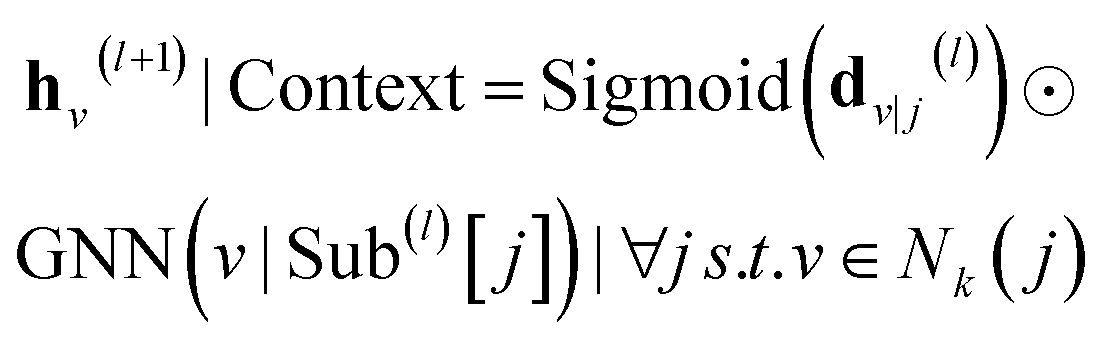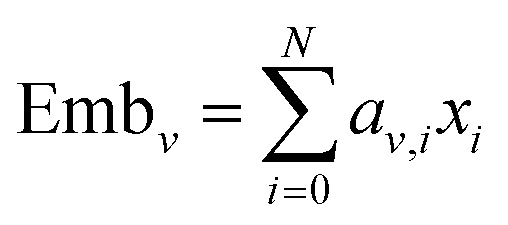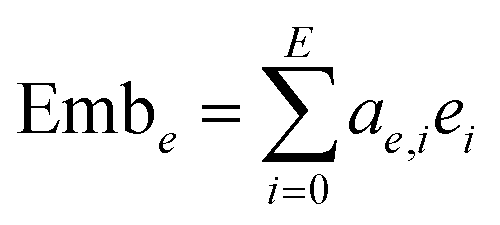DOI:
10.1039/D4SC02781G
(Edge Article)
Chem. Sci., 2024,
15, 17533-17546
Enhancing chemistry-intuitive feature learning to improve prediction performance of optical properties†
Received
26th April 2024
, Accepted 22nd September 2024
First published on 26th September 2024
Abstract
Emitters have been widely applied in versatile fields, dependent on their optical properties. Thus, it is of great importance to explore a quick and accurate prediction method for optical properties. To this end, we have developed a state-of-the-art deep learning (DL) framework by enhancing chemistry-intuitive subgraph and edge learning and coupling this with prior domain knowledge for a classic message passing neural network (MPNN) which can better capture the structural features associated with the optical properties from a limited dataset. Benefiting from technical advantages, our model significantly outperforms eight competitive ML models used in five different optical datasets, achieving the highest accuracy to date in predicting four important optical properties (absorption wavelength, emission wavelength, photoluminescence quantum yield and full width at half-maximum), showcasing its robustness and generalization. More importantly, based on our predicted results, one new deep-blue light-emitting molecule PPI-2TPA was successfully synthesized and characterized, which exhibits close consistency with our predictions, clearly confirming the application potential of our model as a quick and reliable prediction tool for the optical properties of diverse emitters in practice.
1 Introduction
Emitters have been widely applied in a variety of fields like organic light-emitting diodes (OLEDs), organic dyes, organic solar cells and bio-sensors.1–3 Absorption wavelength, emission wavelength, photoluminescence quantum yield (PLQY) and full width at half-maximum (FWHM) are four key optical properties required by versatile applications. However, it is time-consuming and complex to conduct experiments to develop new emitters with desired properties, due to a trial-and-error strategy.4 The quantum mechanical (QM) method plays important roles in supplementing experimental research, but its high requirement in computation resources limits its application in probing large unknown chemical spaces.5 Furthermore, the computational conditions of QM are hardly the same as the experimental ones, generally leading to a relatively large difference from the experimental values.
Data-driven machine learning (ML), as a core technique of artificial intelligence, has exhibited great success in various fields.6–8 ML can mine the structure–property relationship underlying complex data, through which it can quickly predict the properties of unseen compounds. Some attempts have already utilized ML methods to develop various models for predicting optical properties, including random forest (RF), support vector machine (SVM), multi-layer perceptron (MLP), light gradient boosting machine (LightGBM), and gradient boost regression tree (GBRT)9,10 models, which present relatively high accuracy for wavelengths (R2 of ∼0.92) and moderate accuracy for PLQY (R2 of ∼0.70), but on a small dataset. It is known that traditional ML algorithms are generally limited in capturing complex causality due to their relatively simple architectures and dependence on hand-selected feature engineering.11 Deep learning (DL) shows stronger learning capacity benefiting from its more complicated model architecture, and it can extract features automatically by an end-to-end learning method so that it can avoid labor-intensive feature engineering.11–13 These advantages have driven the wide use of DLs in practice, including the prediction of optical properties.13–17 For example, Joung et al. for the first time constructed a DL model based on a message passing neural network (MPNN).14 Hung et al. used a modified DL framework to predict absorption wavelength, emission wavelength and PLQY based on the Deep4Chem database.15,18 Greenman et al. developed an MPNN-based DL model to predict the absorption wavelengths of Deep4Chem.13 Shao et al. adopted a fully connected neural network (FCNN) to predict the absorption wavelengths of the SMFluo1 database.16 Ksenofontov et al. built a deep neural network (DNN) to predict the absorption wavelength of boron-dipyrromethene (BODIPY) dyes.17 Jung et al. employed deep residual convolutional neural networks (DR-CNN) to predict the absorption wavelengths of a combined dataset.19
Collectively, these DL models exhibited relatively high accuracy for the wavelengths (R2 of ∼0.89–0.95) and moderate accuracy with R2 of ∼0.71 for PLQY and FWHM on larger datasets (Deep4chem and BODIPYs). There remains a lot of room for further improvement to provide a more reliable prediction tool for developing diverse organic emitters with desired properties. In addition, it should be noted that most existing models were tested on only one type of dataset,9,10,14,16 which cannot ensure their generalization and robustness to diverse optical material fields. However, improvement in generalization and robustness has been considered to be one of the most difficult challenges for MLs.20,21
Among various DL algorithms, graph neural networks (GNNs) have been widely applied in many domains,12,22,23 particularly the MPNN paradigm of GNN, which has been adopted in previous prediction of optical properties.13,14 However, conventional MPNN only updates the state of node features representing atoms in the molecular graph, but ignored the updating of edge features that represent chemical bond features.24 Furthermore, the semantic information in the molecular structure associated with functional groups and the long-range interaction at the intramolecular level are overlooked.22 The limitations existing in the classic MPNN paradigm weaken the learning capacity of MPNN in extracting chemical structure features from the molecular graph.22,24,25 In addition, despite the theoretical advantage that DL can avoid hand-engineering, it needs sufficient data to support the feature of self-learning.26 However, in the real world, the data are generally limited, especially for material science.27 In this case, coupling prior knowledge from domains into the end-to-end learning will be beneficial to improving feature representation for DLs.12,28
Motivated by urgent need and technical challenges, we explored a novel GNN-based deep learning framework to improve performance in predicting optical properties, which is named Subgraph Optical Graph (SubOptGraph). To address the limitations of conventional MPNN architecture, we improved the feature learning on the molecular graph by adding edge and subgraph learning. In addition, for limited optical data, we further integrated empirical knowledge closely associated with optical properties into the molecular graph learning to further enhance its feature representation. To sufficiently validate the technical advantages of our model, eight competitive models were adopted for comparison: two traditional ML models and six DL models. In addition, unlike previous ML work that used a single dataset to test their performance, the generalization of SubOptGraph is strongly validated against five different datasets (Deep4Chem,18 BODIPYs database,17 ChemFluor,10 SMFluo1,16 JCIM_Abs19). Our model significantly outperforms all the competitive models in the five different datasets. To further test the generalization of our model to unseen compounds, we applied the model to emitters of OLEDs by a transfer learning strategy, given the importance of OLED in commercial application while the current optical databases rarely include the emitters of OLED. Based on the prediction results, we designed and synthesized a new deep-blue emitter PPI-2TPA. The experimental characterization exhibits close consistency with our prediction results, strongly validating the application potential of our model in practice.
2 Data and model construction
2.1 Data collection and sample representation
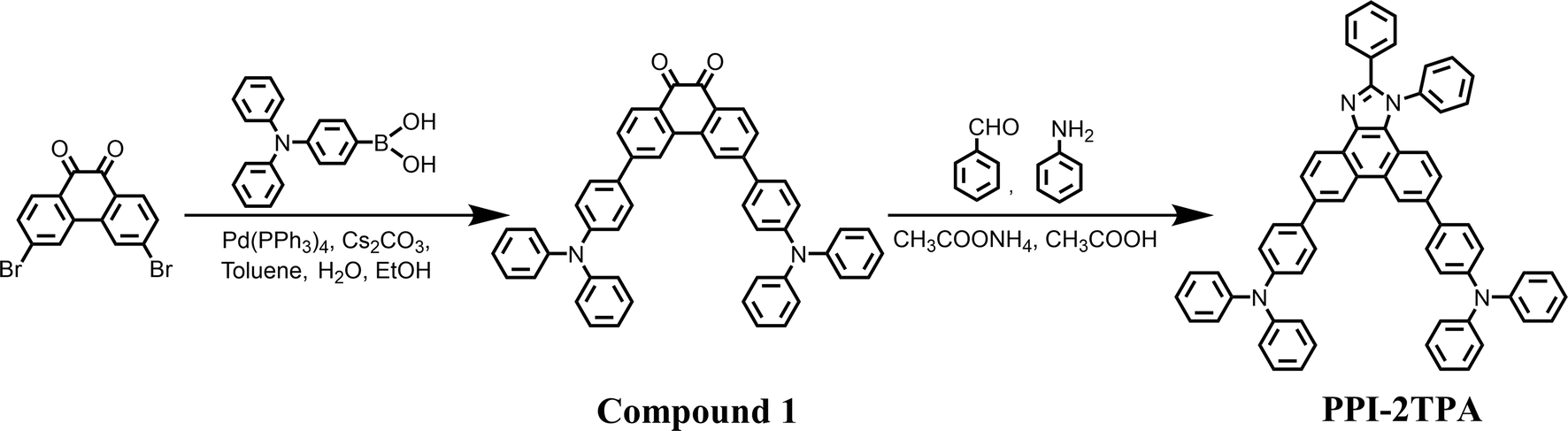
Deep4Chem is the current largest experimental optical database established by Joung et al.,18 including 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 295 absorption wavelengths, 18142 emission wavelengths, 13837 PLQY and 7198 FWHM. As shown in Fig. 1a, we extracted the simplified molecular input line entry system (SMILES) and labels of emitters in different solvents from Deep4Chem. All the properties were normalized to follow the standard normal distribution.
295 absorption wavelengths, 18142 emission wavelengths, 13837 PLQY and 7198 FWHM. As shown in Fig. 1a, we extracted the simplified molecular input line entry system (SMILES) and labels of emitters in different solvents from Deep4Chem. All the properties were normalized to follow the standard normal distribution.
 |
| | Fig. 1 Overview of SubOptGraph framework. (a) The collection of samples from Deep4Chem. The gas-phase samples were removed. (b) Representation of the emitter and solvent, including node features V (i.e., N atoms with C-dimensional atom features), edge features A (i.e., E edges with A-dimensional bond features) and state features U representing the prior domain knowledge. (c) The framework of SubOptGraph. SubConv (light green) is the message passing phase. Readout (light blue) is the readout phase. Full-connect (light grey) is a multi-layer perceptron (MLP) used to predict optical properties. (d) The message passing phase of SubOptGraph, including three MPNNs, i.e., Subgraph MPNN (to extract subgraph features), node-centered MPNN (to extract node features) and edge-centered MPNN (to extract edge features). The original features, U, A and V are transformed to U′, A′ and V′ in this stage. (e) Readout phase. The updated features are transformed to be embeddings by the method of global attention in order to make predictions. vi and ei denote the node and edge features, respectively. αi is the attention coefficient for each node and edge. | |
It is accepted that feature representation is a key factor for ML performance. According to some previous GNN studies,12,29,30 we used 37 atom features and 6 bond features to characterize the molecular graph, as listed in Table 1. Unlike a conventional MPNN that characterizes the samples only by the molecular graph, we coupled prior domain knowledge as a state feature into the molecular graph, in order to alleviate the limitation in the feature mining of DL from a small-size dataset. Experimental findings revealed that a spiral structure, aromaticity and molecular rings are highly related to optical properties.31–33 Herein, we selected five molecular descriptors associated with spiral structures (RotatableBond), aromaticity (Fr_NO and Fr_AromAtoms), and molecular rings (AliphaticRings and AromaticRings) as the state features (Table 2). Thus, these state features are inferred from the prior domain knowledge. All the features were calculated with RDKit (https://rdkit.org/docs/index.html). In addition, unlike existing MPNN-based DL models that separately conduct message passing for the emitter and solvent,13,14 we fused the molecular graphs of the emitter and solvent through vertical concatenation to become a full one that can feed the features into the model more conveniently and more quickly (as depicted in Fig. 1b).
Table 1 Molecular graph features used in the work
| Features |
Description |
|
Atom
|
| Atom type |
The type of the atom (one-hot) |
| Hydrogens |
The number of hydrogens (integer) |
| Hybridization |
The types of hybridization (one-hot) |
| ElectroNegativity |
Pauling electronegativity (floating) |
| Donor |
Accepts electron (binary) |
| Acceptor |
Donates electron (binary) |
| Is in a ring |
Atom in a ring (binary) |
| Is aromatic |
Atom is aromatic (binary) |
| Atomic number |
The atomic number (integer) |
| Vdw radius |
Van der Waals radius (floating) |
| Formal charge |
Formal charge (integer) |
| ExplicitValence |
The explicit valence (integer) |
| ImplicitValence |
The implicit valence (integer) |
| ExplicitHs |
Number of explicit Hs (integer) |
| RadicalElectrons |
Number of radical electrons (integer) |
|
|
Bond
|
| Bond type |
The hybridization type (one-hot) |
| Is in a ring |
Bond is in a ring (binary) |
| Is conjugated |
Bond is conjugated (binary) |
Table 2 Five important molecular descriptors inferred from prior knowledge as state features in the models
| Descriptors |
Description |
| Fr_NO |
(n_N + n_O)/n_heavy (floating) |
| Fr_AromAtoms |
n_aromatic/n_heavy (floating) |
| RotatableBond |
Number of rotatable bonds (integer) |
| AliphaticRings |
Number of aliphatic rings (integer) |
| AromaticRings |
Number of aromatic rings (integer) |
2.2 Modification of the MPNN architecture by enhancing feature learning on the subgraph and edge as well as coupling prior domain knowledge
As outlined above, a conventional MPNN does not update the edge feature and neglects the semantic information involving functional groups in its learning process.22,34 To address these limitations, we modified the MPNN framework by updating the edge feature, and incorporating subgraph learning and prior knowledge to enhance feature extraction.24 As shown in Fig. 1c, our SubOptGraph includes mainly the message passing phase and the readout phase. To conduct the above modifications, SubOptGraph uses three MPNNs in the message passing phase, namely Subgraph MPNN, node-centered MPNN and edge-centered MPNN to extract features, as shown in Fig. 1d. Accordingly, the message passing phase includes three kinds of feature updating: subgraph feature, node feature and edge feature, through which U, V and A are transformed to U′, V′ and A′. In the V transformation, the state feature representing prior knowledge is incorporated.
In the readout phase, we adopt the global attention method to read out V′ and A′.35 As illustrated by Fig. 1e, the feature vectors of every node and edge would be multiplied by the attention coefficient instead of simply summing or averaging; this assigns different scores to each node and edge in order to further optimize hidden embedding. Consequently, two embeddings from the node feature and edge feature are obtained, and then we concatenate them to become the final embedding. Finally, the full-connect layers are applied to predict different optical properties, as highlighted in grey in Fig. 1c.
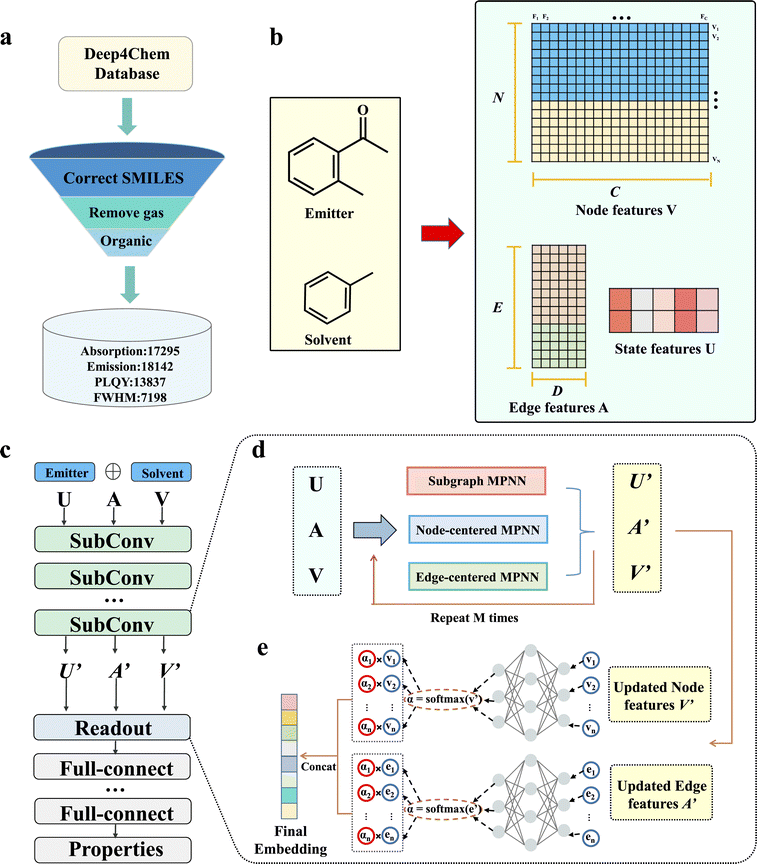
To more clearly exhibit key techniques in the architecture modification, Fig. 2 further illustrates subgraph and edge feature extraction as well as the complementary feature we have proposed. As shown in Fig. 2a, Subgraph MPNN includes three stages: subgraph extraction, subgraph embedding and subgraph aggregation. Subgraphs H[Nk(v)] are extracted from the original molecular graph. And then a function Fsub is used to describe the embedding and aggregation of the subgraph features (see Methods for more details). After iterating the process L times, the aggregated feature is obtained, which is called the graph embedding. Herein, L is determined by hyperparameter optimization. To update the node and edge features, three trainable nonlinear functions, namely Fu, Fv, and Fe, are adopted, as shown in Fig. 2b. Fu represents an MLP that is used to compute the hidden representation of the state feature U derived from prior knowledge to become U′. Then U′ is fused with the node features V through a concatenation operation. Fv and Fe act as graph convolutional functions that propagate and update information from neighbouring nodes/edges to the central nodes/edges. After that, the graph embedding is fused with the node features V′ through element-wise addition, ready for the next updating. After updating the node and edge features M times in both the node-centered MPNN and the edge-centered MPNN, the final node and edge features V′ and A′ are read out by the global attention method to obtain the final embedding, which will be used to predict the optical properties. The details regarding the subgraph function, state feature fusion, graph convolutional functions, and global attention readout function are described in Methods.
 |
| | Fig. 2 Key details of SubOptGraph's message passing phase. (a) The message passing of Subgraph MPNN. The model extracts k-hop graphs as subgraphs first (i.e., H[Nk(v)], see Methods for details), and then obtains three kinds of embedding from every subgraph. After L updates, the three kinds of embedding will be aggregated to become the graph embedding. (b) The message passing of node-centered MPNN and edge-centered MPNN. Fu is the state function realized by an MLP to calculate U′ while Fv and Fe are convolution functions to propagate and update neighboring node/edge features to the central node/edge. U′ is fused with V through a concatenation operation. In the node-centered MPNN, graph embedding from subgraph features is fused with V′ through an addition operation. Thus, after M updates, the original V fused with U′ and A are transformed to V′ and A′. After the readout phase, the final embedding is sent to the MLP to make predictions. | |
3 Results and discussion
3.1 Ablation experiments
In order to evaluate the effectiveness of the proposed modifications, we conducted a series of ablation experiments for the model architecture and the complementary feature based on the Deep4Chem dataset, including edge feature updating (labelled MPNN-Edge), subgraph learning (labelled MPNN-Sub), and state feature coupling (labelled MPNN-State). Table 3 shows results of the ablation experiments. In this work, we use three main metrics to evaluate the model performance: coefficient of determination (R2), mean absolute error (MAE) and root mean square error (RMSE). Details of their calculation are described in ESI.†
Table 3 Results of ablation experiments for the model architecture and the state featurea
| Property |
Models |
R
2
|
MAE |
RMSE |
|
The results are derived from 10-fold validation. For the absorption, emission, and FWHM tasks, the units of MAE and RMSE are nm. The best results are shown in bold. MPNN-State, MPNN-Sub and MPNN-Edge denote MPNN coupled with the five state features from the domain knowledge, subgraph feature learning and edge feature updating, respectively.
|
|
λ
Abs
|
SubOptGraph |
0.977 ± 0.004
|
8.343 ± 0.334
|
15.872 ± 1.471
|
|
|
MPNN-State |
0.961 ± 0.007 |
12.709 ± 1.010 |
20.809 ± 1.776 |
|
|
MPNN-Sub |
0.970 ± 0.010 |
10.006 ± 0.808 |
18.058 ± 2.931 |
|
|
MPNN-Edge |
0.965 ± 0.004 |
12.020 ± 0.506 |
19.761 ± 1.131 |
|
|
MPNN |
0.957 ± 0.008 |
13.641 ± 0.768 |
21.943 ± 2.122 |
|
λ
Emi
|
SubOptGraph |
0.948 ± 0.005
|
12.609 ± 0.415
|
21.435 ± 1.057
|
|
|
MPNN-State |
0.923 ± 0.004 |
17.253 ± 0.294 |
26.197 ± 0.630 |
|
|
MPNN-Sub |
0.940 ± 0.007 |
14.548 ± 0.403 |
22.853 ± 1.036 |
|
|
MPNN-Edge |
0.926 ± 0.005 |
16.362 ± 0.343 |
25.528 ± 0.959 |
|
|
MPNN |
0.916 ± 0.006 |
17.968 ± 0.432 |
27.213 ± 0.900 |
| PLQY |
SubOptGraph |
0.734 ± 0.019
|
0.105 ± 0.003
|
0.159 ± 0.006
|
|
|
MPNN-State |
0.671 ± 0.017 |
0.121 ± 0.003 |
0.175 ± 0.005 |
|
|
MPNN-Sub |
0.683 ± 0.062 |
0.120 ± 0.012 |
0.172 ± 0.016 |
|
|
MPNN-Edge |
0.669 ± 0.023 |
0.121 ± 0.004 |
0.177 ± 0.006 |
|
|
MPNN |
0.658 ± 0.022 |
0.125 ± 0.005 |
0.180 ± 0.006 |
| FWHM |
SubOptGraph |
0.735 ± 0.022
|
9.614 ± 0.380
|
14.787 ± 1.176
|
|
|
MPNN-State |
0.685 ± 0.025 |
10.772 ± 0.595 |
16.133 ± 1.358 |
|
|
MPNN-Sub |
0.699 ± 0.043 |
10.367 ± 0.713 |
15.752 ± 1.364 |
|
|
MPNN-Edge |
0.682 ± 0.023 |
10.815 ± 0.674 |
16.200 ± 1.259 |
|
|
MPNN |
0.663 ± 0.031 |
11.368 ± 0.609 |
16.668 ± 1.426 |
It can be seen from Table 3 that MPNN-Edge, MPNN-Sub and MPNN-State achieve better performance than the conventional MPNN, supporting their effectiveness. In addition, given that the state function includes five state features, we also conducted an ablation experiment for each state feature and the results demonstrate the effectiveness of each state feature in improving the model prediction performance (Table S1†). After fusing all five state features into the MPNN, the model achieves the best performance with respect to any single state feature (Table S1†), showcasing the necessity of simultaneously considering the five state features. In all four tasks, MPNN-Sub exhibits greater improvement than MPNN-Edge or MPNN-State, indicating that the subgraph feature can more effectively enhance feature learning. When our SubOptGraph integrates the edge-updating, subgraph learning and the state feature into the MPNN, the best performance across all the four tasks is achieved (Table 3), in which the improvement is relatively slight for wavelength prediction while the improvement is relatively larger for PLQY and FWHM. Significant improvements can be observed when we compare the conventional MPNN with only molecular graph representation to our SubOptGraph. For example, the absorption wavelength prediction exhibits an obvious increase in R2 from 0.957 to 0.977, a drop in MAE from 13.641 nm to 8.343 nm and a drop in RMSE from 21.943 nm to 15.872 nm. For the emission wavelength, the prediction presents an increase in R2 from 0.916 to 0.948, a drop in MAE from 17.968 nm to 12.609 nm and a drop in RMSE from 27.213 nm to 21.435 nm. The PLQY prediction exhibits an increase in R2 from 0.658 to 0.734, a drop in MAE from 0.125 to 0.105 and a drop in RMSE from 0.180 to 0.159. Similar improvements are observed for the FWHM prediction, as reflected in Table 3. These ablation experiments clearly confirm the effectiveness of our proposed improvement strategy in enhancing the edge and subgraph feature learning and coupling the prior domain knowledge.
3.2 Performance of SubOptGraph and comparison with competitive models on five different datasets
Unlike previous work that tested performance by using only a single dataset, we tested SubOptGraph against five datasets: Deep4Chem, JCIM_Abs, ChemFluor, SMFluo1, and BODIPYs. As evidenced by ESI Table S2,† the five datasets are significantly different. Meanwhile, in order to verify the advantages of our model, our SubOptGraph was compared with existing ML and DL models applied to the five datasets. We also selected random forest (RF) as a representative traditional machine learning algorithm for comparison, as it has been widely used in molecular property prediction with good performance.36–38 Sufficient comparisons and verifications can effectively demonstrate the robustness and generalization of our SubOptGraph. All the results are presented in Table 4.
Table 4 Comparisons of SubOptGraph with competitive models for five different optical databasesa
|
|
Property |
Methods |
Metrics |
|
R
2/rb |
MAE |
RMSE |
|
For absorption, emission and FWHM, the units of MAE and RMSE are nm.
For the Deep4Chem, ChemFluor, and BODIPYs databases, the metric uses the same R2 as the competitive models. For the SMFluo1 database, the metric used is the Pearson coefficient (r), the same as in the competitive model.
The GNN model is established according to the main parameters reported by Joung et al.14
RF used Morgan fingerprints as feature descriptors.
|
| Deep4Chem |
λ
Abs
|
Ours |
0.983 |
8.9 |
14.0 |
|
|
|
GNNc |
0.955 |
13.4 |
22.3 |
|
|
|
Bondstep15 |
0.946 |
12.3 |
27.4 |
|
|
|
RFd |
0.963 |
10.3 |
20.4 |
|
|
λ
Emi
|
Ours |
0.952 |
13.2 |
21.2 |
|
|
|
GNNc |
0.908 |
17.8 |
27.5 |
|
|
|
Bondstep15 |
0.906 |
18.2 |
29.3 |
|
|
|
RFd |
0.928 |
16.2 |
24.6 |
|
|
PLQY |
Ours |
0.763 |
0.106 |
0.150 |
|
|
|
GNNc |
0.650 |
0.110 |
0.179 |
|
|
|
Bondstep15 |
0.718 |
0.263 |
0.398 |
|
|
|
RFd |
0.730 |
0.112 |
0.163 |
|
|
FWHM |
Ours |
0.767 |
9.3 |
13.6 |
|
|
|
GNNc |
0.672 |
10.5 |
16.0 |
|
|
|
RFd |
0.719 |
9.9 |
15.1 |
|
|
λ
Abs
|
Ours |
0.94 |
14.64 |
23.41 |
|
|
|
Chemprop13 |
0.90 |
18.72 |
27.47 |
| BODIPYs |
λ
Abs
|
Ours |
0.97 |
7.2 |
14.6 |
|
|
|
DNN17 |
0.95 |
10.0 |
18.4 |
|
|
|
RFd |
0.94 |
9.1 |
20.0 |
| JCIM_Abs |
λ
Abs
|
Ours |
0.95 |
12.7 |
24.6 |
|
|
|
DR-CNN19 |
0.91 |
14.6 |
31.3 |
|
|
|
RFd |
0.90 |
16.6 |
33.6 |
| ChemFluor |
λ
Abs
|
Ours |
0.962 |
10.33 |
19.34 |
|
|
|
GBRT10 |
0.954 |
10.47 |
23.18 |
|
|
|
RFd |
0.926 |
13.77 |
27.21 |
|
|
λ
Emi
|
Ours |
0.938 |
13.69 |
22.44 |
|
|
|
GBRT10 |
0.925 |
14.31 |
24.77 |
|
|
|
RFd |
0.856 |
20.99 |
34.25 |
|
|
PLQY |
Ours |
0.71 |
0.11 |
0.16 |
|
|
|
GBRT10 |
0.71 |
0.11 |
0.16 |
|
|
|
RFd |
0.65 |
0.13 |
0.18 |
| SMFluo1 |
λ
Abs
|
Ours |
0.992 |
9.23 |
15.62 |
|
|
|
FCNN16 |
0.989 |
9.54 |
17.93 |
|
|
|
RFd |
0.978 |
14.08 |
25.27 |
3.2.1 Comparisons with competitive models on Deep4Chem.
Joung et al.14 first used a conventional MPNN framework to construct their optical property prediction models based on Deep4Chem, which separately conducted the message passing for the chromophore and solvent. Unfortunately, they did not give the model code. To perform the comparison, we established a GNN model following the main parameters provided in their work. We also optimized the GNN model with the Deep4Chem dataset. To compare them as fairly as possible, we adopted the same data splitting method as the competitive work to train and test our model. As shown in Table 4, our model (SubOptGraph) achieves R2 of 0.983, 0.952, 0.763, and 0.767 for absorption wavelength, emission wavelength, PLQY and FWHM, respectively, significantly superior to the GNN model in Joung's work (0.955, 0.908, 0.650 and 0.672) and the RF model (0.963, 0.928, 0.730, 0.719). Our SubOptGraph model also significantly reduces MAE and RMSE with respect to the GNN and RF models, as reflected in Table 4. Hung et al.15 developed a new DL model called Schnet-bondstep (labelled Bondstep in Table 4), which exhibited good performance for predicting three optical properties of Deep4Chem. Their R2 values are 0.946 for absorption wavelength, 0.908 for emission wavelength and 0.718 for PLQY, also lower than our SubOptGraph, along with significantly larger MAE and RMSE than ours (see Table 4). In addition, Greenman et al. developed Chemprop D-MPNN (labelled Chemprop in Table 4) to predict absorption wavelength for the Deep4Chem dataset. They first utilized computational data from TD-DFT for initial training. Then, the model was further trained and tested on Deep4Chem to predict absorption wavelength, which achieved an R2 of 0.90, MAE of 18.72 nm and RMSE of 27.47 nm for the test set. Utilizing the same training and test dataset as them,13 our SubOptGraph obtains an R2 of 0.94, MAE of 14.64 nm and RMSE of 23.41 nm for the test set, also superior to the Chemprop D-MPNN model.
3.2.2 Comparisons with competitive models on four other optical databases.
Besides Deep4Chem, there are other four datasets: BODIPYs, JCIM_Abs ChemFluor and SMFluo1. The BODIPYs database curated by Ksenofontov et al., contains 13339 absorption wavelengths for BODIPY dye molecules.17 JCIM_Abs, as named by us, denotes a combined dataset used in Jung et al.‘s work,19 which contains 26395 experimentally measured absorption wavelengths. ChemFluor, constructed by Ju et al., comprises 4252 absorption wavelengths, 4386 emission wavelengths, and 3090 PLQY.10 SMFluo1, recently developed by Shao et al., consists of 1181 small-molecule fluorophores covering the ultraviolet–visible–near-infrared absorption window.16
For the BODIPYs dataset, Ksenofontov et al. used DNN coupled with consensus descriptors to predict the adsorption wavelength and achieved an R2 of 0.95, MAE of 10 nm and RMSE of 18.4 nm for the 5-fold cross-validation set.17 When applied to BODIPYs our model exhibits higher performance with an R2 of 0.97, MAE of 7 nm and RMSE of 14.5 nm, also superior to the RF model with Morgan fingerprints. For the absorption wavelength of the JCIM_Abs dataset, our SubOptGraph still exhibits better performance than DR-CNN19 or RF, as evidenced by Table 4. For the ChemFluor database, Ju et al. adopted the GBRT algorithm to predict three optical properties, which used a combination feature of the functionalized structure descriptors and comprehensive general solvent descriptors collected from the literature.10 Using the same data splitting method, our model exhibits higher prediction accuracy than the GBRT or RF models for wavelength prediction. For example, the R2 comparisons are 0.962 vs. 0.954 for the absorption wavelength, and 0.938 vs. 0.925 for the emission wavelength. For the PLQY prediction, our model and GBRT exhibit the same R2. However, it should be noted that Ju et al.‘s GBRT model needs laborious feature engineering:10 for example, calculating the descriptors for the organic fluorescent molecule and selecting the solvent descriptors from the literature. While our SubOptGraph mainly uses the molecular graph as the input and automatically updates features, thus being more convenient in practical application.
As for the SMFluo1 database, Shao et al. used FCNN coupled with Morgan and MACCS fingerprints to achieve high accuracy with a Pearson coefficient (r) of 0.989 on the test set for absorption wavelength.16 Using the same data, our SubOptGraph further improves the prediction accuracy with an r of 0.992 and it outperforms RF (0.978). Despite the slight improvement compared to FCNN (which is not unexpected, as their accuracy was already very high), it should be noted that SMFluo1 contains only ∼1200 samples and ChemFluor has only ∼4300 samples. It is known that a small dataset is generally not suitable for DL training and learning.39 However, even in this case, our SubOptGraph can still achieve higher prediction accuracy than traditional descriptor-based methods. Collectively, benefiting from our improvements in feature learning and the integration of prior domain knowledge, our model significantly outperforms competitive models in five different optical datasets, of either large or small size, strongly confirming its robustness and generalization.
3.3 Application and experimental validation for emitters of OLEDs
OLED emitters have attracted tremendous research attention in flat-panel displays and solid-state lighting sources.40 OLED emitters are rich in heavy atoms, and aromatic and heteroaromatic rings with complicated π-conjugated structures, which not only means great difficulty in synthesis, but also affects control over optical properties.41,42 So, it is of great importance to develop new OLED emitters for commercial application, especially for the narrow-emission and highly efficient blue emitters.43,44 Thus, we collected 238 blue OLED emitter/solvent combinations consisting of 179 unique emitters in 48 solvents from the extensive experimental literature as an external test set to verify our model performance for unseen samples. The 238 combinations contain 38 absorption wavelengths, 100 emission wavelengths, 82 PLQYs and 18 FWHMs. These data were loaded as ESI† in the MS Excel format. Unfortunately, the prediction performance was very poor, as shown in Table 5. The reason is mainly the significant differences in the structure between these blue OLED emitters in the external test set and those molecules in the training set of the Deep4Chem database, as evidenced by the similarity comparison in Fig. S1.† Thus, a model trained only on Deep4Chem cannot learn effective knowledge related to blue OLED emitters, leading to very poor performance. Given this issue, we collected another 1114 blue OLED emitter/solvent combinations consisting of 391 unique emitters in 84 solvents from more than 100 pieces of literature as the training set for blue OLED emitters. The training set contains 211 absorption wavelengths, 414 emission wavelengths, 416 PLQYs, and 73 FWHMs, which are also listed in the Excel file as ESI.† We used the blue OLED training set to directly train SubOptGraph and then predict the external test set of blue OLED emitters. It can be seen from Train-OLED in Table 5 that the performance has improved with respect to the pretrained result, but it is still poor. This should be attributed to the fact that the deep learning model requires a large amount of data to support training due to its complex architecture. To tackle this problem, we finally utilized the transfer learning strategy, which has proved to be effective in improving the performance on target domains different from the source domain.45 Specifically, we froze the message passing and readout layers while the parameters of the full-connect layers were initialized randomly. Then, the model was trained on the blue OLED training set. After transfer learning, the performance on the external test set of the blue OLED emitters was remarkably improved, as evidenced by Table 5. The result also showcases the flexibility of our model, which can also utilize the transfer learning strategy to apply it to other emitter fields with significantly different structures from the training dataset. Although the superior performance of our model is sufficiently demonstrated at the computational level, the final goal of the computational work is to play an effective role in practical application. Thus, to gauge the reliability of our model in practice, we conducted further experimental comparison and validation, which has generally been overlooked in previous work on the prediction of optical properties.10,13,15,16
Table 5 The prediction performance of SubOptGraph for the external test set of blue OLED emittersa
| Property |
Conditionsb |
R
2
|
MAE |
RMSE |
|
For absorption wavelength, emission wavelength and FWHM, the units of MAE and RMSE are nm.
Pretrained, Train-OLED, and transfer learning denote the SubOptGraph model trained on Deep4Chem, SubOptGraph directly trained on the training set of blue OLED emitters, and SubOptGraph trained by transfer learning, respectively.
|
|
λ
Abs
|
Pretrained |
−6.60 |
58.67 |
67.09 |
|
|
Train-OLED |
0.48 |
11.44 |
17.56 |
|
|
Transfer learning |
0.92 |
5.73 |
6.95 |
|
λ
Emi
|
Pretrained |
−1.01 |
40.26 |
48.97 |
|
|
Train-OLED |
0.19 |
24.53 |
31.02 |
|
|
Transfer learning |
0.90 |
8.23 |
10.74 |
| FWHM |
Pretrained |
−0.48 |
23.07 |
27.79 |
|
|
Train-OLED |
0.51 |
13.15 |
15.97 |
|
|
Transfer learning |
0.84 |
7.20 |
9.09 |
| PLQY |
Pretrained |
−1.65 |
0.36 |
0.42 |
|
|
Train-OLED |
0.66 |
0.12 |
0.15 |
|
|
Transfer learning |
0.76 |
0.11 |
0.13 |
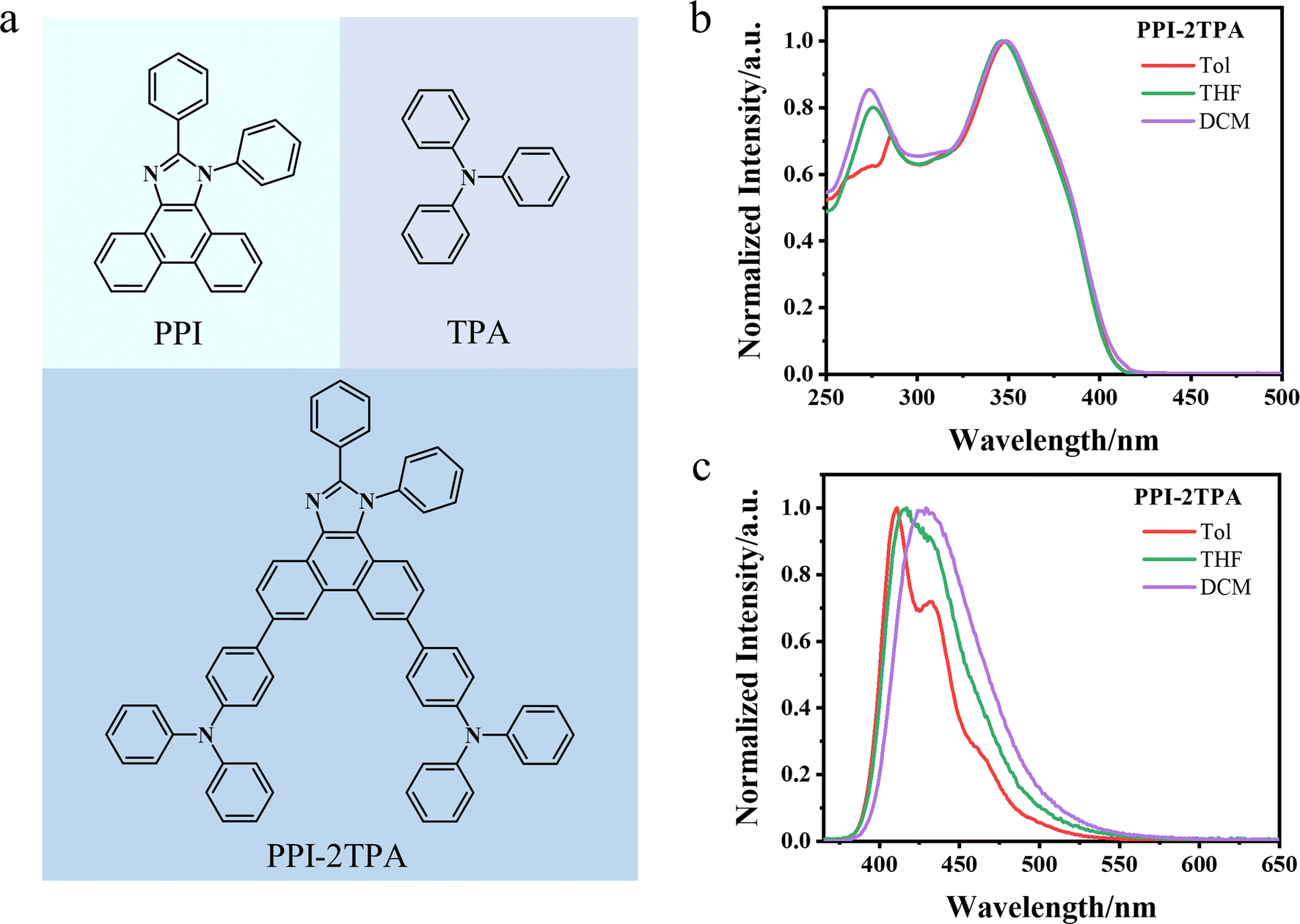
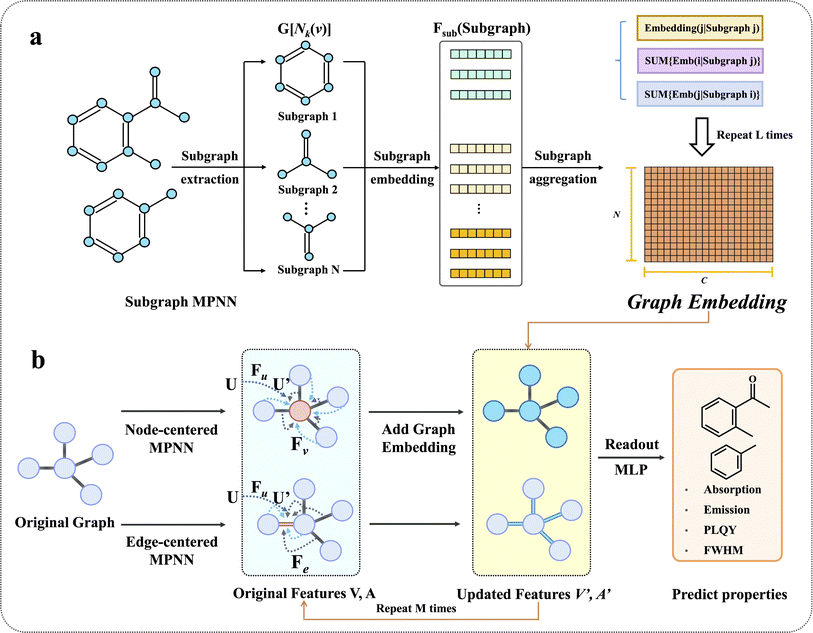
Given the practical need to develop deep blue emitters with high performance in the OLED field,40,46,47 we designed a new potential blue-light-emitting molecule. Specifically, we selected PPI as an acceptor (A) and TPA as a donor (D) to construct a potential blue-light-emitting molecule (PPI-2TPA) with a D–A–D structure (see Fig. 3a), as the two fragments were demonstrated to be associated with deep-blue-light-emission.48,49 As evidenced by the similarity comparison (Fig. S2†), the newly designed compound is different in structure from the existing blue OLED molecules under study. We first used the optimized SubOptGraph to rapidly predict its optical properties in different solvents. Given that the experimental characterization of the absorption wavelength, emission wavelength and FWHM is typically done in dilute solvents while PLQY is generally characterized in the solid state, we finally chose three solvents (toluene, tetrahydrofuran, and dichloromethane) for absorption wavelength, emission wavelength and FWHM while PLQY was determined in a poly(methyl methacrylate) (PMMA) matrix. As shown in Table 6, the emission wavelengths predicted by our model are in the range of 419–428 nm in the three different solvents, showing a deep blue light range.50 The FWHM values predicted by SubOptGraph range from 48 to 57 nm while the PLQY value predicted is 100% in PMMA, suggesting that PPI-2TPA is a potential high-performance deep-blue-light-emitting molecule. Inspired by the prediction results, we successfully synthesized PPI-2TPA and characterized it experimentally. Details of the experimental synthesis details are placed in the Methods section. Fig. 3b and c show the experimental absorption and photoluminescence spectra in the three solvents. The experimentally determined PLQY and FWHM data are also listed in Table 6. The molecular structure of the PPI-2TPA we synthesized was confirmed by a combination of 1H and 13C nuclear magnetic resonance (NMR) as well as high resolution mass spectrometry (HRMS) (Fig. S3–S5†). The details of all the experimental characterizations are described in ESI.† It can be seen from Table 6 that our predicted values are very close to the experimental ones, with 2–6 nm errors for the absorption wavelengths, 1–8 nm errors for the emission wavelengths and 1–4 nm errors for the FWHM. The predicted PLQY value differs from the experimental one by 0.10, which is an acceptable level. The experimental results confirm that PPI-2TPA is a deep blue OLED emitter with good PLQY and acceptable FWHM, which provides a new emitter candidate for the development of deep blue OLEDs. Also, it strongly validates the reliability of our ML model in practical application, indicating that our model can serve as a quick and reliable tool for predicting optical properties in OLED emitter fields. For comparison, we also employed the quantum mechanical (QM) method to calculate the absorption and emission wavelengths of PPI-2TPA (see ESI† for QM calculation details). Table 6 indicates that the QM-calculated wavelengths are smaller than the ML-predicted ones, presenting a larger difference from the experimental ones. In addition, we compared correlation coefficients between the computational values (SubOptGraph and QM) and the experimental ones for PPI-2TPA, as shown in Table S3.† The values predicted by SubOptGraph present much higher correlation coefficients with the experimental ones than the values calculated by QM, further confirming the better prediction performance of our ML model than QM for the new molecule PPI-2TPA. Furthermore, it should be noted that the QM calculations take several hours on 64 CPU cores for each molecule, while our model prediction is on the scale of seconds with the NVIDIA GeForce RTX 4090 GPU, showcasing the much greater speed in application, suitable for high-throughput screening.
 |
| | Fig. 3 The structure and spectra of PPI-2TPA. (a) The structure of PPI-2TPA. PPI is the acceptor and TPA is the donor. (b) The absorption spectra of PPI-2TPA in three solvents. (c) The photoluminescence spectra of PPI-2TPA in three solvents. | |
Table 6 The experimental values and predicted values of PPI-2TPAa
| Property |
Solventsb |
Experimental |
Calculatedc |
Predictedd |
|
For absorption wavelength, emission wavelength and FWHM, the unit is nm.
The absorption wavelength, emission wavelength and FWHM are measured in three solvents (the concentration is 10−5 M), and the PLQY is measured with 1 wt%-doped thin film in a PMMA matrix.
The data are derived from QM calculation.
The data are predicted by our SubOptGraph.
|
|
λ
Abs
|
Toluene |
349 |
332 |
355 |
|
|
Tetrahydrofuran |
346 |
332 |
343 |
|
|
Dichloromethane |
348 |
332 |
346 |
|
λ
Emi
|
Toluene |
411 |
396 |
419 |
|
|
Tetrahydrofuran |
417 |
406 |
423 |
|
|
Dichloromethane |
429 |
407 |
428 |
| FWHM |
Toluene |
44 |
— |
48 |
|
|
Tetrahydrofuran |
54 |
— |
55 |
|
|
Dichloromethane |
61 |
— |
57 |
| PLQY |
PMMA |
0.90 |
— |
1.0 |
4 Conclusions
To improve the accuracy of prediction for optical properties and to address the limitations of a widely used paradigm (MPNN) of a graph neural network in feature learning, which focuses mainly on node features, our SubOptGraph embeds chemistry-intuitive feature learning into the MPNN architecture by adding subgraph learning, updating edge learning and coupling prior domain knowledge into the end-to-end molecular graph learning. With these improvements, the structural features associated with the optical properties can be better extracted from a limited dataset. Consequently, SubOptGraph achieves the highest accuracy to date for four important optical properties, in which the R2 values of the independent test set are 0.983 for the absorption wavelength, 0.952 for the emission wavelength, 0.763 for PLQY and 0.767 for FWHM in the largest optical experiment database (Deep4Chem). Unlike previous ML work that mainly used a single dataset to validate the model performance, we used five different optical datasets to verify the robustness and generalization of SubOptGraph. Benefiting from these technical advantages, our SubOptGraph greatly outperforms the eight competitive models in the five different optical databases. Furthermore, our SubOptgraph is also flexible, and can be applied to other emitter fields significantly different from the training data by transfer learning, further confirming its generalization. More importantly, we also conducted experimental synthesis and characterization to verify the reliability of our model in practical application, which has generally been overlooked in previous ML work. The experimental results strongly validate the application potential of our SubOptGraph in practice. All source codes and blue OLED emitter data under study are freely available at https://github.com/Jo3690/SOG. We expect that they will become useful tools for aiding the design of new emitters with desired optical properties for a variety of fields.
5 Methods
5.1 Subgraph function Fsub
The subgraph function Fsub is used for subgraph feature extraction. First, subgraphs are extracted from the original graph, which can be expressed as eqn (1):| | | Sub(l)[v] = G(l)[Nk(v)] | (1) |
where G(l)[Nk(v)] represents the l-th layer k-hop subgraphs centered at node v. The k-hop subgraph is a graph consisting of the central node v and its neighboring nodes, while k-hop means the steps from the central node v to the farthest node.
Then, for subgraph embedding, a GNN (i.e., MPNN) is used to extract the subgraph features, including three encoding processes: subgraph encoding, centroid encoding and context encoding. F subgraph encoding can be described by eqn (2):
| | | h(l+1)|Subgraph = GNN(i|Sub(l)[j]) | (2) |
where GNN(
i|Sub
(l)[
j]) represents the embedding of node
i in the
l-th layer subgraph centered at node
j (
i.e., Sub
(l)[
j],
i ≠
j).
Secondly, for Sub(l)[j], the embedding of node j is also extracted. Thus, centroid encoding is defined as eqn (3):
| | | h(l)|Centroid = GNN(j|Sub(l)[j]) | (3) |
where GNN(
j|Sub
(l)[
j]) represents the embedding of node
j in the
l-th layer subgraph centered at node
j.
Finally, to capture the information about node v in different subgraphs (i.e., context encoding), the information about node v is condensed in terms of eqn (4):
| | | h(l+1)v|Context = GNN(v|Sub(l)[j])|∀j s.t. v ∈ Nk(j) | (4) |
where ∀
j s.t.
v ∈
Nk(
j) means that node
v is in the different subgraphs centered at node
j.
Considering that the distance-to-centroid of the k-hop subgraph has been calculated and proved to be essential for augmenting the node features, we have fused this feature with the eventual embedding, and made a gate mechanism to subgraph encoding and context encoding, in order to control the contributions of different nodes, as described by eqn (5) and (6):
| | | h(l+1)|Subgraphv = Sigmoid(d(l)v|j)⊙GNN(i|Sub(l)[j]) | (5) |
| | 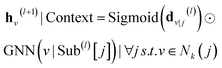 | (6) |
where Sigmoid and ⊙ denote the activation function and element-wise multiplication, respectively.
d(l)v|j represents the distance between node
v and node
j in the
l-th layer.
For subgraph aggregation, the three kinds of features are condensed to graph-level embedding, which is described by eqn (7):
| | | h(l+1)sub_v = FUSE(d(l+1)i|j, h(l+1)v|Centroid, h(l+1)v|Subgraph, h(l+1)v|Context) | (7) |
where FUSE indicates the concatenation operation.
Finally, the node features and subgraph features are fused in terms of eqn (8):
| | | x(l+1)i,out = x(l+1)i + h(l+1)sub_v | (8) |
where
x(l+1)i,out are the updated node features that are fused with the subgraph features.
5.2 State feature fusion
The state features are derived from experimental findings for organic emitters that reveal important structural factors contributing to the optical properties, thus representing prior domain knowledge. They are different from the conventional molecular descriptors, which are calculated according to some common rules to represent the overall structural features of a molecule, rather than focusing on important structural factors. In addition, the state features are integrated with node features during the message passing phase to conduct feature extraction while the descriptor-based neural network updates only the descriptors, and does not involve integration with feature updating of other types. Herein, MLP is used as the state function Fu to make the nonlinear transformation for the state features of the emitters and solvents. It is defined by eqn (9):where u is the state features and uout denotes the updated state feature. MLP is a neural network with the Relu activation function. Before the message passing phase of node-centered MPNN and edge-centered MPNN, the node features are fused with the state feature through a concatenation operation, as expressed by eqn (10)where xi and ui,out refer to the node features and state features of node i, respectively. ⊕ denotes the concatenation operation.
5.3 Graph convolutional functions Fv and Fe
Herein, Fv and Fe are used for the message passing of the node-centered and edge-centered MPNN. The message passing phase of node-centered MPNN can be described as eqn (11):| | | x(l+1)i = ϕ(l)(x(l)i, f(l)(x(l)i, x(l)j, eji)) | (11) |
where i is the central node and j is the neighboring node. xi and xj represent the node features of node i and node j, respectively. eji is the edge feature between node j and i. l is the number of layers. ϕ(l) and f(l) are the l-th layer update and aggregation functions, respectively.
For the edge feature, several studies have focused on edge learning to harness the graph edge information. The weave model proposed by Kearnes et al. utilized multiple MLPs to incorporate the edge feature with the node feature,51 but not for the final prediction. EGNN constructed by Gong and Cheng mainly used double stochastic normalization of graph edge features to realize edge updating,52 but its architecture is complex. CD-MVGNN developed by Ma et al.24 used an edge-central encoder to conduct message passing for edge features with a relatively simple architecture, achieving an improvement in prediction of drug properties. Thus, similar to CD-MVGNN, we also adopted the edge-centered MPNN by aggregating the messages from all the neighboring edges to the central edge, as defined by eqn (12):
| | | e(l+1)ij = ϕ(l)(e(l)ij, f(l)(x(l)i, x(l)j, eki)) | (12) |
where
k represents the neighboring nodes of node
i, and
eki is the edge feature between node
k and
i.
5.4 Global attention readout function
For the readout phase, we adopted the global attention mechanism to extract the embeddings from the node-centered MPNN and edge-centered MPNN. Global attention can calculate the coefficients of each node and edge based on the node and edge features, as shown by eqn (13) and (14):| | | αv,i = Softmax(MLP(vi)) | (13) |
| | | αe,i = Softmax(MLP(ei)) | (14) |
where Softmax denotes the activation function. αv,i and αe,i are the coefficients of node v and edge e of the i-th dimension, respectively.
Then the embeddings are obtained by summing the product of the coefficient and the corresponding feature, which can be expressed as eqn (15) and (16):
| | 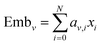 | (15) |
| | 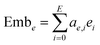 | (16) |
where N is the dimension of the node features and
E is the dimension of the edge features.
xi and
ei are the node and edge features of the
i-th dimension, respectively. Emb
e and Emb
v are then concatenated to become the final embedding, as shown by
eqn (17):
5.5 Learning curve for the training set
The data were divided in a ratio of 8:2 for training and testing. We performed a learning curve for all four optical properties of the Deep4Chem dataset on the five models, including our SubOptGraph and its four ablated versions (MPNN, MPNN-State, MPNN-Edge, MPNN-Sub). All the results are shown in Fig. S6.† It can be seen that all five models perform better with an increasing amount of training data, in which SubOptGraph achieves the best performance. In addition, it should be noted that the performances of all five models approach convergence when using all the training data, suggesting that the size of the training dataset should be reasonable for all the tasks under study.
5.6 The synthesis of PPI-2TPA
As illustrated by Fig. 4, a mixture of (4-(diphenylamino)phenyl)boronic acid (875 mg, 3.03 mmol), 3,6-dibromophenanthrene-9,10-dione (500 mg, 1.36 mmol), Pd(PPh3)4 (75 mg, 0.06 mmol) and Cs2CO3 (1.56 g, 4.80 mmol) were dissolved in toluene (9 mL), water (3 mL) and ethanol (3 mL). The reaction mixture was degassed with argon and stirred at 100 °C for 12 h. When the reaction was complete, the mixture was poured into water. After extraction with CH2Cl2 (15 mL × 3), the resultant organic phase was washed with brine and dried over anhydrous Na2SO4. After removing the solvent, the residue was purified using column chromatography on silica gel employing CH2Cl2/petroleum ether (PE) (1/1) as an eluent to afford a deep red powdery solid (Compound 1) with a yield of 53%. Then, benzaldehyde (297 mg, 2.8 mmol), Compound 1 (2.01 g, 2.9 mmol), aniline (119 mg, 12.8 mmol) and ammonium acetate (1.19 g, 10.2 mmol) were dissolved in acetic acid (30 mL) and refluxed at 120 °C for 2 hours. After completion of the reaction, the mixture was poured into water. After extraction with CH2Cl2 (30 mL × 3), the resultant organic phase was washed with brine and dried over anhydrous Na2SO4. After removing the solvent, the residue was purified using column chromatography on silica gel employing CH2Cl2/PE (1/3) as an eluent to afford a white powdery solid (PPI-2TPA) with a yield of 60%. The structure of the synthesized PPI-2TPA was confirmed by 1H NMR, 13C NMR and HRMS, as shown in Fig. S3–S5 in ESI.†1H NMR (400 MHz, DMSO-d6) δ(ppm): 9.22 (s, 1H), 9.18 (s, 1H), 8.74 (d, J = 8.0 Hz, 1H), 8.07 (dd, J1 = 8.4 Hz, J2 = 1.6 Hz, 1H), 7.94 (m, 2H), 7.87 (m, 2H), 7.75–7.69 (m, 5H), 7.65 (dd, J1 = 8.8 Hz, J2 = 1.6 Hz, 1H), 7.60 (m, 2H), 7.39–7.32 (m, 11H), 7.16–7.05 (m, 17H). 13C NMR (200 MHz, DMSO-d6) δ(ppm): 150.6, 147.0, 146.9, 146.7, 146.6, 138.0, 137.0, 136.3, 134.4, 133.6, 130.3, 130.2, 130.1, 129.5, 129.1, 129.0, 128.3, 128.1, 127.8, 126.2, 125.6, 125.1, 124.1, 124.0, 123.4, 123.2, 123.12, 123.05, 122.6, 121.7, 121.3, 121.0, 120.7. HRMS (ESI): calcd.: 857.3639 [M + H]+; found: 857.3640.
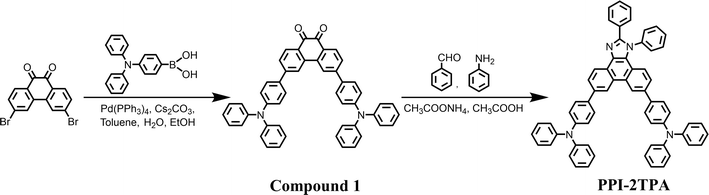 |
| | Fig. 4 The synthesis route of PPI-2TPA. | |
5.7 Model implementation
We implemented our graph neural network model using the PyTorch 1.10 DL framework and training the models on the NVIDIA GeForce RTX 4090 GPU.53 The model was optimized by a grid search method. All codes and related data are available at the Github repository.
Data availability
Collected OLED emitter data can be found in ESI.† The codes and related data are available in the Github repository at https://github.com/Jo3690/SOG.
Author contributions
Ming Sun and Caixia Fu contributed equally to this work. Ming Sun: conceptualization, formal analysis, investigation, methodology, model, writing – original draft; Caixia Fu: synthesis, characterization; Haoming Su: formal analysis, investigation; Rueyue Xiao: data curation, methodology. Chaojie Shi: data curation, investigation. Zhiyun Lu: funding acquisition, project administration, review & editing. Xuemei Pu: funding acquisition, project administration, writing – review & editing, conceptualization, supervision.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This project was supported by National Natural Science Foundation of China (No. 62475177), Sichuan International Science and Technology Innovation Cooperation Project (No. 24GJHZ0431) and the National Natural Science Foundation of China (No. 22075187). Dr Pengchi Deng of Analytical & Testing Centre, Sichuan University is gratefully acknowledged for NMR characterizations.
References
- J. Shinar and R. Shinar, Organic light-emitting devices (OLEDs) and OLED-based chemical and biological sensors: an overview, J. Phys. D: Appl. Phys., 2008, 41, 133001 CrossRef.
- H. Li, Y. Kim, H. Jung, J. Y. Hyun and I. Shin, Near-infrared (NIR) fluorescence-emitting small organic molecules for cancer imaging and therapy, Chem. Soc. Rev., 2022, 51, 8957–9008 RSC.
- J. Munshi, W. Chen, T. Chien and G. Balasubramanian, Transfer Learned Designer Polymers For Organic Solar Cells, J. Chem. Inf. Model., 2021, 61, 134–142 CrossRef CAS PubMed.
- R. Gómez-Bombarelli, J. Aguilera-Iparraguirre, T. D. Hirzel, D. Duvenaud, D. Maclaurin, M. A. Blood-Forsythe, H. S. Chae, M. Einzinger, D.-G. Ha, T. Wu, G. Markopoulos, S. Jeon, H. Kang, H. Miyazaki, M. Numata, S. Kim, W. Huang, S. I. Hong, M. Baldo, R. P. Adams and A. Aspuru-Guzik, Design of efficient molecular organic light-emitting diodes by a high-throughput virtual screening and experimental approach, Nat. Mater., 2016, 15, 1120–1127 CrossRef PubMed.
- B. Bauer, S. Bravyi, M. Motta and G. K.-L. Chan, Quantum Algorithms for Quantum Chemistry and Quantum Materials Science, Chem. Rev., 2020, 120, 12685–12717 CrossRef CAS PubMed.
- C. Gao, X. Min, M. Fang, T. Tao, X. Zheng, Y. Liu, X. Wu and Z. Huang, Innovative Materials Science via Machine Learning, Adv. Funct. Mater., 2022, 32, 2108044 CrossRef CAS.
- M. Sajjan, J. Li, R. Selvarajan, S. H. Sureshbabu, S. S. Kale, R. Gupta, V. Singh and S. Kais, Quantum machine learning for chemistry and physics, Chem. Soc. Rev., 2022, 51, 6475–6573 RSC.
- C. W. Coley, W. Jin, L. Rogers, T. F. Jamison, T. S. Jaakkola, W. H. Green, R. Barzilay and K. F. Jensen, A graph-convolutional neural network model for the prediction of chemical reactivity, Chem. Sci., 2019, 10, 370–377 RSC.
- Z.-R. Ye, I.-S. Huang, Y.-T. Chan, Z.-J. Li, C.-C. Liao, H.-R. Tsai, M.-C. Hsieh, C.-C. Chang and M.-K. Tsai, Predicting the emission wavelength of organic molecules using a combinatorial QSAR and machine learning approach, RSC Adv., 2020, 10, 23834–23841 RSC.
- C.-W. Ju, H. Bai, B. Li and R. Liu, Machine Learning Enables Highly Accurate Predictions of Photophysical Properties of Organic Fluorescent Materials: Emission Wavelengths and Quantum Yields, J. Chem. Inf. Model., 2021, 61, 1053–1065 CrossRef CAS PubMed.
- L. Zhang, J. Tan, D. Han and H. Zhu, From machine learning to deep learning: progress in machine intelligence for rational drug discovery, Drug Discovery Today, 2017, 22, 1680–1685 CrossRef PubMed.
- Y. Jiang, Z. Yang, J. Guo, H. Li, Y. Liu, Y. Guo, M. Li and X. Pu, Coupling complementary strategy to flexible graph neural network for quick discovery of coformer in diverse co-crystal materials, Nat. Commun., 2021, 12, 5950 CrossRef CAS PubMed.
- K. P. Greenman, W. H. Green and R. Gómez-Bombarelli, Multi-fidelity prediction of molecular optical peaks with deep learning, Chem. Sci., 2022, 13, 1152–1162 RSC.
- J. F. Joung, M. Han, J. Hwang, M. Jeong, D. H. Choi and S. Park, Deep Learning Optical Spectroscopy Based on Experimental Database: Potential Applications to Molecular Design, JACS Au, 2021, 1, 427–438 CrossRef CAS PubMed.
- S.-H. Hung, Z.-R. Ye, C.-F. Cheng, B. Chen and M.-K. Tsai, Enhanced Predictions for the Experimental Photophysical Data Using the Featured Schnet-Bondstep Approach, J. Chem. Theory Comput., 2023, 19, 4559–4567 CrossRef CAS PubMed.
- J. Shao, Y. Liu, J. Yan, Z.-Y. Yan, Y. Wu, Z. Ru, J.-Y. Liao, X. Miao and L. Qian, Prediction of Maximum Absorption Wavelength Using Deep Neural Networks, J. Chem. Inf. Model., 2022, 62, 1368–1375 CrossRef CAS PubMed.
- A. A. Ksenofontov, M. M. Lukanov, P. S. Bocharov, M. B. Berezin and I. V. Tetko, Deep neural network model for highly accurate prediction of BODIPYs absorption, Spectrochim. Acta, Part A, 2022, 267, 120577 CrossRef CAS PubMed.
- J. F. Joung, M. Han, M. Jeong and S. Park, Experimental database of optical properties of organic compounds, Sci. Data, 2020, 7, 295 CrossRef CAS PubMed.
- S. G. Jung, G. Jung and J. M. Cole, Automatic Prediction of Peak Optical Absorption Wavelengths in Molecules Using Convolutional Neural Networks, J. Chem. Inf. Model., 2024, 64, 1486–1501 CrossRef CAS PubMed.
- C. Shorten and T. M. Khoshgoftaar, A survey on Image Data Augmentation for Deep Learning, J. Big Data, 2019, 6, 60 CrossRef.
- F. H. Sinz, X. Pitkow, J. Reimer, M. Bethge and A. S. Tolias, Engineering a Less Artificial Intelligence, Neuron, 2019, 103, 967–979 CrossRef CAS PubMed.
- J. Wu, Y. Wan, Z. Wu, S. Zhang, D. Cao, C.-Y. Hsieh and T. Hou, MF-SuP-pKa: Multi-fidelity modeling with subgraph pooling mechanism for pKa prediction, Acta Pharm. Sin. B, 2023, 13, 2572–2584 CrossRef CAS PubMed.
-
J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals and G. E. Dahl, Neural message passing for quantum chemistry, arXiv, 2017, preprint, arXiv:1704.01212, DOI:10.48550/arXiv.1704.01212.
- H. Ma, Y. Bian, Y. Rong, W. Huang, T. Xu, W. Xie, G. Ye and J. Huang, Cross-dependent graph neural networks for molecular property prediction, Bioinformatics, 2022, 38, 2003–2009 CrossRef CAS PubMed.
-
L. Zhao, W. Jin, L. Akoglu and N. Shah, From stars to subgraphs: uplifting any GNN with local structure awareness, arXiv, 2022, preprint, arXiv:2110.03753, DOI:10.48550/arXiv.2110.03753.
- D. Bzdok, M. Krzywinski and N. Altman, Machine learning: a primer, Nat. Methods, 2017, 14, 1119–1120 CrossRef CAS PubMed.
- P. Xu, X. Ji, M. Li and W. Lu, Small data machine learning in materials science, npj Comput. Mater., 2023, 9, 42 CrossRef.
- J. Guo, M. Sun, X. Zhao, C. Shi, H. Su, Y. Guo and X. Pu, General Graph Neural Network-Based Model To Accurately Predict Cocrystal Density and Insight from Data Quality and Feature Representation, J. Chem. Inf. Model., 2023, 63, 1143–1156 CrossRef CAS PubMed.
- Z. Xiong, D. Wang, X. Liu, F. Zhong, X. Wan, X. Li, Z. Li, X. Luo, K. Chen, H. Jiang and M. Zheng, Pushing the Boundaries of Molecular Representation for Drug Discovery with the Graph Attention Mechanism, J. Med. Chem., 2020, 63, 8749–8760 CrossRef CAS PubMed.
- Z. Wu, B. Ramsundar, E. N. Feinberg, J. Gomes, C. Geniesse, A. S. Pappu, K. Leswing and V. Pande, MoleculeNet: a benchmark for molecular machine learning, Chem. Sci., 2018, 9, 513–530 RSC.
- S. Guo, L. Wang, Q. Deng, G. Wang, X. Tian, X. Wang, Z. Liu, M. Zhang, S. Wang, Y. Miao, J. Zhu and H. Wang, Exploiting heterocycle aromaticity to fabricate new hot exciton materials, J. Mater. Chem. C, 2023, 11, 6847–6855 RSC.
- T. Jousselin-Oba, M. Mamada, K. Wright, J. Marrot, C. Adachi, A. Yassar and M. Frigoli, Synthesis, Aromaticity, and Application of peri-Pentacenopentacene: Localized Representation of Benzenoid Aromatic Compounds, Angew. Chem., Int. Ed., 2022, 61, e202112794 CrossRef CAS PubMed.
- Y.-P. Zhang, X. Liang, X.-F. Luo, S.-Q. Song, S. Li, Y. Wang, Z.-P. Mao, W.-Y. Xu, Y.-X. Zheng, J.-L. Zuo and Y. Pan, Chiral Spiro-Axis Induced Blue Thermally Activated Delayed Fluorescence Material for Efficient Circularly Polarized OLEDs with Low Efficiency Roll-Off, Angew. Chem., Int. Ed., 2021, 60, 8435–8440 CrossRef CAS PubMed.
-
D. Chen, L. O'Bray and K. Borgwardt, Structure-aware Transformer for graph representation learning, Proceedings of the 39th International Conference on Machine Learning, 2022, pp. 3469–3489 Search PubMed.
-
Y. Li, D. Tarlow, M. Brockschmidt and R. S. Zemel, Gated graph sequence neural networks, arXiv, 2015, preprint, arXiv:1511.05493, DOI:10.48550/arXiv.1511.05493.
- S. Xu, X. Liu, P. Cai, J. Li, X. Wang and B. Liu, Machine-Learning-Assisted Accurate Prediction of Molecular Optical Properties upon Aggregation, Advanced Science, 2022, 9, 2101074 CrossRef CAS PubMed.
- X. Li, S. Zhang, L. Xu and X. Hong, Predicting Regioselectivity in Radical C–H Functionalization of Heterocycles through Machine Learning, Angew. Chem., Int. Ed., 2020, 59, 13253–13259 CrossRef CAS PubMed.
- Y. Lu, S. Anand, W. Shirley, P. Gedeck, B. P. Kelley, S. Skolnik, S. Rodde, M. Nguyen, M. Lindvall and W. Jia, Prediction of pKa Using Machine Learning Methods with Rooted Topological Torsion Fingerprints: Application to Aliphatic Amines, J. Chem. Inf. Model., 2019, 59, 4706–4719 CrossRef CAS PubMed.
-
L. Brigato and L. Iocchi, A close look at deep learning with small data, arXiv, 2020, preprint, arXiv:2003.12843, DOI:10.48550/arXiv.2003.12843.
- M. Zhu and C. Yang, Blue fluorescent emitters: design tactics and applications in organic light-emitting diodes, Chem. Soc. Rev., 2013, 42, 4963 RSC.
- D. Chen, W. Li, L. Gan, Z. Wang, M. Li and S.-J. Su, Non-noble-metal-based organic emitters for OLED applications, Mater. Sci. Eng., R, 2020, 142, 100581 CrossRef.
- S. Liu, X. Zhang, C. Ou, S. Wang, X. Yang, X. Zhou, B. Mi, D. Cao and Z. Gao, Structure–Property Study on Two New D–A Type Materials Comprising Pyridazine Moiety and the OLED Application as Host, ACS Appl. Mater. Interfaces, 2017, 9, 26242–26251 CrossRef CAS PubMed.
- R. K. Konidena and K. R. Naveen, Boron-Based Narrowband Multiresonance Delayed Fluorescent Emitters for Organic Light-Emitting Diodes, Adv. Photonics Res., 2022, 3, 2200201 CrossRef CAS.
- H. Tan, G. Yang, Y. Deng, C. Cao, J. Tan, Z. Zhu, W. Chen, Y. Xiong, J. Jian, C. Lee and Q. Tong, Deep-Blue OLEDs with Rec.2020 Blue Gamut Compliance and EQE Over 22% Achieved by Conformation Engineering, Adv. Mater., 2022, 34, 2200537 CrossRef CAS.
- F. Zhuang, Z. Qi, K. Duan, D. Xi, Y. Zhu, H. Zhu, H. Xiong and Q. He, A Comprehensive Survey on Transfer Learning, Proc. IEEE, 2021, 109, 43–76 Search PubMed.
- J. U. Kim, I. S. Park, C.-Y. Chan, M. Tanaka, Y. Tsuchiya, H. Nakanotani and C. Adachi, Nanosecond-time-scale delayed fluorescence molecule for deep-blue OLEDs with small efficiency rolloff, Nat. Commun., 2020, 11, 1765 CrossRef CAS PubMed.
- H. Lim, H. J. Cheon, S. Woo, S. Kwon, Y. Kim and J. Kim, Highly Efficient Deep-Blue OLEDs using a TADF Emitter with a Narrow Emission Spectrum and High Horizontal Emitting Dipole Ratio, Adv. Mater., 2020, 32, 2004083 CrossRef CAS.
- W. Li, D. Liu, F. Shen, D. Ma, Z. Wang, T. Feng, Y. Xu, B. Yang and Y. Ma, A Twisting Donor-Acceptor Molecule with an Intercrossed Excited State for Highly Efficient, Deep-Blue Electroluminescence, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef CAS.
- H. Liu, Q. Bai, L. Yao, H. Zhang, H. Xu, S. Zhang, W. Li, Y. Gao, J. Li, P. Lu, H. Wang, B. Yang and Y. Ma, Highly efficient near ultraviolet organic light-emitting diode based on a meta-linked donor–acceptor molecule, Chem. Sci., 2015, 6, 3797–3804 RSC.
- F. Yuan, Y.-K. Wang, G. Sharma, Y. Dong, X. Zheng, P. Li, A. Johnston, G. Bappi, J. Z. Fan, H. Kung, B. Chen, M. I. Saidaminov, K. Singh, O. Voznyy, O. M. Bakr, Z.-H. Lu and E. H. Sargent, Bright high-colour-purity deep-blue carbon dot light-emitting diodes via efficient edge amination, Nat. Photonics, 2020, 14, 171–176 CrossRef CAS.
- S. Kearnes, K. McCloskey, M. Berndl, V. Pande and P. Riley, Molecular Graph Convolutions: Moving Beyond Fingerprints, J. Comput. Aided Mol. Des., 2016, 30, 595–608 CrossRef CAS.
-
L. Gong and Q. Cheng, Exploiting Edge Features for Graph Neural Networks, IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), 2019, pp. 9203–9211 Search PubMed.
- A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury, G. Chanan, T. Killeen, Z. Lin, N. Gimelshein, L. Antiga, A. Desmaison, A. Kopf, E. Yang, Z. DeVito, M. Raison, A. Tejani, S. Chilamkurthy, B. Steiner, L. Fang, J. Bai and S. Chintala, PyTorch: An Imperative Style, High-Performance Deep Learning Library, Adv. Neural Inf. Process. Syst., 2019, 32, 8026–8037 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Model evaluation metrics, experimental characterization details, 1H NMR, 13C NMR, and HRMS spectra, QM computational details, supporting tables and figures, collected OLED emitter data. See DOI: https://doi.org/10.1039/d4sc02781g |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Xuemei
Pu
* and
Xuemei
Pu