
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnprecedented selectivity for homologous lectin targets: differential targeting of the viral receptors L-SIGN and DC-SIGN†
Clara
Delaunay‡
a,
Sara
Pollastri‡
 b,
Michel
Thépaut
a,
Gianluca
Cavazzoli
b,
Laura
Belvisi
b,
Clémentine
Bouchikri
a,
Nuria
Labiod
c,
Fatima
Lasala
c,
Ana
Gimeno
d,
Antonio
Franconetti
d,
Jesús
Jiménez-Barbero
def,
Ana
Ardá
de,
Rafael
Delgado
cg,
Anna
Bernardi
*b and
Franck
Fieschi
*ah
b,
Michel
Thépaut
a,
Gianluca
Cavazzoli
b,
Laura
Belvisi
b,
Clémentine
Bouchikri
a,
Nuria
Labiod
c,
Fatima
Lasala
c,
Ana
Gimeno
d,
Antonio
Franconetti
d,
Jesús
Jiménez-Barbero
def,
Ana
Ardá
de,
Rafael
Delgado
cg,
Anna
Bernardi
*b and
Franck
Fieschi
*ah
aUniversité Grenoble Alpes, CNRS, CEA, Institut de Biologie Structurale, Grenoble, France. E-mail: franck.fieschi@ibs.fr
bUniversità degli Studi di Milano, Dipartimento di Chimica, via Golgi 19, Milano, Italy. E-mail: anna.bernardi@unimi.it
cInstituto de Investigacion Hospital Universitario 12 de Octubre, Universidad Complutense, School of Medicine, Madrid, Spain
dChemical Glycobiology Lab, Center for Cooperative Research in Biosciences (CIC bioGUNE), Basque Research and Technology Alliance (BRTA), 48160 Derio, Bizkaia, Spain
eIkerbasque, Basque Foundation for Science, Bilbao, Spain
fCentro de Investigacion Biomedica En Red de Enfermedades Respiratorias, 28029 Madrid, Spain
gSchool of Medicine, Universidad Complutense, Madrid, Spain
hInstitut Universitaire de France (IUF), Paris, France
First published on 27th August 2024
Abstract
DC-SIGN (CD209) and L-SIGN (CD209L) are two C-type lectin receptors (CLRs) that facilitate SARS-CoV-2 infections as viral co-receptors. SARS-CoV-2 manipulates both DC-SIGN and L-SIGN for enhanced infection, leading to interest in developing receptor antagonists. Despite their structural similarity (82% sequence identity), they function differently. DC-SIGN, found in dendritic cells, shapes the immune response by recognizing pathogen-associated carbohydrate patterns. In contrast, L-SIGN, expressed in airway epithelial endothelial cells, is not directly involved in immunity. COVID-19's primary threat is the hyperactivation of the immune system, potentially reinforced if DC-SIGN engages with exogenous ligands. Therefore, L-SIGN, co-localized with ACE2-expressing cells in the respiratory tract, is a more suitable target for anti-adhesion therapy. However, designing a selective ligand for L-SIGN is challenging due to the high sequence identity of the Carbohydrate Recognition Domains (CRDs) of the two lectins. We here present Man84, a mannose ring modified with a methylene guanidine triazole at position 2. It binds L-SIGN with a KD of 12.7μM ± 1 μM (ITC) and is the first known L-SIGN selective ligand, showing 50-fold selectivity over DC-SIGN (SPR). The X-ray structure of the L-SIGN CRD/Man84 complex reveals the guanidinium group's role in achieving steric and electrostatic complementarity with L-SIGN. This allows us to trace the source of selectivity to a single amino acid difference between the two CRDs. NMR analysis confirms the binding mode in solution, highlighting Man84's conformational selection upon complex formation. Dimeric versions of Man84 achieve additional selectivity and avidity in the low nanomolar range. These compounds selectively inhibit L-SIGN dependent trans-infection by SARS-CoV-2 and Ebola virus. Man84 and its dimeric constructs display the best affinity and avidity reported to date for low-valency glycomimetics targeting CLRs. They are promising tools for competing with SARS-CoV-2 anchoring in the respiratory tract and have potential for other medical applications.
Introduction
To detect pathogens and thus potential danger, antigen presenting cells of our immune system possess a battery of receptors, called pathogen recognition receptors (PRRs), that are dedicated to their surveillance and identification.1 Among PRRs, C-type lectin receptors (CLRs) are dedicated to the recognition of carbohydrate molecular patterns associated with pathogens, and can then contribute to shape an immune response. This family of receptors shares a common domain, known as the Carbohydrate Recognition Domain (CRD), containing a conserved calcium binding site used for the recognition of carbohydrates. The diverse CLRs differ in size, oligomerization state and signaling pathways, and will therefore have a different impact on the immune system depending on the panels of glycans they recognize.2,3 However, not all CLRs are strictly dedicated to immunity and some of them are even expressed in other cell types and tissues such as hepatocytes or endothelial cells from blood capillaries to airway epithelia.4,5 These lectin receptors, therefore, have other functions such as physiological clearance mechanisms (e.g. asialoglycoprotein receptors6) and cell adhesion molecules (e.g. selectins7), for example.Some pathogens have found strategies to bypass the role of CLRs in immunity activation and even hijack CLRs for their own benefit during the infection process. Thus, subversion of CLRs has been reported to turn these lectins into alternate receptors or attachment factors, notably by HIV,8 Ebola virus9 and SARS-CoV virus,10,11 responsible for the severe acute respiratory syndrome (SARS) in 2002. In the context of the 2020 world-scale coronavirus outbreak, we,12 and others,13–15 tested the potential role of several CLRs toward the viral transmission process and found that only two of them, DC-SIGN and L-SIGN (CD209 and CD209L, respectively), are exploited by SARS-CoV-2 to enhance its infection. SARS-CoV-2 can interact with both lectins through its highly glycosylated Spike protein and uses them as anchor points at the cell surface. There is still a debate on the detail of this process: some groups have suggested DC/L-SIGN involvement as direct alternative receptors,13,15 others only as promoters of a trans-infection mode, whereby the two CLRs play the role of attachment factors, enabling binding and concentration of viruses onto cell surfaces, before transferring it to its fusion receptor ACE2 on permissive cells along the respiratory tract.12,14
DC-SIGN and L-SIGN (also called DC-SIGNR for DC-SIGN Related) have a very high sequence similarity (82% for the whole protein,16 and up to 72% for the sole Carbohydrate Recognition Domain). Both are tetrameric proteins and bind to mannosylated oligosaccharides.17 The tetramers have similar, but not identical, topology and dynamics and this impacts on some aspects of their recognition profile of multivalent glycoconjugates.18,19 Finally, a major difference between the two lectins is their expression in different cell types and tissues. DC-SIGN is expressed on immature dendritic cells, L-SIGN is expressed in human liver sinusoidal endothelial cells, human lung in type II alveolar cells and in endothelial cells and is co-expressed with ACE2 on respiratory tract cells.15,20–22
Because of its known role in various infections, DC-SIGN has already been the focus of intense efforts as a target for drug design, mostly employing glycomimetic ligands.23–29 Glycomimetics are structural and/or functional mimics of carbohydrates used to replace the template molecules, typically as the ligand of a protein target.30–32 We and others have developed monovalent and multivalent glycomimetic ligands of DC-SIGN and have used them as antagonists to block DC-SIGN-mediated infections of HIV, dengue fever and Ebola.23–29 In a recent work, we demonstrated that using previously developed polyvalent glycomimetics we were able to block the trans-infection of SARS-CoV-2 mediated by DC-SIGN.12 This suggested that antagonist glycomimetics could be efficient additions in the tool-box against SARS-CoV-2 spreading.
Despite the fact that L-SIGN is known to be the preferential attachment factor for West Nile Virus,33 there was no report of antagonist development or simply of L-SIGN targeting in anti-infective strategy before the recent COVID 19 outbreak and the identification of L-SIGN as a potential target together with DC-SIGN. COVID 19 has been associated with hyperactivation of the immune system as a major threat for patients.22 DC-SIGN is a dendritic cell PRR and it is not yet known whether its engagement by recognition of exogenous ligands could reinforce this hyperactivation. In contrast, L-SIGN is not involved in the mucosal immune response and colocalizes with ACE2-expressing cells in the lungs. Thus, in the case of SARS-CoV-2, for both reasons above, L-SIGN appears as a more relevant target than DC-SIGN for host-targeted antiviral therapies. We have recently reported a set of C2 triazole-modified mono- and pseudo-di-mannosides that inhibit both DC-SIGN and L-SIGN binding to SARS-CoV-2 spike, up to the low micromolar level.34 However, all these molecules have more or less the same impact on the two receptors, or are slightly DC-SIGN selective. No selective ligand has yet been described for L-SIGN. This is not too surprising, considering the 72% identity of the two CRDs,35 nonetheless, an L-SIGN selective ligand would be welcomed, both as a probe of the CLR’ role in viral infections and for development as secure antiadhesive antiviral. To further pursue this goal, we moved from the aminotriazole derivative 1 (Man79), which in our previous study34 displayed an IC50 value 278 ± 7 μM for L-SIGN and 318 ± 2 μM for DC-SIGN (SPR experiment, binding inhibition to immobilized Spike). The binding mode of this molecule in DC-SIGN can be inferred from recently reported X-ray structures that show the existence of an ammonium binding site in the vicinity of the canonical Ca2+ ion.24,29 This area is conserved in L-SIGN, which can explain the good affinity of 1 for this target. Comparison of the two lectins CRDs suggests that the ammonium binding area may be less hindered in L-SIGN than in DC-SIGN, as noted in early structural studies. This motivated us to prepare the guanidinotriazole derivative 2 (Man84) and study its interaction with both lectins. Here we report that Man84 (2) is the first L-SIGN selective ligand. It binds to L-SIGN with μM affinity and has a surprising 50-fold selectivity for L-SIGN over DC-SIGN. The structure of the L-SIGN/Man84 complex was obtained by X-ray crystallography and helped to explain the source of this selectivity. NMR studies allowed us to examine the binding process in solution for both lectins, confirming the binding selectivity and highlighting the structural differences between the two complexes. Dimeric constructs bearing two copies of Man84 were also prepared. This low level of multivalency allowed nM affinity to be reached for L-SIGN and increased the selectivity up to 3 orders of magnitude against DC-SIGN. Finally, the ability of the divalent constructs to block DC-/L-SIGN mediated infection of host cells by Ebola and SARS-CoV-2 viruses was investigated, revealing that they are powerful inhibitors of the process.
Results and discussion
Synthesis
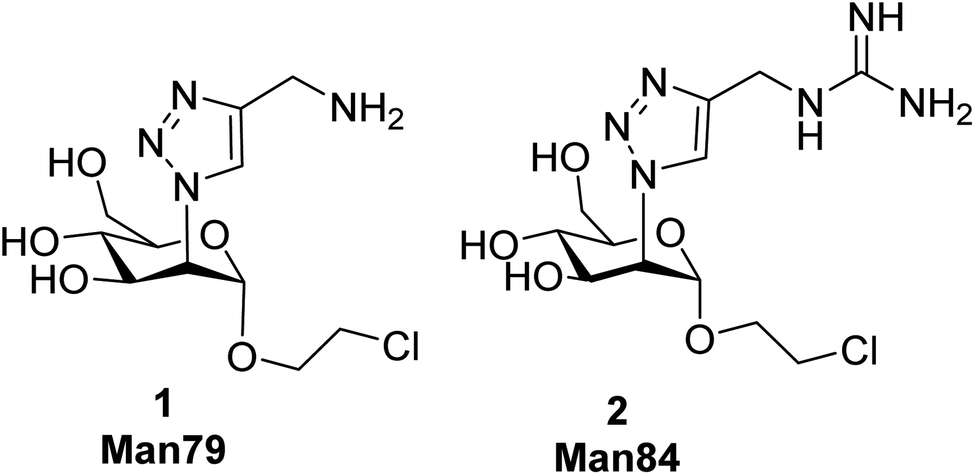 | ||
| Fig. 1 Monovalent ligands analyzed in this study. | ||
The guanidine analogue 2 (Man84) was prepared by CuAAC reaction of the 2-azido-mannoside 3 (ref. 24) with alkyne 4, in turn prepared from propargylamine 5 and Goodman reagent 6, using a slightly adapted protocol36,37 (Scheme 1, upper panel). The triazole product 7 was obtained with high yield (91%) and Zemplén deacetylation (0.02 M MeONa, 94%) followed by removal of the Boc protecting groups with either 20% TFA in DCM (quant) or 1 M aq. HCl in CH2Cl2 (3 d, 88%) afforded 2 (Man84) as either the TFA or chloride salt, in high purity.
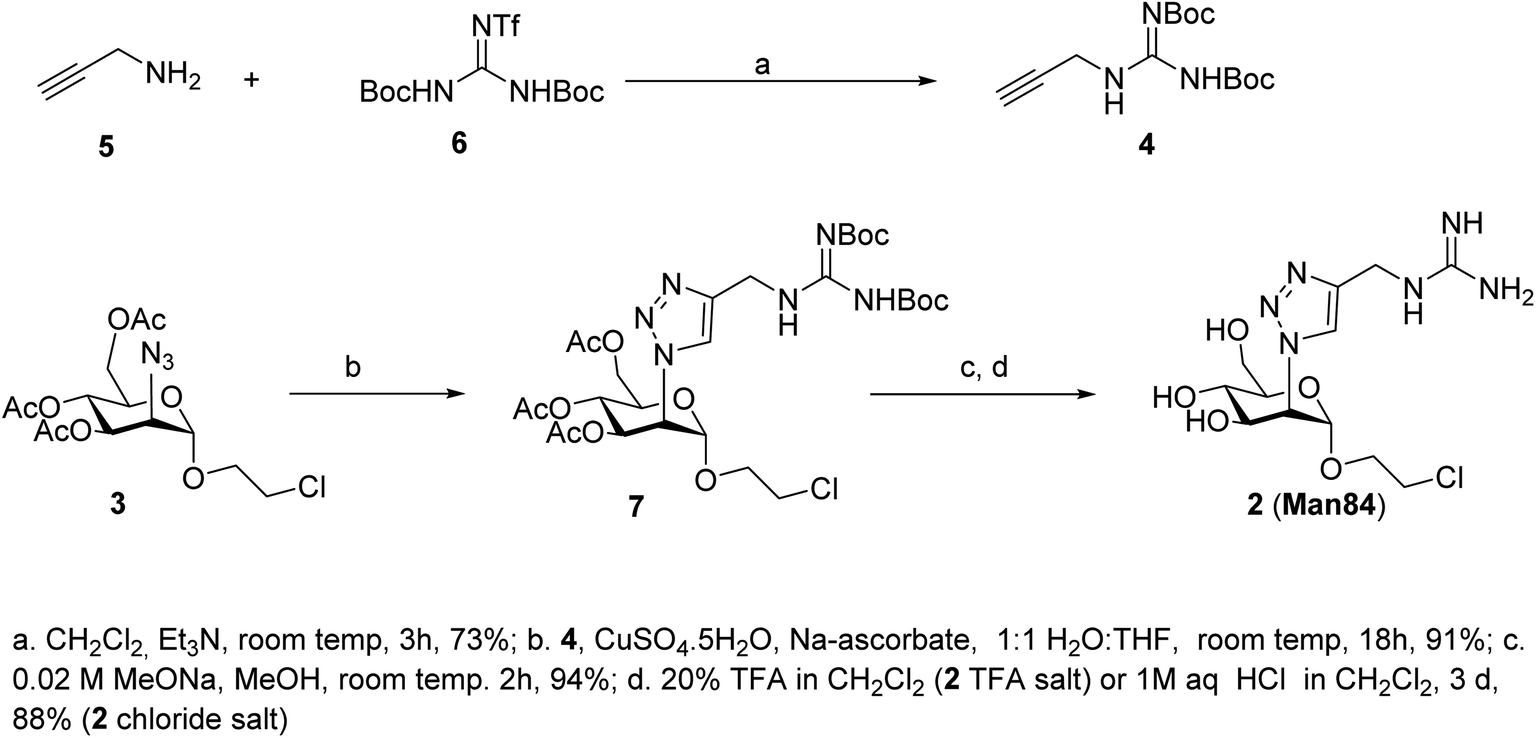 | ||
| Scheme 1 Synthesis of 2 (Man84). | ||
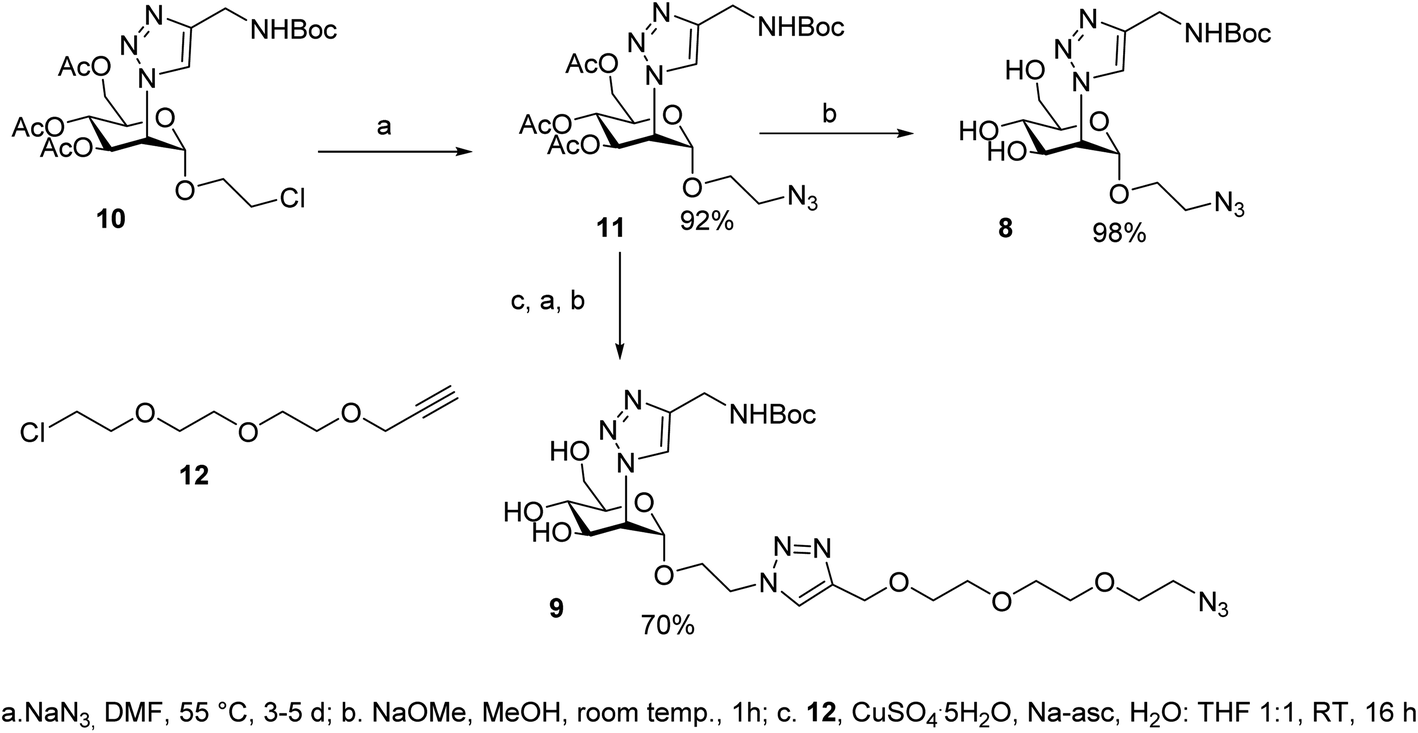 | ||
| Scheme 2 Synthesis of the monovalent ligands 8 and 9. | ||
The synthesis of the dimers was adapted from the previously reported one39 and adjusted for the solubility properties and size of the targeted ligands, which had an impact on the purification methods that were viable in this case (Scheme 3). Thus, TIPS-Rod3 (ref. 38) was desilylated as described (TBAF, THF) and the bis-alkyne Rod-3 was chromatographically isolated to remove the tetrabutylammonium salts. CuAAC ligation of 8 was performed at 60 °C, under microwave irradiation, until complete consumption of Rod3 was observed by TLC (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH2Cl2:MeOH). Crude 13 was purified via reverse-phase automated chromatography (76%). Boc-removal was performed at 10 mM in a 9:1 DCM:TFA solution, the crude was purified by RP HPLC (water/CH3CN gradient with of 0.1% HCOOH) and PM68 was isolated in 92% yield as the double formate salt.
:1 CH2Cl2:MeOH). Crude 13 was purified via reverse-phase automated chromatography (76%). Boc-removal was performed at 10 mM in a 9:1 DCM:TFA solution, the crude was purified by RP HPLC (water/CH3CN gradient with of 0.1% HCOOH) and PM68 was isolated in 92% yield as the double formate salt.
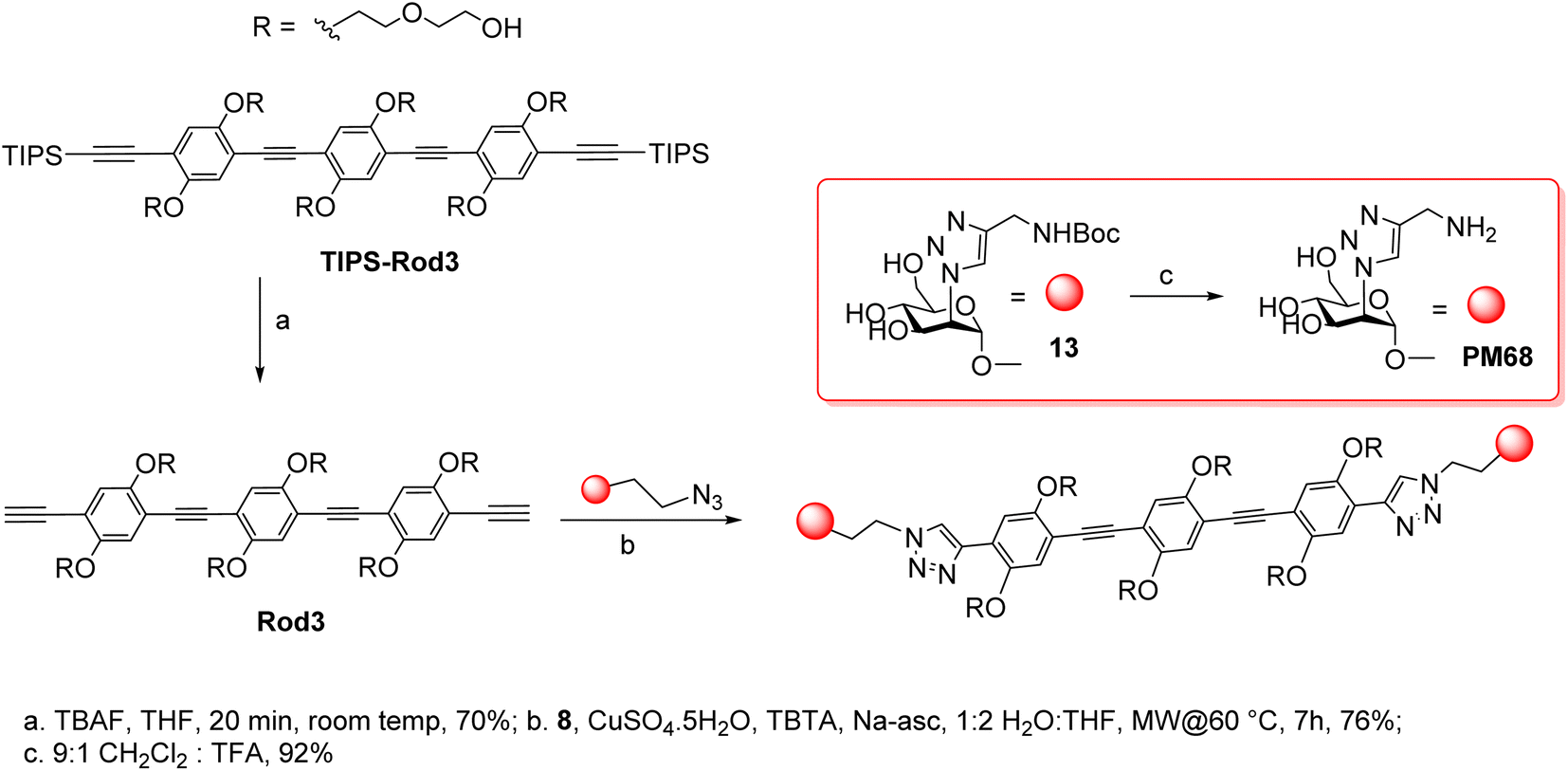 | ||
| Scheme 3 Synthesis of PM68. | ||
The synthesis of the corresponding guanidine derivative PM69 was attempted by direct guanidinylation of PM68 by Goodman reagent 6 (CH2Cl2, Et3N), which afforded 14 in 41% yield, after gel filtration on LH-20 Sephadex (Scheme 4, upper panel). As an alternative approach, reaction of Rod3 with azide 15, obtained from 8 as detailed in the ESI section,† afforded 14 in 38% yield upon gel filtration (Scheme 4, lower panel). Deprotection of the guanidine group (9:1 CH2Cl2:TFA) and isolation by RP HPLC afforded PM69.
 | ||
| Scheme 4 Synthesis of PM69 was achieved both by direct guanidinylation of PM68 (upper panel), or via CuAAC of 15 with Rod 3 (lower panel), with similar results. | ||
With a similar approach, the long-linker dimers PM70 and PM74 (Fig. 2) were prepared as detailed in the ESI section.†
 | ||
| Fig. 2 The four divalent ligands analyzed in this study: PM68 and PM70 are a divalent presentation of the amino-substituted ligand Man79; PM69 and PM74 are divalent presentations of the guanidine-substituted ligand Man84. For each monovalent spearhead, a short linker and a long linker presentation were prepared. | ||
Surface plasmon resonance determination of the inhibitory power of Man84 and Man79 towards DC-SIGN and L-SIGN
The entire extracellular domain (ECD), a soluble form of both receptors, was used to maintain their tetramerized state for ligand evaluation. Given the size of the ligands and their potential low affinity for lectins, monovalent ligands were evaluated using a competition assay for binding to the SARS-CoV-2 spike protein. In this SPR assay recently described,34 increasing concentrations of the glycomimetics were used to inhibit DC/L-SIGN binding to a surface functionalized with SARS-CoV-2 Spike glycoprotein (Fig. 3A). IC50 were obtained from the resulting inhibition curves (Fig. 3B and C) for both DC-SIGN and L-SIGN.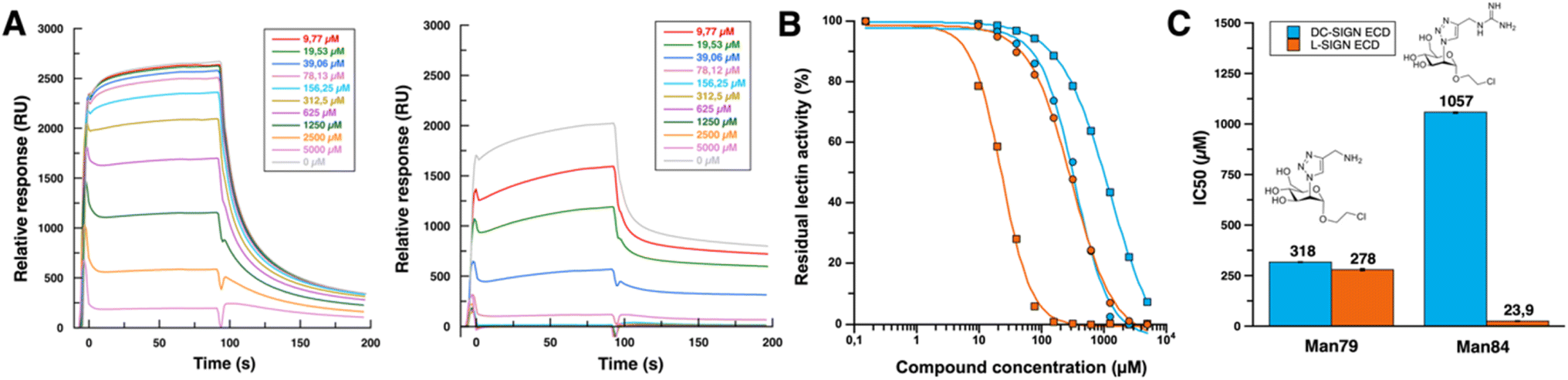 | ||
| Fig. 3 (A) Sensorgrams of DC-SIGN binding inhibition (left panel) and L-SIGN binding inhibition (right panel) by Man84. Range of Man84 concentrations goes from 5 mM to 10 μM by serial dilution by a factor of 2 with same color code for both DC-SIGN and L-SIGN inhibition. (B) SPR inhibition curves for Man79 (circle) and Man84 (square) (see ESI† for all sensorgrams). Inhibition curves concerning DC-SIGN are represented in blue and L-SIGN in orange. Man79 data, were already described in Pollastri et al.,34 and are shown here for direct comparison with Man84 data. (C) IC50 values of Man79 and Man84 for DC-SIGN (in blue) and L-SIGN (in orange). Values represented are the corresponding IC50. | ||
Comparison of the inhibition curves (Fig. 3B) indicates that there is no difference in the inhibitory potential of Man79 towards either of the lectins, given the similarity of the IC50 values obtained towards DC-SIGN and L-SIGN (318 and 278 μM respectively). However, these values are 10 times lower than the natural mannose residue (IC50 ∼ 2–3 mM), confirming that the addition of this 2-triazol-amino group at C2 is highly efficient in raising affinity for both lectin sites, while it does not induce any selectivity, as already described.34 This compound is a monosaccharide derivative of a previously characterized disaccharide glycomimetic where the amine has been shown to reach a specific pocket, in proximity to the calcium binding site of DC-SIGN,24 made of the F313, E358 and S360 binding triad in DC-SIGN. These residues are strictly conserved in L-SIGN (F325, E370 and S372) suggesting that Man79 will bind exactly the same way in both lectins. These compounds, together with a large series of other ligands previously tested,34 illustrate the difficulty to generate a selectivity between these two highly homologous targets.
However, one related compound seems to have achieved this feat. Sensorgrams derived from inhibition of DC-SIGN and L-SIGN by Man84 (Fig. 3A) show a high relative response without ligand (2500 RU for DC-SIGN and 2000 RU for L-SIGN), which decreases as the glycomimetic concentration increases, reflecting an inhibitory effect on lectins activity. For the same ligand concentration, we observe a huge difference in signal between the two lectins, where the L-SIGN response is inhibited much more rapidly. Above a ligand concentration of 156 μM, the L-SIGN binding response is close to 0, compared with over 2000 RU for the same concentration to inhibit DC-SIGN interaction. The addition of a guanidium group on the triazole moiety (Man84 compound) resulted in enhancement of inhibitory potential towards L-SIGN (IC50 = 23,9 μM) by a factor of 81 relative to mannose (IC50 = 1.94 mM),34 but only by a factor of 3 towards DC-SIGN (IC50 = 1.06 mM vs. IC50 = 3 mM for mannose).34 Thus, Man84 exhibits a binding affinity for L-SIGN that is over 40 times greater than that for DC-SIGN. The IC50 value in the low micromolar range firmly establishes Man84 among the most potent monovalent glycomimetics reported so far for a C-type lectin target. Additionally, these data suggest that the guanidinium group establishes interactions with L-SIGN that cannot be formed with DC-SIGN, suggesting a distinct binding mode compared to Man79.
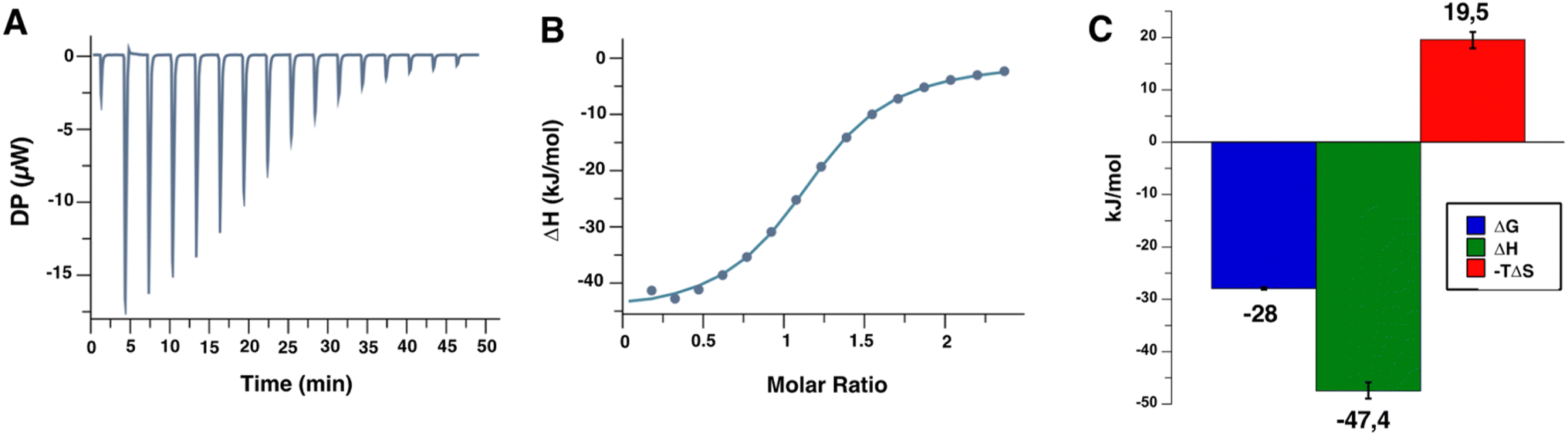 | ||
| Fig. 4 Titration of the Man84 ligand at 2 mM to L-SIGN ECD (172 μM) by ITC. (A) Representative data among a series of 3 of the titration thermograms obtained (see ESI† for all ITC titration experiments). (B) Data integration with fitted curve, using 1:1 binding model. (C) Average thermodynamic parameters values obtained following the L-SIGN CRD/Man84 complex formation. | ||
Triplicate measurements by ITC determined an equilibrium dissociation constant KD of 12.7 μM ± 1 μM with a 1:1 stoichiometry of binding. This is in full agreement with the IC50 evaluated above from the SPR competition experiments. It is striking to find such an increase in affinity by a factor of 230, compared to the natural mannose residue, resulting from this unique modification in C2.34
The determination of the thermodynamic parameters of the L-SIGN/Man84 complex formation reveals an average variation of enthalpy (ΔH) of −47.4 ± 1.5 kJ mol−1, and a variation of entropy (−TΔS) of 19.5 ± 1.6 kJ mol−1, leading to a ΔG of −28 ± 0.2 kJ mol−1. It suggests an enthalpy driven complex formation as the major driving force, with a strong contribution to the interactions of the triazole-guanidium group added in position 2 within the L-SIGN active site. The observed entropy variation is likely due to loss of conformational freedom upon binding and solvent contribution.
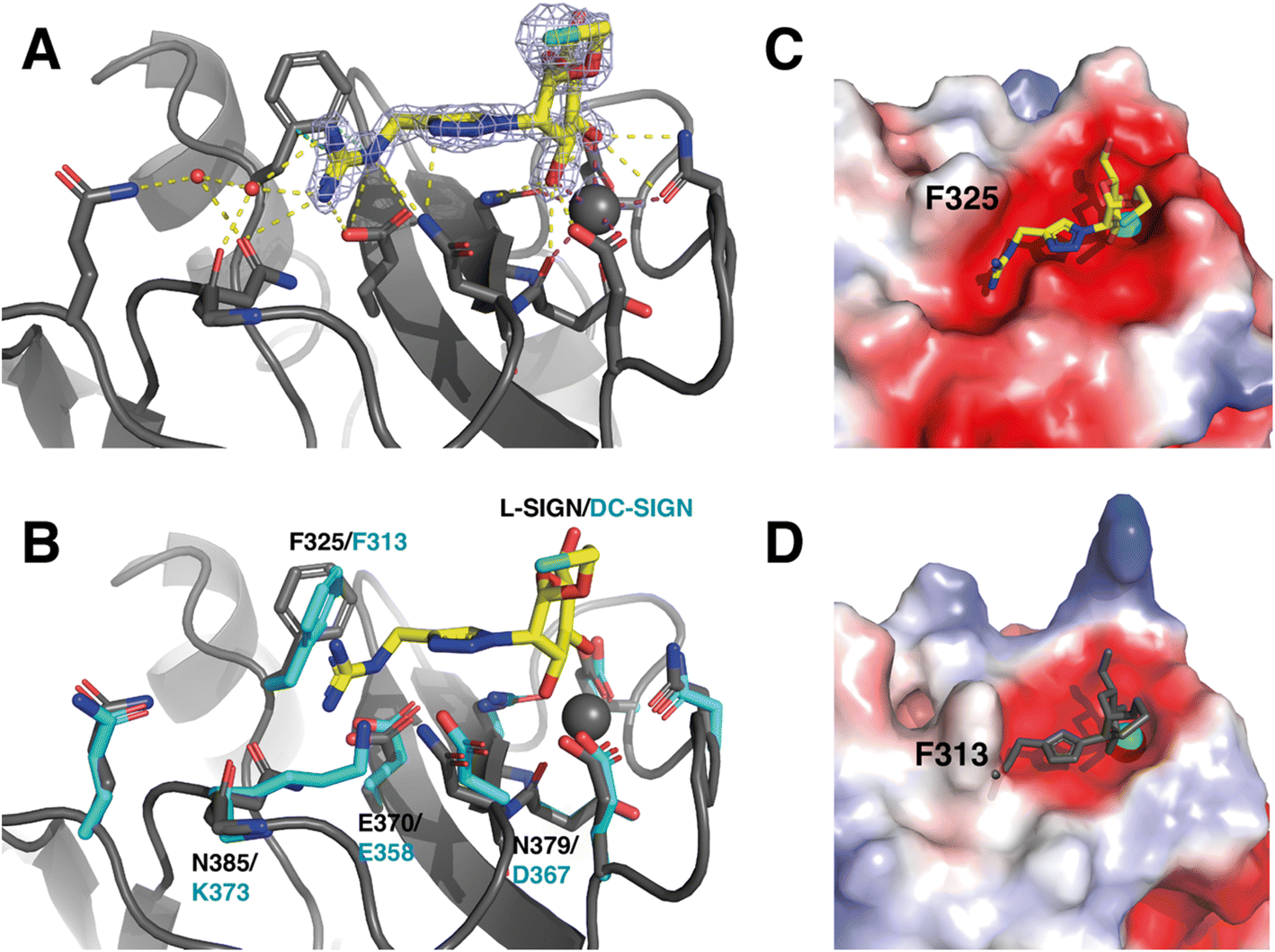 | ||
| Fig. 5 3D structural binding mode of Man84 within L-SIGN CRD and mechanism of selectivity. (A) Structure of the L-SIGN CRD/Man84 complex (PDB: 8RCY). Man84 is shown superimposed over the Fo − Fc electron density map (light blue, 2σ contour) within L-SIGN CRD. Side chain of residues involved in the binding are represented as sticks. H-Bonds are represented as yellow dashed lines, Ca2+ coordination bond as magenta dashed line and π-cation interaction by green dashed lines. Water molecules are represented as red spheres. (B) Alignment with the CRD of DC-SIGN (PDB: 2IT6) for comparative purposes. Side chain of corresponding residue from DC-SIGN CRD are presented as cyan sticks and labelled. Electrostatic surfaces of L-SIGN CRD (C) and DC-SIGN CRD (D) were calculated via the PyMol software, with complexed Man84 represented in yellow in L-SIGN CRD or in grey in DC-SIGN CRD where it has been added by structural alignment for comparison. The Ca2+ ion in the binding site is represented by a grey sphere (see Table S1† for data collection and structure refinement statistics). | ||
The ligand interacts in the active site of L-SIGN with its mannose moiety coordinating the Ca2+ ion in the canonical interaction site through its C3 and C4 hydroxyl groups (Fig. 5).44 In addition, the guanidinium end of the ligand makes a salt bridge with Glu370 in the cleft close to the active site (Fig. 5A). This group also forms a network of electrostatic interactions and hydrogen bonds with adjacent residues in the active site such as Asn385 and Asn379, which is consistent with the thermodynamic profile obtained with the ITC assays. There is a π-cation interaction between the electronegative dipole induced by the aromatic ring of Phe325 and the positive charge of the Man84 guanidine. As this guanidinium group is positively charged in solution, it is thus perfectly complementary to the electronegative pocket close to the calcium site and extending under Phe325 in L-SIGN (Fig. 5C). To note, the guanidinium group also contributes to the stabilization of two water molecules within the structure that contribute additional H-Bonds to the whole complex (Fig. 5A). These crystallographic water molecules, stabilized within the complex due to Man84, could participate to entropy contribution observed upon binding. Finally, comparison with the active site of DC-SIGN (PDB: 2IT6), revealed the atomic details that determine the selectivity of Man84 between the lectins (Fig. 5B). It lies in differences within the guanidium-binding pocket: all the side chains interacting with Man84 are exactly identical or equivalent in DC-SIGN and in L-SIGN except for N385, which is replaced by K373 in DC-SIGN. Thus, the electronegativity of the cleft present in L-SIGN, critical to accommodate the guanidium group, is first cancelled by the positive side chain of K373 but also filled by this larger side chain (Fig. 5D). Thus, Man84 cannot interact similarly with DC-SIGN, given the steric hindrance and the electrostatic repulsive effect with the guanidinium. Here a single amino acid difference between two very conserved sites, explains a 40-fold selectivity mechanism.
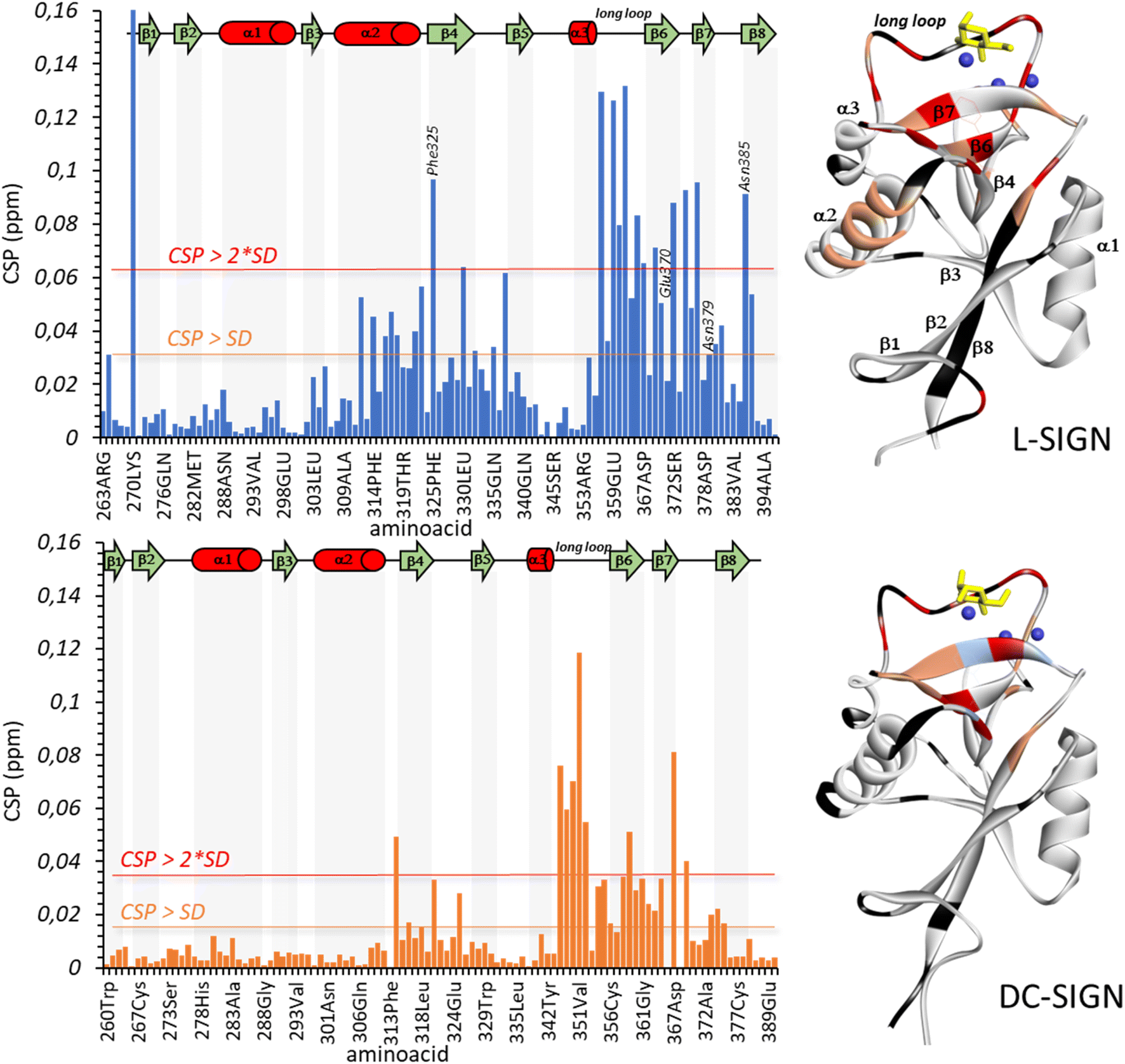 | ||
| Fig. 6 The binding of Man84 to L-SIGN (top) and DC-SIGN (bottom) in solution, by monitoring NMR signals of the corresponding protein in the absence and presence of the ligand (30 equivalents in the case of L-SIGN, and 100 equivalents for DC-SIGN). On the left, plots for the CSP analysis (average chemical shift difference between protein free and bound states) and on the right 3D cartoon representation of the corresponding protein highlighting the most affected residues in the CSP analysis: in red residues with CSP above twice the standard deviation (SD) of the whole data set, and orange residues with CSP above the SD. Residues in black are not assigned or are prolines. Residues in light blue disappear in the bound state. The ligands are in yellow and Ca2+ ions in dark blue. For L-SIGN, residues involved in direct intermolecular interactions with Man84 as found by X-ray crystallography are annotated in the CSP plots. | ||
To obtain information of the recognition process from the ligand perspective, trNOESY experiments were performed. First, a NOESY spectrum of the ligand alone was acquired, which showed positive NOEs. The most interesting piece of information in this case are the NOEs of the proton of the triazole ring (H_Tz), which define its orientation with respect to the pyranose (Man) ring (Fig. 7A). H_Tz showed strong (S) NOEs with H4Man and H1Man, and medium (M) with H2Man and H6Man. Since H4Man and H1Man/H2Man are on different sides of the sugar ring relative to the C2(Man)–N(Triazol) bond, this set of NOEs indicates a high degree of conformational averaging around this bond (Fig. 7D).
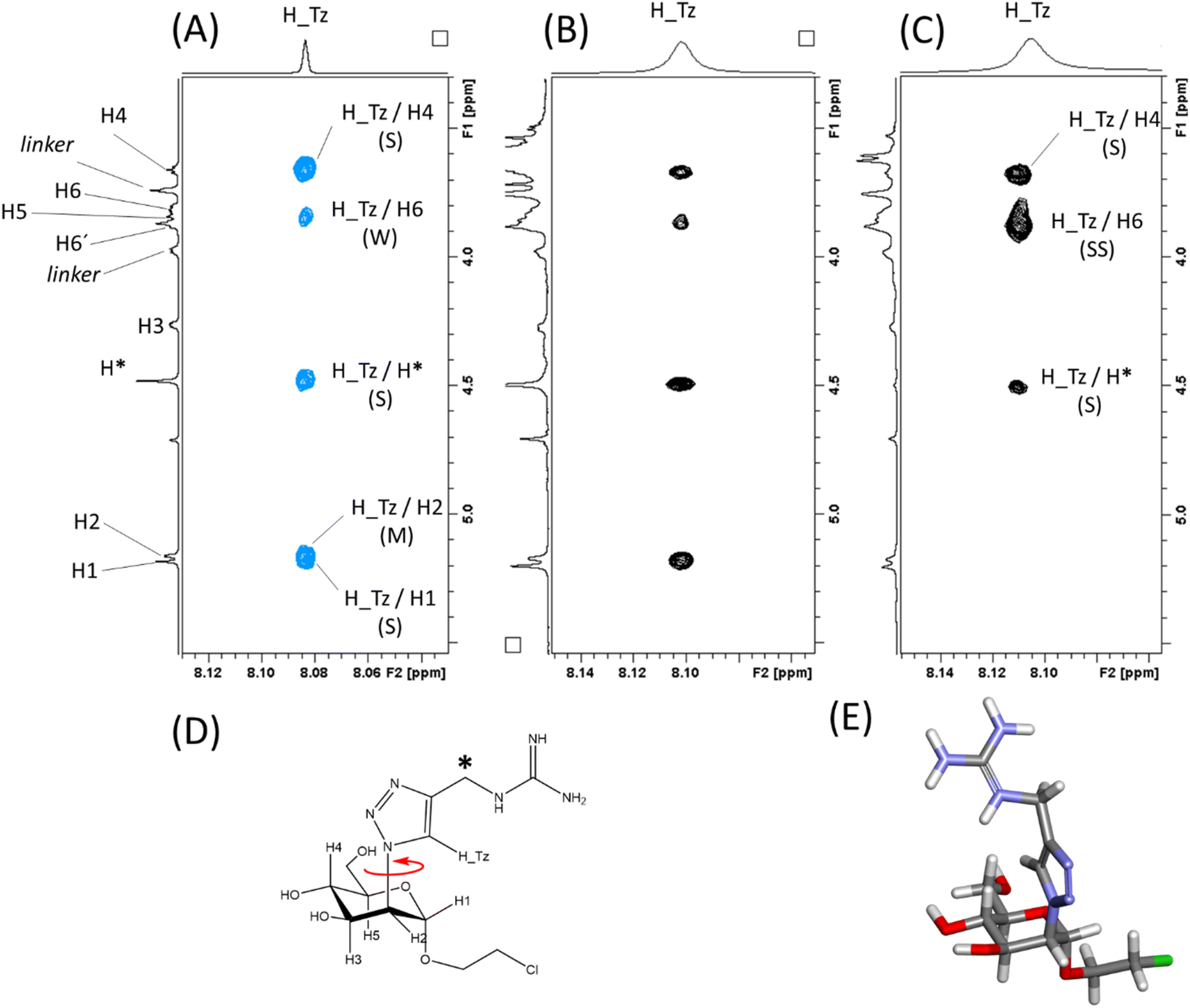 | ||
| Fig. 7 The binding of Man84 to DC-SIGN and L-SIGN in solution from the ligand perspective. Blue contours correspond to positive NOE, while black contours correspond to negative NOE. (A) NOESY spectrum of Man84 showing the positive NOE correlations of H_Tz. (B) The same region of the trNOESY spectrum of Man84 in the presence of DC-SIGN (1:17 protein:ligand molar ratio). (C) The same region of the trNOESY spectrum of Man84 in the presence of L-SIGN (1:10 protein:ligand molar ratio). (D) Structure of Man84 showing the free rotation around the C2(Man)–N(Triazol) bond. (E) The L-SIGN-bound conformation of Man84, as derived by NMR interaction data. This conformation corresponds to the one observed in the X-ray structure (Fig. 5A). | ||
The same experiment acquired under the same conditions but in the presence of DC-SIGN (1:17 protein:ligand molar ratio), showed the same set of NOEs with the same relative intensity, but with a different sign, changing for positive (fast tumbling small molecule) to negative (Fig. 6B), as expected for a DC-SIGN-bound ligand. This change in the sign indicates that the observed signals are indeed exchange-transferred NOE (trNOE) cross peaks.46 The fact that the NOE pattern is exactly the same as in the free form (Fig. 6A and B) reveals that Man84 bound to DC-SIGN conserves the same conformational behavior around the C2(Man)–N(Triazol) as in the free form. The same experiment performed in the presence of L-SIGN (1:10 protein:ligand molar ratio) (Fig. 7C) showed again negative NOE correlations (and thus trNOE), but the experimental observations were dramatically different from the previous ones. Now, the H_Tz/H1 and H_Tz/H2 correlations are lost, while those for H_Tz/H4 and H_Tz/H6 are strong and very strong, respectively. This cross-peak pattern reveals a conformational selection process upon binding to L-SIGN. The experimental data show that Man84 is bound to L-SIGN in a particular, well-defined conformation, shown in Fig. 7E, in which H_Tz points towards H6Man and H4Man and corresponding to the one observed in the X-ray structure (Fig. 5A). These NMR data, therefore, are in full agreement with the X-ray crystallographic structure of the Man84/L-SIGN complex, and strongly support the rationale for the L-SIGN versus DC-SIGN selectivity. For DC-SIGN, the aglycon in Man84 keeps the same conformational flexibility as in the free state, corroborating that this moiety is not involved in direct and persistent intermolecular interactions with the lectin. In contrast, a single conformation is selected upon binding to L-SIGN, which is in agreement with the X-ray crystallography data. There are additional contacts with the protein that strengthens the interaction from the enthalpy point of view. Moreover, this conformational selection process also accounts for the observed entropy penalty upon binding.
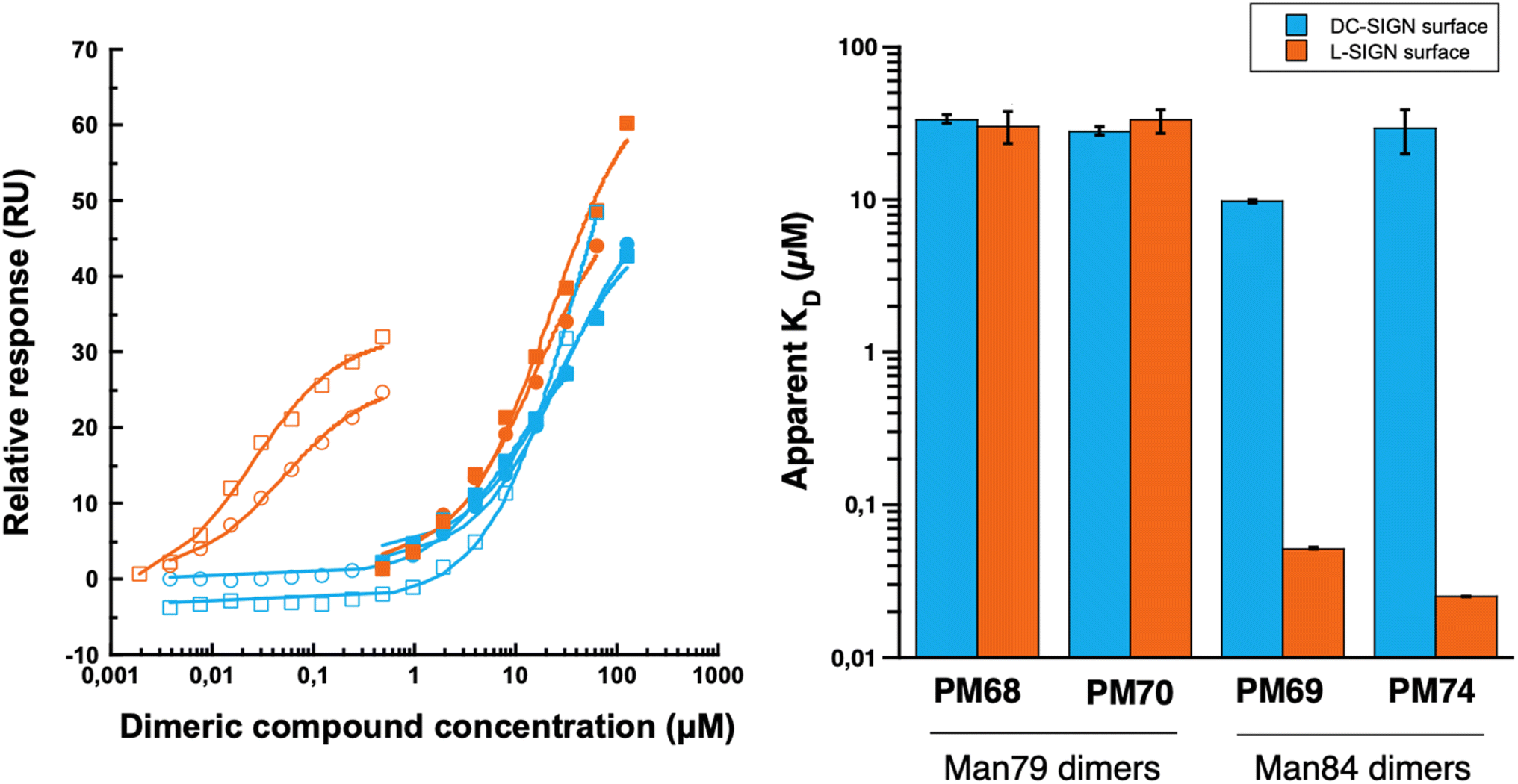 | ||
| Fig. 8 SPR interaction tests with titration of PM68 (filled circles), PM69 (empty circles), PM70 (filled squares) and PM74 (empty squares) on DC-SIGN ECD (blue curves) and L-SIGN ECD (orange curves) oriented surfaces. Range of interactions were performed in duplicates. | ||
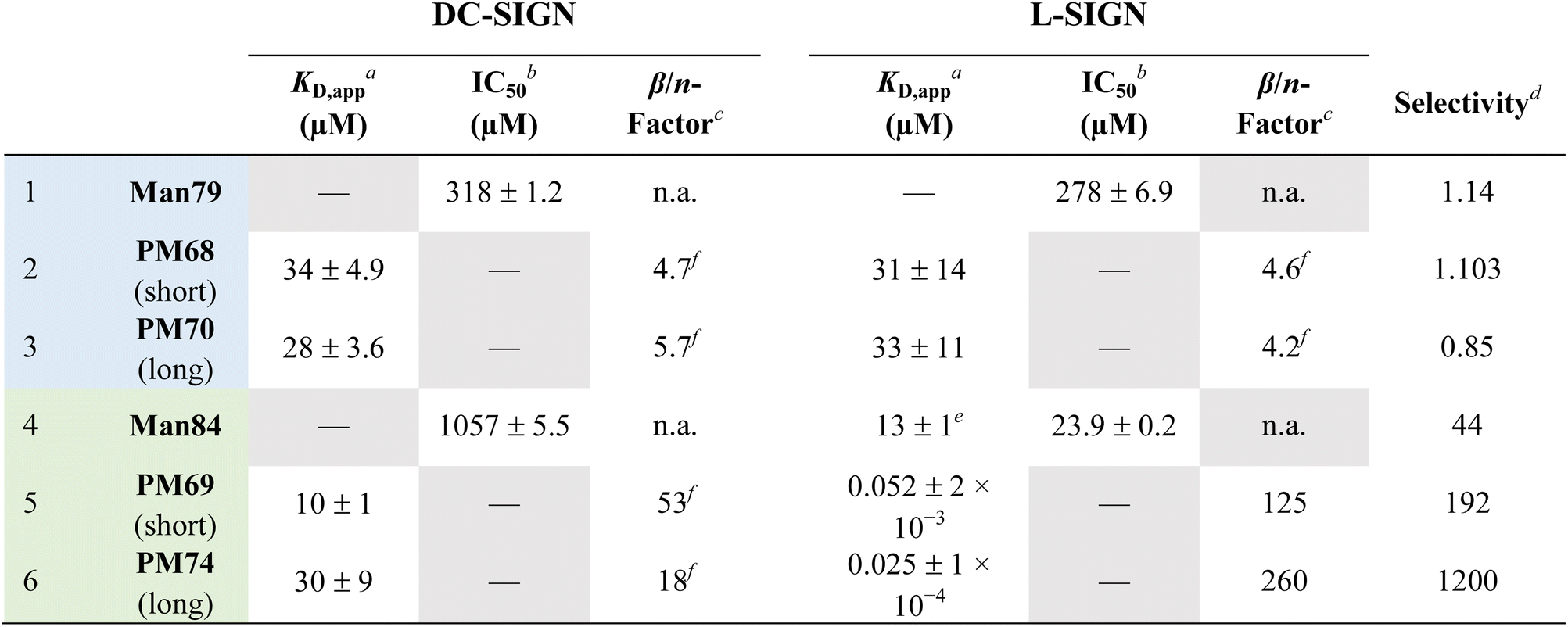
Dimerization of Man79 and Man84 ligands on the rod-like scaffold appears to be very efficient and significantly improves the affinity towards the lectins (Table 1). For Man79, which is a weak non selective binder of both CLRs (IC50 318 μM and 278 μM for DC-SIGN and L-SIGN, respectively, Table 1 entry 1 and Fig. 2C) the affinity increases by an order of magnitude in the dimers PM68 (Table 1, entry 2) and PM70 (Table 1, entry 3), for both lectins and independent of the length of the flexible linker. Similarly, the dimers of Man84, PM69 and PM74 (Table 1, entries 5 and 6) bind to DC-SIGN in the low μM range, with an increase by up to 2 orders of magnitude relative to the monovalent spearhead (10 μM and 30 μM, respectively). The same two dimers display an apparent KD of 52 nM (PM69) and 25 nM (PM74) for L-SIGN, confirming a strong effect of the dimerization on the affinity (β/n-factor48 of 125 and 250, respectively) and achieving an impressive selectivity for L-SIGN vs. DC-SIGN, that reaches 3 orders of magnitude with the long linker dimer PM74. The effect of selectivity is almost 10 times lower for short linker PM69 (192 for “short” PM69 and 1200 for “long” PM74), clearly suggesting that distance matters.
| a SPR direct interaction (Fig. 8). b SPR inhibition experiments (Fig. 3C). c β factor is as defined in ref. 48 and normalized here by the valency n (β/n = KD,monovalent/(n × KD,polyvalent)). d K D,app or IC50 ratio of DC-SIGN over L-SIGN. e As measured by ITC (Fig. 4). f K D of the monovalent ligand not determined. β-Factor calculated using the IC50 associated with the monovalent ligand and the KD,apps determined for the dimeric PM compounds. |
|---|
|
Overall, these data suggest an important avidity phenomenon for these divalent compounds, and a chelating effect in the binding to the two lectins. Contrary to PM26, an hexavalent rod-based construct that we described as a potent DC-SIGN ligand and characterized recently,41 no cumulative avidity effect coming from statistical rebinding can be expected here, since a unique spearhead is presented at each extremity of the rod core. Rather, in the present case, there is probably an optimization of chelation properties with an improved access to two adjacent CRDs within the L-SIGN tetramer. However, potential clustering effect between two adjacent tetramers on the SPR chip cannot be excluded, since a single surface density was used for this characterization in the SPR assay. The higher efficiency of the ligand with the longer linker could be explained by the topology of DC-SIGN and L-SIGN tetramers as showed in Fig. 9.
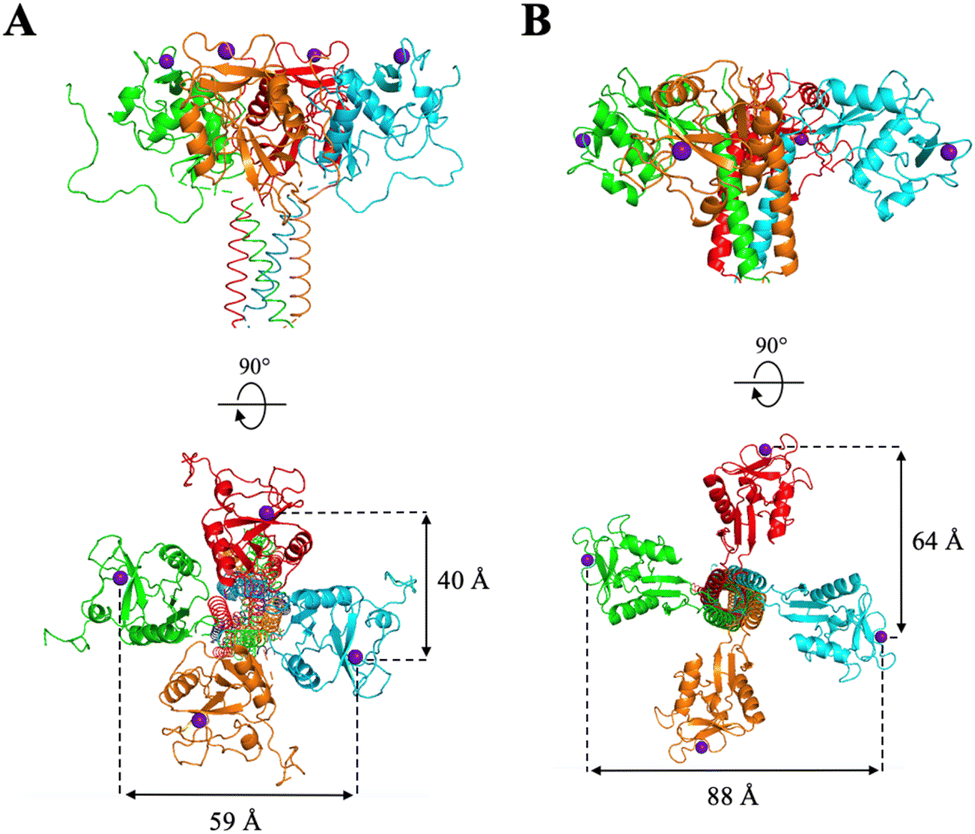 | ||
| Fig. 9 Topology of DC-SIGN (panel A) and L-SIGN (panel B) active sites. Tetrameric representation issued form SAXS, X-ray crystallography studies and molecular modelling from ref. 19 and 41–43. Figures made with PyMol. | ||
Despite the high overall homology between the two lectins (77% identity16), their CRDs, and thus binding sites, are not oriented in the same direction of space: while those of DC-SIGN are oriented upwards, those of L-SIGN are rather turned to the lateral side, increasing the gap between adjacent and opposite sites (between 60 and 80 Å for L-SIGN versus 40 to 60 Å for DC-SIGN). Such differential spacing and topology of the L-SIGN tetramer, with respect to DC-SIGN has been recently documented experimentally with results consistent with a larger tetramer and more outwardly exposed CRDs.49,50 A linker that is too short may cover only a single portion of the side and not connect two sites that are facing each other, thus limiting chelation properties. Moreover, in a detailed molecular dynamic study performed recently, we have shown, for DC-SIGN multivalent binders, that distances between two internal sites can vary, due to internal flexibility between CRDs, and that the size of the linker helps to keep chelation-binding available and buffers distance fluctuation between sites.41 That effect might be at work here also with PM74vs.PM69 increasing the dynamic situations where the chelation binding is still operative.
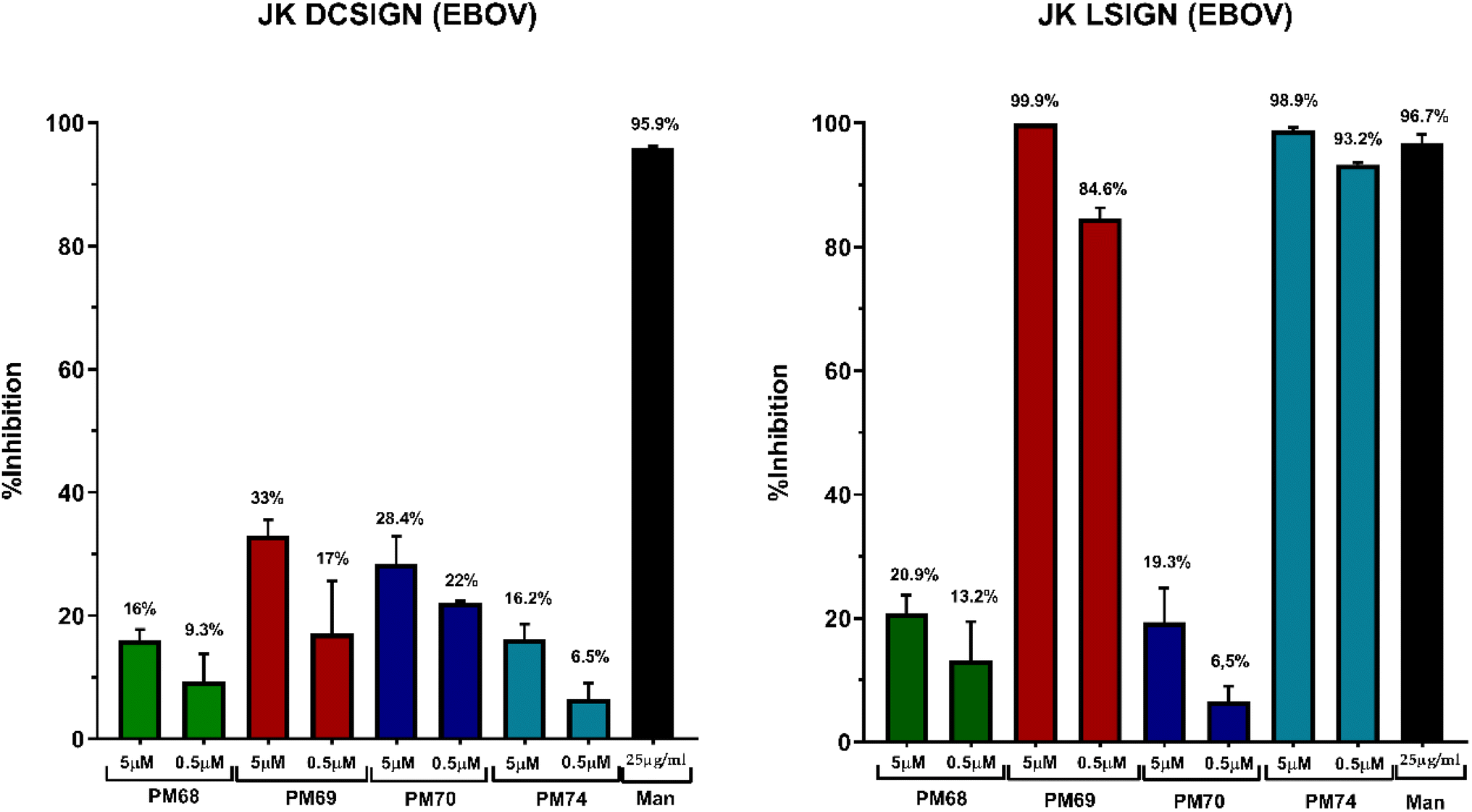 | ||
| Fig. 10 Trans-infection assays of EBOV pseudotyped rVSV-luc in VeroE6 mediated by Jurkat DC-SIGN (left panel) and Jurkat L-SIGN (right panel). Results are presented as percentage of EBOV trans-infection control in the presence of compounds: PM ligands and mannan (Man) as compared to trans-infection of EBOV in the absence of inhibitors. The results were analyzed using GraphPad Prism v8. | ||
The results are shown in Fig. 10 as % of trans-infection inhibition compared to the assay conducted in the absence of ligands. Mannan (Man in Fig. 10) was used as a positive control. The data indicate that all dimers at the tested concentrations are modest inhibitors of DC-SIGN mediated EBOV trans-infection (Fig. 10, left panel) and show a dose-dependent inhibition, slightly greater at 5 μM than at 500 nM. Similarly, the Man79-dimers PM68 and PM70 only partially inhibit L-SIGN mediated trans-infection of the Vero cells (Fig. 10, right panel). L-SIGN mediated trans-infection was blocked efficiently by PM69 and PM74 up to 99.9% at 5 μM, confirming the activity and selectivity of these ligands in a cellular model. The poor inhibition provided with PM68 and PM70 shows that the efficacy of Man84 dimers is not only linked to the multivalency of the compounds but also to their affinity for L-SIGN. The long-linker PM74 has a slight advantage, as previously observed in the SPR interaction studies, since it provides stronger inhibition at a 500 nM concentration than PM69 (94% vs. 84%).
The same series of competitive experiments were carried out to study the inhibition of the trans-infection phenomenon with SARS-CoV-2 (Fig. 11). The inhibition of DC-SIGN mediated trans-infection is even lower than in the EBOV experiments for all ligands at both the tested concentrations (here between 1% and 19% inhibition vs. 7% to 33% for EBOV), which is not the case for L-SIGN, further emphasizing the selectivity of Man084-derived PMs (PM69 and PM74).
 | ||
| Fig. 11 Trans-infection assays of SARS-CoV-2 pseudotyped rVSV-luc in VeroE6 mediated by Jurkat DC-SIGN (left panel) and Jurkat L-SIGN (right panel). Results are presented as percentage of EBOV trans-infection control in the presence of compounds: PMs and mannan (Man) as compared to trans-infection of EBOV in the absence of inhibitors. The results were analyzed using GraphPad Prism v8. | ||
The dose-dependent effect is less pronounced for this panel of experiments: while PM69 and PM74 respectively inhibit infection by 98.5 and 94.6% at 5 μM, they still achieve an inhibition of 97 and 92% for a 10-fold lower dose, showing the tool's efficacy. In addition, for the SARS-CoV-2 experiments, we did not observe any differences in selectivity, whether the linker is short or long. However, given the topology of the active sites (Fig. 9), this factor should not be ruled out for future uses of these compounds. Assuming PM74 as the most promising compound, on the basis of its molecular properties (Table 1), a wider range of concentrations of PM74 were used in trans-infection assays with SARS-CoV-2 to derive its IC50. Thus, while PM74 IC50 is estimated to be >100 μM for trans-infection of SARS-CoV-2 mediated by Jurkat DC-SIGN cells, its IC50 is determined to be of 65 nM when using Jurkat L-SIGN cells (with 95% confidence interval 28–154 nM) (see Fig. S4†). A strong selectivity towards L-SIGN dependent SARS CoV2 trans-infection, vs. DC-SIGN, is demonstrated for this compound with an IC50 value in agreement with its KD,app determined on purified L-SIGN receptor (Fig. 8 and Table 1).
These inhibitions were carried out directly with the biological target of interest, the Spike protein, and therefore enabled inhibition measurements to be obtained that could be directly extrapolated to the biological interaction of interest.
Conclusion
We have described here the development of a glycomimetic ligand, Man84; that exclusively binds to L-SIGN with affinity in the low μM range and barely interacts with DC-SIGN. The compound selectivity has been established by several biophysical methods at the molecular and cellular level, demonstrating the viability of the tool. A structural characterization of the L-SIGN CRD/Man84 complex allowed to determine that the origin of this selectivity lies mainly in the presence of a triazole-guanidine group, which introduces both steric and electrostatic complementarity with the L-SIGN active site, while producing unfavorable interactions in the corresponding site of DC-SIGN. To the best of our knowledge, this level of selectivity is unprecedented for glycomimetic structures targeting CLRs with similar specificity and is particularly striking because the CRDs of DC-SIGN and L-SIGN share 72% of their primary structure. On the other hand, X-ray analysis of the Man84/L-SIGN complex and comparison with the same region of DC-SIGN allows to locate the origin of the selectivity in a single amino acid difference (Asn385 in L-SIGN vs. Lys373 in DC-SIGN). This reinforces the notion that rational differential design that we have used in a different context to obtain selectivity towards DC-SIGN and against langerin51 can be a powerful tool towards selective lectin ligands.Through the modification at position 2 with a methylene guanidino triazole moiety, this mannose derived Man84 showed an IC50 for the receptor in the micromolar scale, gaining a 100-fold affinity compared to natural mannose (IC50 ∼ 2 mM (ref. 34)). This potential was further exploited with the dimerization of the compound on a rod-like scaffold, whose multivalency enabled to reach a nanomolar scale-affinity, most likely through chelating phenomenon. Divalency also increased the L-SIGN vs. DC-SIGN selectivity ratio, reaching a 1200-fold value toward L-SIGN with the most efficient ligand PM74.
We also showed that PM74 can block L-SIGN mediated trans infection by SARS-CoV-2 in a cellular model. In view of the hijacking of L-SIGN by SARS-CoV-2 as a co-receptor in the respiratory tract, this tool could therefore be beneficial by competing with the anchoring of the virus. Additional applications can be foreseen for a selective L-SIGN ligand in the medical field, particularly in the prevention and treatment of viral infections and in the immunotherapy of liver tumors where L-SIGN is also abundantly expressed. In addition to this ligand, a panel of Man84 analogues carrying guanidine isosters is currently under development. This optimization could be beneficial in order to further increase the selectivity ratio, but also to tune the basicity of the system, while maintaining the complementarity with the highly electronegative site of L-SIGN. These studies are ongoing and the results will be reported in due course.
Data availability
Crystallographic data for L-SIGN/Man84 complex (PDB: 8RCY) has been deposited at the Protein Data Bank (PDB: https://www.rcsb.org).Author contributions
C. Delaunay; investigation, formal analysis, visualization, writing – original draft. S. Pollastri; investigation, visualization, writing – original draft. M. Thépaut; investigation, formal analysis. G Cavazzoli, C. Bouchikri, F. Lasala, A. Franconetti; investigation. L. Belvisi; conceptualization, supervision. N. Labiod, A. Gimeno; investigation, visualization. J. Jiménez-Barbero; funding acquisition. A. Arda, R. Delgado; supervision, writing – original draft. A. Bernardi, F. Fieschi; conceptualization, funding acquisition, project administration, validation, supervision, writing – original draft, writing – review and editing.Conflicts of interest
The authors declare the following competing financial interest(s): S. P., C. D., M. T., F. F., and A. B. declare the filing of a patent covering the use of glycomimetic L-SIGN ligands as antagonist, anti-viral adhesion, and for targeting human L-SIGN-expressing cells.Acknowledgements
This work used the platforms of the Grenoble Instruct-ERIC center (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL) within the Grenoble Partnership for Structural Biology (PSB), supported by FRISBI (ANR-10-INBS-05-02) and GRAL, within the University Grenoble Alpes graduate school CBH-EUR-GS (ANR-17-EURE0003). F. F. acknowledges the French Agence Nationale de la Recherche PIA for Glyco@Alps (ANR-15-IDEX-02). C. D. thanks Ministry of Higher Education and Research for her PhD Fellowship. F. F. and C. D. thank also GRAL (see above) for GRAL PhD Operating costs. Research in R.D. lab is supported by grants from the Instituto de Investigación Carlos III, ISCIII, CIBERINFEC CB21/13/00039 and FIS PI2100989, the Program for Biomedicine Research of the Comunidad de Madrid. P2022/BMD-7274 and the European Commission: Project e-FabRIC, HORIZON-HLTH-2023-DISEASE-03-04, ID: 101137157 and Ebola-PREP-TBOX, HORIZON-JU-GH-EDCTP3, ID 101145709. Research in A.B. lab was supported by EU funding within the NextGeneration EU-MUR PNRR Extended Partnership initiative on Emerging Infectious Diseases (Project no. PE00000007, INF-ACT). HRMS analyses were obtained through the COSPECT Unitech (University of Milano). The group at CIC bioGUNE thanks Agencia Estatal de Investigación of Spain for the Severo Ochoa Center of Excellence Accreditation CEX2021-001136-S, funded by MCIN/AEI/10.13039/501100011033 and CIBERES, an initiative of Instituto de Salud Carlos III (ISCIII, Madrid, Spain).References
- O. Takeuchi and S. Akira, Cell, 2010, 140, 805–820 CrossRef CAS PubMed.
- T. B. H. Geijtenbeek and S. I. Gringhuis, Nat. Rev. Immunol., 2009, 9, 465–479 CrossRef CAS PubMed.
- S. J. Van Vliet, J. D. Dunnen, S. I. Gringhuis, T. B. Geijtenbeek and Y. Van Kooyk, Curr. Opin. Immunol., 2007, 19, 435–440 CrossRef CAS PubMed.
- G. A. Kinberger, T. P. Prakash, J. Yu, G. Vasquez, A. Low, A. Chappell, K. Schmidt, H. M. Murray, H. Gaus, E. E. Swayze and P. P. Seth, Bioorg. Med. Chem. Lett., 2016, 26, 3690–3693 CrossRef CAS PubMed.
- D. R. Dorscheid, A. E. Conforti, K. J. Hamann, K. F. Rabe and S. R. White, Histochem. J., 1999, 31, 145–151 CrossRef CAS PubMed.
- L. Dini, F. Autuori, A. Lentini, S. Oliverio and M. Piacentini, FEBS Lett., 1992, 296, 174–178 CrossRef CAS PubMed.
- J.-G. Geng, M. Chen and K.-C. Chou, Curr. Med. Chem., 2004, 11, 2153–2160 CrossRef CAS PubMed.
- T. B. H. Geijtenbeek, D. S. Kwon, R. Torensma, S. J. Van Vliet, G. C. F. Van Duijnhoven, J. Middel, I. L. M. H. A. Cornelissen, H. S. L. M. Nottet, V. N. KewalRamani, D. R. Littman, C. G. Figdor and Y. Van Kooyk, Cell, 2000, 100, 587–597 CrossRef CAS PubMed.
- C. P. Alvarez, F. Lasala, J. Carrillo, O. Muñiz, A. L. Corbí and R. Delgado, J. Virol., 2002, 76, 6841–6844 CrossRef CAS PubMed.
- H. Hofmann and S. Pöhlmann, Trends Microbiol., 2004, 12, 466–472 CrossRef CAS PubMed.
- T. Gramberg, H. Hofmann, P. Möller, P. F. Lalor, A. Marzi, M. Geier, M. Krumbiegel, T. Winkler, F. Kirchhoff, D. H. Adams, S. Becker, J. Münch and S. Pöhlmann, Virology, 2005, 340, 224–236 CrossRef CAS PubMed.
- M. Thépaut, J. Luczkowiak, C. Vivès, N. Labiod, I. Bally, F. Lasala, Y. Grimoire, D. Fenel, S. Sattin, N. Thielens, G. Schoehn, A. Bernardi, R. Delgado and F. Fieschi, PLoS Pathog., 2021, 17, e1009576 CrossRef PubMed.
- R. Amraei, W. Yin, M. A. Napoleon, E. L. Suder, J. Berrigan, Q. Zhao, J. Olejnik, K. B. Chandler, C. Xia, J. Feldman, B. M. Hauser, T. M. Caradonna, A. G. Schmidt, S. Gummuluru, E. Mühlberger, V. Chitalia, C. E. Costello and N. Rahimi, ACS Cent. Sci., 2021, 7, 1156–1165 CrossRef CAS PubMed.
- F. A. Lempp, L. B. Soriaga, M. Montiel-Ruiz, F. Benigni, J. Noack, Y.-J. Park, S. Bianchi, A. C. Walls, J. E. Bowen, J. Zhou, H. Kaiser, A. Joshi, M. Agostini, M. Meury, E. Dellota, S. Jaconi, E. Cameroni, J. Martinez-Picado, J. Vergara-Alert, N. Izquierdo-Useros, H. W. Virgin, A. Lanzavecchia, D. Veesler, L. A. Purcell, A. Telenti and D. Corti, Nature, 2021, 598, 342–347 CrossRef CAS PubMed.
- Y. Kondo, J. L. Larabee, L. Gao, H. Shi, B. Shao, C. M. Hoover, J. M. McDaniel, Y.-C. Ho, R. Silasi-Mansat, S. A. Archer-Hartmann, P. Azadi, R. S. Srinivasan, A. R. Rezaie, A. Borczuk, J. C. Laurence, F. Lupu, J. Ahamed, R. P. McEver, J. F. Papin, Z. Yu and L. Xia, JCI Insight, 2021, 6, e148999 CrossRef PubMed.
- E. J. Soilleux, R. Barten and J. Trowsdale, J. Immunol., 2000, 165, 2937–2942 CrossRef CAS PubMed.
- Y. Guo, H. Feinberg, E. Conroy, D. A. Mitchell, R. Alvarez, O. Blixt, M. E. Taylor, W. I. Weis and K. Drickamer, Nat. Struct. Mol. Biol., 2004, 11, 591–598 CrossRef CAS PubMed.
- Y. Guo, I. Nehlmeier, E. Poole, C. Sakonsinsiri, N. Hondow, A. Brown, Q. Li, S. Li, J. Whitworth, Z. Li, A. Yu, R. Brydson, W. B. Turnbull, S. Pöhlmann and D. Zhou, J. Am. Chem. Soc., 2017, 139, 11833–11844 CrossRef CAS PubMed.
- G. Tabarani, M. Thépaut, D. Stroebel, C. Ebel, C. Vivès, P. Vachette, D. Durand and F. Fieschi, J. Biol. Chem., 2009, 284, 21229–21240 CrossRef CAS PubMed.
- U. Svajger, M. Anderluh, M. Jeras and N. Obermajer, Cell. Signalling, 2010, 22, 1397–1405 CrossRef CAS PubMed.
- V. S. F. Chan, K. Y. K. Chan, Y. Chen, L. L. M. Poon, A. N. Y. Cheung, B. Zheng, K.-H. Chan, W. Mak, H. Y. S. Ngan, X. Xu, G. Screaton, P. K. H. Tam, J. M. Austyn, L.-C. Chan, S.-P. Yip, M. Peiris, U.-S. Khoo and C.-L. S. Lin, Nat. Genet., 2006, 38, 38–46 CrossRef CAS PubMed.
- Q. Lu, J. Liu, S. Zhao, M. F. Gomez Castro, M. Laurent-Rolle, J. Dong, X. Ran, P. Damani-Yokota, H. Tang, T. Karakousi, J. Son, M. E. Kaczmarek, Z. Zhang, S. T. Yeung, B. T. McCune, R. E. Chen, F. Tang, X. Ren, X. Chen, J. C. C. Hsu, M. Teplova, B. Huang, H. Deng, Z. Long, T. Mudianto, S. Jin, P. Lin, J. Du, R. Zang, T. T. Su, A. Herrera, M. Zhou, R. Yan, J. Cui, J. Zhu, Q. Zhou, T. Wang, J. Ma, S. B. Koralov, Z. Zhang, I. Aifantis, L. N. Segal, M. S. Diamond, K. M. Khanna, K. A. Stapleford, P. Cresswell, Y. Liu, S. Ding, Q. Xie and J. Wang, Immunity, 2021, S1074761321002120 Search PubMed.
- N. Varga, I. Sutkeviciute, C. Guzzi, J. McGeagh, I. Petit-Haertlein, S. Gugliotta, J. Weiser, J. Angulo, F. Fieschi and A. Bernardi, Chem.–Eur. J., 2013, 19, 4786–4797 CrossRef CAS PubMed.
- L. Medve, S. Achilli, J. Guzman-Caldentey, M. Thépaut, L. Senaldi, A. Le Roy, S. Sattin, C. Ebel, C. Vivès, S. Martin-Santamaria, A. Bernardi and F. Fieschi, Chem.–Eur. J., 2019, 25, 14659–14668 CrossRef CAS PubMed.
- S. Sattin, A. Daghetti, M. Thépaut, A. Berzi, M. Sánchez-Navarro, G. Tabarani, J. Rojo, F. Fieschi and A. Bernardi, ACS Chem. Biol., 2010, 5, 301–312 CrossRef CAS PubMed.
- I. Sutkeviciute, M. Thépaut, S. Sattin, A. Berzi, J. McGeagh, S. Grudinin, J. Weiser, A. Le Roy, J. J. Reina, J. Rojo, M. Clerici, A. Bernardi, C. Ebel and F. Fieschi, ACS Chem. Biol., 2014, 9, 1377–1385 CrossRef CAS PubMed.
- M. Andreini, D. Doknic, I. Sutkeviciute, J. J. Reina, J. Duan, E. Chabrol, M. Thépaut, E. Moroni, F. Doro, L. Belvisi, J. Weiser, J. Rojo, F. Fieschi and A. Bernardi, Org. Biomol. Chem., 2011, 9, 5778–5786 RSC.
- J. Cramer, RSC Med. Chem., 2021, 12, 1985–2000 RSC.
- D. D. Nemli, X. Jiang, R. P. Jakob, L. M. Gloder, O. Schwardt, S. Rabbani, T. Maier, B. Ernst and J. Cramer, J. Med. Chem., 2024, 67, 13813–13828 CrossRef CAS PubMed.
- S. Leusmann, P. Ménová, E. Shanin, A. Titz and C. Rademacher, Chem. Soc. Rev., 2023, 52, 3663–3740 RSC.
- V. C. Damalanka, A. R. Maddirala and J. W. Janetka, Expert Opin. Drug Discovery, 2021, 16, 513–536 CrossRef CAS PubMed.
- A. Tamburrini, C. Colombo and A. Bernardi, Med. Res. Rev., 2020, 40, 495–531 CrossRef CAS PubMed.
- C. W. Davis, H.-Y. Nguyen, S. L. Hanna, M. D. Sánchez, R. W. Doms and T. C. Pierson, J. Virol., 2006, 80, 1290–1301 CrossRef CAS PubMed.
- S. Pollastri, C. Delaunay, M. Thépaut, F. Fieschi and A. Bernardi, Chem. Commun., 2022, 58, 5136–5139 RSC.
- U.-S. Khoo, K. Y. K. Chan, V. S. F. Chan and C. L. S. Lin, J. Mol. Med., 2008, 86, 861–874 CrossRef CAS PubMed.
- K. Feichtinger, C. Zapf, H. L. Sings and M. Goodman, J. Org. Chem., 1998, 63, 3804–3805 CrossRef CAS.
- K. Feichtinger, H. L. Sings, T. J. Baker, K. Matthews and M. Goodman, J. Org. Chem., 1998, 63, 8432–8439 CrossRef CAS.
- F. Pertici, N. Varga, A. Van Duijn, M. Rey-Carrizo, A. Bernardi and R. J. Pieters, Beilstein J. Org. Chem., 2013, 9, 215–222 CrossRef CAS PubMed.
- S. Ordanini, N. Varga, V. Porkolab, M. Thépaut, L. Belvisi, A. Bertaglia, A. Palmioli, A. Berzi, D. Trabattoni, M. Clerici, F. Fieschi and A. Bernardi, Chem. Commun., 2015, 51, 3816–3819 RSC.
- A. Berzi, S. Ordanini, B. Joosten, D. Trabattoni, A. Cambi, A. Bernardi and M. Clerici, Sci. Rep., 2016, 6, 35373 CrossRef CAS PubMed.
- V. Porkolab, M. Lepšík, S. Ordanini, A. St John, A. Le Roy, M. Thépaut, E. Paci, C. Ebel, A. Bernardi and F. Fieschi, ACS Cent. Sci., 2023, 9, 709–718 CrossRef CAS PubMed.
- H. Feinberg, Y. Guo, D. A. Mitchell, K. Drickamer and W. I. Weis, J. Biol. Chem., 2005, 280, 1327–1335 CrossRef CAS PubMed.
- H. Feinberg, C. K. W. Tso, M. E. Taylor, K. Drickamer and W. I. Weis, J. Mol. Biol., 2009, 394, 613–620 CrossRef CAS PubMed.
- H. Feinberg, R. Castelli, K. Drickamer, P. H. Seeberger and W. I. Weis, J. Biol. Chem., 2007, 282, 4202–4209 CrossRef CAS PubMed.
- F. Probert, S. B.-M. Whittaker, M. Crispin, D. A. Mitchell and A. M. Dixon, J. Biol. Chem., 2013, 288, 22745–22757 CrossRef CAS PubMed.
- A. Poveda and J. Jiménez-Barbero, Chem. Soc. Rev., 1998, 27, 133–144 RSC.
- V. Porkolab, C. Pifferi, I. Sutkeviciute, S. Ordanini, M. Taouai, M. Thépaut, C. Vivès, M. Benazza, A. Bernardi, O. Renaudet and F. Fieschi, Org. Biomol. Chem., 2020, 18, 4763–4772 RSC.
- M. Mammen, S.-K. Choi and G. M. Whitesides, Angew Chem. Int. Ed. Engl., 1998, 37, 2754–2794 CrossRef PubMed.
- J. Hooper, D. Budhadev, D. L. Fernandez Ainaga, N. Hondow, D. Zhou and Y. Guo, ACS Appl. Nano Mater., 2023, 6, 4201–4213 CrossRef CAS PubMed.
- D. Budhadev, E. Poole, I. Nehlmeier, Y. Liu, J. Hooper, E. Kalverda, U. S. Akshath, N. Hondow, W. B. Turnbull, S. Pöhlmann, Y. Guo and D. Zhou, J. Am. Chem. Soc., 2020, 142, 18022–18034 CrossRef CAS PubMed.
- V. Porkolab, E. Chabrol, N. Varga, S. Ordanini, I. Sutkeviciute, M. Thépaut, M. J. García-Jiménez, E. Girard, P. M. Nieto, A. Bernardi and F. Fieschi, ACS Chem. Biol., 2018, 13, 600–608 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods. Synthesis of monovalent and divalent compounds, protein production, surface plasmon resonance and isothermal calorimetry analysis, crystallogenesis and crystal structure determination, SARS-CoV-2 an Ebola trans infection assays. See DOI: https://doi.org/10.1039/d4sc02980a |
| ‡ These two authors have contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2024 |