
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccurate construction of monolayer, bilayer, sandwich bilayer, four-layer, multi-layer and chiral bilayer 2D pillararene-type supramolecular networks†
Zhao-Nian
Chen‡
a,
Le-Ping
Zhang‡
a,
Huai-Li
Wu
a,
Qiao-Yan
Qi
b,
Meng
Yan
c,
Jia
Tian
 b,
Guan-Yu
Yang
a,
Zhan-Ting
Li
*bd and
Bo
Yang
*a
b,
Guan-Yu
Yang
a,
Zhan-Ting
Li
*bd and
Bo
Yang
*a
aCollege of Chemistry, Zhengzhou University, 100 Kexue Street, Zhengzhou, Henan 450001, China. E-mail: yangbohy@zzu.edu.cn
bKey Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, Shanghai, 200032, China
cSchool of Chemistry and Chemical Engineering, Henan University of Technology, Zhengzhou, Henan 450001, China
dDepartment of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Fudan University, 2205 Songhu Road, Shanghai, 200438, China. E-mail: ztli@fudan.edu.cn
First published on 5th July 2024
Abstract
The accurate construction of mono-, bi- and multi-layer networks has been an important challenge, especially for bi- and multi-layer networks. Monolayer, bilayer, sandwich bilayer, four-layer, and multi-layer two-dimensional pillararene-type metal–organic coordination networks have been constructed from functionalized pillar[5]arene and pillar[6]arene by utilizing the coordination interaction of cobalt and copper ions and combining with temperature control and guest induction. These two-dimensional coordination networks exhibit the excellent plasticity of pillararenes and structural variety, which are characterized by X-ray single crystal diffraction and PXRD, confirming that pillararenes units can function as excellent tunable scaffolds for structural regulation. Two-dimensional chiral double-layer structure products are also constructed from R- and S-pillar[6]arene, which are obtained by high-performance liquid chromatography. Atomic force microscopic imaging confirms the thicknesses of these networks. Moreover, these networks also exhibit high iodine adsorption capacity in aqueous environments at ambient temperature. The monolayer, bilayer, sandwich bilayer, four-layer and multi-layer structures of the pillararene-type networks represent a new facile supramolecular self-assembly strategy and platform for designing more mono-, bi- and multi-layer two-dimensional nanomaterials and chiral two-dimensional double-layer structures provide a new method for the construction of more two-dimensional chiral polymers.
Introduction
Since the emergence of graphene monolayer materials,1 research towards developing more types of monolayer two-dimensional (2D) materials has exploded.2,3 These 2D materials usually exhibit exceptional performance in diverse applications due to their distinct structural characteristics of ultrathin thicknesses, enormously high surface areas, and well-defined 2D extended skeletons. Controlling the arrangement, thickness, or number of monolayers also played a crucially important role, especially when the designed function involves electron or energy transfer between the adjacent monolayers,4,5 so direct access to mono-, bi- and multi-layer structures remains an important challenge in constructing 2D materials. At the same time, it is more challenging to use one compound to accurately control monolayers and bilayers, or construct adjustable double-layer, four-layer and multi-layer structures by adjusting their structure, especially for bilayer, four-layer and multilayer structures. Supramolecular host macrocycles are a special class of ring structures,6–9 which can be modified on the ring to adjust the size of the cavity according to the size of the guest molecule, have been used in constructing bilayer sheets.10–12 Kian Ping Loh and co-workers introduced a viologen threading unit based on crown ethers and reported that the formation of a host–guest complex facilitates the self-exfoliation of covalent organic frameworks (COFs) into crystalline monolayers or bilayers.10 Zhao et al. constructed a unique Janus bilayer 2D supramolecular organic framework through self-assembly of amphiphilic tritopic molecules and cucurbit[8]uril.11 We also realized the hierarchical self-assembly of a bilayer 2D supramolecular organic framework in a controllable manner through cucurbit[8]uril-encapsulation-promoted dimerization of 4-phenylpyridinium subunits and the coordination of rod-like bipyridine ligands with zinc porphyrin subunits.12Pillararenes, a relatively new class of macrocycles, have shown great promise in host–guest molecular recognition and the fabrication of various materials for divergent applications, since Ogoshi and co-workers reported the first pillararene in 2008.13–23 The rims of pillararenes are highly customizable, enabling them to be fitted with different substituents for different functions, which significantly facilitates their hybridization into metal–organic networks or pillars.24–30 Meanwhile the existing several metal–organic networks or pillars are A1/A2-difunctionalized or five para-substituted pillar[5]arenes and the rigid pillar architectures of pillararenes are all maintained. To the best of our knowledge, bi- and multi-layer 2D polymeric architectures by using the cavity of pillararenes as tunable scaffolds by deforming or adjusting the rigid pillar cavity have not been accessed to date. Herein, considering the versatile potential of modification on the rims of pillararenes and the tunability of cavities of pillararenes, we have incorporated pillar[5]arene and pillar[6]arene units as excellent tunable scaffolds into the construction of monolayer, bilayer, and multi-layer coordination networks. In this system, we report the controlled self-assembly of 2D monolayer and 2D bilayer supramolecular coordination networks from a tetrapyridine functionalized pillar[5]arene with Co(II) through controlling the temperature, and the accurate construction of 2D bilayer, 2D sandwich bilayer, 2D four-layer and 2D multi-layer supramolecular coordination polymers from a tetrapyridine functionalized pillar[6]arene with Cu(II) by temperature control and guest induction, and precise chirality control of a 2D R-bilayer and a 2D S-bilayer supramolecular coordination network by high-performance liquid chromatography.
Results and discussion
The published literature showed that A1/A2-difunctionalized pillar[5]arene is an excellent assembly motif for constructing metal–organic hybrid materials.24–30 We also found that the dipyridine-functioned pillar[5]arene ligand P5PhPy2 (ref. 27), which exhibits a compact packing manner in the solid state, could be coordinated with transition metals Cu(II) and Co(II) to afford 1D single-line (P5PhPy2Cu-1 and P5PhPy2Cu-2) at room temperature and parallel double-line (P5PhPy2Cu-3) and ladder-like double-line coordination polymeric arrays (P5PhPy2Cu-4 and P5PhPy2Co-1) at 80 °C with good crystallinity under enclosed conditions (Fig. S1–S4†). To extend 1D coordination polymeric arrays to various 2D structures, a symmetrical tetrapyridine functionalized pillar[5]arene ligand (P5PhPy4) and pillar[6]arene ligand (P6PhPy4) were designed and prepared (Fig. 1), to investigate their coordination with Co(II) and Cu(II), respectively, to afford 2D monolayer, bilayer and multi-layer pillararene-type metal–organic coordination networks (PMOCNs). For the preparation of P5PhPy4 (Scheme S1†), 2 (ref. 31) was first reduced with NaBH4 and then reacted with triflic anhydride to produce 3 and then further coupled with 4-(4-pyridyl)phenylboric acid to give rise to P5PhPy4 in 48% yield. The compound P6PhPy4 was prepared through coupling of 4 (ref. 32) and 4-(4-pyridyl)phenylboric acid in 40% yield (Scheme S2†).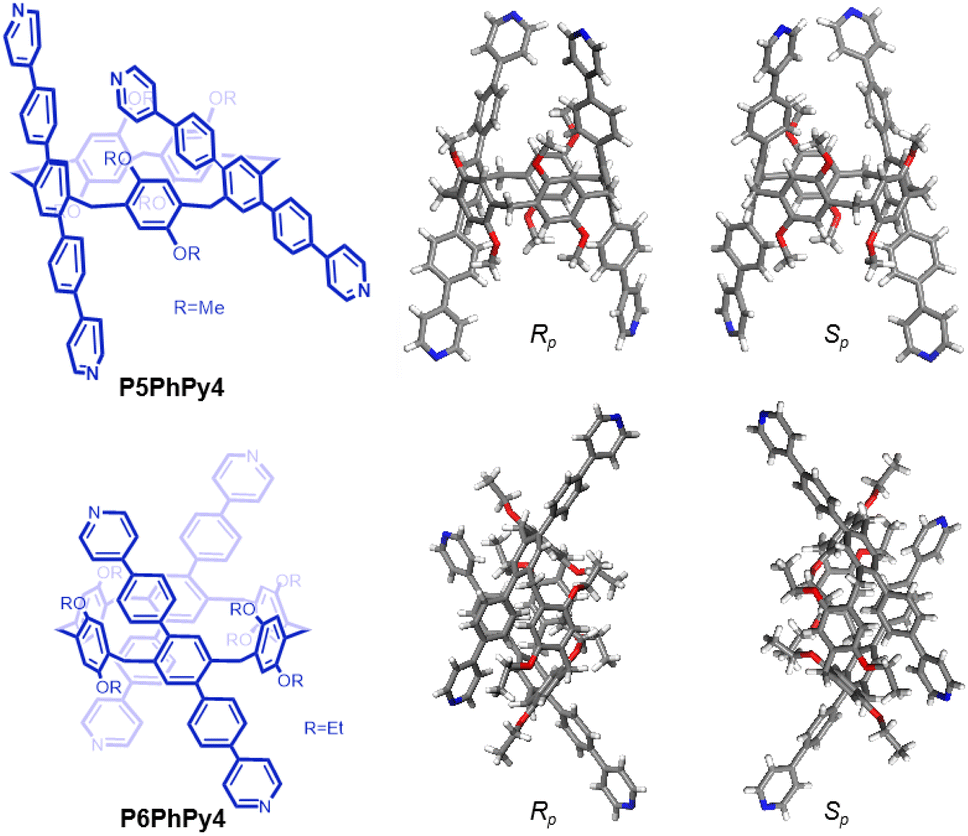 | ||
| Fig. 1 Compounds P5PhPy4 and P6PhPy4 and the crystal structure of P5PhPy4 (Rp, Sp) and P6PhPy4 (Rp, Sp). | ||
Crystals of free P5PhPy4 were obtained as enantiomers (Rp, Sp) via slow evaporation from the solution of dichloromethane (Fig. 1) and the cavity of P5PhPy4 was a perfect well-maintained pentagonal macrocycle. The molecular arrangements of P5PhPy4 from the top view demonstrated a compact packing manner in the (1,0,0), (0,1,0), and (0,0,1) directions (Fig. S5†). Meanwhile, the pillar[5]arene units of P5PhPy4 could be used as height adjustable stands to allow for the formation of mono- and bilayer networks (ml-2D-P5MOCNs and bl-2D-P5MOCNs) under the coordination of Co(II).
The coordination of P5PhPy4 with Co(II) nitrate in DMF in the presence of acetic acid at 80 °C afforded crystalline products, a monolayer compact coordination network (ml-2D-P5MOCNs) (Fig. S6†). X-ray diffraction analysis revealed that the rigid cavity of P5PhPy4 collapsed to some extent due to the coordination interaction between cobalt and pyridine. The structure of P5PhPy4 in the crystal of ml-2D-P5MOCNs was different from that in the crystal of free P5PhPy4; the configuration of two 1,4-dimethoxybenzene units was reversed under coordination at 80 °C, while they were still enantiomers. The two straight ligands of P5PhPy4 adopted a misaligned parallel orientation, and the torsional angle of the two straight ligands of free P5PhPy4 changed from 43.26° to close to 0° under coordination interaction (Fig. 2a). There were two types of monolayer structures with three coordinated forms of cobalt in the single crystal of ml-2D-P5MOCN: six-coordinate, five-coordinate, and four-coordinate. One type of monolayer network showed Co(II) atoms take six-coordinate and five-coordinate, Co(II) atoms linked two P5PhPy4 ligands through Co–N bonds (2.102–2.165 Å) to form a –Co–P5PhPy4–Co–P5PhPy4– rail and such parallel rails were cross-linked by Co–O contacts (1.894–2.222 Å) as “rungs”, O atoms from CH3COO− (Fig. 2b). The other type of monolayer network showed Co(II) atoms take five-coordinate and four-coordinate form (Fig. S7†), Co(II) atoms linked two P5PhPy4 ligands through Co–N bonds (2.076–2.199 Å), one type Co–O contacts (1.914–2.312 Å) as “rungs”, O atoms from CH3COO− and HCOO−, another type Co–O contacts (1.692–2.331 Å) independent, O atoms from CH3COO− and H2O. To be exact, it was the four parallel line group formed by four parallel pillararenes and Co(II), which then formed a 2D structure through hydrogen bonds (Fig. S7b–d†). All these results revealed that two types of independent 2D monolayer structures with Co–N bonds as longitudinal linkers along with a collapsed macrocycle, and Co–O bonds or Co–O bonds and hydrogen bonds (2.105–2.306 Å) as transverse linkers, and the pillar[5]arene units exhibited an alternate –Rp–Sp–Rp–Sp– arrangement (Fig. 2b and c and S7a–c†). Longitudinal side views indicated that the monolayer structures were stacked in the form of dislocation and back to back, due to the outer-surface action of the pillar[5]arene (Fig. 2d). The lateral side view showed that the pillar[5]arene units in the laminated structure were arranged in a micro-helix-like S-shaped curve (Fig. S8†). This result confirmed that pillar[5]arene units could be used as excellent tunable scaffolds to accurately control the monolayer by adjusting the rigid pillar cavity.
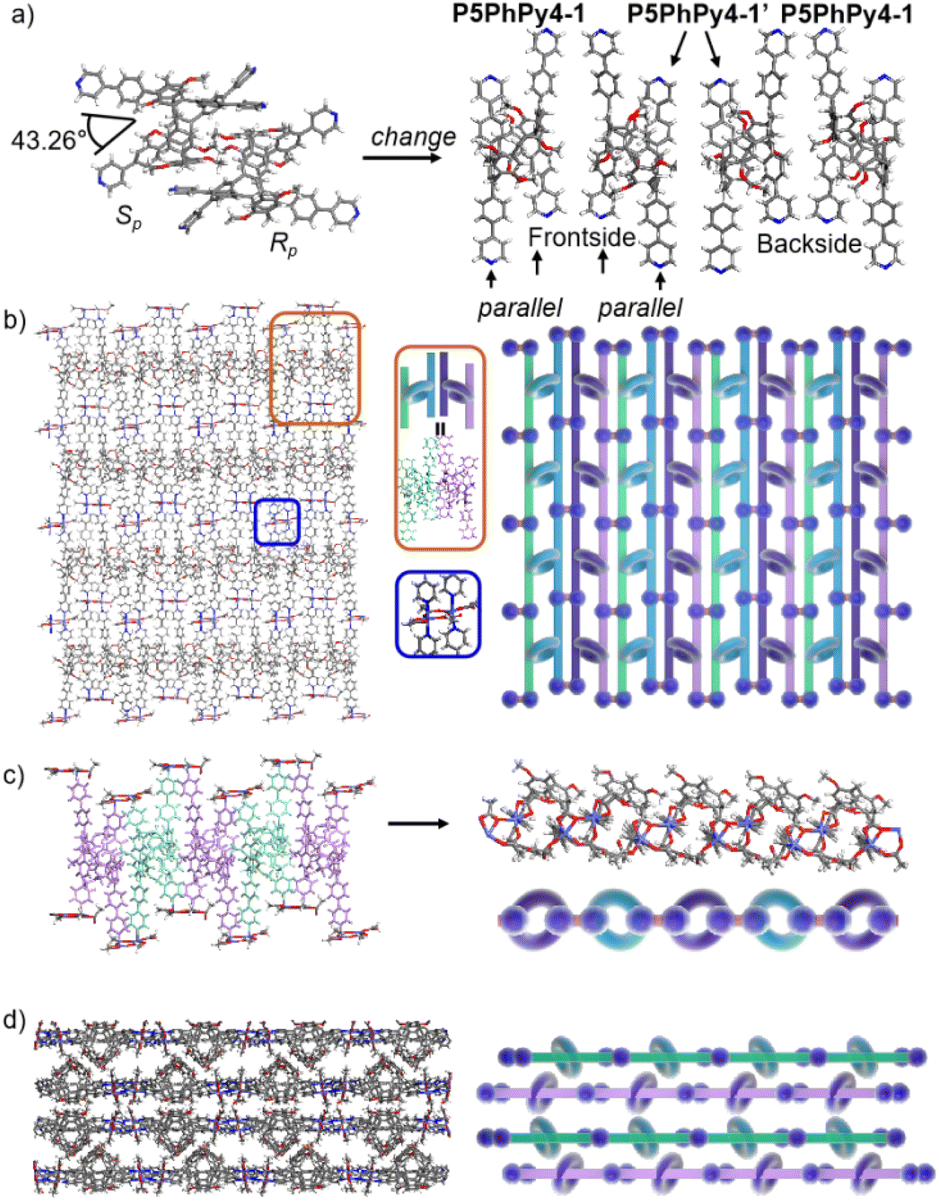 | ||
| Fig. 2 (a) The transformation of P5PhPy4 (Rp, Sp) in ml-2D-P5MOCN. (b) The top view of a single layer 2D coordination of the polymer ml-2D-P5MOCN. (c) The side view of the alternate –Rp–Sp–Rp–Sp– arrangement of the polymer ml-2D-P5MOCN. (d) The stacking structure of ml-2D-P5MOCN. Noncoordinated solvent molecules are omitted. | ||
As excellent tunable scaffolds, the deformation of the cavity of pillar[5]arene may vary, so we cultured P5PhPy4 and cobalt nitrate under the same conditions at room temperature. The reaction of P5PhPy4 with Co(II) nitrate in DMF in the presence of acetic acid at room temperature afforded another different crystalline product, a double-layer metal–organic network bl-2D-P5MOCN (Fig. S9†). The X-ray single-crystal structure of bl-2D-P5MOCN revealed that this rigid cavity of P5PhPy4 was greatly deformed due to the coordination interaction between cobalt and pyridine. The torsional angle of the two straight ligands of P5PhPy4 was changed from 43.26° to 100.78° (Fig. 3a). Due to the existence of the deformed cavity, the two straight ligands of P5PhPy4 showed a form of parallel up and down crossing, and then a bilayer structure was formed under the coordination of Co(II) (Fig. 3b and S10†). In the bilayer 2D network of bl-2D-P5MOCN, the top view presented a quadrilateral grid structure with two layers in the (1,1,0) direction, the upper and lower square-grid windows presented a parallel dislocation form, and the dimensions of the upper and lower square-grid windows were all 24.35 × 24.01 Å. Each of these square-grid windows was constructed from one side ligands of two enantiomers (Rp, Sp) (Fig. 3a). The Co(II) atom was six-coordinate, bonded by four pyridine N atoms from four different P5PhPy4 ligands (Co–N, 2.156–2.163 Å), one oxygen donor from H2O (Co–O, 2.096 Å), and another oxygen donor from nitrate ions (Co–O, 2.153 Å) (Fig. 3b). H2O and NO3− were located above and below the cobalt, and the upper layer was NO3− (up) and H2O (down), while the lower layer was H2O (up) and NO3− (down). The side views presented a clear double-layer form in the (0,0,1) direction. This bilayer structure was an independent 2D bilayer structure with deformed macrocycles as scaffolds. It is worth noting that 2D bilayer networks stacked in a pile-up of dislocations manner (Fig. 3c), presenting a staggered overlapping arrangement of bilayers. The bilayer network further confirmed that pillararene units were excellent tunable scaffolds and could form 2D monolayer and bilayer supramolecular coordination polymers by controlling the temperature.
 | ||
| Fig. 3 (a) The transformation of P5PhPy4 (Rp, Sp) in bl-2D-P5MOCN. (b) The crystal structure and diagram of a bilayer 2D coordination of the polymer bl-2D-P5MOCN. (c) The stacking structure of a bilayer 2D coordination of the polymer bl-2D-P5MOCN. Noncoordinated solvent molecules are omitted. | ||
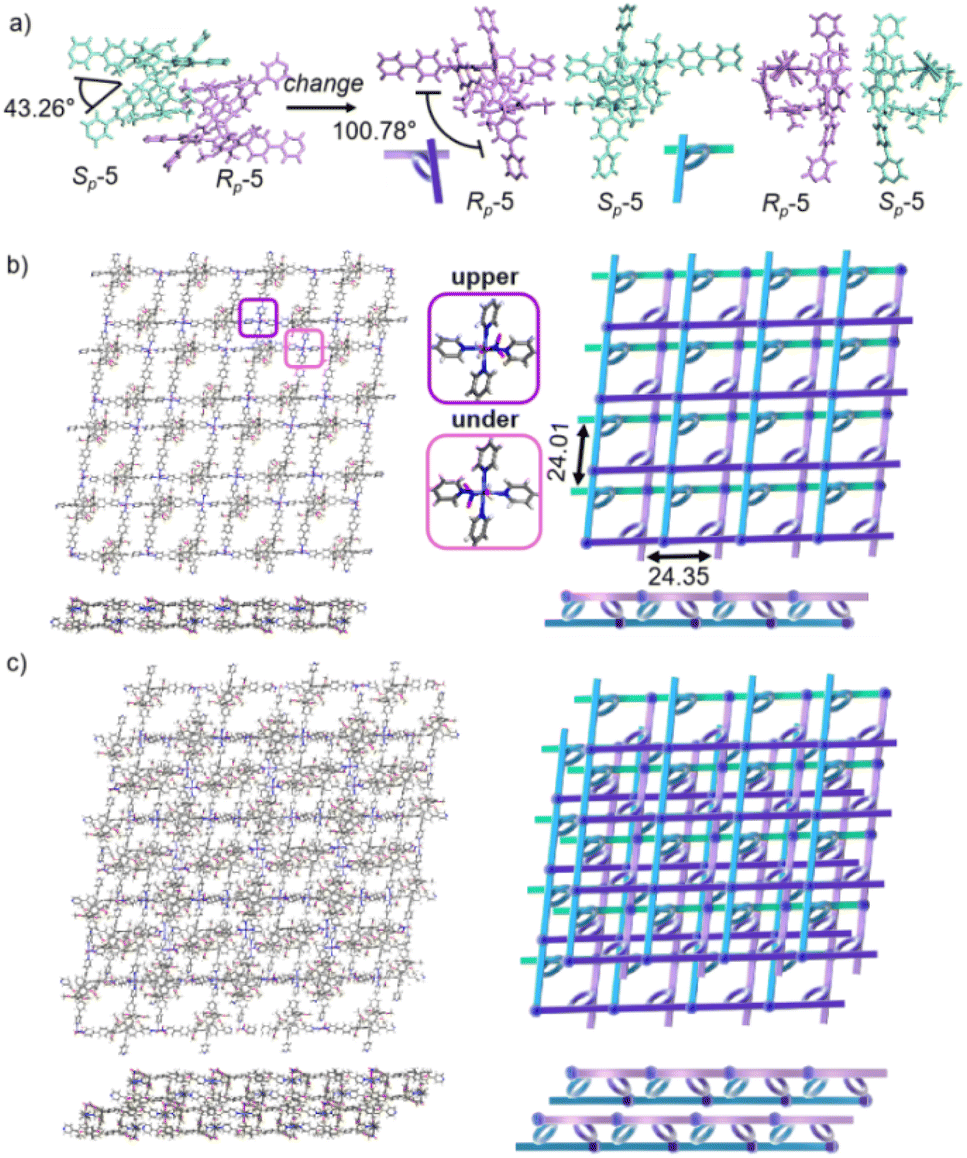
Crystals of free P6PhPy4 were obtained as enantiomers (Rp, Sp) via slow evaporation from the solution of dichloromethane/methanol (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), while P6PhPy4 showed a twisted hexagonal structure (Fig. 1), and the molecular arrangements of P6PhPy4 were similar to those of P5PhPy4, from the top view demonstrating a compact packing manner in the (1,0,0), (0,1,0), and (0,0,1) directions (Fig. S11†). Following the single crystal culture method, P5PhPy2 with Cu(II) or Co(II), P5PhPy4 with Co(II), and P6PhPy4 with Cu(II) nitrate in DMF in the presence of acetic acid at 80 °C for 24 h afforded the crystalline product mtl-2D-P6MOCN (Fig. 4a and S12†). The crystal structure of mtl-2D-P6MOCN revealed that the cavity of P6PhPy4 was changed from a twisted hexagonal structure to a perfect well-maintained hexagonal macrocycle under the coordination interaction between copper and pyridine and the modulation of disordered water molecules (Fig. 4a). Due to the perfect well-maintained hexagonal cavity, the two straight ligands of P6PhPy4 showed a form of parallel up and down crossing, and P6PhPy4 (Rp, Sp) showed a regular up and down arrangement under coordination interaction, and then a multi-layer structure with staggered and parallel connections of pillararenes was formed (Fig. 4b and S12†). The top view of the structure presented a regular diamond structure in the (1,0,0) direction and each of these diamond windows was constructed from one side ligands of two enantiomers (Rp, Sp) (Fig. 4b). The Cu(II) atom was six-coordinate, bonded by two pyridine N atoms from two different P6PhPy4 ligands (Co–N, 2.020–2.023 Å), three oxygen donors from CH3COO− ions, and one oxygen donor from H2O (Co–O, 1.788–2.336 Å) (Fig. 4b). The side view showed a parallel multi-layer with a parallel staggered arrangement of P6PhPy4 in the (0,1,0) and (0,0,1) directions (Fig. 4b). Although this multi-layer structure was formed with perfect well-maintained hexagonal macrocycles as scaffolds, the multi-layer structure stacked in a four-fold interpenetrating form, and the stacking of pillararenes presented multiple metal–organic pillar arrangement forms (Fig. 4c and S13†). The reasonable explanation of the occurrence of such structural interpenetration of mtl-2D-P6MOCN was driven by the minimization of the systematic energy through the optimal filling of the void space,33 which reflected the law that nature abhors a vacuum.
:1), while P6PhPy4 showed a twisted hexagonal structure (Fig. 1), and the molecular arrangements of P6PhPy4 were similar to those of P5PhPy4, from the top view demonstrating a compact packing manner in the (1,0,0), (0,1,0), and (0,0,1) directions (Fig. S11†). Following the single crystal culture method, P5PhPy2 with Cu(II) or Co(II), P5PhPy4 with Co(II), and P6PhPy4 with Cu(II) nitrate in DMF in the presence of acetic acid at 80 °C for 24 h afforded the crystalline product mtl-2D-P6MOCN (Fig. 4a and S12†). The crystal structure of mtl-2D-P6MOCN revealed that the cavity of P6PhPy4 was changed from a twisted hexagonal structure to a perfect well-maintained hexagonal macrocycle under the coordination interaction between copper and pyridine and the modulation of disordered water molecules (Fig. 4a). Due to the perfect well-maintained hexagonal cavity, the two straight ligands of P6PhPy4 showed a form of parallel up and down crossing, and P6PhPy4 (Rp, Sp) showed a regular up and down arrangement under coordination interaction, and then a multi-layer structure with staggered and parallel connections of pillararenes was formed (Fig. 4b and S12†). The top view of the structure presented a regular diamond structure in the (1,0,0) direction and each of these diamond windows was constructed from one side ligands of two enantiomers (Rp, Sp) (Fig. 4b). The Cu(II) atom was six-coordinate, bonded by two pyridine N atoms from two different P6PhPy4 ligands (Co–N, 2.020–2.023 Å), three oxygen donors from CH3COO− ions, and one oxygen donor from H2O (Co–O, 1.788–2.336 Å) (Fig. 4b). The side view showed a parallel multi-layer with a parallel staggered arrangement of P6PhPy4 in the (0,1,0) and (0,0,1) directions (Fig. 4b). Although this multi-layer structure was formed with perfect well-maintained hexagonal macrocycles as scaffolds, the multi-layer structure stacked in a four-fold interpenetrating form, and the stacking of pillararenes presented multiple metal–organic pillar arrangement forms (Fig. 4c and S13†). The reasonable explanation of the occurrence of such structural interpenetration of mtl-2D-P6MOCN was driven by the minimization of the systematic energy through the optimal filling of the void space,33 which reflected the law that nature abhors a vacuum.
 | ||
| Fig. 4 (a) The crystal structure and diagram of P6PhPy4 (Rp, Sp) in mtl-2D-P6MOCN. (b) The crystal structure and diagram of a multi-layer 2D coordination of the polymer mtl-2D-P6MOCN. (c) The stacking structure of a multi-layer 2D coordination of the polymer mtl-2D-P6MOCN. Noncoordinated solvent molecules were omitted. | ||
We redissolved the mtl-2D-P6MOCN crystals and recultured them under the same conditions at 80 °C for 72 h, affording a new bilayer structure product bl-2D-P6MOCN. X-ray diffraction analysis revealed that the pillar[6]arene units of P6PhPy4 in bl-2D-P6MOCN showed a twisted hexagonal structure similar to that of free P6PhPy4 in the solid state. The two straight ligands of P6PhPy4 showed a form of parallel up and down crossing similar to bl-2D-P5MOCN, and the torsional angle of the two straight ligands approached 120° (Fig. 5a), and then a bilayer structure was formed under the coordination of Cu(II) (Fig. 5b and S14†). The Cu(II) atoms linked P6PhPy4 ligands through Cu–N (2.162–2.171 Å), Cu–O (1.958–1.992 Å), and Cu–Cu bonds (2.640 Å) to form a –P6PhPy4–Cu–Cu–P6PhPy4– rail, with O atoms from acetate. Each Cu(II) atom was six-coordinate, with Cu(II) bound to one nitrogen donor from one P6PhPy4 ligand and four CH3COO− ions in a monodentate manner. The Cu–Cu contact completed the six-coordinate Cu center. It is worth noting that this bilayer structure featured an ordered cross arrangement of two sets of parallel lines and the metal coordination was the junction between the ligands, whereas pillar[6]arene played an important role in isolation and linkage. Although bl-2D-P6MOCN was also an independent 2D bilayer structure with a deformed macrocycle as scaffolds similar to bl-2D-P5MOCN, each independent 2D bilayer network was constructed by single chiral pillararenes (R- or S-) (Fig. 5c), one 2D bilayer network was constructed by R-P6PhPy4 and another 2D bilayer network was formed through S-P6PhPy4, presented an antiparallel dislocation stacking arrangement of R-bl-2D-P6MOCN and S-bl-2D-P6MOCN. This behaviour may provide more ideas for the construction of 2D single chirality bilayer planar structures (2D-R and 2D-S) by chiral separation.
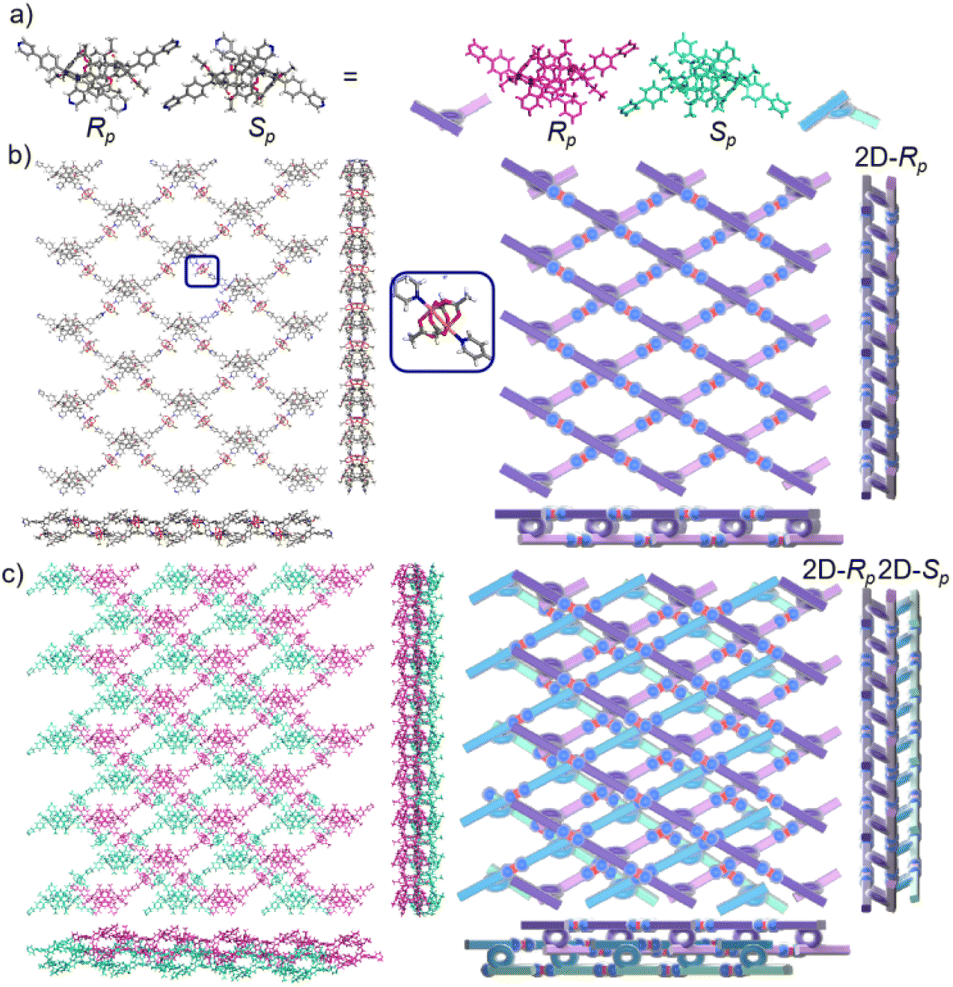 | ||
| Fig. 5 (a) The crystal structure and diagram of P6PhPy4 (Rp, Sp) in bl-2D-P6MOCN. (b) The crystal structure and diagram of a bilayer 2D coordination of the polymer bl-2D-P6MOCN (2D-Rp). (c) The stacking structure of a bilayer 2D coordination of the polymer bl-2D-P6MOCN. Noncoordinated solvent molecules were omitted. | ||
To further investigate the possible effect of reaction time, we cultured P6PhPy4 and copper nitrate under the same conditions at 80 °C for 10 days, which afforded a new four-layer structure product fl-2D-P6MOCN. The crystal structure revealed that pillar[6]arene units in fl-2D-P6MOCN also showed a twisted hexagonal structure in the solid state, while the torsional angle of the two straight ligands of P6PhPy4 appeared nearly 90° (Fig. 6a and S15†). We have drawn a slightly exaggerated schematic for ease of presentation (Fig. 6b). The two straight ligands of each P6PhPy4 in fl-2D-P6MOCN showed a form of parallel up and down crossing similar to bl-2D-P5MOCN, and two P6PhPy4 molecules, one R-P6PhPy4 and the other S-P6PhPy4, were arranged in an antiparallel order under the coordination of Cu(II), and then a four-layer structure was formed. There were two link models of Cu(II) in this crystal structure; one link model of Cu(II) atoms and P6PhPy4 was the same as that of bl-2D-P6MOCN, in which Cu(II) atoms linked P6PhPy4 ligands through Cu–N (2.170–2.183 Å), Cu–O (1.776–1.916 Å), and Cu–Cu (2.654 Å) bonds to form a –P6PhPy4–Cu–Cu–P6PhPy4– rail, with O atoms from acetate, and there were two such parallel rails at the top and the bottom of the four-layer structure. The other link model of Cu(II) atoms and P6PhPy4 was a ladder-like model in the middle of the four-layer network, in which the Cu(II) atoms linked P6PhPy4 ligands through Cu–N bonds (2.010–2.019 Å) to form a –Cu–P6PhPy4–Cu–P6PhPy4– rail and two such parallel rails of different P6PhPy4 were cross-linked by Cu–O contacts as “rungs” (1.989–2.328 Å), with O atoms from the acetate ion (Fig. 6b). The Cu(II) atom of the –Cu–P6PhPy4–Cu–P6PhPy4– rail was also six-coordinate, with Cu(II) bound to two nitrogen donors from two P6PhPy4 ligands, and the Cu–O contact (1.965–2.711 Å) of Cu(II) and four oxygen donors of three CH3COO− ions completed the six-coordinate Cu center. It is worth noting that two four-layer structures formed a more dense four-layer braided two-dimensional structure through interpenetration (Fig. 6c), and the four-layer braided two-dimensional network was stacked in the form of dislocation stacking arrangement layer by layer (Fig. S16†). The molecular woven network adopted the overlying weaving mode with the up-and-down entanglement pattern and innovative interlocking pattern at the same time due to the presence of P6PhPy4,34 which was extremely significant for enriching the weaving types and entanglement patterns of molecular woven structures. Similar to mtl-2D-P6MOCN, the structures of fl-2D-P6MOCN could also transform into bl-2D-P6MOCN through redissolving the crystals of fl-2D-P6MOCN and reculturing them under the same conditions at 80 °C for 7 days, while bl-2D-P6MOCN could not be returned to fl-2D-P6MOCN and mtl-2D-P6MOCN and maintained the same structure, meaning that bl-2D-P6MOCN may be a more stable form, which may be attributed to the structure of P6PhPy4 in bl-2D-P6MOCN being closest to the free P6PhPy4 without solvent molecules, representing its lowest energy state.
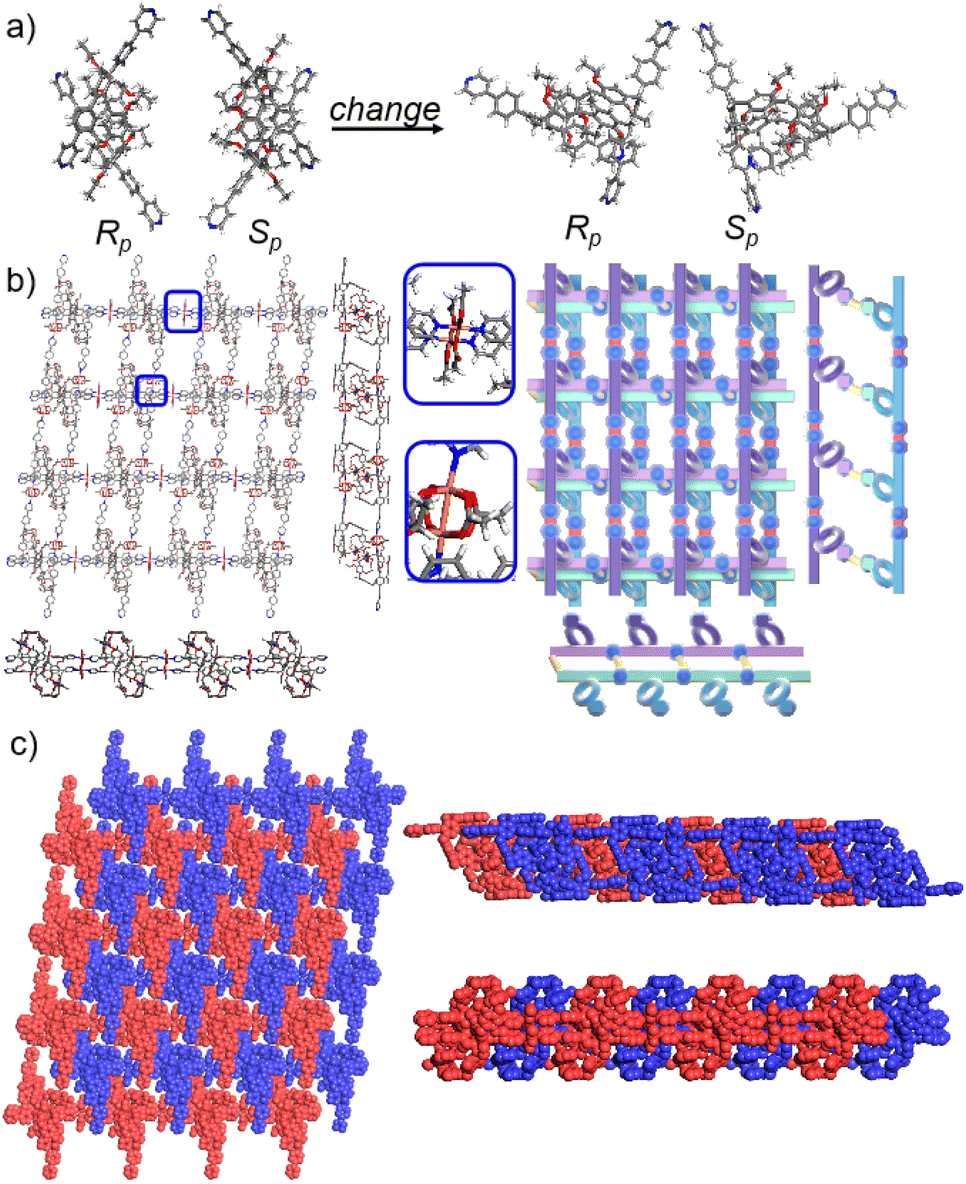 | ||
| Fig. 6 (a) The crystal structure and diagram of P6PhPy4 (Rp, Sp) in fl-2D-P6MOCN. (b) The crystal structure and diagram of a four-layer 2D coordination of the polymer fl-2D-P6MOCN. (c) The four-layer braided two-dimensional network of fl-2D-P6MOCN with the up and down entanglement pattern and interlocking pattern. Noncoordinated solvent molecules were omitted. | ||
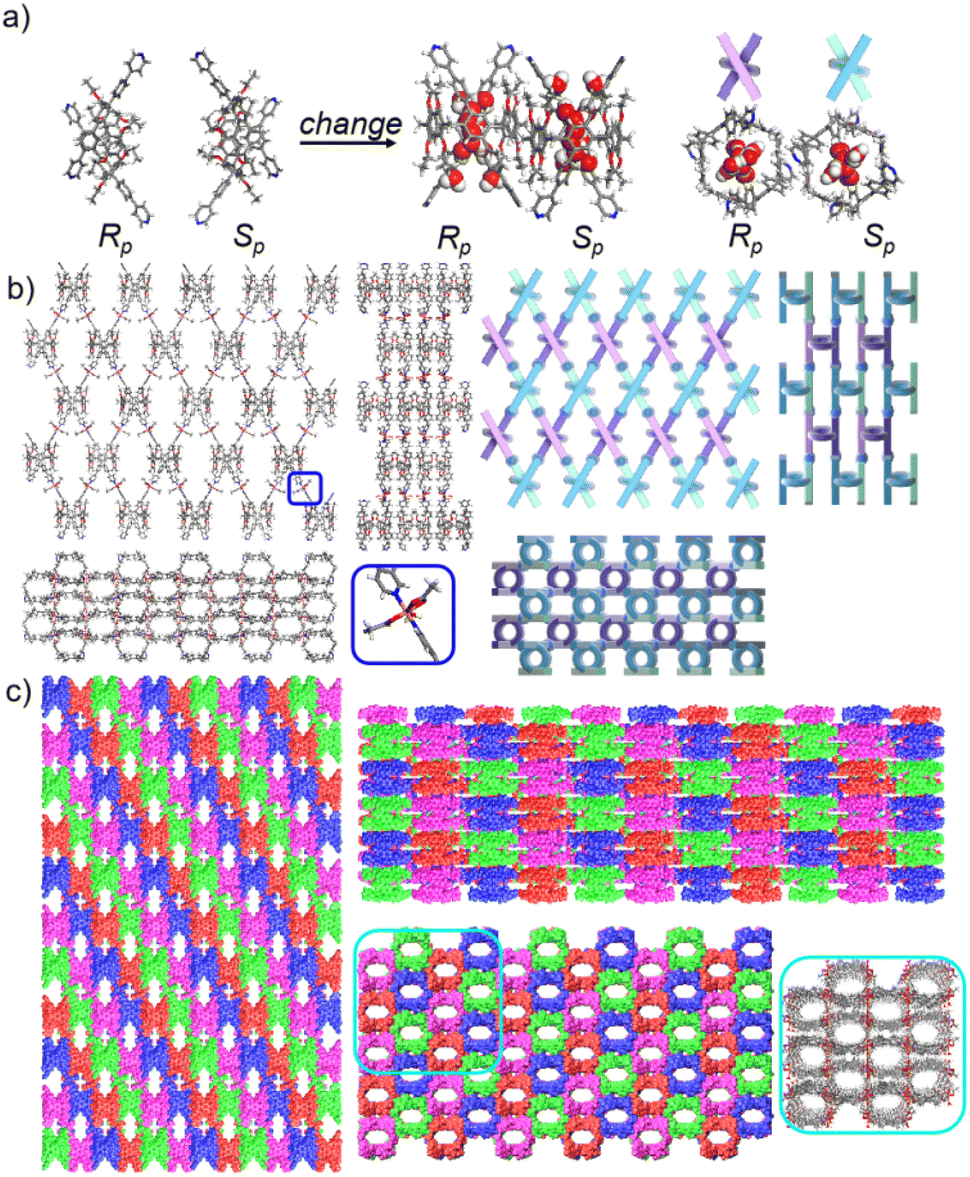
The pillar[6]arene units of P6PhPy4 in bl-2D-P6MOCN and fl-2D-P6MOCN all exhibited twisted hexagonal structures and to transform them into a perfect well-maintained hexagonal macrocycle, we modulated the coordination network by adopting guest induction.35 We cultured P6PhPy4, adiponitrile and copper nitrate under the same conditions at 80 °C for 3–10 days and then afforded an amazing sandwich bilayer structure product sbl-2D-P6MOCN (Fig. 7a and S17†). Unlike fl-2D-P6MOCN and mtl-2D-P6MOCN effected by reaction time, from 3 days to 10 days, it is still a sandwich bilayer structure, except that the molecules DMF or disordered adiponitrile in the cavity of pillar[6]arene units had changed (Fig. 7 and S18†). X-ray diffraction analysis revealed that the two straight ligands of P6PhPy4 showed a form of parallel up and down crossing and the coordination pattern of Cu(II) in sbl-2D-P6MOCN was similar to that of mtl-2D-P6MOCN, a –Cu–P6PhPy4–Cu–P6PhPy4– model, where the Cu(II) atom was six-coordinate or five-coordinate, bonded by two pyridine N atoms from two different P6PhPy4 ligands (Cu–N, 1.973–2.066 Å) and four or three oxygen donors from CH3COO− ions or CH3COO−, COO− and H2O (Cu–O, 2.045–2.660 Å). While the arrangement of P6PhPy4 in sbl-2D-P6MOCN was different from that of mtl-2D-P6MOCN, P6PhPy4 showed a coplanar parallel arrangement under coordination interaction, and then an unusual sandwich bilayer structure was formed. Similar to bl-2D-P6MOCN, each independent 2D sandwich bilayer structure was constructed from single chiral pillararenes; one was constructed from R-P6PhPy4 and the other was formed from S-P6PhPy4, and this coordination network also presented an alternating dislocation stacking arrangement of R-sbl-2D-P6MOCN and S-sbl-2D-P6MOCN (Fig. 7c, S17 and S18†). The behaviors of sbl-2D-P6MOCN and bl-2D-P6MOCN not only represented the adjustability of pillar[6]arene, but also further confirmed the feasibility of forming 2D single chirality bilayer planar polymer structures (2D-R and 2D-S).
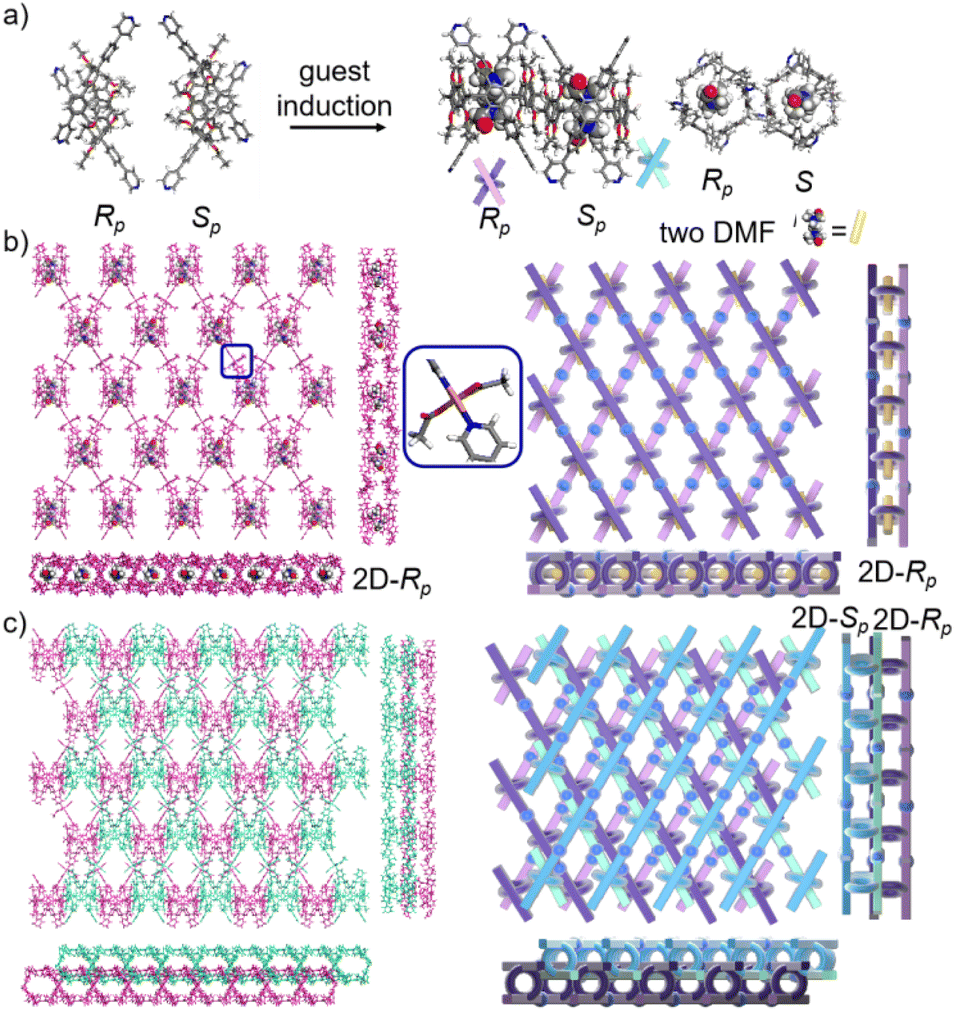 | ||
| Fig. 7 (a) The transformation of P6PhPy4 (Rp, Sp) in sbl-2D-P6MOCN. (b) The crystal structure and diagram of a sandwich bilayer 2D coordination of the polymer sbl-2D-P6MOCN (2D-Rp). (c) The stacking diagram of a sandwich bilayer 2D coordination of the polymer sbl-2D-P6MOCN (2D-Rp and 2D-Sp). Noncoordinated solvent molecules were omitted. | ||
The experimental powder X-ray diffraction (PXRD) patterns of ml-2D-P5MOCN, bl-2D-P5MOCN, bl-2D-P6MOCN, sbl-2D-P6MOCN, fl-2D-P6MOCN and mtl-2D-P6MOCN matched well with the simulated data satisfactorily, confirming their crystallinity (Fig. S19†). An interesting phenomenon is that the configurations of four 1,4-dimethoxybenzene units of P5PhPy2 in P5PhPy2Cu-3, two 1,4-dimethoxybenzene units of P5PhPy4 in ml-2D-P5MOCN and two 1,4-diethoxybenzene units of P6PhPy4 in fl-2D-P6MOCN were reversed under the coordination at 80 °C. Pillararene host structures in P5PhPy2Cu-3, ml-2D-P5MOCN and fl-2D-P6MOCN were also relatively rare partially reversed crystal examples of pillararene derivatives at high temperatures.36–381H NMR variable-temperature experiments were carried out to get insight into the transformation of P5PhPy2 and P5PhPy4 in DMSO-d6 and P6PhPy4 in DMF-d7 by changing temperature (Fig. S20–S22†). 2D NOESY and COSY experiments were recorded for P5PhPy2 and P5PhPy4 in DMSO-d6 and P6PhPy4 in DMF-d7 to assign the signals at room temperature (Fig. S23–S28†). The changes in the signals of 1,4-dimethoxybenzene or 1,4-diethoxybenzene units and methylene groups were very obvious, while the signal change of the phenylpyridine groups was relatively small, which may be rationalized by considering that 1,4-dimethoxybenzene and 1,4-diethoxybenzene units in part were rotated sharply and alternately, and the phenylpyridine units of P5PhPy2, P5PhPy4 and P6PhPy4 were shaking violently with increasing temperature. 1H NMR variable-temperature experiments for P5PhPy4 in CDCl3 and P6PhPy4 in CDCl3 and DMSO-d6 (low solubility) were also performed (Fig. S29–S31†). The variable-temperature results showed that whether in CDCl3, DMSO-d6 or DMF, P5PhPy4 and P6PhPy4 showed similar chemical shift changes. The adiponitrile@P6PhPy4 in DMF-d7 was also tested at room temperature and 80 °C (Fig. S32†), and the experiment results showed that the introduction of guests can inhibit the rotation of 1,4-diethoxybenzene units. The variable-temperature experiments confirmed that the mechanism of this inversion was related to factors such as temperature and guest molecules. While with the introduction of coordination, guests and solvents, as the number of bulky conjugated moieties on pillararenes increases, these rotations may be limited, fewer repeat units turned over at high temperatures, and some did not even turn over, which was why the pillar[6]arene units of bl-2D-P6MOCN, sbl-2D-P6MOCN and mtl-2D-P6MOCN did not undergo partial switching at 80 °C.
The crystal structure of bl-2D-P6MOCN and sbl-2D-P6MOCN showed that a single chiral two-dimensional plane could be formed through a single chiral pillararene (Rp or Sp), and the symmetrical tetra-functionalized pillar[6]arene may also provide one platform for the construction of multiple 2D chiral bilayer polymers (2D-R and 2D-S). Due to the bulky conjugated moiety on P6PhPy4, the interconversion of enantiomers of P6PhPy4 was inhibited and then the enantiomers R-P6PhPy4 and S-P6PhPy4 were obtained by chiral high-performance liquid chromatography. Two peaks with nearly equal areas were observed by chiral HPLC, and these two fractions were collected as optically pure P6 enantiomers (Fig. 8a and b and S33–S35†). The P6 enantiomers had a consistent NMR spectrum and similar UV and fluorescence spectra (Fig. S36 and S37†). We cultured R-P6PhPy4 or S-P6PhPy4 with Cu(II) nitrate in DMF under the same conditions as bl-2D-P6MOCN, and then we got two chiral two-dimensional bilayer structure products R-bl-2D-P6MOCN and S-bl-2D-P6MOCN (Fig. 8c and d), respectively. The arrangement patterns of R-bl-2D-P6MOCN and S-bl-2D-P6MOCN were similar to that of bl-2D-P6MOCN, and the torsional angle of the two straight ligands approached 120° with a form of parallel up and down crossing (Fig. 8c–f) and pillar[6]arene units showed a twisted hexagonal structure, while the coordination pattern of Cu(II) in R-bl-2D-P6MOCN or S-bl-2D-P6MOCN was similar to that of sbl-2D-P6MOCN and mtl-2D-P6MOCN, a –Cu–P6PhPy4–Cu–P6PhPy4– model, where the Cu(II) atom was six-coordinate, bonded by two pyridine N atoms from two different P6PhPy4 ligands (Cu–N, 1.986–2.008 Å) and four oxygen donors from two COO− ions (Cu–O, 2.006–2.565 Å). Two bilayer structures formed a denser bilayer braided two-dimensional structure by interpenetration, which means that the molecular woven network adopted the overlying weaving mode with the up-and-down entanglement mode, and the bilayer braided two-dimensional network was stacked in the form of AB stacking arrangement layer by layer (Fig. S38†). The π⋯π interaction between the benzene ring of pillar[6]enes in the upper layer and the pyridine ring of the ligand in the lower layer and the C–H⋯π interaction between methylene of pillar[6]enes in the upper layer and the benzene ring of the ligand in the lower layer were the main reasons to form the AB stacking arrangement (Fig. S38†). The experimental powder X-ray diffraction (PXRD) patterns of R-bl-2D-P6MOCN and S-bl-2D-P6MOCN not only had the same diffraction peaks but also matched with the simulated data satisfactorily, confirming their crystallinity (Fig. S39†). The CD spectra of R-bl-2D-P6MOCN (red line) and S-bl-2D-P6MOCN (blue line) in the solid state further confirmed the single chiral 2D structure (Fig. S40†). The assembly of two-dimensional chiral pillararene-type structures provided a new method for the construction of 2D mono-, bi-, and multi-layer chiral planar polymers and chiral molecular woven structures.
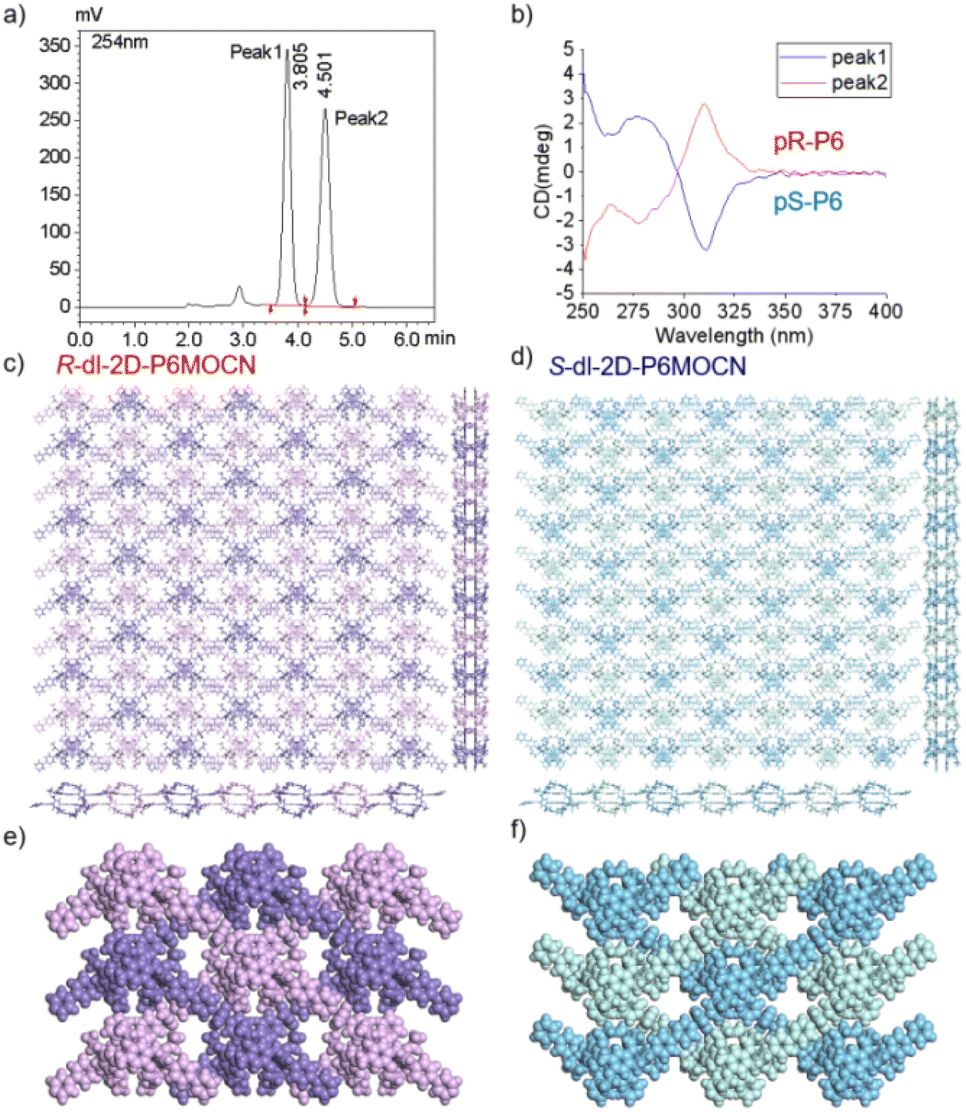 | ||
| Fig. 8 (a) Resolution of the enantiomers of P6PhPy4 by chiral HPLC. (b) CD spectra of S-P6PhPy4 (blue line) and R-P6PhPy4 (red line) in dichloromethane. (c and d) The crystal structure bilayer 2D coordination of the polymers R-bl-2D-P6MOCN and S-bl-2D-P6MOCN. (e and f) The up and down entanglement pattern of R-bl-2D-P6MOCN and S-bl-2D-P6MOCN. Noncoordinated solvent molecules were omitted. | ||
The morphologies of ml-2D-P5MOCN, bl-2D-P5MOCN, bl-2D-P6MOCN, sbl-2D-P6MOCN, fl-2D-P6MOCN, mtl-2D-P6MOCN, R-bl-2D-P6MOCN and S-bl-2D-P6MOCN, which were prepared by using the ultrasonic diffusion method, were further investigated by transmission electron microscopy (TEM) and by tapping-mode atomic force microscopy (AFM). Interestingly, the TEM images of these 2D PMOCNs showed clear sheet morphology structures in the form of multilayer stacking and the clear lattice fringes further indicated that these 2D PMOCNs had high crystallinity (Fig. 9a–h and S41–S48†). Aliquots (10 μL) of the micellar solution prepared by the ultrasonic diffusion method as described were deposited on a single-face-polished silicon wafer substrate and dried at room temperature in vacuo (Fig. 9i–p and S49–S56†). Height-vs-distance analysis indicated that the flakes of ml-2D-P5MOCN, bl-2D-P5MOCN, bl-2D-P6MOCN, sbl-2D-P6MOCN, fl-2D-P6MOCN, R-bl-2D-P6MOCN and S-bl-2D-P6MOCN exhibited a thickness of around 0.9 nm, 0.9 nm, 1.0 nm, 1.5 nm, 2.5 nm, 0.9 nm and 0.9 nm, respectively, which well matched with the calculated height (1.0 nm, 1.0 nm, 1.2 nm, 1.3 nm, 2.1 nm, 1.1 nm and 1.1 nm) of the set of PMOCNs. Due to the multilayer structure of mtl-2D-P6MOCN, the thickness (10.0–17.0 nm) was significantly higher than that of other networks. The thickness of these coordination polymeric arrays also validated that the structure of the networks constructed from macrocycles provided an excellent method to control the thickness of the flakes.
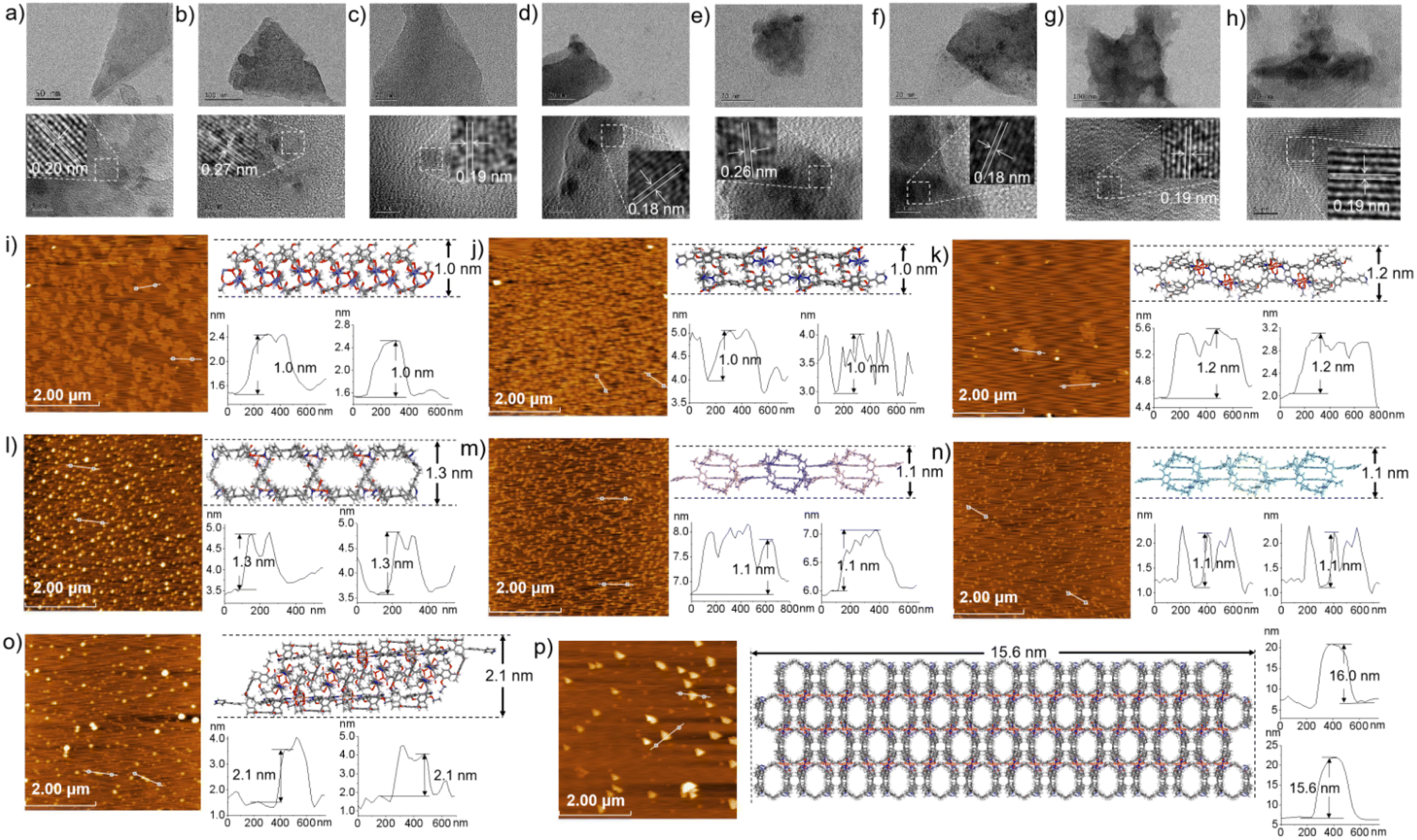 | ||
| Fig. 9 TEM images of ml-2D-P5MOCN, bl-2D-P5MOCN, bl-2D-P6MOCN, sbl-2D-P6MOCN, fl-2D-P6MOCN, mtl-2D-P6MOCN, R-bl-2D-P6MOCN and S-bl-2D-P6MOCN (a–h). AFM images of ml-2D-P5MOCN, bl-2D-P5MOCN, bl-2D-P6MOCN, sbl-2D-P6MOCN, R-bl-2D-P6MOCN, S-bl-2D-P6MOCN, fl-2D-P6MOCN, and mtl-2D-P6MOCN flakes with a thickness of around 1.0 nm, 1.0 nm, 1.2 nm, 1.3 nm, 1.1 nm, 1.1 nm, 2.1 nm and 15.6 nm, respectively (i–p). | ||
The networks of ml-2D-P5MOCN, bl-2D-P5MOCN, bl-2D-P6MOCN, sbl-2D-P6MOCN, fl-2D-P6MOCN and mtl-2D-P6MOCN changed easily with temperature in the thermal analysis experiment (Fig. S57†) and showed no adsorption effect of nitrogen at 77 K (Fig. S58†). The adsorption capacity of carbon dioxide of ml-2D-P5MOCN, bl-2D-P5MOCN, bl-2D-P6MOCN, sbl-2D-P6MOCN, fl-2D-P6MOCN and mtl-2D-P6MOCN was 14.46, 10.99, 24.66, 5.58, 9.56, and 6.38 cm3 g−1 at 273 K, respectively (Fig. S59†). Because these networks were able to maintain order in H2O, acetone and methanol for 12 h (Fig. S60†) and considering the solubility of P5PhPy4 and P6PhPy4 in methanol, we chose to directly carry out the adsorption experiment of the aqueous phase after obtaining crystals. The crystals of ml-2D-P5MOCN, bl-2D-P5MOCN, bl-2D-P6MOCN, sbl-2D-P6MOCN, fl-2D-P6MOCN and mtl-2D-P6MOCN were all subjected to iodine adsorption experiments in water at room temperature (Fig. S61–S64†). A time-dependent UV/vis experiment showed that upon the addition of 1.0 mg of ml-2D-P5MOCN, bl-2D-P5MOCN, bl-2D-P6MOCN, sbl-2D-P6MOCN, fl-2D-P6MOCN and mtl-2D-P6MOCN, the aqueous solution of I2 (1.0 mM, 2.5 mL) changed from yellowish brown to colourless transparent, and the crystals changed from pink and green or blue to reddish-brown, the adsorption rates of I2 were 96.59%, 93.36%, 95.42%, 93.59%, 93.32% and 97.12%, respectively, and the final adsorption capacity was 1.83 mg mg−1, 1.80 mg mg−1, 1.73 mg mg−1, 1.84 mg mg−1, 1.53 mg mg−1 and 1.90 mg mg−1, respectively, demonstrating their excellent iodine adsorption properties (Fig. 10a and S65†). More interesting, the PXRD profiles of I2 ⊂ ml-2D-P5MOCN and I2 ⊂ bl-2D-P5MOCN suggested that the two networks were transformed from a crystalline form to an amorphous form, with no diffraction peak observed at 3–50° (Fig. S66a and b†). These results indicated that the structures of ml-2D-P5MOCN and bl-2D-P5MOCN may have been interrupted upon I2 adsorption, which may be attributed to the deformed cavity of pillar[5]arene and N⋯I, O⋯I and Co⋯I bonding interactions.39,40 The diffraction peak of I2 ⊂ P6MOCNs can still be observed (Fig. S66c–f†), confirming that the crystalline form of these P6MOCNs was maintained with higher stability compared to P5MOCNs, which may be related to the cavity of P6PhPy4, since the twisted hexagonal structure and perfect well-maintained hexagonal structure were the structures in which they could exist stably. Fortunately, we obtained the crystal structure of mtl-2D-P6MOCN after iodine adsorption in water, and the cell parameters were consistent with mtl-2D-P6MOCN (Table S5†). Due to the solvent exchange of the crystal, the main structure of the network had not changed except that the anion coordinated with Cu(II) had become a water molecule (Fig. 10b), and the disordered iodine ions were distributed in the cavities and pores (Fig. 10c and d and S67†), which further confirmed the stability of P6MOCNs relative to P5MOCNs in water. The crystal structure showed that one reason for iodine adsorption may be the host–guest interaction of the cavity of pillararenes. To obtain more information on the interaction between the PMONs and iodine species, FT-IR spectra, X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy were employed. The FT-IR spectra showed an obvious change in the area of the stretching vibration peaks of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds of the coordinate carboxylate group and the peaks of the CC/C–H bonds of the benzene rings in all the PMONs after iodine adsorption, indicating that there are strong interactions between the adsorbed iodine and the CO bonds and aromatic rings within the PMONs (Fig. S68†). The XPS spectra of these PMONs after iodine adsorption showed characteristic peaks, further indicating the interactions between PMONs and the adsorbed iodine species (Fig. S69†).41,42 Two prominent peaks located at 629.0 and 616.9 eV for ml-2D-P5MOCN, 625.9 and 613.9 eV for bl-2D-P5MOCN, 628.9 and 616.9 eV for fl-2D-P6MOCN, and 628.9 and 617.9 eV for mtl-2D-P6MOCN, one weak peak at 614.9 eV for bl-2D-P6MOCN, and two weak peaks located at 628.9 and 617.9 eV for sbl-2D-P6MOCN were observed, which can be assigned to the I 3d3/2 and I 3d5/2 orbitals of iodine molecules, indicating that the captured iodine species in the PMONs were mainly in their molecule form. Raman spectroscopic measurements were conducted to further detect the adsorbed iodine species. No visible peaks were observed in the Raman spectra of these networks before iodine adsorption. Two characteristic peaks at 110 and 168 cm−1 for ml-2D-P5MOCN, bl-2D-P5MOCN, sbl-2D-P6MOCN, fl-2D-P6MOCN and mtl-2D-P6MOCN and 108 and 170 cm−1 for bl-2D-P6MOCN were observed after iodine capture (Fig. S70†). These peaks were attributed to the symmetric and asymmetric stretching vibrations of polyiodine anions (I3− and I5−),41,42 and the generation of iodine polyanions also indicated that charge transfer occurred, because of the strong interactions between the structures of PMONs and iodine. FT-IR, XPS, and Raman spectra demonstrated that there were strong interactions between PMONs and the adsorbed iodine species. More importantly, the set of P6MOCNs could adsorb iodine directly from the aqueous environment at ambient temperature and maintain the same structure, expanding the application and research of pillararene-type metal–organic coordination networks.
O bonds of the coordinate carboxylate group and the peaks of the CC/C–H bonds of the benzene rings in all the PMONs after iodine adsorption, indicating that there are strong interactions between the adsorbed iodine and the CO bonds and aromatic rings within the PMONs (Fig. S68†). The XPS spectra of these PMONs after iodine adsorption showed characteristic peaks, further indicating the interactions between PMONs and the adsorbed iodine species (Fig. S69†).41,42 Two prominent peaks located at 629.0 and 616.9 eV for ml-2D-P5MOCN, 625.9 and 613.9 eV for bl-2D-P5MOCN, 628.9 and 616.9 eV for fl-2D-P6MOCN, and 628.9 and 617.9 eV for mtl-2D-P6MOCN, one weak peak at 614.9 eV for bl-2D-P6MOCN, and two weak peaks located at 628.9 and 617.9 eV for sbl-2D-P6MOCN were observed, which can be assigned to the I 3d3/2 and I 3d5/2 orbitals of iodine molecules, indicating that the captured iodine species in the PMONs were mainly in their molecule form. Raman spectroscopic measurements were conducted to further detect the adsorbed iodine species. No visible peaks were observed in the Raman spectra of these networks before iodine adsorption. Two characteristic peaks at 110 and 168 cm−1 for ml-2D-P5MOCN, bl-2D-P5MOCN, sbl-2D-P6MOCN, fl-2D-P6MOCN and mtl-2D-P6MOCN and 108 and 170 cm−1 for bl-2D-P6MOCN were observed after iodine capture (Fig. S70†). These peaks were attributed to the symmetric and asymmetric stretching vibrations of polyiodine anions (I3− and I5−),41,42 and the generation of iodine polyanions also indicated that charge transfer occurred, because of the strong interactions between the structures of PMONs and iodine. FT-IR, XPS, and Raman spectra demonstrated that there were strong interactions between PMONs and the adsorbed iodine species. More importantly, the set of P6MOCNs could adsorb iodine directly from the aqueous environment at ambient temperature and maintain the same structure, expanding the application and research of pillararene-type metal–organic coordination networks.
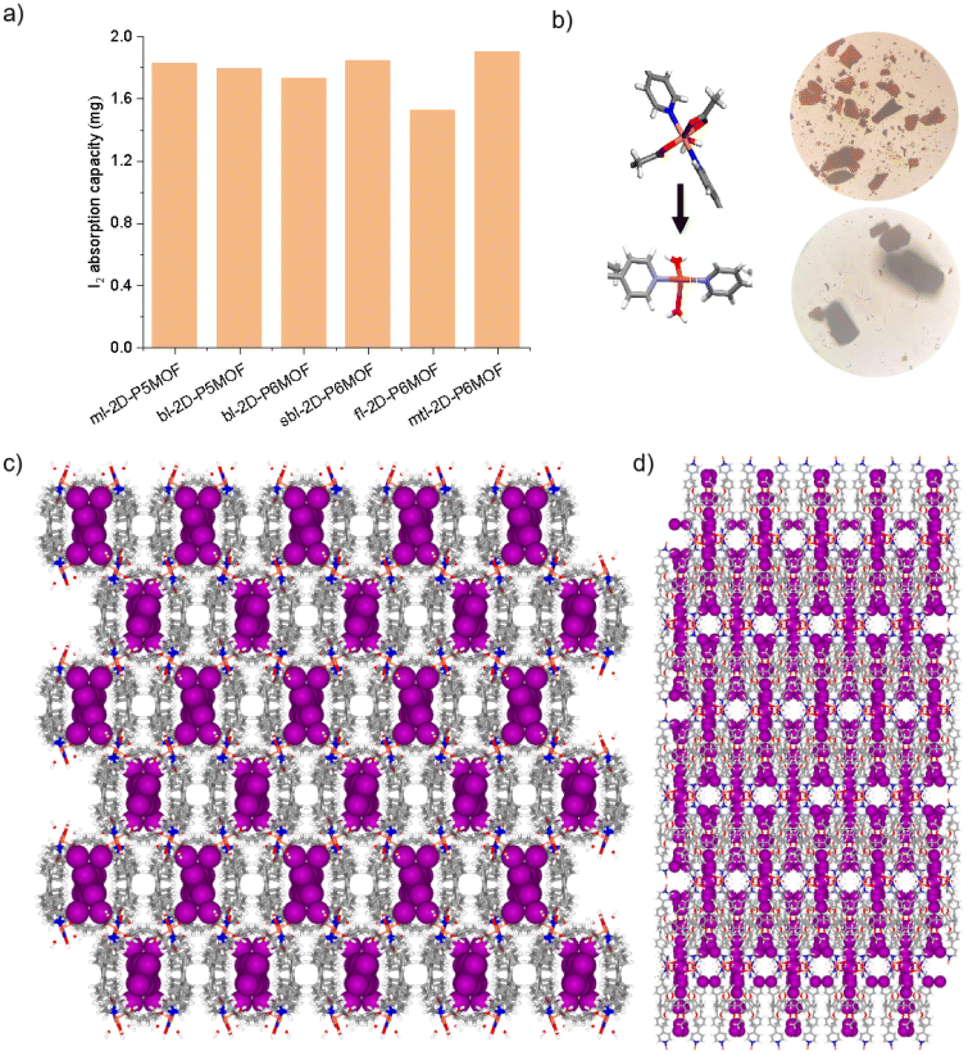 | ||
| Fig. 10 (a) The final iodine adsorption capacity (1.0 mM, 10 mL) of ml-2D-P5MOCN, bl-2D-P5MOCN, bl-2D-P6MOCN, sbl-2D-P6MOCN, fl-2D-P6MOCN and mtl-2D-P6MOCN in aqueous solution (the amount of each network was 1 mg). (b–d) The crystal structure of mtl-2D-P6MOCN after iodine adsorption in water. Noncoordinated solvent molecules were omitted. | ||
Conclusions
In summary, we have achieved 2D monolayer, bilayer, four-layer and multilayer coordination polymeric arrays via facile supramolecular self-assembly processes using two conjugated pillararenes P5PhPy4 and P6PhPy4. Pillar[5]arene and pillar[6]arene units were used as excellent tunable scaffolds for structural regulation to precisely construct ladder-like monolayer, square-grid bilayer, sandwich bilayer, four-layer and multi-layer supramolecular networks. Single crystal structures confirmed changes in the configuration of pillararenes as the network changes. Single crystal structures also confirmed that the repeating units of pillar[5]arene and pillar[6]arene could be partially rotated in the networks under heating conditions and coordination. The enantiomers R-P6PhPy4 and S-P6PhPy4, obtained by high-performance liquid chromatography, could also be used as excellent units to precisely construct a 2D bilayer chiral polymer array. Atomic force microscopy imaging confirmed the ultrathin thicknesses of these 2D materials. The structures of 2D P5MOCNs and P6MCONs exhibited excellent iodine adsorption properties in the aqueous phase at room temperature; especially pillar[6]arene-type networks were able to maintain the frame structure after iodine adsorption. From a crystal engineering and supramolecular chemistry perspective, this work not only provided a new strategy for developing more sophisticated and functional mono-, bi-, three-, four-, and multi-layer 2D materials and chiral two-dimensional materials but also provided reference models for the construction of complex bilayer networks with a specific thickness or number of layers, enriching the studies in supramolecular self-assembly of multiple types of multi-layer 2D nanomaterials. From a practical point of view, in addition to the adsorption function as adsorbents, we also hope that this field further develops towards the integration of pillararenes and functional ligands or modules into frameworks that can be responsive to pH, ions, light and electrons and then make progress in the fields of catalysis, energy conversion and biological functions.Data availability
All characterization data and experimental protocols are provided in this article and the ESI Appendix. Crystallographic data have been deposited in the Cambridge Crystallographic Data Centre (CCDC) under accession numbers CCDC: 2244320 (for P5PhPy4), 2244321 (for P5PhPy2), 2244322 (for P5PhPy2Cu-1), 2244323 (for P5PhPy2Cu-2), 2244324 (for P5PhPy2Cu-3), 2244325 (for P5PhPy2Cu-5), 2244326 (for P5PhPy2Co-1), 2244327 (for ml-2D-P5MOCN), 2244328 (for bl-2D-P5MOCN), 2326170 (for P6PhPy4), 2326171 (for bl-2D-P6MOCN), 2342430 (for sbl-2D-P6MOCN), 2326172 (for sbl-2D-P6MOCN), 2326174 (for fl-2D-P6MOCN), 2326175 (for mtl-2D-P6MOCN), 2326165 (for I2@mtl-2D-P6MOCN), 2326166 (for R-bl-2D-P6MOCN), and 2326167 (for S-bl-2D-P6MOCN). These data can be obtained free of charge from the Cambridge Crystallographic Date Centre viahttps://www.ccdc.cam.ac.uk/datarequest/cif.† Extra data are available from the corresponding author upon request.Author contributions
B. Y. and Z. T. L. conceptualized the ideas and supervised the investigations; Z. N. C. and L. P. Z. performed the synthesis and X-ray single crystal diffraction, TEM, AFM, PXRD and iodine adsorption experiments; H. L. W. and M. Y. performed 1H NMR variable-temperature, 2D NOESY and COSY experiments. Z. N. C., L. P. Z., and B. Y. collected the data; Q. Y. Q. and B. Y. drew the schematic diagram; Z. N. C., L. P. Z., B. Y., J. T., G. Y. Y. and Z. T. L. analysed the data; B. Y. and Z. T. L. finished the writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (No. 22201268) for financial support.Notes and references
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- M. Xu, T. Liang, M. Shi and H. Chen, Chem. Rev., 2013, 113, 3766–3798 CrossRef CAS PubMed.
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G.-H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed.
- P. You, G. Tang and F. Yan, Mater. Today Energy, 2019, 11, 128–158 CrossRef CAS.
- J. Liang, H. Yu, J. Shi, B. Li, L. Wu and M. Wang, Adv. Mater., 2023, 35, 2209814 CrossRef CAS PubMed.
- G. Yu, K. Jie and F. Huang, Chem. Rev., 2015, 115, 7240–7303 CrossRef CAS PubMed.
- Y. Zhou, K. Jie, R. Zhao and F. Huang, Adv. Mater., 2020, 32, 1904824 CrossRef CAS PubMed.
- D. Xia, P. Wang, X. Ji, N. M. Khashab, J. L. Sessler and F. Huang, Chem. Rev., 2020, 120, 6070–6123 CrossRef CAS PubMed.
- X.-Q. Wang, W.-J. Li, W. Wang and H.-B. Yang, Acc. Chem. Res., 2021, 54, 4091–4106 CrossRef CAS PubMed.
- X. Li, H.-S. Xu, K. Leng, S. W. Chee, X. Zhao, N. Jain, H. Xu, J. Qiao, Q. Gao, I.-H. Park, S. Y. Quek, U. Mirsaidov and K. P. Loh, Nat. Chem., 2020, 12, 1115–1122 CrossRef CAS PubMed.
- Z.-J. Yin, S.-Y. Jiang, N. Liu, Q.-Y. Qi, Z.-Q. Wu, T.-G. Zhan and X. Zhao, CCS Chem., 2022, 4, 141–150 CrossRef CAS.
- B. Yang, S. B. Yu, P. Q. Zhang, Z. K. Wang, Q. Y. Qi, X. Q. Wang, X. H. Xu, H. B. Yang, Z. Q. Wu, Y. Liu, D. Ma and Z.-T. Li, Angew. Chem., Int. Ed., 2021, 60, 26268–26275 CrossRef CAS PubMed.
- T. Ogoshi, S. Kanai, S. Fujinami, T.-A. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed.
- T. Ogoshi, T. Kakuta and T.-A. Yamagishi, Angew. Chem., Int. Ed., 2018, 58, 2197–2206 CrossRef PubMed.
- H. Zhang, Z. Liu and Y. Zhao, Chem. Soc. Rev., 2018, 47, 5491–5528 RSC.
- N. Song, T. Kakuta, T.-A. Yamagishi, Y.-W. Yang and T. Ogoshi, Chem, 2018, 4, 2029–2053 CAS.
- T. Ogoshi, T.-A. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937–8002 CrossRef CAS PubMed.
- L. Liu, Y. Hu, S. Huang, Y. Jin, J. Cui, W. Gong and W. Zhang, Chem. Sci., 2021, 12, 13316–13320 RSC.
- W.-J. Li, W.-T. Xu, X.-Q. Wang, Y. Jiang, Y. Zhu, D.-Y. Zhang, X.-Q. Xu, L.-R. Hu, W. Wang and H.-B. Yang, J. Am. Chem. Soc., 2023, 145, 14498–14509 CrossRef CAS PubMed.
- M.-H. Li, C.-L. Xu and Y.-W. Yang, Coord. Chem. Rev., 2024, 512, 215894 CrossRef CAS.
- X. Li, M.-L. Shen, J. Yang, L.-L. Liu and Y.-W. Yang, Adv. Mater., 2024, 36, 2313317 CrossRef CAS PubMed.
- Z.-Q. Wang, X. Wang and Y.-W. Yang, Adv. Mater., 2024, 36, 2301721 CrossRef CAS PubMed.
- X.-Y. Lou, S.-Y. Zhang, Y. Wang and Y.-W. Yang, Chem. Soc. Rev., 2023, 52, 6644 RSC.
- N. L. Strutt, D. Fairen-Jimenez, J. Iehl, M. B. Lalonde, R. Q. Snurr, O. K. Farha, J. T. Hupp and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 17436–17439 CrossRef CAS PubMed.
- N. L. Strutt, H.-C. Zhang and J. F. Stoddart, Chem. Commun., 2014, 50, 7455–7458 RSC.
- E. Lee, I. H. Park, H. Ju, S. Kim, J. H. Jung, Y. Habata and S. S. Lee, Angew. Chem., Int. Ed., 2019, 58, 11296–11300 CrossRef CAS PubMed.
- H. Zhu, Q. Li, B. Shi, H. Xing, Y. Sun, S. Lu, L. Shangguan, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 17340–17345 CrossRef CAS PubMed.
- X.-Y. Lou and Y.-W. Yang, J. Am. Chem. Soc., 2021, 143, 11976–11981 CrossRef CAS PubMed.
- Y. Wu, M. Tang, Z. Wang, L. Shi, Z. Xiong, Z. Chen, J. L. Sessler and F. Huang, Nat. Commun., 2023, 14, 4927 CrossRef CAS PubMed.
- X. Wan, S. Li, Y. Tian, J. Xu, L.-C. Shen, H. Zuilhof, M. Zhang and A. C. H. Sue, Chem, 2022, 8, 2136–2147 CAS.
- M. Pan and M. Xue, Eur. J. Org. Chem., 2013, 4787–4793 CrossRef CAS.
- H. Zeng, P. Liu, H. Xing and F. Huang, Angew. Chem., Int. Ed., 2022, 61, e202115823 CrossRef CAS PubMed.
- B. Yang, J.-W. Zhang, S.-B. Yu, Z.-K. Wang, P.-Q. Zhang, X.-D. Yang, Q.-Y. Qi, G.-Y. Yang, D. Ma and Z.-T. Li, Sci. China: Chem., 2021, 64, 1228–1234 CrossRef CAS.
- Q.-Y. Huang, W.-L. Li, Z. Mao, H. Zhang, Y. Li, D.-Y. Ma, H.-Y. Wu, J. Zhao, Z.-Y. Yang, Y. Zhang, L. Gong, M. P. Aldred and Z.-G. Chi, Chem, 2021, 5, 1321–1332 Search PubMed.
- M. Li, B. Hua, H. Liang, J. Liu, L. Shao and F. Huang, J. Am. Chem. Soc., 2020, 142, 20892–20901 CrossRef CAS PubMed.
- J.-B. Yao, W.-H. Wu, W.-T. Liang, Y.-J. Feng, D.-Y. Zhou, J. J. Chruma, G. Fukuhara, T. Mori, Y. Inoue and C. Yang, Angew. Chem., Int. Ed., 2017, 56, 6869–6873 CrossRef CAS PubMed.
- S. Fa, K. Egami, K. Adachi, K. Kato and T. Ogoshi, Angew. Chem., Int. Ed., 2020, 59, 20353–20356 CrossRef CAS PubMed.
- J. Yao, W.-H. Wu, C. Xiao, D. Su, Z.-H. Zhong, T. Mori and C. Yang, Nat. Commun., 2021, 12, 2600 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- Y. Lin, X. Jiang, S. T. Kim, S. B. Alahakoon, X. Hou, Z. Zhang, C. M. Thompson, R. A. Smaldone and C. Ke, J. Am. Chem. Soc., 2017, 139, 7172–7175 CrossRef CAS PubMed.
- K. Cheng, H.-L. Li, Z.-Y. Li, P.-Z. Li and Y. L. Zhao, ACS Mater. Lett., 2023, 5, 1546–1555 CrossRef CAS.
- X.-F. Li, Z.-M. Jia, J. Zhang, Y.-D. Zou, B. Jiang, Y.-D. Zhang, K.-W. Shu, N. Liu, Y. Li and L.-J. Ma, Chem. Mater., 2022, 34, 11062–11071 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2244320–2244328, 2326165–2326167, 2326170–2326172, 2326174, 2326175, and 2342430. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03022b |
| ‡ Z. N. Chen and L. P. Zhang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |