
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect observation of β-alkynyl eliminations from unstrained propargylic alkoxide Cu(I) complexes by C–C bond cleavage†
Ba L.
Tran
 *,
Jack T.
Fuller
III
,
Jeremy D.
Erickson
,
Bojana
Ginovska
* and
Simone
Raugei
*
*,
Jack T.
Fuller
III
,
Jeremy D.
Erickson
,
Bojana
Ginovska
* and
Simone
Raugei
*
Institute for Integrated Catalysis, Pacific Northwest National Laboratory, Richland, WA 99352, USA. E-mail: ba.tran@pnnl.gov; bojana.ginovska@pnnl.gov; simone.raugei@pnnl.gov
First published on 23rd September 2024
Abstract
β-Carbon eliminations of aryl, allylic, and propargylic alkoxides of Rh(I), Pd(II), and Cu(I) are key elementary reactions in the proposed mechanisms of homogeneously catalysed cross-coupling, group transfer, and annulation. Besides the handful of studies with isolable Rh(I)-alkoxides, β-carbon eliminations of Pd(II)- and Cu(I)-alkoxides are less definitive. Herein, we provide a comprehensive synthetic, structural, and mechanistic study on the β-alkynyl eliminations of isolable secondary and tertiary propargylic alkoxide Cu(I) complexes, LCuOC(H)(Ph)C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh and LCuOC(ArF)2CCPh (L = N-heterocyclic carbene (NHC), dppf, S-BINAP), to produce monomeric (NHC)CuCCPh, dimeric [(diphosphine)CuCCPh]2, and the corresponding carbonyl. Selective β-alkynyl over β-hydrogen elimination was observed for NHC- and diphosphine-supported secondary propargylic alkoxide complexes. The mechanism for the first-order reaction of β-carbon elimination of (IPr*Me)CuOC(ArF)2CCPh is proposed to occur through an organized four-centred transition state via a Cu-alkyne π complex based on Eyring analysis of variable-temperature reaction rates by UV-vis kinetic analysis to provide ΔH‡ = 24(1) kcal mol−1, ΔS‡ = −8(3) e.u., and ΔG‡ (25 °C) = 27 kcal mol−1 over a temperature range of 60–100 °C. Additional quantitative UV-vis kinetic studies conclude that the electronic and steric properties of the NHC ligands engendered a marginal effect on the elimination rate, requiring 2–3 h at 100 °C for completion, whereas complete β-alkynyl eliminations of diphosphine-supported propargylic alkoxides were observed in 1–2 h at 25 °C.
CPh and LCuOC(ArF)2CCPh (L = N-heterocyclic carbene (NHC), dppf, S-BINAP), to produce monomeric (NHC)CuCCPh, dimeric [(diphosphine)CuCCPh]2, and the corresponding carbonyl. Selective β-alkynyl over β-hydrogen elimination was observed for NHC- and diphosphine-supported secondary propargylic alkoxide complexes. The mechanism for the first-order reaction of β-carbon elimination of (IPr*Me)CuOC(ArF)2CCPh is proposed to occur through an organized four-centred transition state via a Cu-alkyne π complex based on Eyring analysis of variable-temperature reaction rates by UV-vis kinetic analysis to provide ΔH‡ = 24(1) kcal mol−1, ΔS‡ = −8(3) e.u., and ΔG‡ (25 °C) = 27 kcal mol−1 over a temperature range of 60–100 °C. Additional quantitative UV-vis kinetic studies conclude that the electronic and steric properties of the NHC ligands engendered a marginal effect on the elimination rate, requiring 2–3 h at 100 °C for completion, whereas complete β-alkynyl eliminations of diphosphine-supported propargylic alkoxides were observed in 1–2 h at 25 °C.
Introduction
The elucidation of mechanisms and selectivities for reactions catalysed by Earth abundant first-row metals is critical to the development of sustainable organotransition chemistry.1–5 The elementary reactions of β-carbon elimination from metal-alkyl and -alkoxide intermediates are prominent in olefin polymerization and catalytic C–C bond formations.6–9 Specifically, the β-eliminations of allyl,9 alkynyl,10–20 and aryl groups17,21–23 from unstrained alkoxides of Pd and Rh to generate the corresponding organo-Pd and -Rh intermediates are implicated in the mechanism of cross-coupling, annulation, and aryl shuttling catalysis.8,9 To date, only a handful of synthetic and mechanistic studies of β-carbon elimination from alkoxide Rh complexes have been reported.21–24 Besides Pd and Rh, Cu-mediated β-allyl25 and β-alkynyl eliminations26–28 of allylic alcohols and propargylic alcohols to generate allyl and acetylide intermediates in catalytic allyl group transfer and annulation reactions have been proposed, yet definitive spectroscopic, structural or mechanistic evidence remains unreported to our knowledge.In contrast to the vast literature on the related mechanism of β-hydrogen elimination, experimental examples and mechanistic knowledge of β-carbon elimination are less established.6–9 The formation of π complexes with allyl, alkynyl, or aryl ligands can be favorable compared to that of M–H–C σ-complexes. However, the reorganization of the larger carbon groups at a metal centre compared to the small spherical H atom of the C–H group is kinetically disfavored. Because of the mechanistic similarities of these two elimination pathways, selective promotion of β-carbon elimination over β-hydrogen elimination is not fully understood, yet is of synthetic interest.8
We direct our studies towards the β-alkynyl eliminations of propargylic alcohols at Cu(I) centres because of the broad interest in Cu-acetylide chemistry in catalysis and inorganic materials.29–32 Moreover, the stability of Cu-acetylide is ideal for product isolation in combination with spectroscopic, structural, and mechanistic investigation.33–35 Based on experimental and computational mechanistic analysis of β-carbon eliminations of unstrained alkoxide Rh complexes,17,22,23,36 we envision a coordinatively unsaturated Cu(I) centre can coordinate and activate the alkyne group at the propargylic alkoxide via a Cu(I)-alkyne π complex to promote β-alkynyl elimination. Herein, we report a rare comprehensive study detailing the synthesis, structure–reactivity relationship, and mechanism of β-alkynyl eliminations from two- and three-coordinate propargylic alkoxide Cu complexes supported by electronically and sterically varied NHCs and diphosphines (Scheme 1).
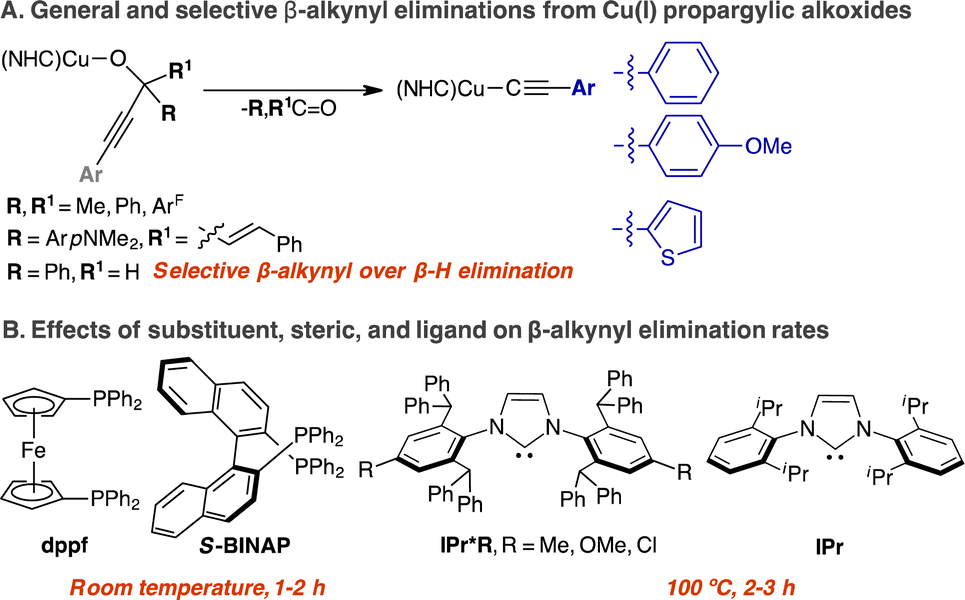 | ||
| Scheme 1 This work details a comprehensive study on the general and selective β-alkynyl eliminations of secondary and tertiary propargylic alkoxides of Cu(I). | ||
Results and discussion
Synthesis, characterisation, and β-alkynyl elimination of (NHC)Cu(I) propargylic alkoxides
A series of tertiary propargylic alkoxide complexes, (IPr*R)CuOC(ArF)2CCPh (R = Me, 1-Me; OMe, 1-OMe; Cl, 1-Cl), were isolated in 73–80% by treating the corresponding (IPr*R)CuCl with 1.05 equiv. of NaN(SiMe3)2 in toluene for 0.5 h, followed by addition of solid HOC(ArF)2CCPh for another 1 h at 25 °C (Scheme 2). Subsequent removal of NaCl by filtration and layering of pentane over the toluene reaction mixture at 25 °C over 24–48 h gave analytically clean, off-white crystalline products of 1-Me, 1-OMe, and 1-Cl. Synthetic procedures with accompanying 1H, 13C{1H}, and 19F{1H} NMR data are provided in the ESI (pg. S3–S8†). The propargylic alkoxide complexes exhibit similar spectroscopic and structural properties; we therefore discuss the characterisation of (IPr*Me)CuOC(ArF)2CCPh (1-Me) as a representative example.
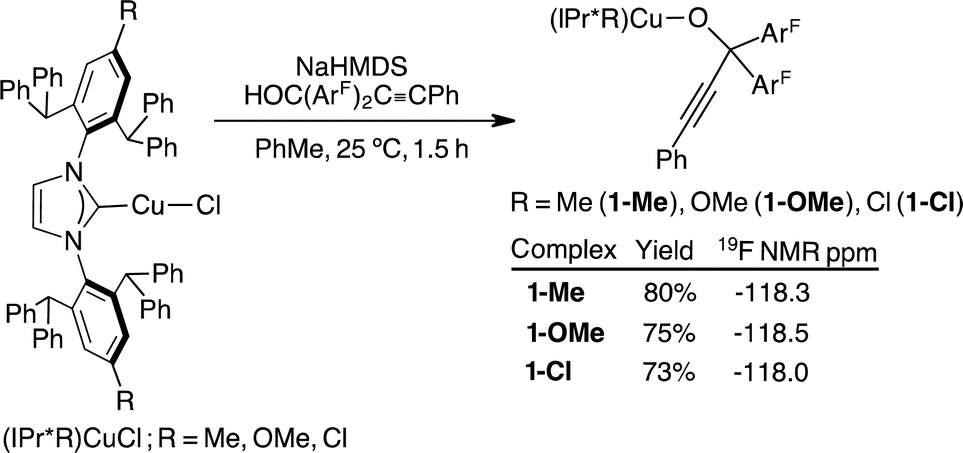 | ||
| Scheme 2 Synthesis and 19F{1H} NMR spectroscopic data of (IPr*R)CuOC(ArF)2CCPh. | ||
The 19F{1H} NMR spectrum of 1-Me contains a sharp singlet at −118 ppm, shifting upfield upon complexation from free HOC(ArF)2CCPh at −115 ppm. The −118 ppm signal is diagnostic for the formation of CuOC(ArF)2CCPh and is insensitive to the identity of IPr*R ligands (Scheme 2). The 1H NMR spectrum shows a multiplet at 8.03 ppm corresponding to the C–H group ortho to the C–O group of the propargyl alkoxide. The alkynyl and C–O functionalities are identified by 13C NMR resonances at 101, 83 and 77 ppm, respectively.
The single crystal X-ray diffraction (scXRD) structure of 1-Me, shown in Fig. 2, verified its linear geometry. Standard bond distances of 1.8586(16) Å and 1.8094(13) Å were observed for Cu1–C1 and Cu1–O1, respectively. The C1–Cu1–O1 angle of 163.46(7)° of 1-Me is notably bent compared to those of related IPr*Me-supported alkoxides and allylic alkoxides at 172–178°.37,38 The distances from Cu1 to C3 and C4 of the alkyne group of 3.11 and 3.64 Å, respectively, do not support a Cu-alkyne π-complex, which exhibits a range of 1.95–1.99 Å for Cu(I)-alkyne.39 The scXRD structure of 1-Cl also contains a bent C1–Cu1–O1 angle at 162.77(5)° whereas the C1–Cu1–O1 angle for 1-OMe is at 172.46(7)° (Fig. 2).
(IPr*Me)CuOC(ArF)2CCPh (1-Me) is stable in C6D6 at room temperature during the monitoring time of 24 h by 1H, 19F{1H} NMR spectroscopy. Subsequent thermolysis at 100 °C for 3 h quantitatively produced (IPr*Me)Cu(CCPh) (2-Me) and ArF2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O by 1H, 19F{1H} NMR spectroscopy in the presence of a 1,3,5-trimethoxybenzene internal standard. 19F{1H} NMR spectroscopic analysis over time shows the consumption of 1-Me at −118 ppm with concomitant growth of ArF2CO at −107 ppm, supporting the extrusion of ArF2CO from 1-Me (Fig. S5B†). The product of 2-Me was identified by the two doublets at 7.69 ppm (J = 7.5 Hz), 7.51 ppm (J = 7.7 Hz) and a triplet at 7.25 ppm (J = 7.7 Hz) by 1H NMR spectroscopy (Fig. 1), consistent with reported 1H NMR data for 2-Me (ref. 33) and independently synthesized 2-Me (Fig. S24A†). The elimination of ArF2CO from 1-Me proceeds sluggishly at lower temperature. Thermolysis of 1-Me at 50 °C for 1 h and 24 h produced 5% and 27% of 2-Me, respectively.
O by 1H, 19F{1H} NMR spectroscopy in the presence of a 1,3,5-trimethoxybenzene internal standard. 19F{1H} NMR spectroscopic analysis over time shows the consumption of 1-Me at −118 ppm with concomitant growth of ArF2CO at −107 ppm, supporting the extrusion of ArF2CO from 1-Me (Fig. S5B†). The product of 2-Me was identified by the two doublets at 7.69 ppm (J = 7.5 Hz), 7.51 ppm (J = 7.7 Hz) and a triplet at 7.25 ppm (J = 7.7 Hz) by 1H NMR spectroscopy (Fig. 1), consistent with reported 1H NMR data for 2-Me (ref. 33) and independently synthesized 2-Me (Fig. S24A†). The elimination of ArF2CO from 1-Me proceeds sluggishly at lower temperature. Thermolysis of 1-Me at 50 °C for 1 h and 24 h produced 5% and 27% of 2-Me, respectively.
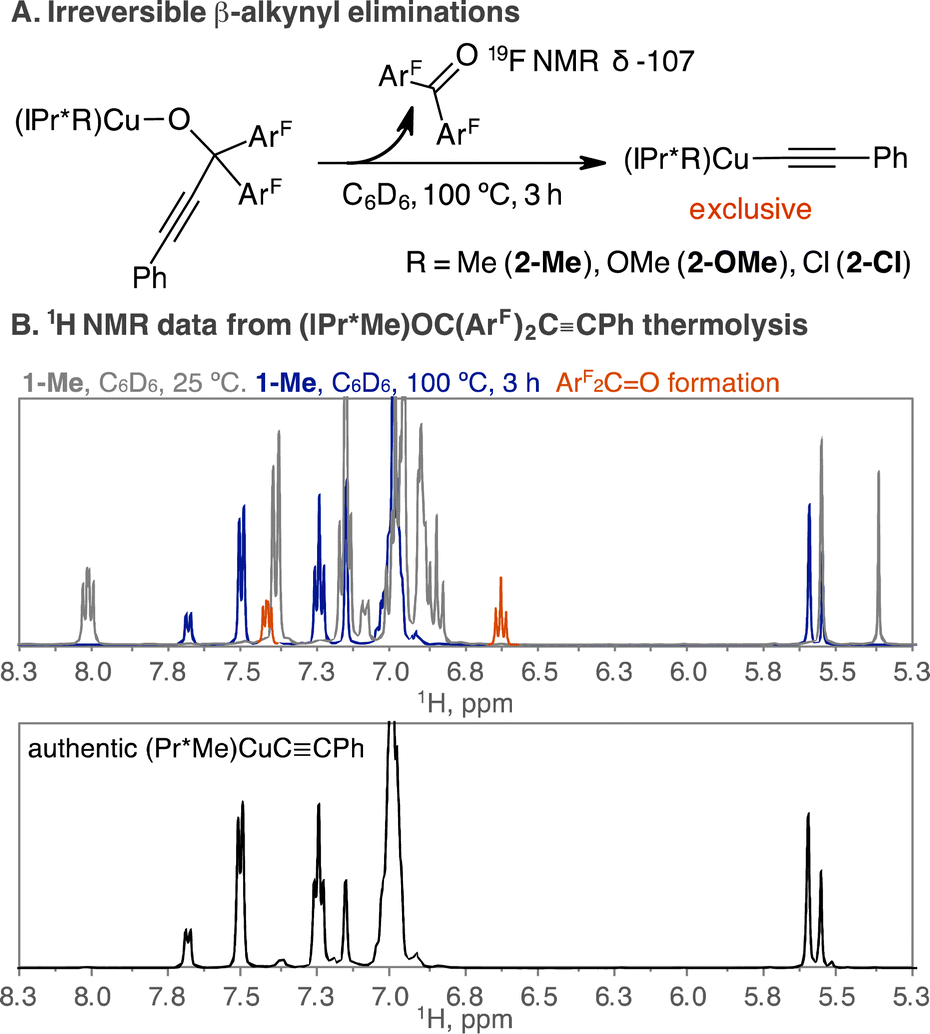 | ||
| Fig. 1 (A) Thermolysis of (IPr*R)CuOC(ArF)2CCPh produced the products (IPr*R)CuCCPh and ArF2CO. (B) The formation of ArF2CO and (IPr*Me)CuCCPh (2-Me) was identified (top) and verified using independently synthesized 2-Me by 1H NMR spectroscopy (bottom). | ||
The β-alkynyl elimination is irreversible as the insertion reaction of ArF2CO and 2-Me to regenerate 1-Me was not observed after 48 h at 25 °C by 1H and 19F{1H} NMR spectroscopy.40 The scXRD structural determination of 1-Me (Fig. 2), from the preparative-scale thermolysis of 1-Me in toluene at 100 °C for 3 h, provided unequivocal evidence of the β-alkynyl elimination reaction. Analogously, the thermolysis of 1-OMe and 1-Cl in C6D6 at 100 °C for 3 h also cleanly produced (IPr*OMe)CuCCPh (2-OMe) and (IPr*Cl)CuCCPh (2-Cl), respectively, and ArF2CO (Fig. S6 and S7†). The spectroscopic assignments of 2-OMe and 2-Cl were verified by independent synthesis along with spectroscopic (Fig. S25 and S26†) and structural characterisation (Fig. 2).
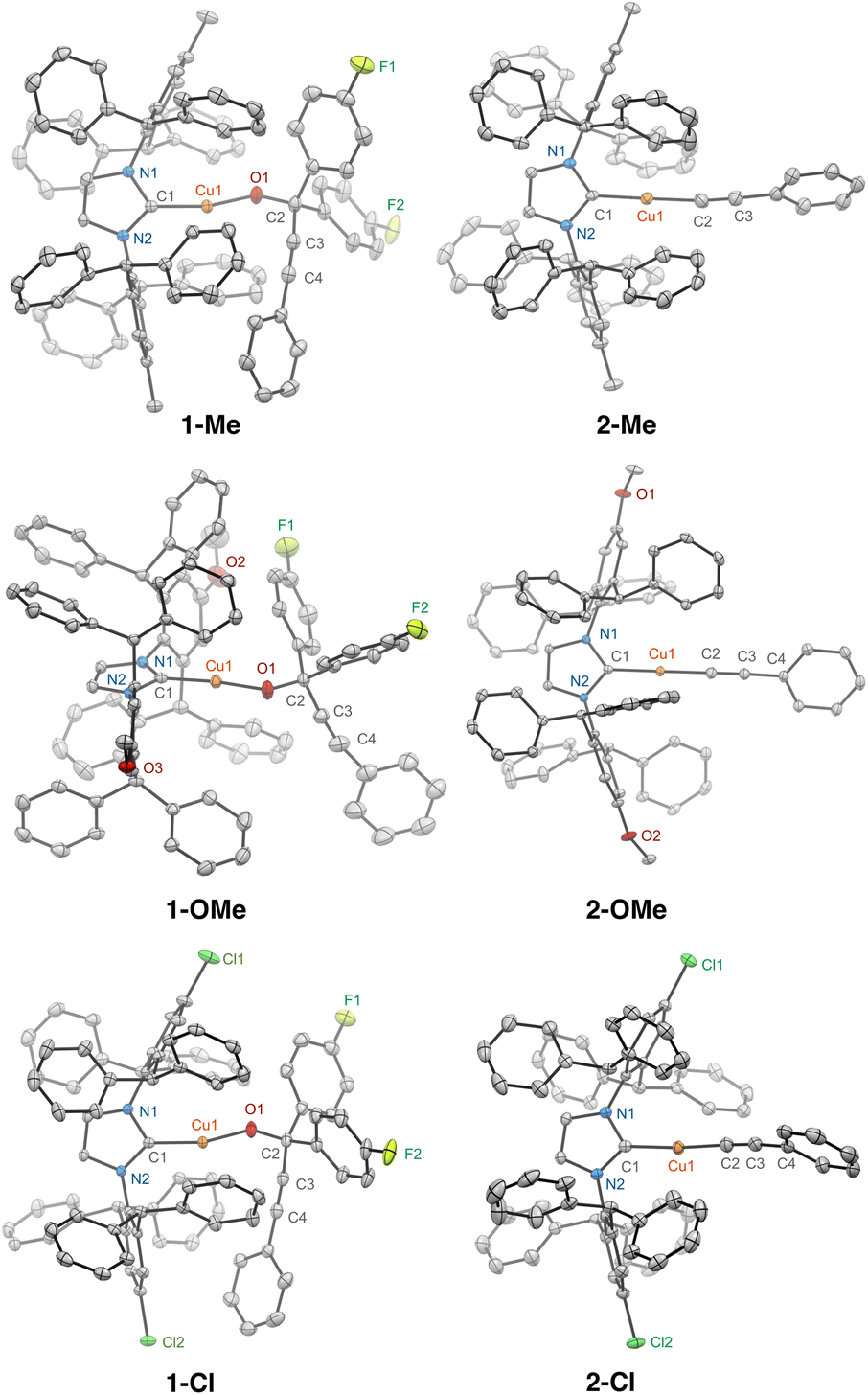 | ||
| Fig. 2 scXRD structures of (IPr*R)CuOC(ArF)2CCPh (1-Me, 1-OMe, 1-Cl) and (IPr*R)CuCCPh (2-Me, 2-OMe, 2-Cl) are shown at a 50% probability thermal ellipsoid. All hydrogen atoms, positional disorders, and solvents are omitted. Metrical parameters of these complexes are mostly similar; therefore, we list selected bonds (Å) and angles (°) of 1-Me and 2-Me: 1-Me, Cu1–C1 = 1.8586(16) and Cu1–O1 = 1.8094(13); C3–C4 = 1.200(3); C1–Cu1–O1 = 163.46(7) and Cu1–O1–C2 = 128.06(12). 2-Me, Cu1–C1 = 1.8869(13), Cu1–C2 = 1.8809(16), and C2–C3 = 1.190(2); C1–Cu1–C2 = 172.07(6) and Cu1–C2–C3 = 171.20(15). | ||
We next evaluated whether reducing the steric congestion at the Cu(I) centre of the propargylic alkoxide complex by employing a less bulky NHC, such as IPr, relative to IPr*Me can facilitate the formation of a Cu(I)-alkyne π complex to lower the energy barrier for β-alkynyl elimination based on the isolation of a Rh(I)-arene π complex from the β-aryl elimination of a Rh(I)-alkoxide.23 To do so, we isolated (IPr)CuOC(ArF)2CCPh (3) in 65% and examined its reactivity (Fig. S4 and S8†). Similar to the (IPr*R)CuOC(ArF)2CCPh series, 3 is stable in C6D6 at 25 °C over 24 h, and complete conversion of 3 to (IPr)CuCCPh and ArF2CO required heating at 100 °C for 3 h based on 1H and 19F{1H} NMR spectroscopic analysis. A quantitative UV-vis kinetic analysis of the steric properties of NHC ligands is reported in a later section.
Diphosphines enabled β-alkynyl eliminations of LCuOC(ArF)2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) CPh (L = dppf, S-BINAP) at room temperature
CPh (L = dppf, S-BINAP) at room temperature
Hartwig and co-workers structurally characterized the complex of (Et3P)2RhOC(Ph)2(η2-Ph), in which one of the phenyls of the triphenylmethoxide ligand contains a Rh-arene interaction, as a key intermediate en route to β-aryl elimination.23 The same authors observed rapid β-alkynyl elimination of in situ generated trigonal planar (Et3P)2RhOC(Ph)2CCPh at −40 °C,17 in which it is plausible that β-alkynyl elimination proceeds by a Rh-alkyne π complex.
From these precedents and the observed stability of linear (IPr*R)CuOC(ArF)2CCPh for β-alkynyl elimination, we postulate that diverting from NHC-supported linear propargylic alkoxides to diphosphine-supported trigonal planar propargylic alkoxide complexes might lower the energy barrier for β-alkynyl elimination by facilitating the formation of the Cu-alkyne π complex. This hypothesis aligns with the qualitative molecular orbital analysis and experimental studies that indicate geometric perturbation of a linear d10 configuration to a bent or trigonal configuration leads to higher reactivity.41–44 Additionally, computation predicts that the pyramidalization of trigonal planar Cu(I) species to accommodate a coordinating ligand (e.g. alkyne group) occurs at lower energy than the bending of linear Cu(I) species.45
Indeed, treating (dppf)Cu(mesityl)46 with 1.0 equiv. of HOC(ArF)2CCPh in C6D6 at 25 °C produced [(μ-dppf)CuCCPh]2 (4), ArF2CO, and a new species by the initial collection of the NMR spectroscopic data. As expected, the 31P{1H} NMR spectrum contains product 4 at −11 ppm, which has been spectroscopically characterized.34 The 31P{1H} NMR signal at −21.8 ppm and the ferrocenyl 1H NMR signals at 4.06 and 3.81 ppm in the 31P{1H} and 1H NMR spectra (Fig. S9A and C†), respectively, indicate the formation of a new species. These signals do not correspond to free dppf, which contains a 31P{1H} NMR signal at −17.5 ppm and 1H NMR signals of the ferrocenyl moiety at 4.14 and 4.03 ppm in C6D6. The 19F{1H} NMR spectrum shows a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio of ArF2CO (−107 ppm) and the new species (−117 ppm) (Fig. S9B†), consistent with a CuOC(ArF)2CCPh species based on the 19F{1H} NMR data of (NHC)CuOC(ArF)2CCPh complexes (see above). From the collective multinuclear NMR data, we therefore proposed the formation of (dppf)CuOC(ArF)2CCPh as the new species.47 After 1 h at 25 °C, only major products of 4 and ArF2CO were detected by 1H, 19F{1H}, and 31P{1H} NMR spectroscopy (Fig. S9†). A preparative-scale reaction of in situ generated (dppf)Cu(mesityl) and HOC(ArF)2CCPh in toluene at 25 °C for 2 h produced 4 in an isolated yield of 60% by recrystallization from THF and pentane at 25 °C over 48 h. The 1H and 31P{1H} NMR data of isolated 4 match those of 4 generated in the above elimination reaction and the reported literature (Fig. S10†).34 Moreover, the scXRD structure of 4, presented in Scheme 3, verifies its dimeric structure.
:3 ratio of ArF2CO (−107 ppm) and the new species (−117 ppm) (Fig. S9B†), consistent with a CuOC(ArF)2CCPh species based on the 19F{1H} NMR data of (NHC)CuOC(ArF)2CCPh complexes (see above). From the collective multinuclear NMR data, we therefore proposed the formation of (dppf)CuOC(ArF)2CCPh as the new species.47 After 1 h at 25 °C, only major products of 4 and ArF2CO were detected by 1H, 19F{1H}, and 31P{1H} NMR spectroscopy (Fig. S9†). A preparative-scale reaction of in situ generated (dppf)Cu(mesityl) and HOC(ArF)2CCPh in toluene at 25 °C for 2 h produced 4 in an isolated yield of 60% by recrystallization from THF and pentane at 25 °C over 48 h. The 1H and 31P{1H} NMR data of isolated 4 match those of 4 generated in the above elimination reaction and the reported literature (Fig. S10†).34 Moreover, the scXRD structure of 4, presented in Scheme 3, verifies its dimeric structure.
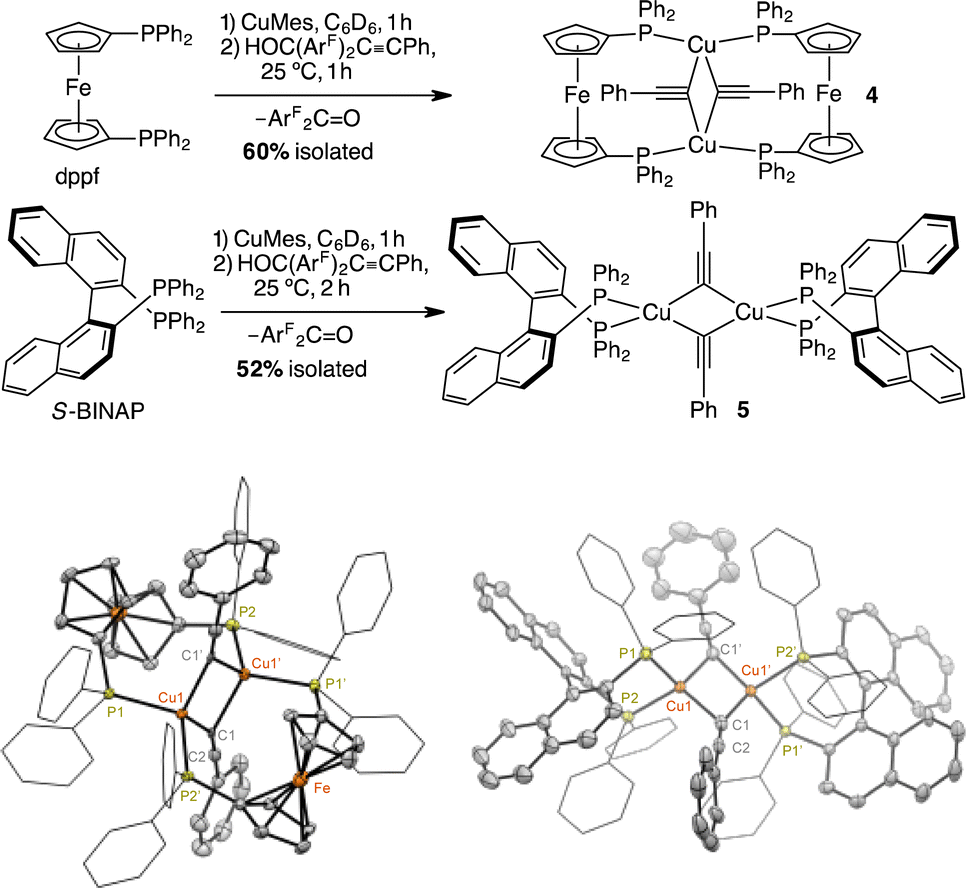 | ||
| Scheme 3 scXRD structures of dimeric [(μ-dppf)Cu(CCPh)]2 (4) and [(S-BINAP)Cu(CCPh)]2 (5) shown at a 50% probability thermal ellipsoid. All hydrogen atoms, positional disorders, and solvents are omitted. | ||
To demonstrate the generality of diphosphines for mediating β-alkynyl elimination at room temperature, we examined the reaction of S-BINAP, CuMes, and HOC(ArF)2CCPh in C6D6. After 2 h at 25 °C, the formation [(S-BINAP)CuCCPh]2 (5) and ArF2CO was evidenced by 1H, 19F{1H}, 31P{1H} NMR spectroscopy (Fig. S11 and S12†). Vapor diffusion of pentane into the reaction mixture at 25 °C afforded yellow crystals of 5 in 52% isolated yields and scXRD structural determination for the dimer of 5 is shown in Scheme 3. Besides the effect of ligands on the rates of β-alkynyl elimination, X-ray crystallographic analysis also revealed the dependence of ligands on the nuclearity of the acetylide complexes in the solid state. Specifically, the preferred formations of monomeric and dimeric phenylacetylide complexes are supported by NHCs33 and diphosphines,35 respectively.
Scope and selectivity of β-alkynyl eliminations
Thus far, we have shown that NHC and diphosphine ligands can promote β-alkynyl eliminations of LCuOC(ArF)2CCPh complexes at different rates and temperatures. We next determined the scope for substituted propargylic alkoxides and the selectivity of β-alkynyl versus β-hydrogen elimination. The scope of propargylic alkoxide Cu(I) complexes for β-alkynyl elimination is summarized in Scheme 4.
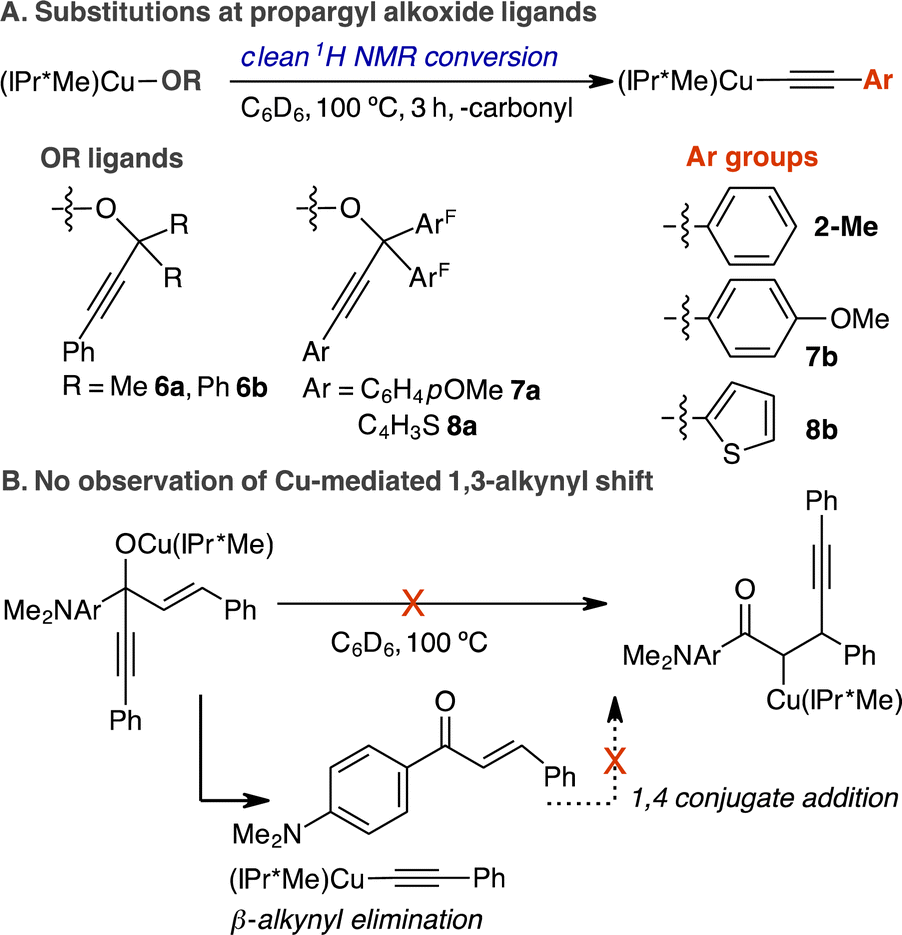 | ||
| Scheme 4 (A) The scope of propargyl alkoxide complexes for β-alkynyl eliminations. (B) Probing for 1,3-alkynyl rearrangement in the reaction of (IPr*Me)CuN(SiMe3)2 and HOC(CCPh)(C6H4-p-NMe2)(–CHCHPh) in C6D6, 100 °C, 3 h. | ||
The steric properties of the dimethyl and diphenyl groups at the β-carbon of the propargylic alkoxide were tolerated as evidenced by the clean conversion of (IPr*Me)CuOC(Me)2CCPh (6a) and (IPr*Me)CuOC(Ph)2CCPh (6b) to 2-Me in C6D6 at 100 °C for 3 h (Scheme 4, Fig. S15 and S18†). Replacing phenylacetylene with electron-rich 4-methoxyphenyl acetylene or 2-thiophene acetylene in complexes of (IPr*Me)CuOC(ArF)2CC(C6H4-p-OMe) (7a) and (IPr*Me)CuOC(ArF)2CC(C4H3S) (8a) also underwent complete elimination to the corresponding acetylide products (IPr*Me)CuCC(C6H4-p-OMe) (7b) and (IPr*Me)CuCC(C4H3S) (8b) (Fig. S19, S20, S27, and S28†). The solid-state structures of 8a and 8b have been verified by scXRD measurement and are shown in Fig. 3. Detailed synthetic, spectroscopic, and structural characterisation of propargylic alkoxide complexes and acetylide products is provided in the ESI.†
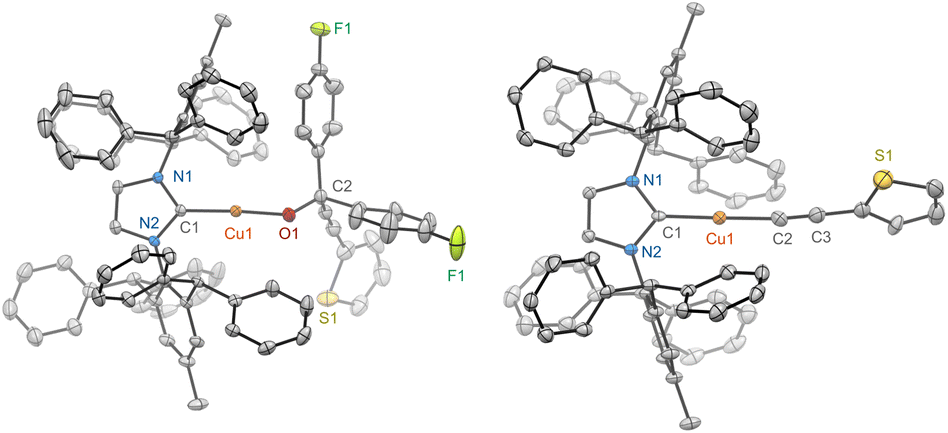 | ||
| Fig. 3 scXRD structures of 8a (left) and 8b (right) are shown at a 50% probability thermal ellipsoid. All hydrogen atoms, positional disorders, and solvents are omitted. | ||
The proposed stepwise reaction sequence of β-alkynyl elimination and 1,4-conjugate addition has enabled (BINAP)Rh-catalysed 1,3-alkynyl rearrangement of alkenyl carbinols to the corresponding β-alkynylketones.14 Therefore, we investigated whether Cu(I) can mediate this 1,3-alkynyl rearrangement by examining the stoichiometric reaction of (IPr*Me)CuN(SiMe3)2, (dppf)CuMes or (S-BINAP)CuMes with 1.0 equiv. HOC(CCPh)(C6H4-p-NMe2)(–CHCHPh) in C6D6 at 25–100 °C for 3–24 h (Scheme 4B). 1H, 31P{1H} NMR spectroscopic analysis of the reaction indicates only the formation of corresponding phenylacetylide complexes of 2-Me, 4, and 5. Control reactions of isolated 2-Me, 4, or 5 with 1.0–2.0 equiv. of 3-(fluorophenyl)-1-phenyl-2-propen-1-one in C6D6 at 25–80 °C for 24–48 h produced no reactions. Collectively, these results indicate that the resulting NHC- or diphosphine-supported phenylacetylide Cu(I) complexes after β-alkynyl eliminations from propargylic alkoxides cannot engage the extruded α,β-unsaturated ketone in 1,4-conjugate addition to complete the 1,3-alkynyl rearrangement as previously observed for Rh chemistry.
To determine the selectivity of β-alkynyl and β-hydrogen elimination, we analyzed the thermolysis of secondary propargylic alkoxide complexes of LCuOC(H)(Ph)CCPh (L = IPr*Me, dppf). We hypothesized that β-alkynyl elimination can be selective over β-hydrogen elimination because the irreversible formation of Cu-acetylides from tertiary propargylic alkoxides hints at a highly exothermic reaction compared to the endothermic reaction of β-hydrogen elimination of Cu(I)-alkoxide to generate Cu–H as suggested by computation studies.48 If β-alkynyl and β-hydrogen elimination traverses a similar four-centred transition state via a Cu-alkyne π or a Cu–H–C σ-complex, respectively, then the π-complex is likely favored over the σ-complex based on the varied reports of Cu-alkyne π complexes29,39 and the computationally predicted repulsive nature of the Cu–H–C interaction.49
Analogous to 1-Me, the secondary propargylic alkoxide complex of (IPr*Me)CuOC(H)(Ph)CCPh (9) is stable at 25 °C in C6D6 for 24 h by 1H NMR spectroscopy.50 Heating the C6D6 solution of 9 at 100 °C for 2 h and 4 h produced (IPr*Me)CuCCPh (2-Me) in 75% and 85%, respectively, against a 1,3,5-trimethoxybenzene internal standard, indicating selective β-alkynyl elimination (Fig. 4). The expected product of PhCHO was detected at 9.63 ppm and verified by spiking the post-reaction mixture with fresh PhCHO, leading to the growth of the 9.63 ppm signal. The presence of [(IPr*Me)CuH]2 or metallic copper from [(IPr*Me)CuH]2 decomposition or Cu-vinyl species from alkyne insertion into the Cu–H bond was not spectroscopically observed (Fig. S22†).
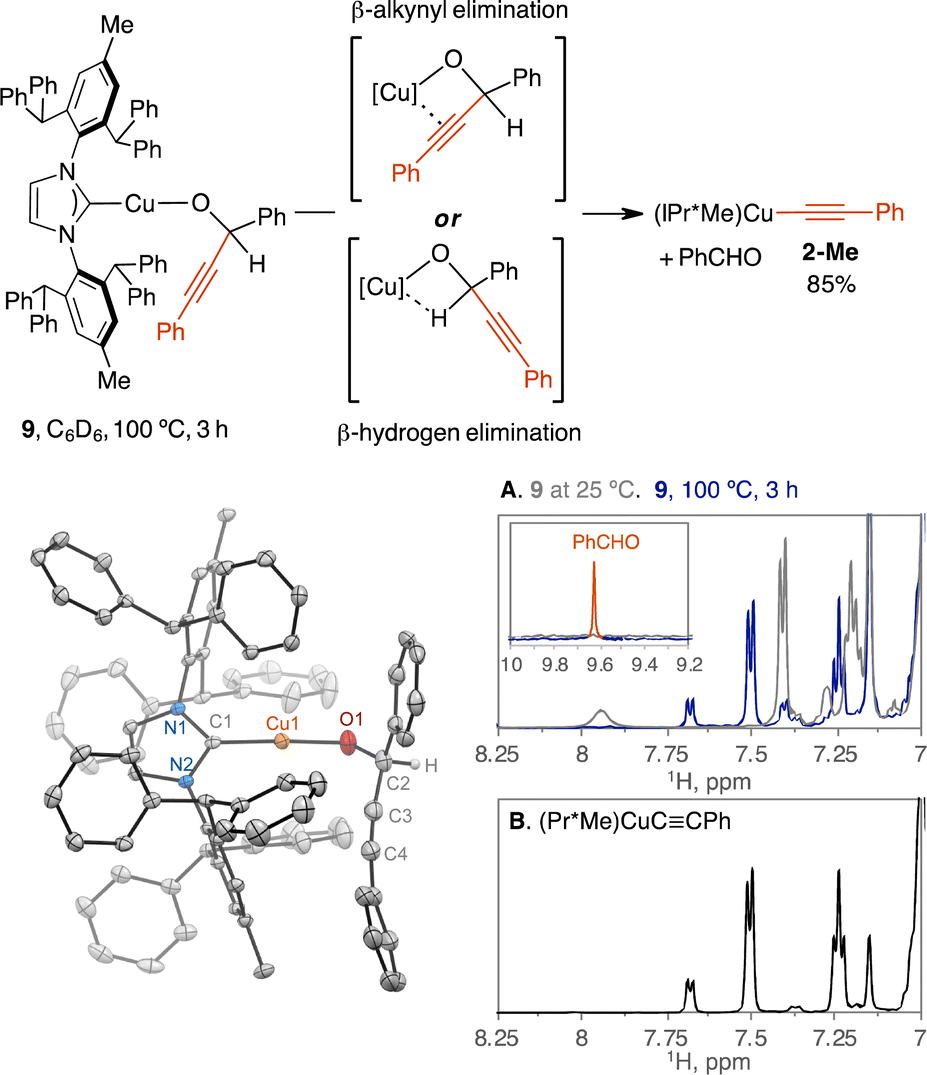 | ||
| Fig. 4 Selective β-alkynyl over β-hydrogen elimination of compound 9 to products 2-Me and PhCHO. (A) 1H NMR data of 9 at 25 °C before heating and at 100 °C after 3 h, showing the presence of 2-Me and PhCHO. (B) Verification of 2-Me in the reaction by referencing the 1H NMR spectrum of authentic 2-Me. The scXRD structure of 9 is shown at a 50% probability thermal ellipsoid and the hydrogen atom on the alkoxide ligand was located and refined without constraint. | ||
Selective β-alkynyl elimination was also achieved in the reaction of dppf, CuMes, and HOC(H)(Ph)CCPh. The initial 1H NMR spectrum (C6D6, 25 °C) after treating in situ generated (dppf)CuMes with HOC(H)(Ph)CCPh contained 58% of PhCHO and [(dppf)Cu(CCPh)]2 (5), and a proposed new species of (dppf)CuOC(H)(Ph)CCPh based on resonances at 6.36 ppm and 4.13, 3.82 ppm corresponding to the C–H group of OC(H)(Ph)CCPh and ferrocenyl C–H of dppf (Fig. S23A†). After 1 h, the major products of PhCHO and 5 were observed in 95% by 1H and 31P{1H} NMR spectroscopy (Fig. S23A and B†). These results demonstrate that the conserved selectivity of β-alkynyl over β-hydrogen elimination in secondary propargyl alkoxide Cu(I) complexes is independent of NHC and diphosphine ligands. Selective β-alkynyl elimination from a secondary propargylic alkoxide of (dppm)2ReOC(H)(Ph)CCPh has been reported (dppm = bis(diphenylphosphino)methane).51 However, the product of (dppm)2ReCCPh was not isolated, as the acetylide complex underwent an acid-mediated secondary reaction to form a vinylidene complex.
Eyring analysis and the effect of NHC ligands on the rates of β-alkynyl elimination by quantitative UV-vis kinetic studies
Based on experimental and computational studies of β-aryl eliminations from Rh alkoxides,22–24,36 β-alkynyl elimination of (NHC)CuOC(ArF)2CCPh likely proceeds through a four-centred transition state via a Cu-alkyne π complex, in which an associative mechanism for rate-limiting C–C bond cleavage should engender a negative entropy of activation ΔS‡. Alternatively, a dissociative mechanism for the rate-limiting extrusion of ArF2CO from (NHC)CuCCPh(ArF2CO) after C–C bond cleavage should yield a positive ΔS‡.
To distinguish these scenarios, we performed an Eyring analysis in a temperature range of 60–100 °C for the thermolysis of (IPr*Me)CuOC(ArF)2CCPh (1-Me) in toluene to produce (IPr*Me)CuOCCPh (2-Me) and ArF2CO. The rates of β-alkynyl elimination, kobs, were determined by UV-vis kinetic studies, in which the reaction progress was monitored at 355 nm for the formation of ArF2CO over 5–6 half-lives (Fig. 5). A representative UV-vis kinetic profile and plot of absorbance versus time for the first-order reaction of β-alkynyl elimination of 3.8 mM 1-Me at 100 °C is shown in Fig. 5. Eyring analysis of the variable-temperature rate data provided activation parameters of ΔH‡ = 24(1) kcal mol−1, ΔS‡ = −8(3) e.u., and ΔG‡ (25 °C) = 27 kcal mol−1 (Fig. 5). A free energy of ΔG‡ (25 °C) = 27 kcal mol−1 is consistent with the lack of β-alkynyl elimination of 1-Me at room temperature. The magnitude and negative ΔS‡ support an associative mechanism for the β-alkynyl elimination of 1-Me and are consistent with reported Eyring analysis for unimolecular β-hydrogen and β-alkyl elimination of metal alkyl complexes that proceed through four-centred transition states.52–55 We further demonstrated the unimolecular β-alkynyl elimination of 1-Me in the solid state. Heating of solid 1-Me at 100 °C under a N2 atmosphere, followed by dissolution of the solid aliquots at the designated time intervals in C6D6 for 1H, 19F{1H} NMR spectroscopic analysis, showed a mixture of 1-Me and 2-Me in ratios of 1.0:2.5, 1.0:8.0, and 1.0:25 after 3 h, 6 h and 9 h, respectively (Fig. S30†).
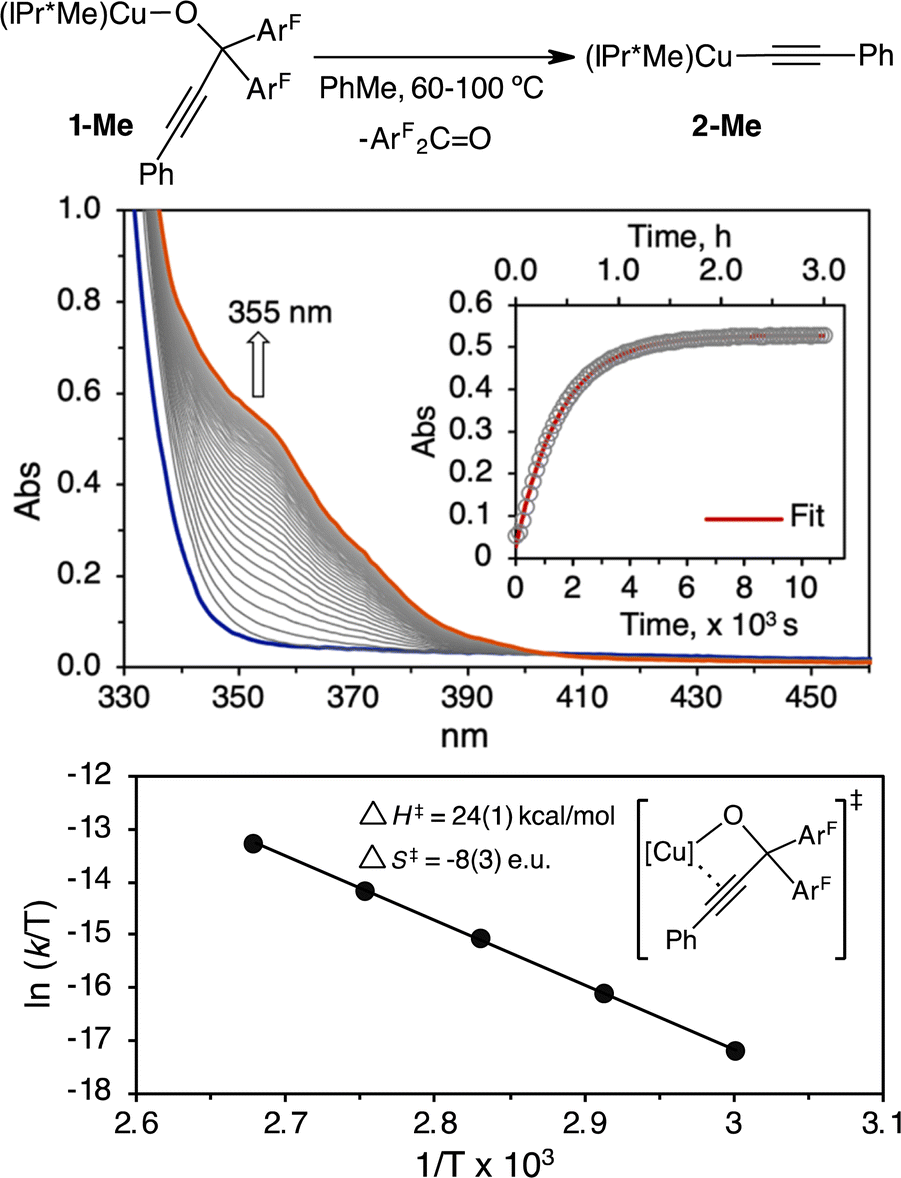 | ||
| Fig. 5 UV-vis kinetic and Eyring analysis for the β-alkynyl elimination of 1-Me (3.8 mM) support an associative mechanism. Representative UV-vis kinetic profile and analysis of 1-Me (3.8 mM) in toluene at 100 °C indicate first-order kinetic behavior and a kobs = 6.2 × 10−4 s−1. A summary of all kobs at 60–100 °C in duplicate runs is provided in Table S1 of the ESI.† | ||
To determine the effect of steric and electronic properties of NHC ligands on β-alkynyl elimination of tertiary propargylic alkoxides, we measured the rates of elimination for 3.8 mM of 1-Me, 1-Me, 1-OMe, 1-Cl and 3 in toluene at 90 °C using the above UV-vis kinetic procedure. The resulting rates of reaction are summarized in Fig. 6. Complexes 1-Me and 3, which contain calculated %volume buried (r = 5.5 Å) of 63.6 for IPr*Me and 46.3 for IPr,56 exhibited similar elimination rates of 2.6(1) × 10−4 s−1 and 2.4(1) × 10−4 s−1, respectively, whereas electronically varied 1-OMe and 1-Cl underwent β-alkynyl elimination 2.2 and 1.3 times faster than that of 1-Me, respectively. Additional studies to elucidate the electronic effects of 1-OMe and 1-Cl on the elimination rate were not pursued since the contribution is minor compared to diphosphines, which can promote β-alkynyl elimination at room temperature. Collectively, propargylic alkoxide complexes supported by diphosphines exhibited faster rates of β-alkynyl elimination at lower reaction temperature than those supported by sterically and electronically modified monodentate NHC ligands.
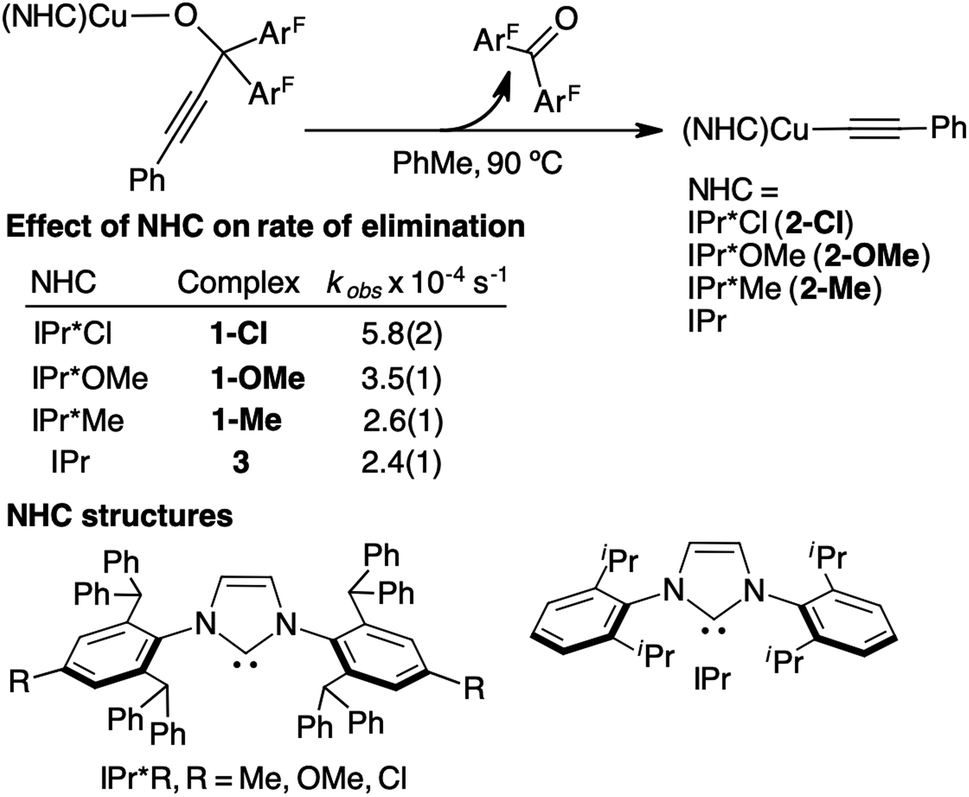 | ||
| Fig. 6 A summary of UV-vis kinetic studies for the β-alkynyl elimination rates of 3.8 mM 1-Me, 1-OMe, 1-Cl, and 3. Additional UV-vis kinetic plots and duplicate or triplicate measurements of rates are provided in Fig. S29A–D and Table S2 of the ESI.† | ||
The accelerated β-alkynyl eliminations of diphosphine-supported propargylic alkoxide Cu(I) complexes at room temperature compared to those of NHC-supported propargylic alkoxide Cu(I) complexes at higher temperature is rationalized by a lowered energy barrier for C–C bond cleavage of the propargylic alkoxide from a three-coordinate propargylic alkoxide complex or a four-coordinate (diphosphine)Cu-alkyne π complex as the ground-state (GS) structure. To distinguish between these mechanistic scenarios, we performed computational analysis on complexes of LCuOCArFCCPh (L = IPr*Me, S-BINAP) using dispersion-corrected DFT57 with an ωB97X-D3 functional and the def2-TZVP basis set.58 Geometry optimizations were conducted in a continuum solvent (benzene).59 Details of the methodology are provided on page S42 of the ESI.†
The computed GS structure of (S-BINAP)CuOC(ArF)2CCPh is a trigonal planar complex rather than a Cu-alkyne π complex (Fig. 7). (S-BINAP)CuOC(ArF)2CCPh undergoes β-alkynyl elimination with a calculated free energy barrier (ΔG‡) of 23.5 kcal mol−1 compared to that of (IPr*Me)CuOC(ArF)2CCPh at 24.8 kcal mol−1. This small energy difference of 1.4 kcal mol−1 is less than the approximate energy difference of 4–5 kcal mol−1 based on the observed rate of β-alkynyl elimination as a function of temperature. Due to the limitations in the accuracy of the method, we also analyzed analogous complexes to establish a trend for ΔG‡ to eliminate systematic errors in the activation free energy.
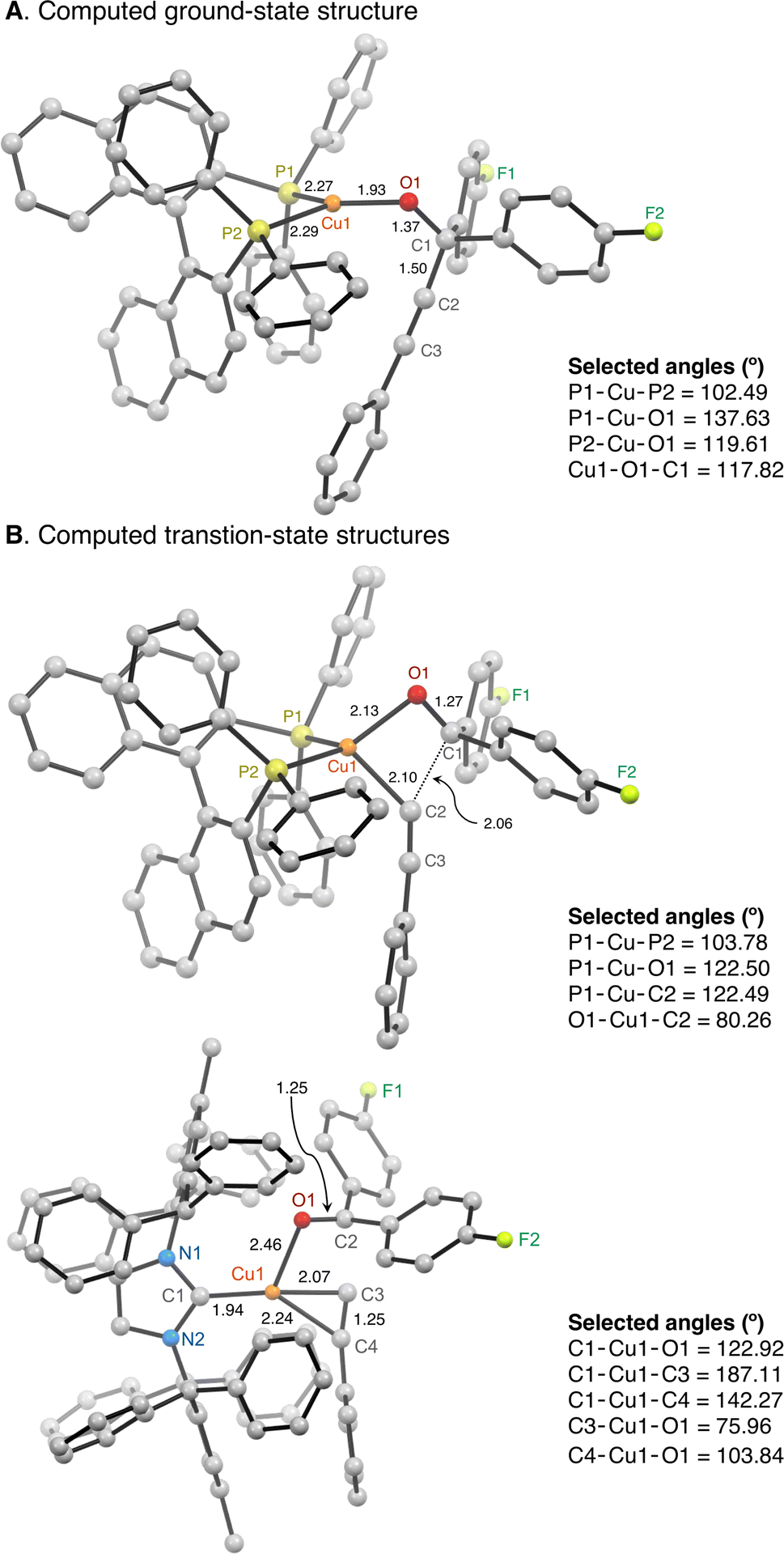 | ||
| Fig. 7 Computational analysis was performed to determine the ground-state structure of (S-BINAP)CuOC(ArF)2CCPh (A) and transition-state structures of LCuOC(ArF)2CCPh complexes (L = S-BINAP, IPr*Me) (B) and their free energy barriers for β-alkynyl elimination. The free energy barrier for β-alkynyl elimination of a trigonal planar (S-BINAP)CuOC(ArF)2CCPh is lowered than that of a linear (IPr*Me)CuOC(ArF)2CCPh. | ||
We therefore calculated the β-alkynyl elimination barrier for (IPr)CuOC(ArF)2CCPh because this complex exhibits a similar β-alkynyl elimination rate to that of (IPr*Me)CuOC(ArF)2CCPh (Fig. 6) and is ligated by an IPr ligand devoid of flexible flanking aryls. A ΔG‡ of 26.3 kcal mol−1 was observed for the β-alkynyl elimination of (IPr)CuOC(ArF)2CCPh. A 2.8 kcal mol−1 increase in the ΔG‡ for the C–C bond cleavage of (IPr)CuOC(ArF)2CCPh compared to that of (S-BINAP)CuOC(ArF)2CCPh approaches the approximate energy difference that is consistent with experimental β-alkynyl elimination rates.
The transition-state (TS) structures for the β-alkynyl elimination of linear (IPr*Me)CuOC(ArF)2CCPh and trigonal planar (S-BINAP)CuOC(ArF)2CCPh complexes show complete C–C bond cleavage of the propargylic alkoxide as evidenced by a bond distance of 1.25–1.27 Å for the CO functionality of ArF2CO (Fig. 7). The presence of monodentate IPr*Me and bidentate S-BINAP ligands also leads to different binding modes of the ruptured phenylacetylide. The TS structure of IPr*Me-supported species resembles a T-shaped geometry based on key angles of C1–Cu1–O1 = 122.92° and C1–Cu1–C3 = 187.11°. The Cu(I) center is additionally coordinated by an η1-OCArF2 and contains a weak Cu–(η2-CCPh) π interaction as evidenced by a C3–C4 distance of 1.25 Å. The computed trigonal planar (S-BINAP)CuOC(ArF)2CCPh complex adopts a distorted tetrahedral complex in the transition state coordinated by η1-OCArF2 and σ-CCPh ligands (Fig. 7).
We also computationally examined the electronic effect of the electron-rich SIPr ligand, which contains a saturated five-membered ring, by calculating the ΔG‡ of β-alkynyl elimination for LCuOC(ArF)2CCPh (L = IPr, SIPr) [SIPr = (1,3-bis(2,6-diisopropylphenyl)dihydroimidazolidin-2-ylidene)]. The calculated ΔG‡ of 26.3 and 26.2 kcal mol−1 for (IPr)CuOC(ArF)2CCPh and (SIPr)CuOC(ArF)2CCPh, respectively, suggests no electronic contribution between IPr and SIPr on the elimination rate at this level of theory. The geometries of the TS structures of LCuOC(ArF)2CCPh (L = IPr, SIPr) are presented in Fig. S32 of the ESI.†
Conclusions
We have provided direct spectroscopic and structural evidence for the general, selective, and irreversible β-alkynyl eliminations of tertiary and secondary propargylic alkoxide Cu(I) complexes supported by various NHC and diphosphine ligands. The transformation tolerates substitutions of aryl, alkyl, hydrogen, aryl alkenyl, –CC(C6H4-p-OMe), and –CC(C4H3S) at the β-carbon of the propargylic alkoxides and exhibits selective β-alkynyl over β-hydrogen elimination for the different secondary propargyl alkoxide Cu(I) complexes. This selectivity is attributed to the favorable formation of a Cu-alkyne π complex over a Cu–H–C σ complex and the stable product of Cu(I)-acetylide.
Eyring analysis of the β-alkynyl elimination of (IPr*Me)CuOC(ArF)2CCPh supports an associative mechanism, in which C–C bond cleavage at the propargylic alkoxide proceeds through a four-centred transition state. Employing diphosphine ligands dramatically influenced the relative rate and reaction temperature for the β-alkynyl elimination of propargylic alkoxide complexes compared to the elimination rates obtained by modulating the steric and electronic properties of the NHC ligands. Computational analysis identified a lower free energy barrier for β-alkynyl elimination at trigonal planar diphosphine-supported propargylic alkoxide Cu(I) species than those of linear NHC-supported analogues. This comprehensive study of C–C bond cleavage at isolable low-coordinate Cu(I)-alkoxides serves to broaden the scope and mechanistic understanding of rarely observed β-carbon elimination of late first-row transition metals beyond that of Pd- and Rh-alkoxides.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.† The computed geometries of the ground-state and transition-state structures reported in this study have been uploaded as XYZ files. All published datasets for the X-ray crystal structures in this study have been uploaded to the Cambridge Structural Database. The assigned CCDC identification numbers and the complexes in the study are as follows: CCDC 23458181-Me, 23458101-OMe, 23458191-Cl, 23458092-Me, 23458112-OMe, 23458142-Cl, 23458174, 23458165, 23458138a, 23458128b, 23458159.†Author contributions
Ba L. Tran: conceptualization, investigation, data analysis, writing, editing, supervision. Jeremy D. Erickson: data analysis. Jack T. Fuller III: investigation, data analysis. Bojana Ginovska: supervision, writing – review & editing. Simone Raugei: supervision, writing – review & editing. All authors approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences and Biosciences Division, Catalysis Science Program, FWP 47319 and the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 using NERSC award BES-ERCAP0026608.Notes and references
- R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Science, 2020, 369, eabc3183 CrossRef CAS PubMed.
- M. R. Friedfeld, H. Zhong, R. T. Ruck, M. Shevlin and P. J. Chirik, Science, 2018, 360, 888–893 CrossRef CAS PubMed.
- C. P. Delaney, E. Lin, Q. Huang, I. F. Yu, G. Rao, L. Tao, A. Jed, S. M. Fantasia, K. A. Püntener, R. D. Britt and J. F. Hartwig, Science, 2023, 381, 1079–1085 CrossRef CAS PubMed.
- P. J. Chirik, K. M. Engle, E. M. Simmons and S. R. Wisniewski, Org. Process Res. Dev., 2023, 27, 1160–1184 CrossRef CAS.
- A. Fürstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef PubMed.
- M. E. O'Reilly, S. Dutta and A. S. Veige, Chem. Rev., 2016, 116, 8105–8145 CrossRef PubMed.
- A. D. Marchese, B. Mirabi and M. Lautens, Synthesis, 2023, 55, 2285–2303 CrossRef CAS.
- M. D. R. Lutz and B. Morandi, Chem. Rev., 2021, 121, 300–326 CrossRef CAS PubMed.
- K. Nogi and H. Yorimitsu, Chem. Rev., 2021, 121, 345–364 CrossRef CAS PubMed.
- H.-F. Chow, C.-W. Wan, K.-H. Low and Y.-Y. Yeung, J. Org. Chem., 2001, 66, 1910–1913 CrossRef CAS PubMed.
- T. Nishimura, H. Araki, Y. Maeda and S. Uemura, Org. Lett., 2003, 5, 2997–2999 CrossRef CAS PubMed.
- A. Funayama, T. Satoh and M. Miura, J. Am. Chem. Soc., 2005, 127, 15354–15355 CrossRef CAS PubMed.
- X. Dou, N. Liu, J. Yao and T. Lu, Org. Lett., 2018, 20, 272–275 CrossRef CAS PubMed.
- T. Nishimura, T. Katoh, K. Takatsu, R. Shintani and T. Hayashi, J. Am. Chem. Soc., 2007, 129, 14158–14159 CrossRef CAS PubMed.
- R. Shintani, K. Takatsu, T. Katoh, T. Nishimura and T. Hayashi, Angew. Chem., Int. Ed., 2008, 47, 1447–1449 CrossRef CAS PubMed.
- K. Yasui, N. Chatani and M. Tobisu, Org. Lett., 2018, 20, 2108–2111 CrossRef CAS PubMed.
- P. Zhao and J. F. Hartwig, Organometallics, 2008, 27, 4749–4757 CrossRef CAS.
- K.-L. Choo and M. Lautens, Org. Lett., 2018, 20, 1380–1383 CrossRef CAS PubMed.
- H. Yuan, Q. Zhou and J. Wang, Org. Chem. Front., 2023, 10, 2081–2094 RSC.
- Y. Zhi, J. Huang, N. Liu, T. Lu and X. Dou, Org. Lett., 2017, 19, 2378–2381 CrossRef CAS PubMed.
- M. D. R. Lutz, V. C. M. Gasser and B. Morandi, Chem, 2021, 7, 1108–1119 CAS.
- M. D. R. Lutz, S. Roediger, M. A. Rivero-Crespo and B. Morandi, J. Am. Chem. Soc., 2023, 145, 26657–26666 CrossRef CAS PubMed.
- P. Zhao and C. D. I. J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 3124–3125 CrossRef CAS PubMed.
- G. Smits, B. Audic, M. D. Wodrich, C. Corminboeuf and N. Cramer, Chem. Sci., 2017, 8, 7174–7179 RSC.
- M. Sai, H. Yorimitsu and K. Oshima, Angew. Chem., Int. Ed., 2011, 50, 3294–3298 CrossRef CAS PubMed.
- T. Li, Z. Wang, M. Zhang, H.-J. Zhang and T.-B. Wen, Chem. Commun., 2015, 51, 6777–6780 RSC.
- T. Li, Z. Wang, W.-B. Qin and T.-B. Wen, ChemCatChem, 2016, 8, 2146–2154 CrossRef CAS.
- X. Yan, R. Ye, H. Sun, J. Zhong, H. Xiang and X. Zhou, Org. Lett., 2019, 21, 7455–7459 CrossRef CAS PubMed.
- H. Lang, A. Jakob and B. Milde, Organometallics, 2012, 31, 7661–7693 CrossRef CAS.
- J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- G. Evano, K. Jouvin, C. Theunissen, C. Guissart, A. Laouiti, C. Tresse, J. Heimburger, Y. Bouhoute, R. Veillard, M. Lecomte, A. Nitelet, S. Schweizer, N. Blanchard, C. Alayrac and A. C. Gaumont, Chem. Commun., 2014, 50, 10008–100018 RSC.
- N. K. Devaraj and M. G. Finn, Chem. Rev., 2021, 121, 6697–6698 CrossRef CAS PubMed.
- I. Ibni Hashim, T. Scattolin, N. V. Tzouras, L. Bourda, K. Van Hecke, I. Ritacco, L. Caporaso, L. Cavallo, S. P. Nolan and C. S. J. Cazin, Dalton Trans., 2021, 51, 231–240 RSC.
- J. Díez, M. P. Gamasa, J. Gimeno, A. Aguirre, S. García-Granda, J. Holubova and L. R. Falvello, Organometallics, 1999, 18, 662–669 CrossRef.
- G. Hattori, K. Sakata, H. Matsuzawa, Y. Tanabe, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2010, 132, 10592–10608 CrossRef CAS PubMed.
- L. Xue, K. C. Ng and Z. Lin, Dalton Trans., 2009, 5841–5850 RSC.
- B. L. Tran, B. D. Neisen, A. L. Speelman, T. Gunasekara, E. S. Wiedner and R. M. Bullock, Angew. Chem., Int. Ed., 2020, 59, 8645–8653 CrossRef CAS PubMed.
- B. L. Tran, J. D. Erickson, A. L. Speelman and R. M. Bullock, Inorg. Chem., 2023, 62, 342–352 CrossRef CAS PubMed.
- A. Noonikara-Poyil, S. G. Ridlen, I. Fernández and H. V. R. Dias, Chem. Sci., 2022, 13, 7190–7203 RSC.
- The reaction of isolated 2-Me with 5 equiv. of ArF2CO in C6D6 at 25–80 °C for 12 h also showed no reaction and no decomposition of 2-Me.
- L. P. Wolters and F. M. Bickelhaupt, ChemistryOpen, 2013, 2, 106–114 CrossRef CAS PubMed.
- T. A. Albright, J. K. Burdett and M.-H. Whangbo, in Orbital Interactions in Chemistry, 2013, pp. 503–526 Search PubMed.
- M. Brendel, C. Braun, F. Rominger and P. Hofmann, Angew. Chem., Int. Ed., 2014, 53, 8741–8745 CrossRef CAS PubMed.
- M. Hackett and G. M. Whitesides, J. Am. Chem. Soc., 1988, 110, 1449–1462 CrossRef CAS.
- M. A. Carvajal, J. J. Novoa and S. Alvarez, J. Am. Chem. Soc., 2004, 126, 1465–1477 CrossRef CAS PubMed.
- Complex (dppf)Cu(mesityl) was generated in situ from the reaction of dppf and Cu-mesityl (CuMes) in C6D6 at 25 °C. [dppf = bis(diphenylphosphino)ferrocene]. A three-coordinate diphosphine-supported Cu(I)-mesityl complex, [PhB(CH2PtBu2)2]Cu-mesityl, has been spectroscopically and structurally characterized: R. C. Handford, L. Zhong and T. D. Tilley, Organometallics, 2022, 41, 2631–2637 CrossRef CAS.
- Examples of spectroscopically and structurally characterized mononuclear (dppf)CuX [X = OAc, SCF3, N(C(O)CF3)(Ar)]: (a) C. B. Khadka, B. K. Najafabadi, M. Hesari, M. S. Workentin and J. F. Corrigan, Inorg. Chem., 2013, 52, 6798–6805 CrossRef CAS PubMed; (b) Y. Yang, L. Xu, S. Yu, X. Liu, Y. Zhang and D. A. Vicic, Chem.–Eur. J., 2015, 22, 858–863 CrossRef PubMed; (c) X. Liu, S. Zhang and Y. Ding, Dalton Trans., 2012, 41, 5897–5902 RSC.
- A. L. Speelman, B. L. Tran, J. D. Erickson, M. Vasiliu, D. A. Dixon and R. M. Bullock, Chem. Sci., 2021, 12, 11495–11505 RSC.
- G. Dos Passos Gomes, G. Xu, X. Zhu, L. M. Chamoreau, Y. Zhang, O. Bistri-Aslanoff, S. Roland, I. V. Alabugin and M. Sollogoub, Chem.–Eur. J., 2021, 27, 8127–8142 CrossRef CAS PubMed.
- The synthesis and spectroscopic characterisation of (IPr*Me)CuOC(H)(Ph)CCPh (9) are provided in the ESI (Fig. S21, pages S27–S28†).
- K. F. Lee, W. Bai, H. H. Y. Sung, I. D. Williams, Z. Lin and G. Jia, Chem.–Eur. J., 2018, 24, 9760–9764 CrossRef CAS PubMed.
- B. J. Burger, M. E. Thompson, W. D. Cotter and J. E. Bercaw, J. Am. Chem. Soc., 1990, 112, 1566–1577 CrossRef CAS.
- H. Xu, C. T. Hu, X. Wang and T. Diao, Organometallics, 2017, 36, 4099–4102 CrossRef CAS.
- L. H. Shultz and M. Brookhart, Organometallics, 2001, 20, 3975–3982 CrossRef CAS.
- C. L. Beswick and T. J. Marks, J. Am. Chem. Soc., 2000, 122, 10358–10370 CrossRef CAS.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- Y.-S. Lin, G.-D. Li, S.-P. Mao and J.-D. Chai, J. Chem. Theory Comput., 2013, 9, 263–272 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, spectroscopic characterisation, and details of kinetics studies (PDF). CCDC 23458181-Me, 23458101-OMe, 23458191-Cl, 23458092-Me, 23458112-OMe, 23458142-Cl, 23458174, 23458165, 23458138a, 23458128b, 23458159. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02982h |
| This journal is © The Royal Society of Chemistry 2024 |