
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Self-correcting mismatches in metastable hydrogen-bonded organic frameworks with an 11-fold interpenetrated array†
Guomin
Xia‡
 bc,
Chunlei
Zhou‡
c,
Xingliang
Xiao
bc,
Yang
Yang
bc,
Fuqing
Yu
ab and
Hongming
Wang
*abc
bc,
Chunlei
Zhou‡
c,
Xingliang
Xiao
bc,
Yang
Yang
bc,
Fuqing
Yu
ab and
Hongming
Wang
*abc
aCollege of Chemistry and Chemical Engineering, Nanchang University, Nanchang 330031, China. E-mail: hongmingwang@ncu.edu.cn
bJiangxi Provincial Key Laboratory of Functional Crystalline Materials Chemistry, Nanchang 330031, China. E-mail: guominxia@ncu.edu.cn
cInstitute for Advanced Study, Nanchang University, Nanchang 330031, China
First published on 6th August 2024
Abstract
The polymorphic self-correction from a metastable phase to a stable one often occurs and plays crucial roles in synthesizing robust hydrogen-bonded organic frameworks (HOFs). However, identifying metastable phases and understanding the self-correcting mechanisms is a challenging venture due to their intrinsic instability. Here, we for the first time introduce 1,8-naphtholactam (Np) as a hydrogen-bonding synthon positioned on the periphery of a bicarbazole to create a versatile molecular unit for 3D HOFs. The as-synthesized NCU-HOF1, analyzed by single-crystal X-ray diffraction (SCXRD), is found to be metastable. It exhibits an 11-fold interpenetrated dia topology with a quarter of the Np units exhibiting monomeric N–H⋯O interactions between adjacent Np link sites, which readily self-correct upon desolvation to form fully dimeric ones. Consequently, the resultant NCU-HOF1a becomes highly robust in polar solvents, strong acid or alkaline aqueous solutions, and has permanent porosity with contracted cavities for selective adsorption and efficient “turn-up” fluorescent sensing of C2H4 gas. This work not only debuts a new hydrogen-bonding synthon but offers more insights into investigating solid-state dynamics in metastable HOFs.
Introduction
Hydrogen-bonded organic frameworks (HOFs) represent a rapidly evolving class of synthetic crystalline porous materials with substantial potential spanning diverse scientific disciplines.1–7 Their distinctive attributes, encompassing porosity, tunability, sustainability, and practicality, render them compelling candidates for a myriad of applications, including gas adsorption and separation,8–10 proton conduction,11,12 heterogeneous catalysis,13,14 biomedical applications,15–17 and beyond. Generally, HOFs are self-assembled by the interaction of organic molecule units through non-covalent hydrogen-bonds.18,19 In this regard, the carboxylic acid,20–22 diaminotriazine (DAT),23,24 pyrazole,25,26 imidazole,27,28 benzimidazolone,29,30 and charge-assisted ionic hydrogen bonding patterns31–33 have been employed in constructing HOF archetypes. In the past decade, careful selection of building blocks and functional group modifications within specific organic molecular units have led to significant advancements in the geometry of HOFs. These advancements encompass aspects such as shape, size, symmetry, and dimensions, as well as surface chemistry1–5,34–40 underscoring the crucial role of pre-design strategies.However, the exploration of HOFs has presented a substantial challenge in implementing reticular chemistry for the self-assembly of hydrogen-bonding units. Hydrogen bonds exhibit significantly weaker (10–40 kJ mol−1), more flexible, and less directional bonding energies and angles compared to coordination (90–350 kJ mol−1) and covalent bonds (300–600 kJ mol−1).1,41 These intrinsic characteristics of hydrogen bonds in HOFs enable mild synthesis conditions,42,43 solution processability,42,44 and ease of regeneration,45,46 yet simultaneously result in the facile formation of metastable structures and complexity.47–49 The arrangement of hydrogen bonds among organic molecule units is susceptible to disruption by solvent disturbances, leading to the formation of (non)porous networks. These networks typically exhibit undesired hydrogen bonding between donor and acceptor groups, as well as fractured hydrogen bonds interacting with solvent molecules.50–54 This situation would be aggravated within highly interpenetrated HOFs, where the interpenetration of frameworks promotes synergistic interactions (π–π stacking, Ar/C–H⋯π interactions, van der Waals interactions)18,55,56 among organic molecule units to further stabilize these mismatched hydrogen bonds. It is thus believed that investigating the solid-state dynamics of mismatched hydrogen bond patterns within metastable HOFs would contribute to deeply deciphering their self-assembly and evolution, yet it remains an ongoing dilemma.
In this study, we fabricated 3D HOFs by the self-assembly of organic molecule units consisting of peripheral 1,8-naphtholactam (Np) and central bicarbazole in solution. The selection of the Np synthon was based on its fluorescence behaviors and its capacity to form stable dimers through intermolecular hydrogen bonding, as recently reported in our laboratory.57,58 Utilizing single-crystal X-ray diffraction (SCXRD) data, we present, for the first time, the self-correcting mismatches of hydrogen bond patterns in an 11-fold interpenetrated array of metastable HOF1 upon desolvation (Fig. 1). This involved a remarkable “cross-door” rotation of nearly 180°between adjacent frameworks at the Np link sites with monomeric N–H⋯O interactions. The ensuing permanent porosity with contracted cavities, coupled with structural robustness and bright green emission of the activated NCU-HOFs were then illustrated, enabling the selective adsorption and efficient “turn-up” fluorescent sensing of ethylene(C2H4) gas.
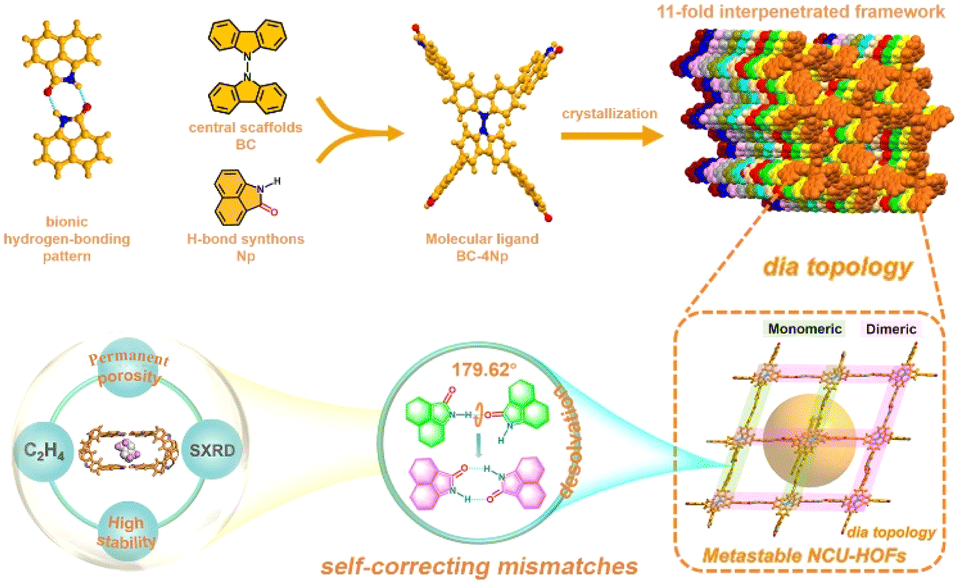 | ||
| Fig. 1 Schematic diagram illustrating the self-correcting mismatches within NCU-HOFs constructed from the BC–4Np molecular ligand. | ||
Results and discussion
The organic ligand BC–4Np was synthesized through a Suzuki coupling reaction involving 3,3′,6,6′-tetrabromo-9,9′-bicarbazole (BC–4Br) and boric acid esterified Np (Np-pin). Comprehensive characterization of its molecular structure was achieved using 1H NMR, 13C NMR, and mass spectroscopy, yielding satisfactory results (details in the ESI†). Block-shaped single crystals were obtained via vapor diffusion of acetone into a solution of BC–4Np in N,N′-dimethylformamide (DMF) at 298 K. SCXRD analysis disclosed that NCU-HOF1 crystallizes in the monoclinic space group I2/a (Table S1†). Within NCU-HOF1, two crystallographically unique BC–4Np molecules exist in the asymmetric unit (Fig. 2a). Distinct hydrogen bonding modes between adjacent Np linkers were observed within each BC–4Np unit, featuring partially monomeric and fully dimeric N–H⋯O hydrogen bonds in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio, with distances ranging from 1.89 to 2.04 Å, forming a single dia 3D network (Fig. 2b). The substantial void space of 70.4 × 49.4 Å2 in this single dia 3D network facilitates the formation of a dia{66} topological structure with an 11-fold interpenetrated array (Fig. 2c and d). This interpenetration is primarily facilitated by multiple Ar–H⋯O, Ar–H⋯π, and π–π interactions between Np and adjacent BC/Np moieties in BC–4Np units (Fig. S1 and S2†). These observations underscore the pivotal role of the large π-planar configuration of the Np synthon in the highly interpenetrated array of NCU-HOF1. Despite the 11-fold interpenetration, serrated 1D pore channels along the c-axis in NCU-HOF1 persist, with cages measuring 9.0 × 9.6 Å2 and necks as small as 5.3 Å (type I, Fig. S3†). Additionally, isolated cavities with cross-sections of approximately 7.6 × 7.0 Å2 were detected in NCU-HOF1 (type II, Fig. S3†). The total solvent-accessible void of NCU-HOF1 was estimated to be around 30% using PLATON.59
:2 ratio, with distances ranging from 1.89 to 2.04 Å, forming a single dia 3D network (Fig. 2b). The substantial void space of 70.4 × 49.4 Å2 in this single dia 3D network facilitates the formation of a dia{66} topological structure with an 11-fold interpenetrated array (Fig. 2c and d). This interpenetration is primarily facilitated by multiple Ar–H⋯O, Ar–H⋯π, and π–π interactions between Np and adjacent BC/Np moieties in BC–4Np units (Fig. S1 and S2†). These observations underscore the pivotal role of the large π-planar configuration of the Np synthon in the highly interpenetrated array of NCU-HOF1. Despite the 11-fold interpenetration, serrated 1D pore channels along the c-axis in NCU-HOF1 persist, with cages measuring 9.0 × 9.6 Å2 and necks as small as 5.3 Å (type I, Fig. S3†). Additionally, isolated cavities with cross-sections of approximately 7.6 × 7.0 Å2 were detected in NCU-HOF1 (type II, Fig. S3†). The total solvent-accessible void of NCU-HOF1 was estimated to be around 30% using PLATON.59
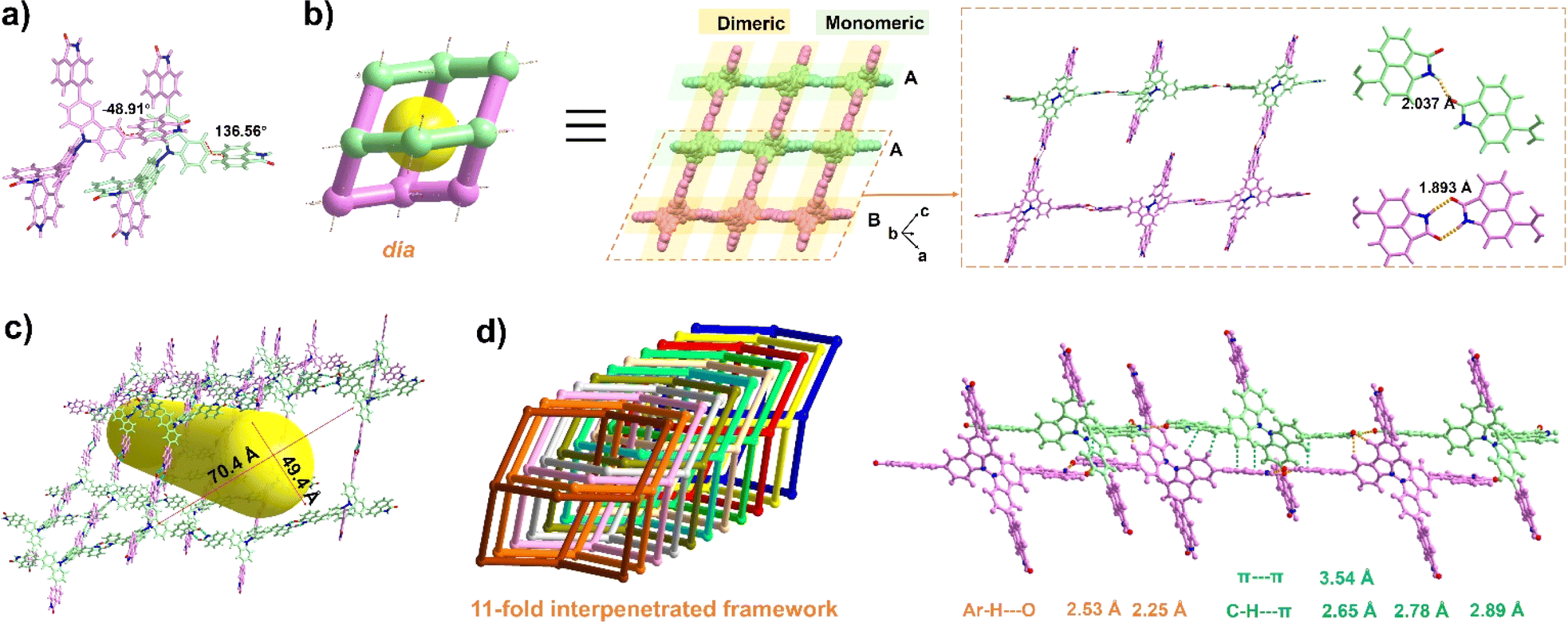 | ||
| Fig. 2 (a) Distinct molecular configurations of BC–4Np. (b) Alternating monomeric and dimeric N–H⋯O hydrogen bonds between adjacent BC–4Np molecules. (c) Void space in the dia topology in NCU-HOF1. (d) The 11-fold interpenetrated topological frameworks (different colors represent different frameworks) and side view of the stacking. The purple structure represents the dimer formed, and the mint green one represents the monomer. | ||
During the activation process, a noteworthy phenomenon was observed in the desolvation of the as-synthesized sample (NCU-HOF1) under high vacuum to generate the activated sample (NCU-HOF1a). The powder X-ray diffraction (PXRD) patterns displayed significant changes, including a shift in the strongest diffraction peak of NCU-HOF1 from 3.1° to 5.2° and a reduction in the number of diffraction peaks, indicative of a structural transformation during desolvation (Fig. S4†). Subsequent tests revealed that elevated temperature and/or vacuum conditions induced a phase transition in NCU-HOF1 (Fig. 3a). Variable-temperature PXRD patterns demonstrated that the phase transition occurred at 313 K, despite being degenerate (Fig. 3b). This observation was corroborated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) data under nitrogen (N2) conditions (Fig. S5†), suggesting the facile escape of disordered solvent molecules from the frameworks. Fourier transform infrared spectroscopy (FTIR) further indicated the structural transformation of NCU-HOF1, as evidenced by the disappearance of the absorption peak at 1713 cm−1, establishing a close correlation with the hydrogen bond receptor C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in Np synthons (Fig. 3c).
O in Np synthons (Fig. 3c).
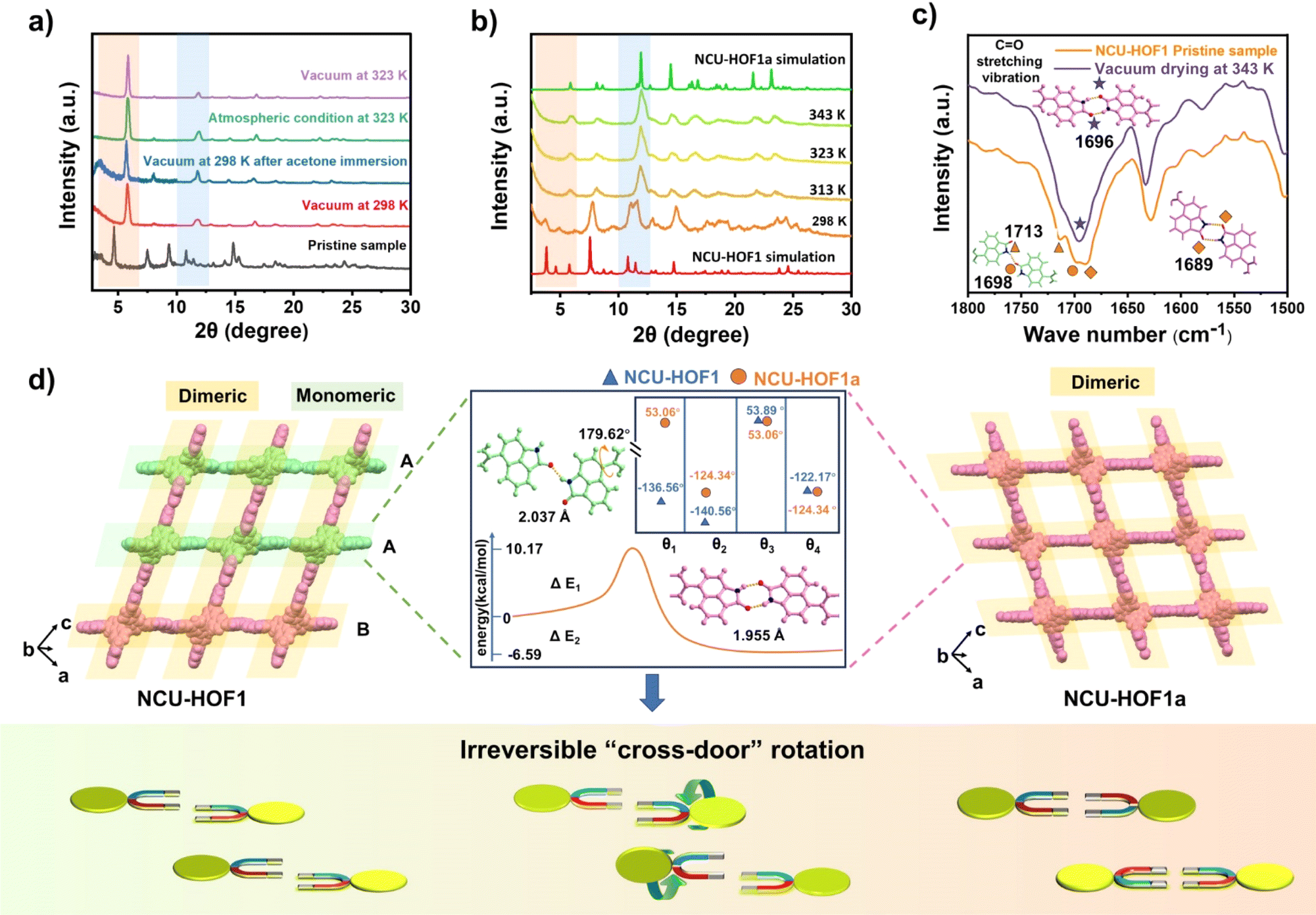 | ||
| Fig. 3 (a) PXRD variations of NCU-HOF1 upon desolvation (vacuum: −1 bar). (b) Variable-temperature PXRD patterns of degraded NCU-HOF1 under a N2 atmosphere. (c) In situ FTIR spectrum of NCU-HOF1. (d) Schematic diagram illustrating the self-correcting N–H⋯O interactions between adjacent Np link sites through an irreversible “cross-door” rotation pathway. Inset: The difference in dihedral angles between the carbazole and Np segments, and the energy profile from monomer to dimer formation of BC–4Np in NCU-HOF1. | ||
Through acetone immersion and vacuum outgassing at 298 K, the guest-free NCU-HOF1a crystal structure is successfully obtained for X-ray diffraction analysis, employing a single-crystal-to-single-crystal (SCSC) transformation approach. SCXRD analysis reveals that NCU-HOF1a crystallizes in the monoclinic space group C2/c, maintaining an 11-fold interpenetrated array with a slight lattice volume contraction in the same dia topology (Table S1 and Fig. S6 and S7†). In contrast to the metastable NCU-HOF1, all BC–4Np molecules in NCU-HOF1a exhibit higher structural symmetry and become crystallographically equivalent. This transformation is associated with the conversion of N–H⋯O interactions between adjacent Np link sites from being partially monomeric to completely dimeric upon activation (Fig. 3d). This observation aligns with the (FTIR) data, underscoring the significance of the hydrogen-bonding pattern in this self-correcting process. A detailed analysis reveals a “cross-door” rotation of two Np units in A–A layers, reaching an angle of nearly 180° (Fig. S8†). Simultaneously, two BC–4Np molecules at the adjacent B layer slide towards each other in opposite directions, maintaining a distance of approximately 4.6 Å. This sliding is primarily induced by the C–H⋯O interaction between the neighboring Np units at layers A and B, generating π-stacking in the frameworks. The self-correction dynamics, characterized by a dramatic rotation and slip of assembly units within the 11-fold interpenetrated HOFs, represent a remarkable and hitherto unreported phenomenon.
In the context of energy considerations, the transformation of monomeric Np units into dimers occurs through rotation around the N–H⋯O bond, exhibiting an energy barrier of 10.17 kcal mol−1 and an exothermic process of 6.57 kcal mol−1 (Fig. 3d middle). When the free N–H of Np units is stabilized by guest acetone molecules, rotation becomes unlikely due to the presence of a higher energy barrier of 28.30 kcal mol−1 and an exothermic energy of 5.79 kcal mol−1 (Fig. S9†). The theoretical analysis concurs with the experimental observation that NCU-HOF1 can be easily activated into NCU-HOF1a through heating and/or vacuum conditions, as it removes guest acetone molecules. Following activation, the serrated 1D pore channel of the HOFs contracts into a dispersive 0D pore, resulting in a reduction of the void pore volume to approximately 20%. Furthermore, NCU-HOF1a crystals exhibit two types of irregular cavities (I and II), primarily surrounded by Np synthons (Fig. 4a). Along the c-axis, cavity I features the largest cages measuring 5.5 × 5.6 Å2 with the smallest necks of 3.7 Å, while cavity II comprises cages of 5.3 × 7.2 Å2 with the smallest necks of 3.5 Å (Fig. S10†). These dimensions align more favorably with the size of small C2H4 molecules (3.3 × 4.2 × 4.8 Å3) than ethane (C2H6) molecules (3.8 × 4.1 × 4.8 Å3),8 providing additional motivation for exploring potential capture and separation within these contracted cavities.
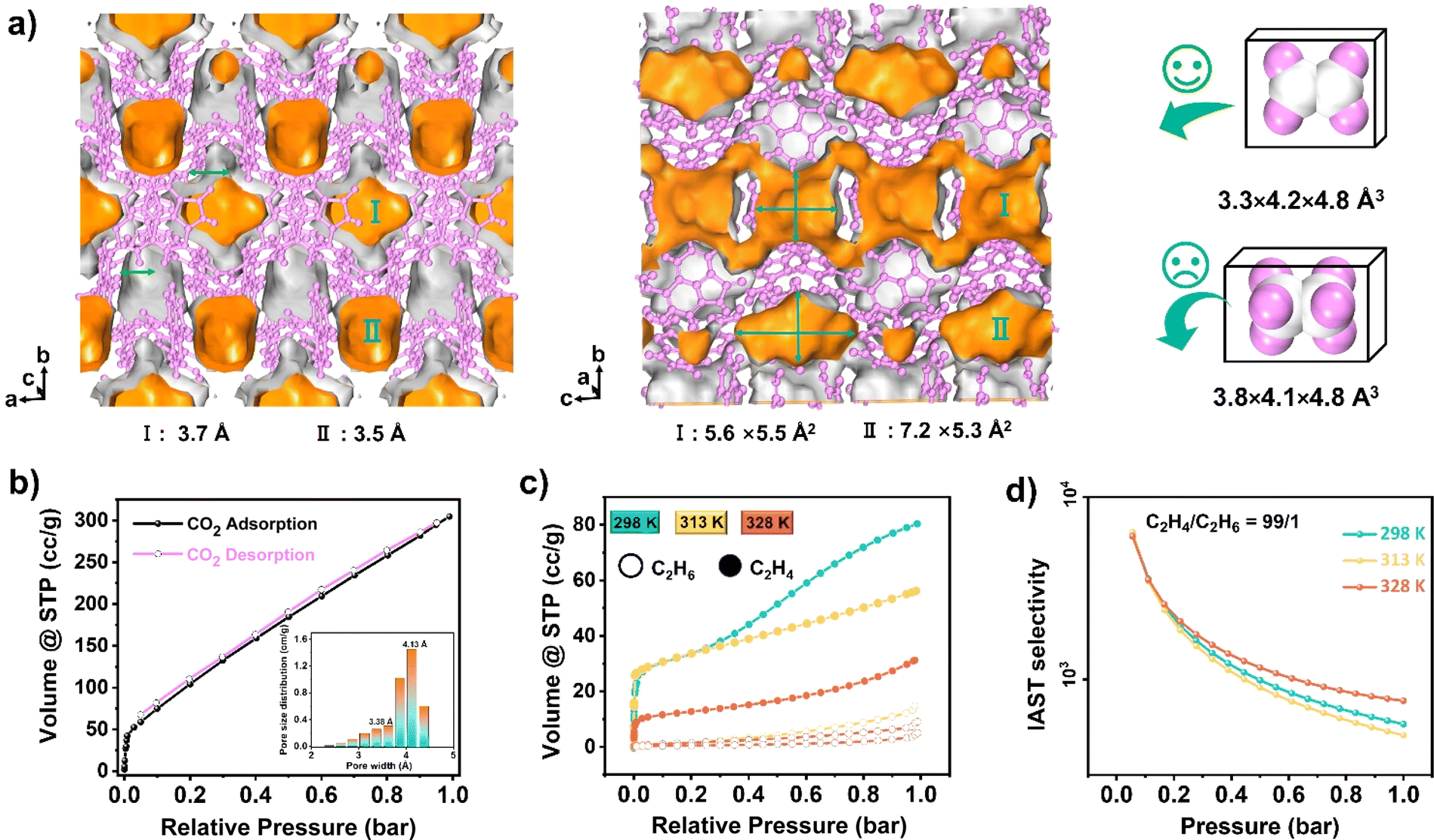 | ||
| Fig. 4 (a) Framework with two pore channels in NCU-HOF1a and the schematic diagram of the size-dependent separation for C2H4 and C2H6 molecules. (b) Gas adsorption and desorption curves for CO2 in NCU-HOF1a at 273 K. (c) Gas adsorption isotherms for C2H4 (hollow circles) and C2H6 (solid circles) in NCU-HOF1a at 298 K (cyan), 318 K (orange), and 333 K (dull red). (d) IAST selectivity of NCU-HOF1a for C2H4 (99:1, v/v) at 298 K (cyan), 318 K (orange), and 333 K (dull red). | ||
To assess chemical stability, NCU-HOF1a underwent treatment in common organic solvents (methanol, acetonitrile, acetone, trichloromethane, and toluene), HCl (pH = 1), and NaOH (pH = 13) solutions at 298 K. After one month, the framework retained its structural integrity, as confirmed by PXRD patterns, scanning electron microscope (SEM) images, and fluorescence photographs (Fig. S11 and S12†), revealing its high structural robustness. Variable-temperature PXRD patterns demonstrated NCU-HOF1a's exceptional thermal stability, with the decomposition temperature confirmed to be above 578 K by thermogravimetric analysis in an N2 atmosphere (Fig. S13†). It is noteworthy that the conversion of NCU-HOF1a to NCU-HOF1 could not be achieved, even when soaked in the mother solution for sample preparation, confirming its thermodynamic stability (Fig. S14†). This irreversibility aligns with crystallographic data both before and after the structural transformation, underscoring the self-correction of the mismatched hydrogen-bonding pattern. Meanwhile, NCU-HOF1a exhibited a maximum emission wavelength (λem) at 526 nm with a fluorescence quantum yield (ΦPL) of up to 36.1% (Fig. S15†). The bright yellow fluorescence of NCU-HOF1a remained unchanged after immersion in common organic solvents and strong acid or alkali water solutions. The fluorescence decay profiles fit well with a double-exponential function, characterized by short lifetimes of approximately 4.79 ns (Fig. S15†). These fluorescence features, mainly inherited from Np synthons, can be sustained and are highly consistent with chemical stability. Notably, NCU-HOF1a represents the first instance of high-efficiency fluorescence derived from hydrogen-bonding synthons, with fluorescence efficiencies comparable to those reported in previous HOF literature (Table S2†).
The permanent porosity of NCU-HOF1a was established through carbon dioxide (CO2) gas sorption experiments at 273 K. In Fig. 4b, the adsorbed CO2 amount in the first step, at P/P0 = 0.001, is approximately 40 cm3 g−1, indicative of a micropore filling process. The total CO2 uptake at 1 bar reaches 300 cm3 g−1, corresponding to a total pore volume of 0.122 cm3 g−1, consistent with the theoretical value calculated from the crystal structure (0.103 cm3 g−1). A small hysteresis loop suggests that HOFs effectively preserve pore characteristics during the adsorption/desorption process. The Brunauer–Emmett–Teller (BET) surface area of NCU-HOF1a was calculated to be 538 m2 g−1 (Fig. S16†), comparable to reported counterparts with an 11-fold interpenetrated array.60 The pore size distribution, calculated using non-localized density functional theory (NL-DFT), exhibited main peaks at 3.38 and 4.13 Å, consistent with values obtained from the crystal structure using the Poreblazer program (Fig. 4b and S17†). N2 isotherms at 77 K revealed a characteristic reversible type-III isotherm for NCU-HOF1a (Fig. S18†), indicating an extremely weak interaction between NCU-HOF1a and N2 gas.
Single-component adsorption isotherms of C2H6 and C2H4 for NCU-HOF1a were examined under ambient conditions (Fig. 4c and S19 and Table S3†). The total C2H4 uptake at 298 K and 1 bar was 80.93 cm3 g−1 (3.61 mmol g−1), with a second adsorption step at 0.4 bar indicating dual adsorption sites for C2H4, one of which exhibits preferential adsorption. Conversely, a type-IV isotherm was detected for C2H6 adsorption, and the total C2H6 uptake was only 14.80 cm3 g−1 (0.66 mmol g−1) under identical conditions. Such a preferential adsorption of C2H4 over C2H6 is accompanied by a large adsorption ratio for C2H4/C2H6 (5.46), suggesting the potential of NCU-HOF1a for separating these two gases. Additionally, the total C2H4 uptake decreased with rising temperature, and the second adsorption step vanished at 313 K and 328 K. Calculations of the adsorption selectivity of NCU-HOF1a for C2H4/C2H6 (50/50, 90/10 and 99/1) mixtures using ideal adsorbed solution theory (IAST) revealed high C2H4 selectivity for mixtures at 298 K and 1 bar (Fig. 4d and S20 and Table S3†). The initial value of the experimental isosteric heat of adsorption (Qst) for C2H4 on NCU-HOF1a was calculated to be 20 kJ mol−1, indicating superior adsorption/desorption processes compared to traditional metals or π coordination gas adsorbing materials.61
The fluorescence behavior of NCU-HOF1a before and after C2H4 uptake was then recorded. As shown in Fig. 5a–c, the as-prepared NCU-HOF1a@C2H4 (for preparation details see the ESI†) maintained bright yellow-green fluorescence at λem = 526 nm, with ΦPL and lifetime gradually increasing from 36.1 to 49.1% and from 4.28 to 8.50 ns upon C2H4 uptake. In comparison, NCU-HOF1a showed a negligible increase in fluorescence intensity upon treatment with C2H2 or C2H6 gas, emphasizing its exceptional selectivity for C2H4 adsorption (Fig. S21†). Variable-temperature fluorescence tests revealed that NCU-HOF1a@C2H4 remains structurally stable under ambient conditions but readily releases adsorbed C2H4 during vacuum drying (Fig. S22†). This fluorescence enhancement can be repeated at least five times with C2H4 pressure ranging from 0 to 2 bar, indicating the excellent reusability of NCU-HOF1a for C2H4 uptake/release (Fig. S23a†). The unchanged PXRD pattern of NCU-HOF1a before and after C2H4 uptake demonstrated that the adsorbed C2H4 gas has no impact on its framework (Fig. S23b†). A summary of all the photophysical data of the as-prepared NCU-HOF1a@C2H4 is provided in Table S4.†
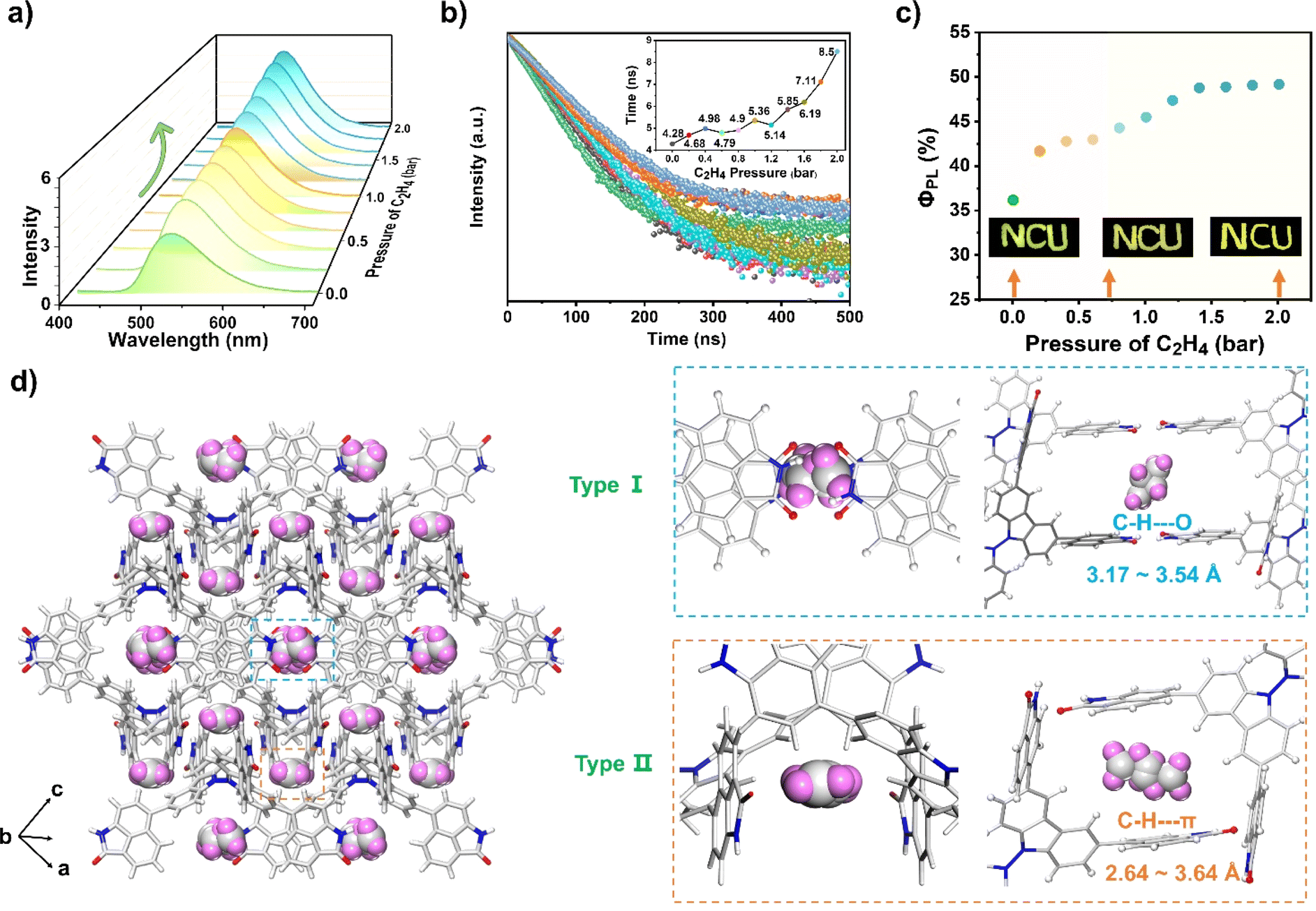 | ||
| Fig. 5 Changes in (a) fluorescence intensity, (b) lifetime profiles, and (c) ΦPL of NCU-HOF1a upon C2H4 uptake (pressure ranging from 0 to 2 bar). (d) Multiple intermolecular interactions between C2H4 gas and Np synthons in the framework of NCU-HOF1a. | ||
Fortunately, the C2H4-adsorbed NCU-HOF1a single crystal, named NCU-HOF1a@1C2H4, was obtained by exposing it to a C2H4 atmosphere at 1 bar and 273 K for approximately 24 hours (Table S1 and Fig. S24 and S25†). With the SCXRD results of NCU-HOF1a@1C2H4, the C2H4-induced fluorescence enhancement behavior of NCU-HOF1a can be further elucidated. Although the location and configuration of the C2H4 molecules within the pores are disordered, two possible modes of C2H4 molecules residing in the NCU-HOF1a framework were observed. In Fig. 5d, the C2H4 molecules were trapped between multiple Np synthons in NCU-HOF1a@1C2H4, forming weak C–H⋯O interactions (3.17–3.54 Å) in the type-I cavity, while forming moderate edge-to-face C–H⋯π interactions (2.64–3.46 Å) in the type-II cavity. These multiple intermolecular interactions between C2H4 molecules and Np synthons were also confirmed by the Hirshfeld surface analysis of the C2H4 molecule within the framework (Fig. S26†), further supporting the crucial role of Np synthons in trapping C2H4 molecules. Moreover, the NCU-HOF1a@1.75C2H4 single crystal was then obtained by raising the C2H4 pressure to 2 bar (Table S1†). The SCXRD analysis revealed a significant increase in the number of intermolecular interactions between C2H4 molecules and Np synthons, accompanied by a decrease in the shortest distance of these intermolecular interactions (Fig. S27 and S28†). This is mainly ascribed to the 1.75 times increase of C2H4 uptake in the NCU-HOF1a@1.75C2H4 compared to that of NCU-HOF1a@1C2H4. It should be noted that the unit cells of both C2H4-adsorbed NCU-HOF1a single crystals remain unchanged compared to NCU-HOF1a, indicating a gradual increase in the internal rigidity of the frameworks in the order of NCU-HOF1a@1.75C2H4 > NCU-HOF1a@1C2H4 > NCU-HOF1a. Precisely, the trapped C2H4 molecules within the pores of C2H4-adsorbed NCU-HOF1a restrict the rotation of the Np synthons, reducing the nonradiative decay pathways and resulting in the fluorescence turn-up of NCU-HOF1a. This is highly consistent with the experimental data, and all of these SCXRD analyses also strongly support the preferential adsorption of C2H4 over C2H6 on NCU-HOF1a under ambient conditions.
Conclusions
In summary, we have demonstrated the facile self-correction of the hydrogen-bonding patterns in 3D NCU-HOFs, self-assembled from molecular units composed of peripheral Np hydrogen-bonded synthons and central bicarbazoles. The as-synthesized metastable NCU-HOF1 has an 11-fold interpenetrated dia topology with a quarter of the Np units exhibiting monomeric N–H⋯O interactions between adjacent Np link sites. These mismatched hydrogen bond patterns readily undergo an inconceivable “cross-door” rotation of nearly 180° upon desolvation, resulting in fully dimeric interactions. Consequently, the activated NCU-HOF1a becomes permanently porous with a BET surface area of 538 m2 g−1 and excellent robustness in highly polar solvents, strong acid, or alkaline aqueous solutions. Moreover, NCU-HOF1a demonstrates biased adsorption of C2H4 over C2H6 due to multiple C–H⋯O/π interactions between Np synthons and C2H4 guests in the contracted cavities. These host–guest interactions enhance the intramolecular rigidity of NCU-HOF1a, enabling efficient “turn-up” fluorescent sensing of C2H4. This work contributes to our understanding of self-correction dynamics in frameworks by nanoconfinement and their influence on the subsequent features and applications of organic porous materials.Data availability
The data are available from the corresponding author on reasonable request.Author contributions
H. W. proposed the research direction and guided the project. G. X. and C. Z. designed and performed the materials synthesis and characterization. X. X., F. Y., and Y. Y. took part in the discussion of experimental data and gave useful suggestions. H. W., G. X., C. Z., and X. X. drafted the manuscript. All authors discussed the results and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22063005), the Natural Science Foundation of Jiangxi Province (No. 20212ACBA203012, 20224BAB214003, 20232BAB203031), the Interdisciplinary Innovation Fund of Natural Science, Nanchang University (No. 9167-27060003-ZD2101, 9167-28220007-YB2113), and the Foundation of Jiangxi Provincial Key Laboratory of Functional Crystalline Materials Chemistry (No. 2024SSY05162).Notes and references
- R.-B. Lin, Y. B. He, P. Li, H. L. Wang, W. Zhou and B. L. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- Z.-J. Lin, S. A. R. Mahammed, T.-F. Liu and R. Cao, ACS Cent. Sci., 2022, 8, 1589–1608 CrossRef CAS PubMed.
- Z. J. Zhang, Y. X. Ye, S. C. Xiang and B. L. Chen, Acc. Chem. Res., 2022, 55, 3752–3766 CrossRef CAS PubMed.
- S. Z. Cai, Z. F. An and W. Huang, Adv. Funct. Mater., 2022, 32, 2207145 CrossRef CAS.
- I. Hisaki, C. Xin, K. Takahashi and T. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 11160–11170 CrossRef CAS PubMed.
- A. E. Amooghin, H. Sanaeepur, M. Ghomi, R. Luque, H. Garcia and B. L. Chen, Coord. Chem. Rev., 2024, 505, 215660 CrossRef.
- J. T. Li and B. L. Chen, Chem. Sci., 2024, 15, 9874–9892 RSC.
- Y. S. Yang, L. B. Li, R.-B. Lin, Y. X. Ye, Z. Z. Yao, L. Yang, F. H. Xiang, S. M. Chen, Z. J. Zhang, S. C. Xiang and B. L. Chen, Nat. Chem., 2021, 13, 933–939 CrossRef CAS PubMed.
- X. Zhang, L. B. Li, J.-X. Wang, H.-M. Wen, R. Krishna, H. Wu, W. Zhou, Z.-N. Chen, B. Li, G. D. Qian and B. L. Chen, J. Am. Chem. Soc., 2020, 142, 633–640 CrossRef CAS PubMed.
- C. H. Jiang, J.-X. Wang, D. Liu, E. Y. Wu, X.-W. Gu, X. Zhang, B. Li, B. L. Chen and G. D. Qian, Angew. Chem., Int. Ed., 2024, 63, e202404734 CrossRef CAS PubMed.
- S. C. Pal, D. Mukherjee, R. Sahoo, S. Mondal and M. C. Das, ACS Energy Lett., 2021, 6, 4431–4453 CrossRef CAS.
- Z. Y. Song, L. Miao, L. Ruhlmann, Y. K. Lv, L. C. Li, L. H. Gan and M. X. Liu, Angew. Chem., Int. Ed., 2023, 61, e202219136 Search PubMed.
- B. Han, H. L. Wang, C. M. Wang, H. Wu, W. Zhou, B. L. Chen and J. Z. Jiang, J. Am. Chem. Soc., 2019, 141, 8737–8740 CrossRef CAS PubMed.
- W. K. Qin, D. H. Si, Q. Yin, X. Y. Gao, Q. Q. Huang, Y. N. Feng, L. Xie, S. Zhang, X. S. Huang, T. F. Liu and R. Cao, Angew. Chem., Int. Ed., 2022, 61, e202202089 CrossRef CAS PubMed.
- D. Q. Yu, H. C. Zhang, Z. Q. Liu, C. Liu, X. B. Du, J. S. Ren and X. G. Qu, Angew. Chem., Int. Ed., 2022, 61, e202201485 CrossRef CAS PubMed.
- J. K. Tang, J. Liu, Q. Z. Zheng, W. T. Li, J. H. Sheng, L. Q. Mao and M. Wang, Angew. Chem., Int. Ed., 2021, 60, 22315–22321 CrossRef CAS PubMed.
- D. Q. Yu, H. C. Zhang, J. S. Ren and X. G. Qu, Chem. Soc. Rev., 2023, 52, 7504–7523 RSC.
- X. Y. Song, Y. Wang, C. Wang, D. Wang, G. W. Zhuang, K. O. Kirlikovali, P. Li and O. K. Farha, J. Am. Chem. Soc., 2022, 144, 10663–10687 CrossRef CAS PubMed.
- R.-B. Lin and B. L. Chen, Chem, 2022, 8, 2114–2135 CAS.
- P. Cui, E. S. Grape, P. R. Spackman, Y. Wu, R. Clowes, G. M. Day, A. K. Inge, M. A. Little and A. I. Cooper, J. Am. Chem. Soc., 2020, 142, 12743–12750 CrossRef CAS PubMed.
- Y.-L. Li, E. V. Alexandrov, Q. Yin, L. Li, Z.-B. Fang, W. B. Yuan, D. M. Proserpio and T.-F. Liu, J. Am. Chem. Soc., 2020, 142, 7218–7224 CrossRef CAS PubMed.
- M. Vicent-Morales, M. Esteve-Rochina, J. Calbo, E. Ortí, I. J. Vitórica-Yrezábal and G. M. Espallargas, J. Am. Chem. Soc., 2022, 144, 9074–9082 CrossRef CAS PubMed.
- S. Feng, Y. X. Shang, Z. K. Wang, Z. X. Kang, R. M. Wang, J. Z. Jiang, L. L. Fan, W. D. Fan, Z. N. Liu, G. D. Kong, Y. Feng, S. Q. Hu, H. L. Guo and D. F. Sun, Angew. Chem., Int. Ed., 2020, 59, 3840–3845 CrossRef CAS PubMed.
- H. L. Wang, B. Li, H. Wu, T.-L. Hu, Z. Z. Yao, W. Zhou, S. C. Xiang and B. L. Chen, J. Am. Chem. Soc., 2015, 137, 9963–9970 CrossRef CAS PubMed.
- T.-H. Chen, I. Popov, W. Kaveevivitchai, Y.-C. Chuang, Y.-S. Chen, O. Daugulis, A. J. Jacobson and O. S. Miljanic, Nat. Commun., 2014, 5, 5131 CrossRef CAS PubMed.
- M. I Hashim, H. T. M. Le, T.-H. Chen, Y.-S. Chen, O. Daugulis, C.-W. Hsu, A. J. Jacobson, W. Kaveevivitchai, X. Liang, T. Makarenko, O. S. Miljanic, I. Popovs, H. V. Tran, X. Q. Wang, C.-H. Wu and J. I. Wu, J. Am. Chem. Soc., 2018, 140, 6014–6026 CrossRef CAS PubMed.
- W. Q. Yan, X. P. Yu, T. Yan, D. F. Wu, E. L. Ning, Y. Qi, Y.-F. Han and Q. W. Li, Chem. Commun., 2017, 53, 3677–3680 RSC.
- A. Maurya, K. Marvaniya, P. Dobariya, M. V. Mane, S. Tothadi, K. Patel and S. Kushwaha, Small, 2023, 2306824 Search PubMed.
- A. Pulido, L. J. Chen, T. Kaczorowski, D. Holden, M. A. Little, S. Y. Chong, B. J. Slater, D. P. McMahon, B. Bonillo, C. J. Stackhouse,, A. Stephenson, C. M. Kane, R. Clowes, T. Hasell, A. I. Cooper and G. M. Day, Nature, 2017, 543, 657–664 CrossRef CAS PubMed.
- M. Mastalerz and I. M. Oppel, Angew. Chem., Int. Ed., 2012, 51, 5252–5255 CrossRef CAS PubMed.
- X.-Y. Chen, L.-H. Cao, X.-T. Bai and X.-J. Cao, Chem. Eur. J., 2024, 30, e202303580 CrossRef CAS PubMed.
- A. Karmakar, R. Illathvalappil, B. Anothumakkool, A. Sen, P. Samanta, A. V. Desai, S. Kurungot and S. K. Ghosh, Angew. Chem. Int. Ed., 2016, 55, 10667 CrossRef CAS PubMed.
- Y. Y. Sun, J. Wei, Z. H. Fu, M. Y. Zhang, S. G. Zhao, G. Xu, C. S. Li, J. Zhang and T. H. Zhou, Adv. Mater., 2023, 35, 2208625 CrossRef CAS PubMed.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- M. A. Little and A. I. Cooper, Adv. Funct. Mater., 2020, 30, 1909842 CrossRef CAS.
- B. Wang, R.-B. Lin, Z. J. Zhang, S. C. Xiang and B. L. Chen, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef CAS PubMed.
- Q. Zhu, L. Wei, C. Zhao, H. X. Qu, B. W. Liu, T. Fellowes, S. Y. Yang, A. Longcake, M. J. Hall, M. R. Probert, Y. B. Zhao, A. I. Cooper and M. A. Little, J. Am. Chem. Soc., 2023, 145, 23352–23360 CrossRef CAS PubMed.
- S. M. Chen, Y. Ju, Y. S. Yang, F. H. Xiang, Z. Z. Yao, H. Zhang, Y. B. Li, Y. F. Zhang, S. C. Xiang, B. L. Chen and Z. J. Zhang, Nat.Commun., 2024, 15, 298 CrossRef CAS PubMed.
- S. C. Wang, Q. S. Zhang, Z. Wang, S. Q. Guan, X. D. Zhang, X. H. Xiong and M. Pan, Angew. Chem., Int. Ed., 2023, 62, e202315382 CrossRef CAS PubMed.
- D. Wang and Y. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202217903 CrossRef CAS PubMed.
- J. C. Jiang, Y. B. Zhao and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 3255–3265 CrossRef CAS PubMed.
- A. K. Mohammed, J. Raya, A. Pandikassala, M. A. Addicoat, S. Gaber, M. Aslam, L. Ali, S. Kurungot and D. Shetty, Angew. Chem. Int. Ed., 2023, 62, e202304313 CrossRef CAS PubMed.
- T. Hashimoto, R. Oketani, M. Nobuoka, S. Seki and I. Hisaki, Angew. Chem., Int. Ed., 2023, 62, e202215836 CrossRef CAS PubMed.
- X. Yang, J. Huang, S. Y. Gao, Y. Q. Zhao, T. Huang, H. F. Li, T. F. Liu, Z. Y. Yu and R. Cao, Adv. Mater., 2023, 35, 2305344 CrossRef CAS PubMed.
- F. L. Hu, C. P. Liu, M. Y. Wu, J. D. Pang, F. L. Jiang, D. Q. Yuan and M. C. Hong, Angew. Chem., Int. Ed., 2017, 56, 2101–2104 CrossRef CAS PubMed.
- M. A. Rahman, C. J. Dionne and A. Giri, Nano Lett., 2022, 22, 8534–8540 CrossRef CAS PubMed.
- J. K. Gao, Y. L. Cai, X. F. Qian, P. X. Liu, H. Wu, W. Zhou, D.-X. Liu, L. B. Li, R.-B. Lin and B. L. Chen, Angew. Chem., Int. Ed., 2021, 60, 20400–20406 CrossRef CAS PubMed.
- P. H. Li, P. Li, M. R. Ryder, Z. C. Liu, C. L. Stern, O. K. Farha and J. F. Stoddart, Angew. Chem., Int. Ed., 2019, 58, 1664–1669 CrossRef CAS PubMed.
- Q. Ji, K. Takahashi, S.-I. Noro, Y. Ishigaki, K. Kokado, T. Nakamura and I. Hisaki, Cryst. Growth Des., 2021, 21, 4656–4664 CrossRef CAS.
- Y. Z. Zhou, C. Chen, R. Krishna, Z. Y. Ji, D. Q. Yuan and M. Y. Wu, Angew. Chem., Int. Ed., 2023, 62, e202305041 CrossRef CAS PubMed.
- X. J. Ding, Z. Y. Liu, Y. S. Zhang, G. Ye, J. F. Jia and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202116483 CrossRef CAS PubMed.
- B. Q. Yu, S. B. Geng, H. L. Wang, W. Zhou, Z. J. Zhang, B. L. Chen and J. Z. Jiang, Angew. Chem., Int. Ed., 2021, 60, 25942–25948 CrossRef CAS PubMed.
- Y. Lv, J. Liang, Z. Xiong, H. Zhang, D. Li, X. Yang, S. Xiang and Z. Zhang, –, Chem.–Eur. J., 2023, 29, e202204045 CrossRef CAS PubMed.
- Y. L. Cai, J. K. Gao, J.-H. Li, P. X. Liu, Y. C. Zheng, W. Zhou, H. Wu, L. B. Li, R.-B. Lin and B. L. Chen, Angew. Chem., Int. Ed., 2023, 62, e202308579 CrossRef CAS PubMed.
- Y. Wang, K. Ma, J. Q. Bai, T. Xu, W. D. Han, C. Wang, Z. X. Chen, K. O. Kirlikovali, P. Li, J. S. Xiao and O. K. Farha, Angew. Chem., Int. Ed., 2022, 61, e202115956 CrossRef CAS PubMed.
- Y. Suzuki, N. Tohnai, A. Saeki and I. Hisaki, Chem. Commun., 2020, 56, 13369–13372 RSC.
- G. G. Ye, Y. G. Wang, L. M. Hong, F. Q. Yu, G. M. Xia and H. M. Wang, J. Mater. Chem. C., 2021, 9, 9142–9146 RSC.
- C. L. Zhou, M. D. Wang, W. H. Guo, G. G. Ye,, Y. G. Wang, Y. Yang, G. M. Xia and H. M. Wang, Dyes Pigm., 2023, 213, 111198 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- W. Yang, J. W. Wang, H. L. Wang, Z. B. Bao, J. C.-G. Zhao and B. L. Chen, Cryst. Growth Des., 2017, 17, 6132–6137 CrossRef CAS.
- D. Sengupta, P. Melix, S. Bose, J. Duncan, X. J. Wang, M. R. Mian, K. O. Kirlikovali, F. Joodaki, T. Islamoglu, T. Yildirim, R. Q. Snurr and O. K. Farha,, J. Am. Chem. Soc., 2023, 145, 20492–20502 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2226443–2226446. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02751e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |