutta, J. E. Erchinger, F. Strieth-Kalthoff, R. Kleinmans and F. Glorius, Chem. Soc. Rev., 2024, 53, 1068–1089 RSC.
- F. Paulus, C. Stein, C. Heusel, T. J. Stoffels, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2023, 145, 23814–23823 CrossRef CAS PubMed.
- L. Wen, J. Ding, L. Duan, S. Wang, Q. An, H. Wang and Z. W. Zuo, Science, 2023, 382, 458–464 CrossRef CAS PubMed.
- A. H. Hu, J. J. Guo, H. Pan and Z. W. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- M. Galeotti, M. Salamone and M. Bietti, Chem. Soc. Rev., 2022, 51, 2171–2223 RSC.
- P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Chem. Rev., 2022, 122, 2017–2291 CrossRef CAS PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- J. Zhang and M. Rueping, Chem. Soc. Rev., 2023, 52, 4099–4120 RSC.
- G. A. Parada, Z. K. Goldsmith, S. Kolmar, B. Rimgard, B. Q. Mercado, L. Hammarström, S. Hammes-Schiffer and J. M. Mayer, Science, 2019, 364, 471–475 CrossRef CAS PubMed.
- J. J. Warren and J. M. Mayer, J. Am. Chem. Soc., 2011, 133, 8544–8551 CrossRef CAS PubMed.
- C. Pratley, S. Fenner and J. A. Murphy, Chem. Rev., 2022, 122, 8181–8260 CrossRef CAS PubMed.
- G. Gryn’ova, D. L. Marshall, S. J. Blanksby and M. L. Coote, Nat. Chem., 2013, 5, 474–481 CrossRef PubMed.
- R. Zhao, Y. Yao, D. Zhu, D. Chang, Y. Liu and L. Shi, Org. Lett., 2018, 20, 1228–1231 CrossRef CAS PubMed.
- R. Zhao, K. Fu, Y. Fang, J. Zhou and L. Shi, Angew. Chem., Int. Ed., 2020, 59, 20682–20690 CrossRef CAS PubMed.
- E. A. Wappes, K. M. Nakafuku and D. A. Nagib, J. Am. Chem. Soc., 2017, 139, 10204–10207 CrossRef CAS PubMed.
- S. Alvi and D. A. Singleton, Org. Lett., 2021, 23, 2174–2177 CrossRef CAS PubMed.
- K. Kwon, R. T. Simons, M. Nandakumar and J. L. Roizen, Chem. Rev., 2022, 122, 2353–2428 CrossRef CAS PubMed.
- E. Tsui, H. Wang and R. R. Knowles, Chem. Sci., 2020, 11, 11124–11141 RSC.
- L. Stella, Angew. Chem., Int. Ed., 1983, 22, 337–350 CrossRef.
- C. Walling and D. Bristol, J. Org. Chem., 1972, 37, 3514–3516 CrossRef CAS.
- A. E. Bosnidou, T. Duhamel and K. Muñiz, Eur. J. Org Chem., 2020, 2020, 6361–6365 CrossRef CAS.
- V. L. Heasley, R. K. Gipe, J. L. Martin, H. C. Wiese, M. L. Oakes, D. F. Shellhamer, G. E. Heasley and B. L. Robinson, J. Org. Chem., 1983, 48, 3195–3199 CrossRef CAS.
- N. M. Atherton, Principles of electron spin resonance, Ellis Horwood, 1993 Search PubMed.
- L. Eberson and O. Persson, J. Chem. Soc., Perkin Trans. 2, 1997, 1689–1696 RSC.
- X.-S. Xue, P. Ji, B. Zhou and J.-P. Cheng, Chem. Rev., 2017, 117, 8622–8648 CrossRef CAS PubMed.
- H. China, Y. Okada and H. Ogino, J. Phys. Org. Chem., 2016, 29, 84–91 CrossRef CAS.
- M. Uyanik, H. Hayashi and K. Ishihara, Science, 2014, 345, 291–294 CrossRef CAS PubMed.
- R. Amorati and L. Valgimigli, Modulation of Biorelevant Radical Reactions by Non-Covalent Interactions, John Wiley & Sons, Inc, Hoboken, NJ, 2016, pp. 361–379 Search PubMed.
- G. Gryn’ova and M. L. Coote, J. Am. Chem. Soc., 2013, 135, 15392–15403 CrossRef PubMed.
- J. S. Murray and P. Politzer, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2011, 1, 153–163 CrossRef CAS.
- P. Politzer, J. S. Murray and F. Bulat, J. Mol. Model., 2010, 16, 1731–1742 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- R. G. Parr, L. V Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- K. Héberger and A. Lopata, J. Org. Chem., 1998, 63, 8646–8653 CrossRef.
- F. De Vleeschouwer, V. Van Speybroeck, M. Waroquier, P. Geerlings and F. De Proft, Org. Lett., 2007, 9, 2721–2724 CrossRef CAS PubMed.
- B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25–35 RSC.
- D. Bím, M. Maldonado-Domínguez, L. Rulíšek and M. Srnec, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 10287–10294 CrossRef PubMed.
- F. Parsaee, M. C. Senarathna, P. B. Kannangara, S. N. Alexander, P. D. E. Arche and E. R. Welin, Nat. Rev. Chem, 2021, 5, 486–499 CrossRef CAS PubMed.
- A. Ruffoni, R. C. Mykura, M. Bietti and D. Leonori, Nat. Synth., 2022, 1, 682–695 CrossRef.
- B. Lu, M. Xu, X. Qi, M. Jiang, W.-J. Xiao and J.-R. Chen, J. Am. Chem. Soc., 2022, 144, 14923–14935 CrossRef CAS PubMed.
- G. Hoffmann, V. Tognetti and L. Joubert, J. Phys. Chem. A, 2020, 124, 2090–2101 CrossRef CAS PubMed.
- C. Morell, A. Grand and A. Toro-Labbé, J. Phys. Chem. A, 2005, 109, 205–212 CrossRef CAS PubMed.
- X. Guo and O. S. Wenger, Angew. Chem., Int. Ed., 2018, 57, 2469–2473 CrossRef CAS PubMed.
- C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Nature, 2017, 547, 79–83 CrossRef CAS PubMed.
- J. C. Siu, G. S. Sauer, A. Saha, R. L. Macey, N. Fu, T. Chauviré, K. M. Lancaster and S. Lin, J. Am. Chem. Soc., 2018, 140, 12511–12520 CrossRef CAS PubMed.
- Z.-M. Xu, H.-X. Li, D. J. Young, D.-L. Zhu, H.-Y. Li and J.-P. Lang, Org. Lett., 2019, 21, 237–241 CrossRef CAS PubMed.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- G. Knizia and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2015, 54, 5518–5522 CrossRef CAS PubMed.
- E. M. N. Klein and G. Knizia, Angew. Chem., Int. Ed., 2018, 57, 11913–11917 CrossRef PubMed.
- M. Mandal, C. E. Elwell, C. J. Bouchey, T. J. Zerk, W. B. Tolman and C. J. Cramer, J. Am. Chem. Soc., 2019, 141, 17236–17244 CrossRef CAS PubMed.
- Q. Shi, Z. Pei, J. Song, S.-J. Li, D. Wei, M. L. Coote and Y. Lan, J. Am. Chem. Soc., 2022, 144, 3137–3145 CrossRef CAS PubMed.
- Y. Wang, W. Chen, Y. Lai and A. Duan, J. Am. Chem. Soc., 2023, 145, 23527–23532 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14(1), 33–38 CrossRef CAS PubMed.
- H. G. Yayla, H. Wang, K. T. Tarantino, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2016, 138, 10794–10797 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and methods, characterization data, NMR spectra, computational methods and energies and Cartesian coordinates. See DOI: https://doi.org/10.1039/d4sc02602k |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |