
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectroreductive deuteroarylation of alkenes enabled by an organo-mediator†
Xinling
Li
a,
Jianfeng
Zhou
a,
Weijie
Deng
a,
Ziliang
Wang
a,
Yating
Wen
a,
Zhenjie
Li
a,
Youai
Qiu
 *b and
Yubing
Huang
*a
*b and
Yubing
Huang
*a
aSchool of Environmental and Chemical Engineering, Wuyi University, Jiangmen, 529090, P. R. China. E-mail: huangyb@wyu.edu.cn
bState Key Laboratory and Institute of Elemento-Organic Chemistry, Frontiers Science Center for New Organic Matter, College of Chemistry, Nankai University, 94 Weijin Road, Tianjin, 300071, People's Republic of China. E-mail: qiuyouai@nankai.edu.cn
First published on 20th June 2024
Abstract
Electroreduction mediated by organo-mediators has emerged as a concise and effective strategy, holding significant potential in the site-specific introduction of deuterium. In this study, we present an environmentally friendly electroreduction approach for anti-Markovnikov selective deuteroarylation of alkenes and aryl iodides with D2O as the deuterium source. The key to the protocol lies in the employment of a catalytic amount of 2,2′-bipyiridine as an efficient organo-mediator, which facilitates the generation of aryl radicals by assisting in the cleavage of the C–X (X = I or Br) bonds in aryl halides. Because its reduction potential matches that of aryl iodides, the organo-mediator can control the chemoselectivity of the reaction and avoid the side reactions of competitive substrate deuteration. These phenomena are theoretically supported by CV experiments and DFT calculations. Our protocol provides a series of mono-deuterated alkylarenes with excellent deuterium incorporation through two single-electron reductions (SER), without requiring metal catalysts, external reductants, and sacrificial anodes.
Introduction
Deuterated compounds hold important application value in nuclear magnetic resonance analysis, the exploration of reaction mechanisms, and research in drug metabolism.1 Incorporating deuterium at specific positions may necessitate the development of new and often intricate synthetic methodologies, leading to challenges in deuteration rate, scalability, and overall synthetic feasibility.2 Regioselective reductive arylation of alkenes with aryl halides emerges as a promising strategy for site-specific introduction of deuterium. In this regard, predominant methods involve transition metal-catalyzed methods achieving high anti-Markovnikov selectivity through the insertion of arylmetal species into alkenes3 (Fig. 1A). Until now, the application of reductive arylation in deuteration is still constrained by several factors, such as the use of stoichiometric external reductants and the high cost of corresponding deuterated reagents. Moreover, the use of transition metal catalysts can lead to the presence of deleterious metal residues in the final products. In comparison, the investigation into metal-free strategies exhibits considerable promise, but the problem of substrate activation needs addressing. Given these challenges and the requirements of green chemistry, there is an urgent need to develop environmentally friendly methods for regioselective deuteroarylation. These methods should utilize cheap and readily available deuterated reagents, without metal catalysts and external reductants, posing extreme challenges.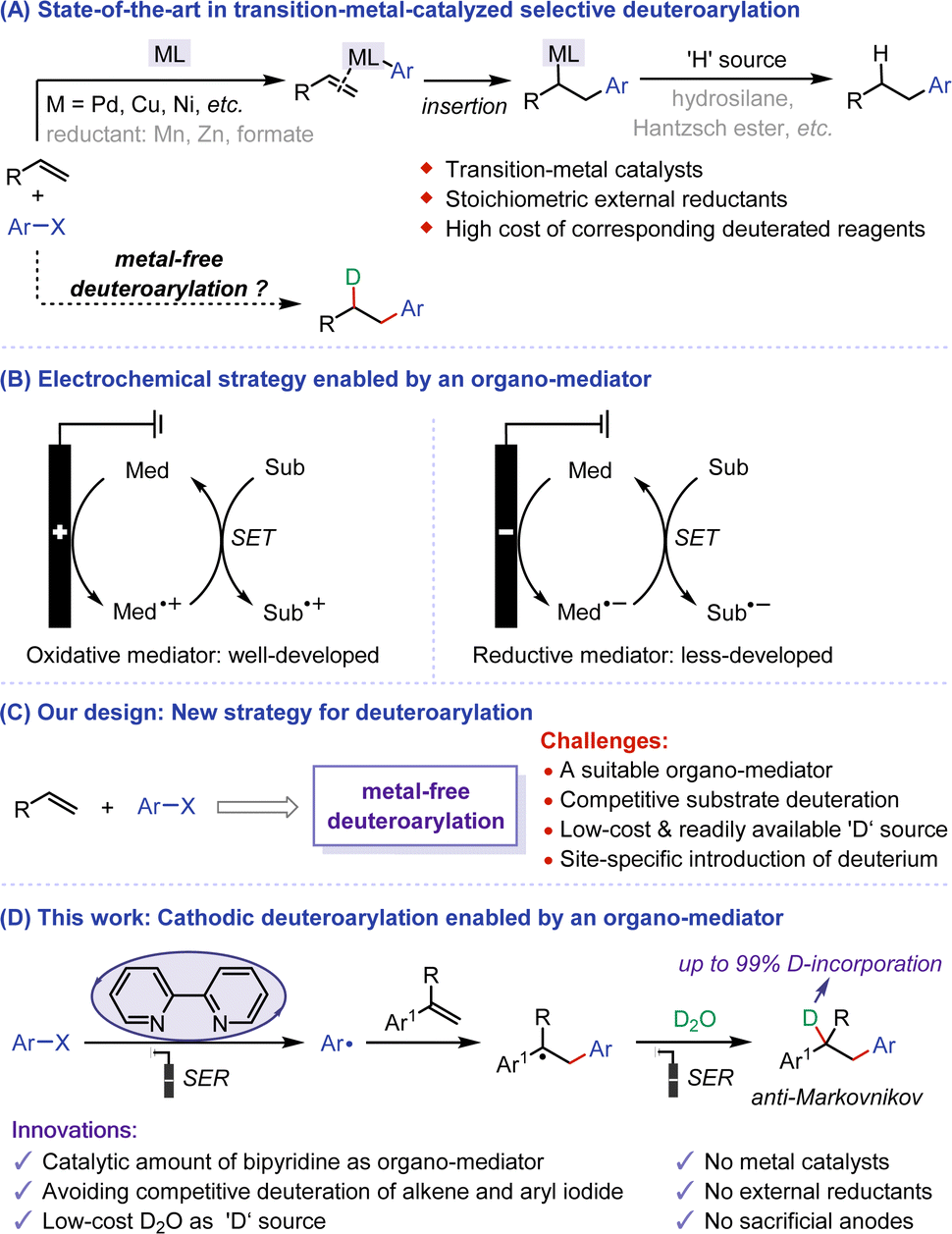 | ||
| Fig. 1 Electroreductive deuteroarylation of alkenes and aryl halides. (A) State-of-the-art in transition-metal-catalyzed selective deuteroarylation. (B) Electrochemical strategy enabled by an organo-mediator. (C) Our design: new strategy for deuteroarylation. (D) This work: cathodic deuteroarylation enabled by an organo-mediator. | ||
Recent years have seen rapid development in organic electrochemistry, driven by its environmental friendliness, high energy efficiency, and no need for additional redox reagents.4,5 Compared to direct electrolysis, indirect electrolysis reactions using organic molecules as redox mediators have their unique advantages, leading to the increasing importance of this transformation.6,7 The reaction process can be effectively promoted by forming active species through direct electrolysis of organo-mediators on the electrode, followed by substrate activation through a single electron transfer (SET) process. Therefore, mediated electrochemical strategies can effectively avoid the intolerance of sensitive substrates or functional groups to high voltages, and control the chemoselectivity in the reaction based on the difference in the redox potential of organo-mediators. Over the years, great efforts have been devoted to mediated electrooxidation reactions,8 with representative studies focusing on N-oxide mediators such as phthalimide N-oxy (PINO) and 2,2,6,6-tetramethylpiperidine N-oxy (TEMPO). In contrast, mediated electroreduction is still in its infancy, and there is a huge room for development9 (Fig. 1B). Electroreduction can provide a highly atom-economic and cost-effective strategy for selective deuteration due to the availability of inexpensive D2O as a direct ‘D’ source, which has achieved remarkable achievements.10 With this in mind, we aim to develop a novel electroreductive deuteroarylation mediated by organic molecules for the site-specific introduction of deuterium in the presence of D2O. However, mediated electroreductive deuteration of alkenes and direct deuteration of aryl halides can lead to undesirable by-products on the cathode. Hence, the primary challenges of this electroreductive deuteroarylation include (a) exploration of suitable organo-mediators to activate the substrate while avoiding competitive side reactions, (b) the use of D2O as a ‘D’ source for site-specific introduction of deuterium, (c) selectivity control of the whole process (Fig. 1C).
Herein, we propose a bipyridine-mediated anti-Markovnikov selective deuteroarylation of alkenes and aryl iodides by using D2O as a deuterium source, providing an environmental-friendly and economical electroreduction approach to access mono-deuterated alkylarenes (Fig. 1D). The reaction involves sequential processes: aryl halide releases aryl radical through bipyridine-mediated single electron reduction (SER), the addition of the aryl radical to the alkene, and the subsequent deuteration. Notable features of this protocol include (a) use of catalytic amounts of dipyridine as an efficient organo-mediator, (b) avoidance of competitive alkene deuteration and the dehalogenative deuteration of aryl iodides11 due to over-reduction by using a suitable mediator with a reduction potential matched that of aryl iodides, (c) utilization of low-cost and readily available D2O as ‘D’ source, (d) elimination of the need for metal catalysts, stoichiometric external reductants, and sacrificial anodes, and (e) achievement of high site selectivity and excellent D-incorporation.
Results and discussion
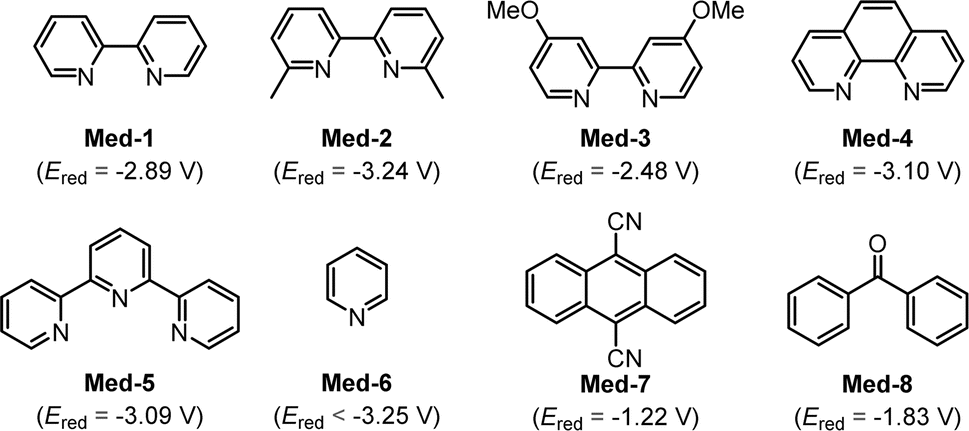
In preliminary attempts, 2,2′-bipyridine was found to be an essential additive that played an important role in electroreductive deuteroarylation. After comparing several organic molecules by cyclic voltammetry (CV) experiments, we speculated that 2,2′-bipyridine might act as a suitable mediator due to its lower reversible reduction potential at −2.89 V vs. Ag/AgCl, which was close to that of iodobenzene (Fig. S5†). To our delight, further CV results showed that the addition of catalytic amounts of 2,2′-bipyridine could indeed effectively facilitate the reduction of iodobenzene and bromobenzene (Fig. 5 and 7). This observation strongly implies that 2,2′-bipyridine should play a role in this reaction as an efficient organo-mediator.As proof of speculation, we evaluated electroreductive anti-Markovnikov selective deuteroarylation of styrene (1) with iodobenzene (2) under different mediators (Table 1). The reaction was performed in an undivided cell equipped with graphite rods as anode and cathode, using D2O as a ‘D’ source, Et4NI as an electrolyte, and Cs2CO3 as a base in DMF. After comparing the reaction effects of different organo-mediators, 2,2′-bipyridine was considered the most suitable mediator (Entries 1–8). The reaction proceeded smoothly under constant current (12 mA) to give 3 in 94% isolated yield with 99% D-incorporation (Entry 1). Control experiments showed that the lack of a mediator, electrolyte, or base would lead to a rapid decrease in yield (Entries 9–11). Among them, the addition of iodine ions could effectively promote this electroreductive deuteroarylation, and it was speculated that iodide ions were oxidized at the anode to provide electrons for the cathode reaction. Subsequently, we investigated the effects of various reaction conditions by changing the electrolyte, base, electrode, solvent, and current. Switching to Et4NBr as the electrolyte resulted in a decrease in yield to 26%, further demonstrating that iodide ions were required in the reaction (Entry 12). Replacing the base with CsCl also gave 3 in good yield, indicating that cesium salt plays an important role in this transformation (Entry 13). The reaction exhibited lower reaction efficiency under the condition with Ni as a cathode (Entry 14). When MeCN was employed as the solvent, a lower yield of the target product (3) was obtained (Entry 15). Increasing the electrolytic current to 30 mA resulted in a reduced yield (Entry 16). When electrolysis was conducted at constant voltages of 4.5 V and 5 V, respectively, the expected product 3 was provided in good yields, which provided further evidence for inferences of electrode reactions (Entry 17). Moreover, no product (3) was obtained in the absence of electricity (Entry 18).
|
|
|||
|---|---|---|---|
| Entry | Variation from conditions | Yield (%)b | D-Inc (%)c |
| a Reaction conditions: undivided cell, graphite rods (ϕ 5 mm) as anode and cathode, constant current = 12 mA, 1 (1.5 equiv.), 2 (0.5 mmol), Med (20 mol%), Et4NI (0.5 equiv.), Cs2CO3 (0.5 equiv.), D2O (30 equiv.), dry DMF (4 mL), air, 18 h. b Isolated yield. Med = mediator. c Deuterium incorporation determined by 1H NMR. | |||
| 1 | None | 94 | 99 |
| 2 | Med-2 instead of Med-1 | 40 | 99 |
| 3 | Med-3 instead of Med-1 | 32 | 99 |
| 4 | Med-4 instead of Med-1 | 35 | — |
| 5 | Med-5 instead of Med-1 | 12 | — |
| 6 | Med-6 instead of Med-1 | <5 | — |
| 7 | Med-7 instead of Med-1 | 25 | — |
| 8 | Med-8 instead of Med-1 | 24 | — |
| 9 | w/o Med-1 | 14 | — |
| 10 | w/o Et4NI | <5 | — |
| 11 | w/o Cs2CO3 | 16 | — |
| 12 | Et4NBr | 26 | — |
| 13 | CsCl as base | 83 | 99 |
| 14 | C (+) | Ni (−) | 40 | 99 |
| 15 | MeCN | <5 | — |
| 16 | 30 mA | 26 | — |
| 17 | CVE = 4.5 V or 5.0 V | 80, 87 | 99, 99 |
| 18 | No electricity | 0 | — |
|
|||
With the optimal conditions in hand, we next investigated the substrate scope for the electroreductive deuteroarylation (Fig. 2). The scope of aryl iodides with different substituents was first examined by reacting with styrene. The reaction of styrene with iodobenzene or 4-iodobiphenyl proceeded smoothly, providing 3-4 in 94% and 54% yields with up to 99% D-incorporation, respectively. Aryl iodides with substituents (bromine, ester, cyano, fluorine, and chlorine) that could be used for late-stage functionalization were well tolerated, affording 5–11 in moderate to good yields with excellent D-incorporation. Dimethoxyiodobenzene was a suitable substrate, affording 12 in 86% yield with 99% D-incorporation. Notably, aryl iodides with large conjugated structures (naphthalene, phenanthrene, and fluorene) were also suitable for reaction with styrene, affording 13–15 in moderate yields with excellent D-incorporation. Styrene also reacted well with 6-iodo-2,3-dihydrobenzo[b][1,4]dioxine, affording 16 in 91% yield with 99% D-incorporation. Subsequently, the reaction of α-methylstyrene with substituted aryl iodides was investigated. To our delight, substrates containing various substituents (tbutyl, methoxy, propylcyclohexyl, halogen, trifluoromethyl, trifluoromethoxy, and difluoromethoxy) at the para- or meta-positions of the benzene ring were well compatible, affording 17–28 in 32–85% yields with excellent D-incorporation. Among them, substrates containing chlorine and bromine atoms were also tolerated in the system, furnishing 22-23, and 28 in moderate yields. Iodobenzene containing ortho-substituents (isopropyl, methoxy, bromine, and fluorine) could also effectively deliver the corresponding product (29–32) in moderate yields. The reaction of α-methylstyrene with dimethyl iodobenzene yielded product 33 with 95% D-incorporation. Naphthalene ring was suitable for this reaction as well, providing the desired product (34) in moderate yield with 99% D-incorporation. Benzo[d][1,3]dioxole and thienyl iodides were also tolerated in electrolysis, and the reaction with α-methylstyrene afforded 35-36 in 45–51% yields with excellent D-incorporation. The reactivity of different alkenes was then studied. Styrenes with tbutyl and methoxy at the para-position of the benzene ring showed good reactivity in the reaction with aryl iodides, affording 37–39 in 76–94% yields with excellent D-incorporation. Alkene derived from benzodioxine was well compatible to give 40 in 97% yield with 99% D-incorporation. α-Methylstyrene substrates with electron-donating (ethyl-, and methoxy-) and electron-withdrawing (chlorine-) substituents were tolerated, affording 41–44 in moderate yields. When two isopropene moieties were attached to the benzene ring, only one of them participated in the reaction with iodobenzene, providing 45 in 64% yield with good D-incorporation. Naphthalene ring and benzo[d][1,3]dioxole were well tolerated to give 46-47 in moderate to good yields. Next, several a-substituted styrenes, including (1-cyclopropylvinyl)benzene, diphenylethylene, and (4-methylpent-1-ene-2,4-diyl)dibenzene, were also applied, furnishing 48–50 in moderate yields. Moreover, selective deuteroarylation of internal alkene also proceeded smoothly to provide the desired product (51) in 39% yield with 97% D-incorporation.
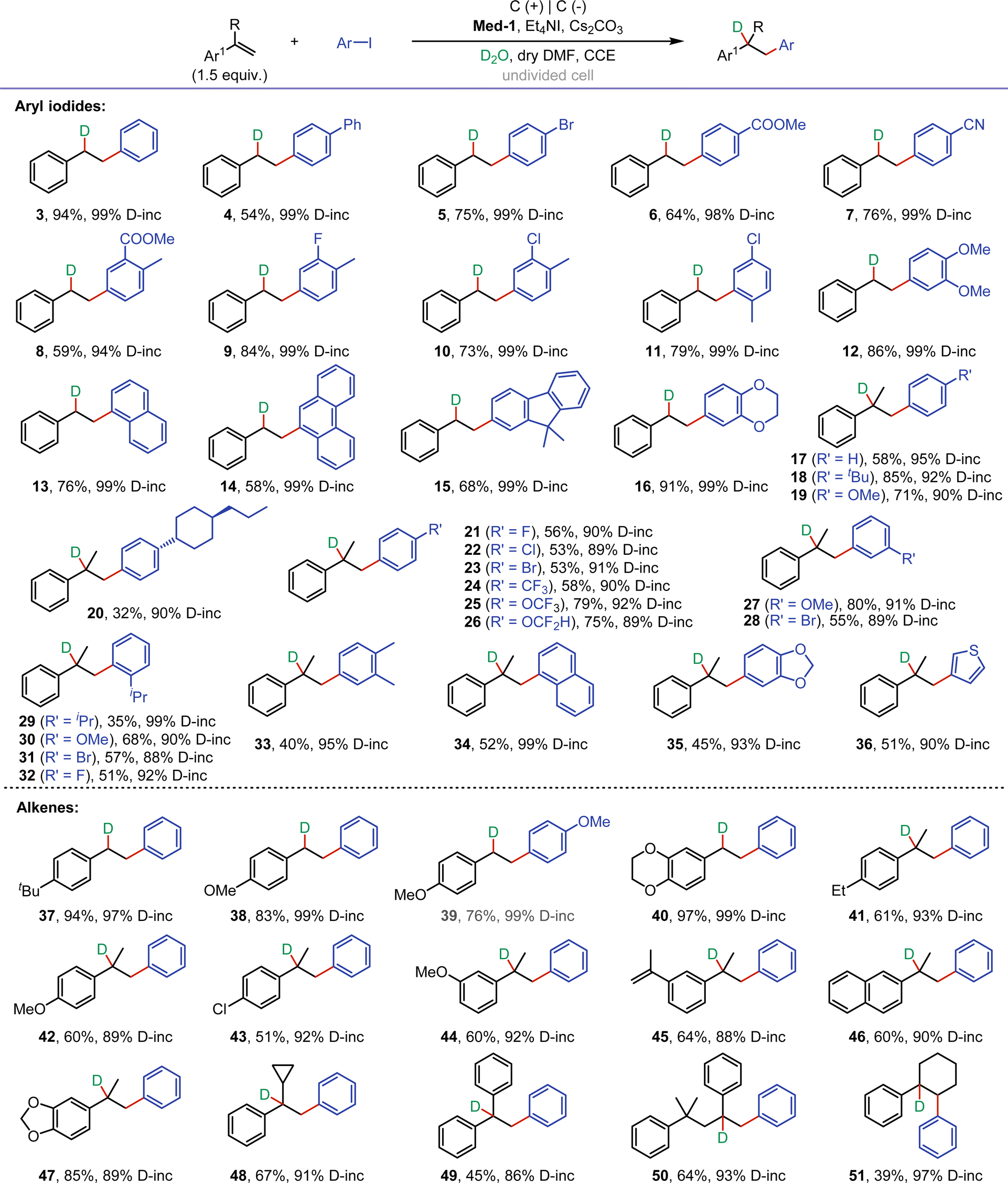 | ||
| Fig. 2 Reaction conditions: undivided cell, graphite rods (ϕ 5 mm) as anode and cathode, constant current = 12 mA, alkenes (1.5 equiv.), aryl iodides (0.5 mmol), 2,2′-bipyridine (20 mol%), Et4NI (0.5 equiv.), Cs2CO3 (0.5 equiv.), D2O (30 equiv.), dry DMF (4 mL), air, 18 h. Isolated yield. Deuterium incorporation determined by 1H NMR. | ||
The synthetic application of our developed deuteroarylation was demonstrated by the applications to other bioactive molecules (Fig. 3a). The aryl iodides derived from several bioactive molecules, including L(−)-borneol, L-menthol, icaridin, N-Boc-L-prolinol, isoborneol, and perillol, could be successfully converted into mono-deuterated products (52–57) in moderate yields with up to 99% D-incorporation. To further illustrate the practicality of this method, scale-up experiments were conducted (Fig. 3b). When the reaction of styrene with iodobenzene was expanded to a 5 mmol scale (constant current = 60 mA, 36 h), the deuterated product (3) was provided stable in 90% yield with 99% D-incorporation. Scaling the reaction of iodobenzene to a 10 mmol scale (72 h) also yielded a satisfactory result. Electrolysis of 1-bromo-4-iodobenzene as the substrate on a 10 mmol scale (constant current = 60 mA, 72 h) gave 5 in 47% yield with 99% D-incorporation, with partial dehalogenation by-product formation. Next, the derivatization of the newly formed deuterated product (23) as a building block was investigated (Fig. 3c). Compound 23 could react smoothly with ethisterone or ethynyl estradiol via Sonogashira coupling, affording the corresponding products 58 and 59 in 78% and 83% yields, respectively. In addition, compound 23 could also undergo Suzuki−Miyaura coupling with trimethoxyphenylboronic acid and be converted into 60 in 63% yield with 91% D-incorporation.
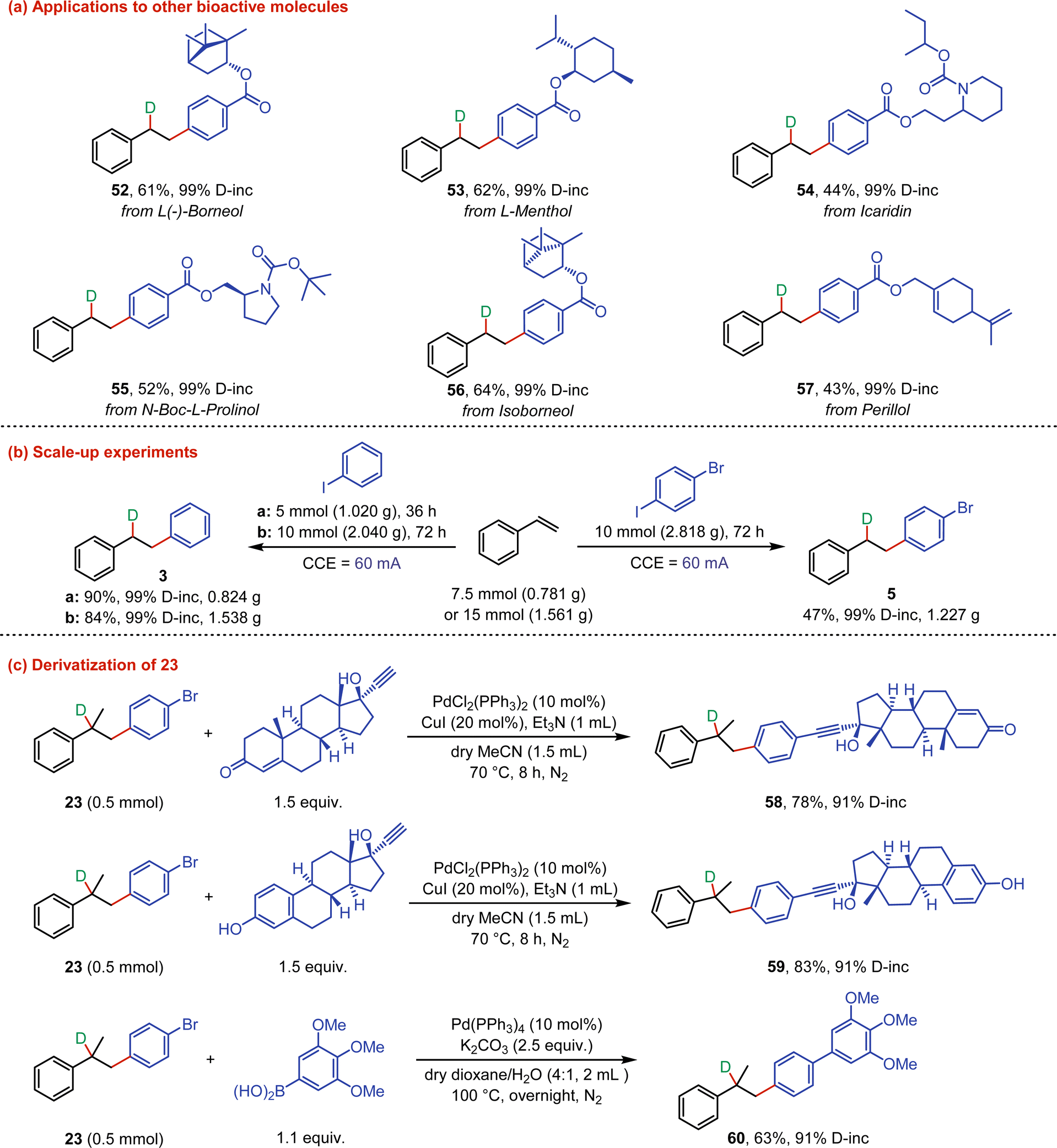 | ||
| Fig. 3 (a) Applications to other bioactive molecules. (b) Scale-up experiments. (c) Derivatization of 23. | ||
Control experiments were then conducted to study the reaction mechanism (Fig. 4). Firstly, the free radical inhibitors (BHT and TEMPO) were added to the model reaction, respectively, and both were found to significantly inhibit the reaction at 2 equiv. The above results indicated that the reaction could undergo a radical process (eqn (a)). An additional radical trap experiment with 9,10-dihydroanthracene confirmed the participation of radicals, as phenyl radicals were captured (61) and the yield of 17 was significantly reduced (eqn (b)). Subsequently, electrolysis of 1,2-diphenylethane under standard conditions showed that no H/D exchange occurred (eqn (c)). A further deuterium-labeling study was performed through the reaction between α-methylstyrene and iodobenzene with DMF-d7 as the solvent, and the result proved that the protons required for the reaction were derived from water rather than DMF (eqn (d)).
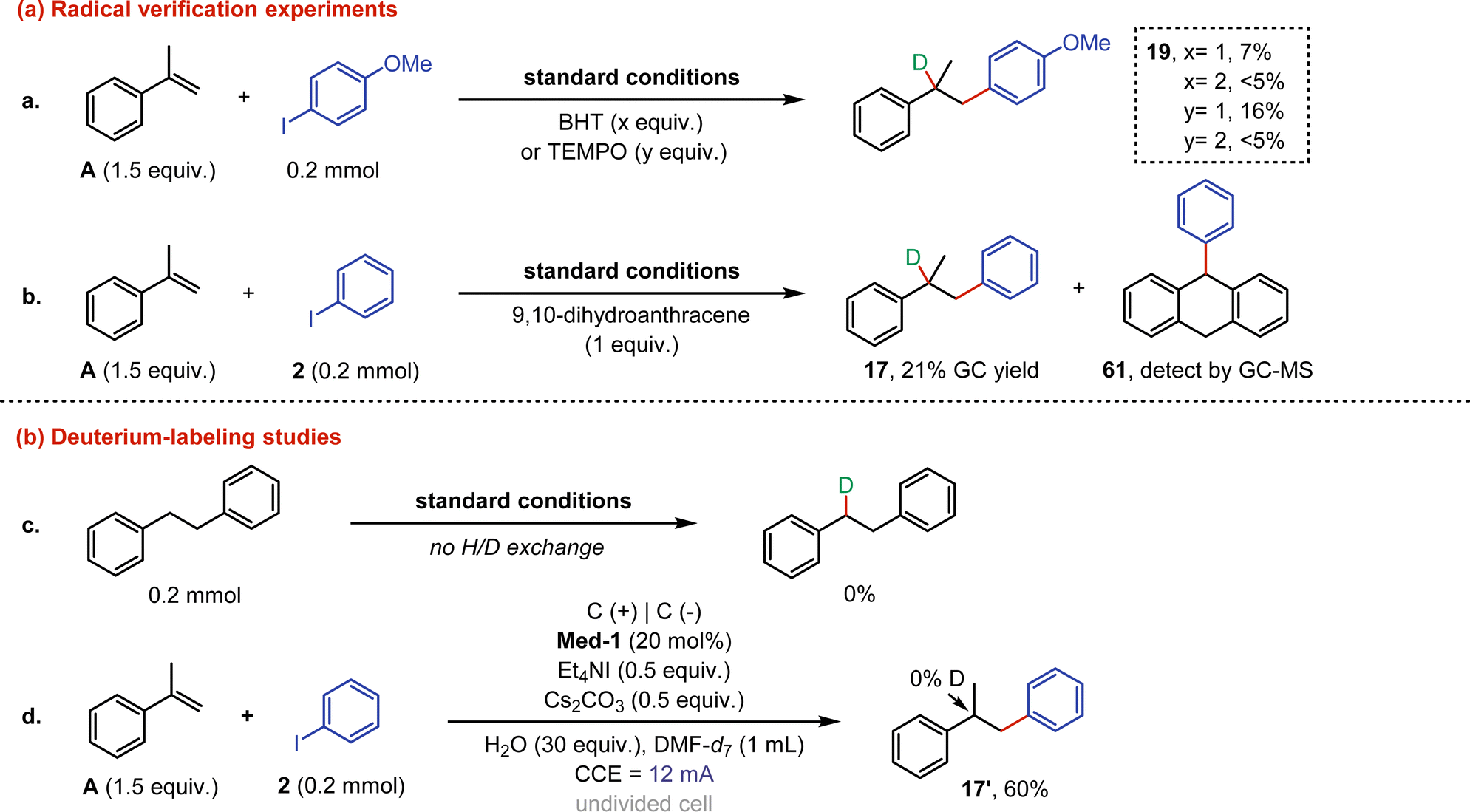 | ||
| Fig. 4 Control experiments. (a) Radical verification experiments. (b) Deuterium-labeling studies. | ||
To elucidate the reaction mechanism in detail, cyclic voltammetry (CV) experiments were next implemented (Fig. 5). With 0.1 M nBu4NPF6 solution in DMF as background, no redox peaks were observed (blank). By comparing the reduction potentials of iodobenzene (−2.64 V vs. Ag/AgCl), bromobenzene (−3.31 V vs. Ag/AgCl), and chlorobenzene (<−3.5 V vs. Ag/AgCl), iodobenzene and bromobenzene were believed to be suitable substrates for electrolysis (curves a–c). The CV of Cs2CO3 displayed no apparent redox potential (curve d). The reduction potential of 2,2′-bipyridine at −2.89 V vs. Ag/AgCl was observed on curve e. The catalytic current of 2,2′-bipyridine increased significantly with the addition of Cs2CO3, demonstrating that Cs2CO3 assisted the electroreduction process mediated by bipyridine (curve f). Meanwhile, two significant oxidation peaks appeared at 1.10 V and 1.46 V vs. Ag/AgCl on curve f, corresponding to the oxidation process of I− to I3− and then to I2. The reduction potential of α-methylstyrene appeared at −3.54 V vs. Ag/AgCl (curve g), which was more difficult to reduce than iodobenzene, indicating that iodobenzene was reduced prior to the alkene and subsequently initiated the reaction. Notably, the CV of the reaction system showed an obvious reduction peak at −2.30 V vs. Ag/AgCl, which was presumed to be the reduction potential of iodobenzene assisted by the mediator (curve h). Mediator-facilitated reduction of iodobenzene can be illustrated by the curves i–k. The addition of Cs2CO3 caused a slight anodic shift in the reduction of iodobenzene (curve i). The addition of 2,2′-bipyridine led to an anodic shift in the reduction potential of iodobenzene to −2.32 V vs. Ag/AgCl (curve j). In addition, the mediator could also promote the reduction of iodobenzene in the presence of D2O, and the reduction potential appeared at −2.23 V vs. Ag/AgCl (curve k). From the CV results, the iodobenzene substrate appeared to be more easily reduced than the mediator. However, the transformation was still dominated by the mediated pathway. This phenomenon can be attributed to the formation of a reaction layer detached from the electrode, resulting in a blockage of substrate access to the electrode.9c
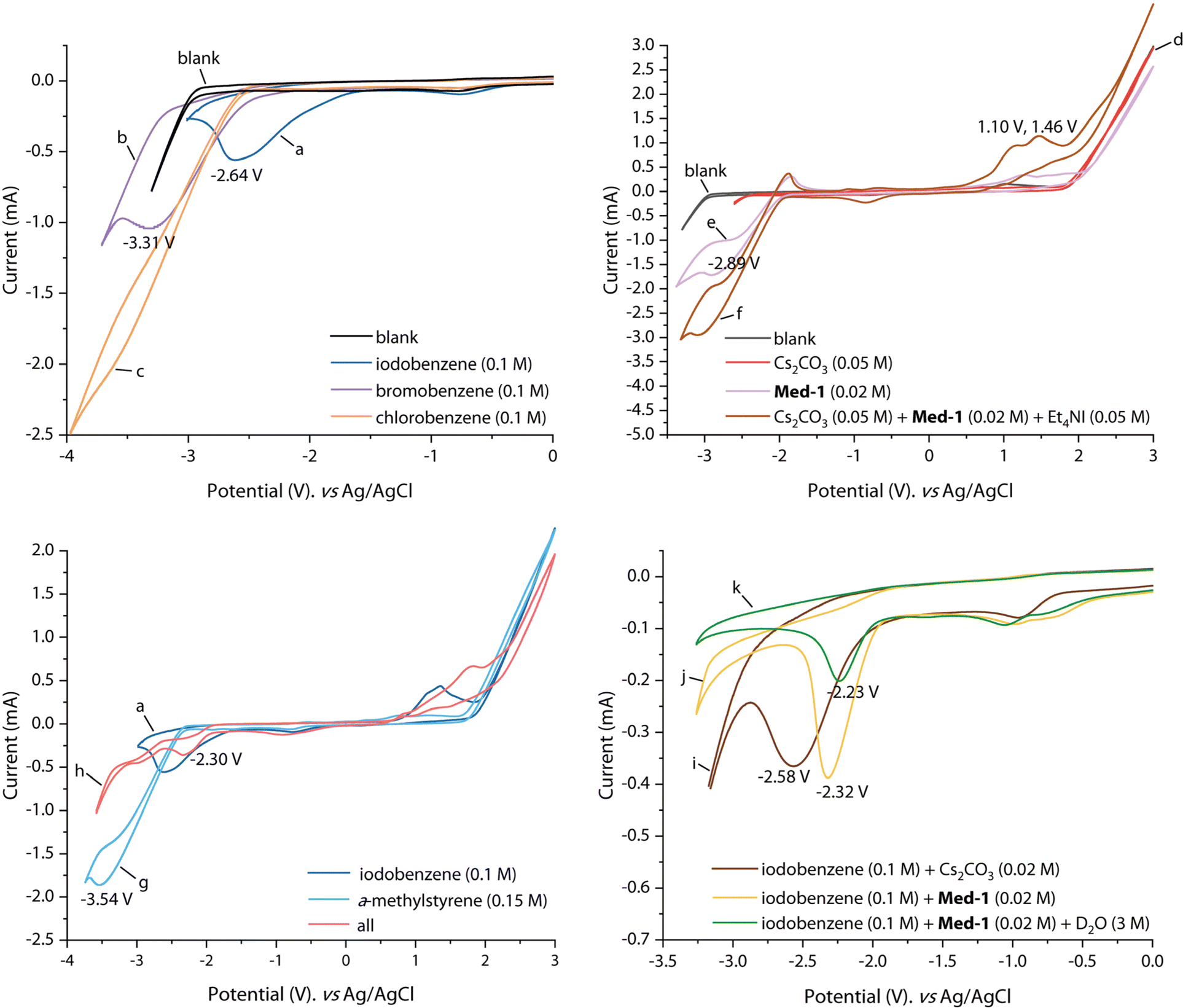 | ||
| Fig. 5 Cyclic voltammetry (CV) experiments. Experiments were conducted by using glassy carbon as the working electrode (6 mm in diameter, BASi), Pt plate, and Ag/AgCl (saturated KCl) as the counter and reference electrode. Scan rate: 100 mV s−1. Solvent: DMF/nBu4NPF6 (0.1 M). For details, please see (ESI).† | ||
Furthermore, density functional theory (DFT) calculations were performed with the G16 program to study the SER (single electron transfer) process in depth. The structures of intermediates and transition states were described and analyzed with the Multiwfn12 and VMD software13 (see the ESI for details†). As shown in Fig. 6a, the bipyridine radical anion can interact with iodobenzene (2) through π–π interaction to form 2-Med adducts. The molecular orbital calculation shows that the energy level of singly occupied molecular orbital (SOMO) in [Med-1]˙− is close to that of the lowest unoccupied molecular orbital (LUMO) in 2, enabling the charge transfer (CT) from [Med-1]˙− to 2 to proceed smoothly and promoting the formation of 2-Med (Fig. 6b). By comparing the energy difference of molecular orbitals, the LUMO(2)–SOMO[Med-1]˙− gap is significantly smaller than the LUMO(A)–SOMO[Med-1]˙− gap, which proves that the dipyridine mediator can effectively control the chemoselectivity and avoid the side reaction of alkene deuteration. After this, 2-Med releases Med-1 to form the iodobenzene radical anion (TS1), in which TS1 appears to be two separate parts: the phenyl radical and the iodide ion. The electrostatic potential (ESP) and electron spin density distribution of TS1 show that the negative charge is mainly distributed in the iodine part and the single electron is mainly located at the single carbon atom of phenyl (Fig. 6c, See the ESI for details†). Following the above experimental results and DFT calculation (Fig. 6d), a reasonable mechanism is proposed for this electroreductive selective deuteroarylation (Fig. 6e). During electrolysis, bipyridine first gains an electron from the cathode to form a radical anion, and then transfers the electron to the iodobenzene (2) through the formation of 2-Med, promoting the formation of the iodobenzene radical anion (TS1). Subsequently, radical anion TS1 releases an iodide ion to form a phenyl radical (B) under the promotion of Cs2CO3, accompanied by a drop in energy (ΔG1 = −3.2 kcal mol−1). The phenyl radical then attacks the unsaturated double bond of the alkene A from the end-site to generate a radical intermediate C, which passes through the transition state TS2 and requires a reaction energy barrier of 10.2 kcal mol−1 (ΔG2). The intermediate C is converted to the anion D after single electron reduction (SER) at the cathode, followed by protonation by D2O to form the final product. In addition, iodide anions are oxidized on the anode to form iodine molecules, a process that provides electrons for the electroreduction reaction.
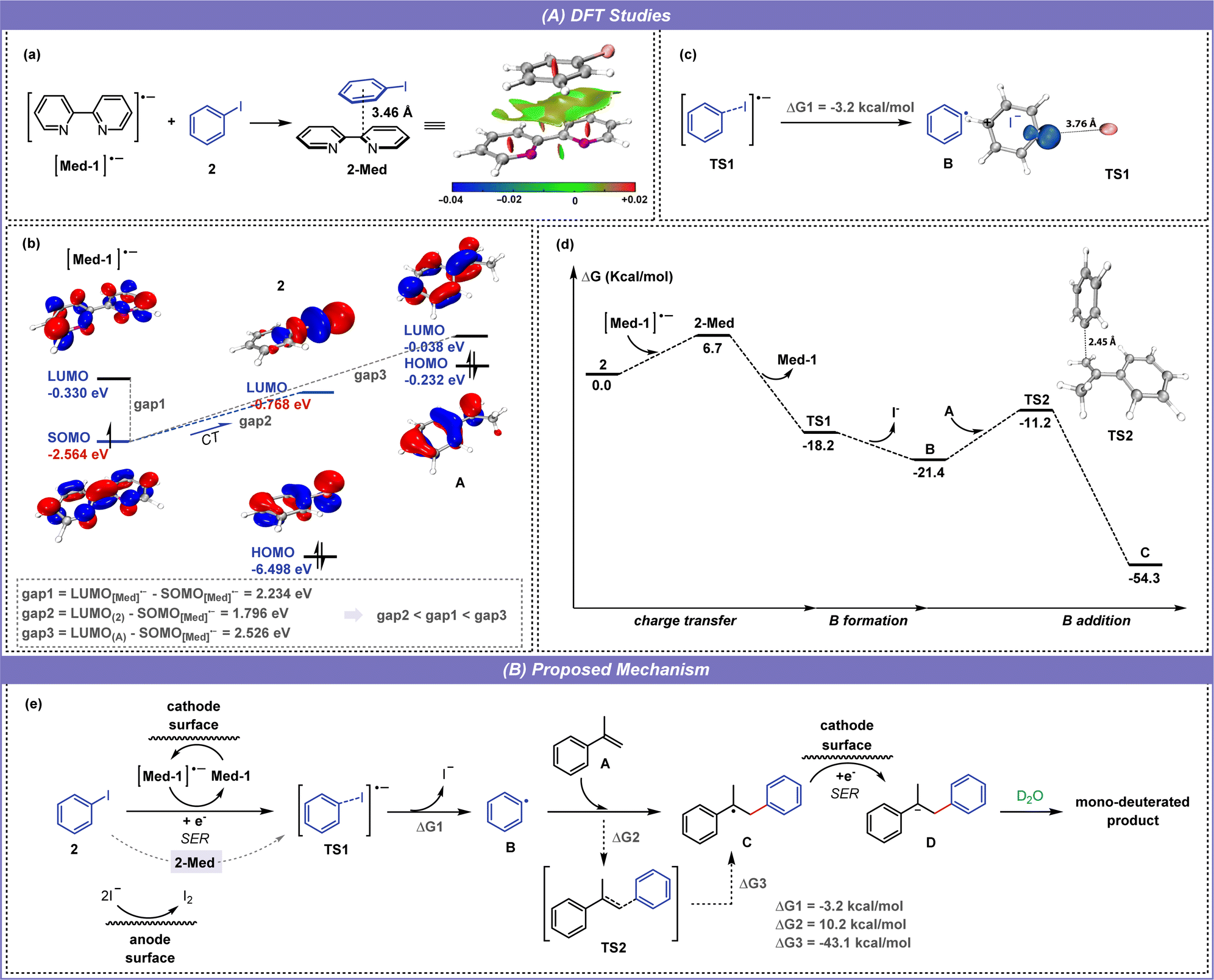 | ||
| Fig. 6 DFT Studies and proposed mechanism. (a) The interaction in 2-Med was visualized using the IRI method, where negative values indicate favorable and positive values indicate unfavorable. (b) The interaction of SOMO orbital of [Med-1]˙− with LUMO orbital of 2. (c) Electron spin density distribution of TS1. (d) Computed Gibbs free energy profile for the formation of C. HOMO: highest occupied molecular orbital. LUMO: lowest unoccupied molecular orbital. SOMO: singly occupied molecular orbital. CT charge transfer. (e) Proposed mechanism. | ||
In the interest of exploring the reactivity of other aryl halides, reactions of alkenes with aryl bromides or chlorides were subsequently performed under standard conditions (Fig. 7A). However, none of the reactions occurred as expected, and no product 19 was formed, but deuterated products (62 and A-d2) of A and aryl halides could be detected in the reaction mixtures (eqn (a)). This suggested that deuteroarylation involving aryl bromides was difficult to carry out in a D2O-containing system. It was noteworthy that alkenes and aryl halides (bromides or iodides) could undergo selective hydroarylation when using ordinary DMF as a solvent. Aryl bromides or iodides containing different substituents (methyl-, n-propyl-, fluorine-, and trifluoromethyl-) reacted well with α-methylstyrene (A) to obtain hydroarylation products (63–67) in 46–96% yields (eqn (b)). The solvent-dependent selectivity enabled electroreductive deuteroarylation to occur with high specificity at the aryl carbon bonded to iodine, effectively preserving the bromine atoms within the substrates. Cyclic voltammetry (CV) experiments were subsequently performed to interpret the above results (Fig. 7B). The reduction potential of bromobenzene initially appeared at −3.31 V vs. Ag/AgCl (curve b), and the anodic shift to −3.04 V vs. Ag/AgCl occurred after the addition of 2,2′-bipyridine (curve l), indicating a mediator-facilitated reduction of bromobenzene. However, no significant reduction potential appeared in the scanning window (−3.5–0 V) after the addition of D2O (curve m), corresponding to the debromodeuteration process of bromobenzene.
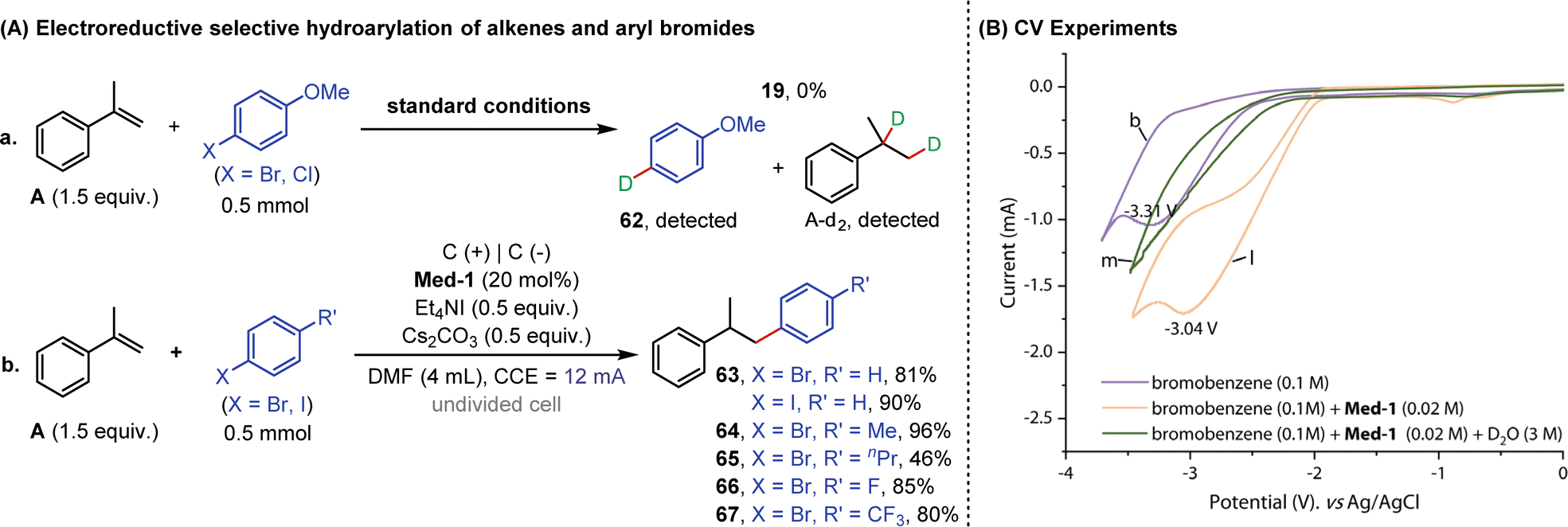 | ||
| Fig. 7 (A) Electroreductive selective hydroarylation of alkenes and aryl bromides. (B) CV experiments. | ||
Conclusions
In conclusion, we have developed a novel anti-Markovnikov selective deuteroarylation of alkenes and aryl iodides in the absence of metal catalysts and external reductants through a mediated electroreduction strategy. This protocol proceeds efficiently under simple electrolysis conditions, using catalytic amounts of bipyridine as an organo-mediator and D2O as the deuterium source. A series of mono-deuterated alkylarenes with excellent deuterium incorporation is provided via the formation of intermolecular C–C and C–D bonds through two single-electron reductions (SER). Our method is distinguished by scalability, broad substrate scope, no sacrificial anodes, and high economic benefits. Cyclic voltammetry experiments have demonstrated the mediator-facilitated reduction of aryl halides. Furthermore, radical verification experiments and DFT studies have confirmed the possible mechanism of aryl radical generation. In addition, scale-up and derivatization experiments have proved the practicality of this protocol. With further exploration, aryl bromides are found to be available for selective hydroarylation under the ordinary solvent. Continued investigations into electroreductive deuteroarylation of alkenes and further applications of this protocol are ongoing in our laboratory.Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the ESI.†Author contributions
XL conducted the experiments and analyzed the results. JZ, WD, ZW, YW, and ZL performed some experimental data analysis. YQ and YH designed the research, supervised the project, and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Founda-tion of China (22001197), the Young Scientific and Technolog-ical Talent Support Project of Jiangmen (31117016), and the Basic and Theoretical Scientific Research Project of Jiangmen (2022030102310003890). This research was supported by Big Data Center for Biomedical Research of Wuyi University.Notes and references
- (a) K. B. Wiberg, Chem. Rev., 1955, 55, 713–743 CrossRef CAS; (b) T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, J. Med. Chem., 2019, 62, 5276–5297 CrossRef CAS PubMed; (c) T. R. Puleo, A. J. Strong and J. S. Bandar, J. Am. Chem. Soc., 2019, 141, 1467–1472 CrossRef CAS PubMed; (d) A. Xia, X. Xie, X. Hu, W. Xu and Y. Liu, J. Org. Chem., 2019, 84, 13841–13857 CrossRef CAS PubMed; (e) A. Palazzolo, S. Feuillastre, V. Pfeifer, S. Garcia-Argote, D. Bouzouita, S. Tricard, C. Chollet, E. Marcon, D.-A. Buisson, S. Cholet, F. Fenaille, G. Lippens, B. Chaudret and G. Pieters, Angew. Chem., Int. Ed., 2019, 58, 4891–4895 CrossRef CAS PubMed; (f) Y. Li, Z. Ye, Y.-M. Lin, Y. Liu, Y. Zhang and L. Gong, Nat. Commun., 2021, 12, 2894 CrossRef CAS PubMed; (g) N. Li, Y. Li, X. Wu, C. Zhu and J. Xie, Chem. Soc. Rev., 2022, 51, 6291–6306 RSC.
- R. M. C. Di Martino, B. D. Maxwell and T. Pirali, Nat. Rev. Drug Discovery, 2023, 22, 562–584 CrossRef CAS PubMed.
- (a) A. J. Boyington, M.-L. Y. Riu and N. T. Jui, J. Am. Chem. Soc., 2017, 139, 6582–6585 CrossRef CAS PubMed; (b) J. A. Gurak and K. M. Engle, ACS Catal., 2018, 8, 8987–8992 CrossRef CAS PubMed; (c) F. Zhou, X. Hu, W. Zhang and C.-J. Li, J. Org. Chem., 2018, 83, 7416–7422 CrossRef CAS PubMed; (d) J. Nguyen, A. Chong and G. Lalic, Chem. Sci., 2019, 10, 3231–3236 RSC; (e) F. Yang, Y. Jin and C. Wang, Org. Lett., 2019, 21, 6989–6994 CrossRef CAS PubMed.
- (a) C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed; (b) W. Zhang and S. Lin, J. Am. Chem. Soc., 2020, 142, 20661–20670 CrossRef CAS PubMed; (c) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed; (d) Q. Wang, C.-H. Xu, Y.-C. Wang, Y.-M. Pan, W.-G. Duan and H.-T. Tang, Green Chem., 2022, 24, 7362–7367 RSC; (e) S. Fang, K. Zhong, S. Zeng, X. Hu, P. Sun and Z. Ruan, Chem. Commun., 2023, 59, 11425–11428 RSC; (f) C. Liu, Y. Wu, B. Zhao and B. Zhang, Acc. Chem. Res., 2023, 56, 1872–1883 CrossRef CAS PubMed; (g) S. Zhang, Y. Liang, K. Liu, X. Zhan, W. Fan, M.-B. Li and M. Findlater, J. Am. Chem. Soc., 2023, 145, 14143–14154 CrossRef CAS PubMed.
- (a) T. Wang, F. He, W. Jiang and J. Liu, Angew. Chem., Int. Ed., 2024, 63, e202316140 CrossRef CAS PubMed; (b) Z. Zhang, X.-J. Meng, F.-H. Cui, H.-T. Tang, Y.-C. Wang, G.-B. Huang and Y.-M. Pan, Org. Lett., 2024, 26, 193–197 CrossRef CAS PubMed; (c) S.-H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2021, 121, 485–505 CrossRef CAS PubMed; (d) W. Deng, X. Li, Z. Li, Y. Wen, Z. Wang, Z. Lin, Y. Li, J. Hu and Y. Huang, Org. Lett., 2023, 25, 9237–9242 CrossRef CAS PubMed; (e) Y. Gao, B. Zhang, J. He and P. S. Baran, J. Am. Chem. Soc., 2023, 145, 11518–11523 CrossRef CAS PubMed; (f) Y. Wang, S. Dana, H. Long, Y. Xu, Y. Li, N. Kaplaneris and L. Ackermann, Chem. Rev., 2023, 123, 11269–11335 CrossRef CAS PubMed; (g) J. Rein, S. B. Zacate, K. Mao and S. Lin, Chem. Soc. Rev., 2023, 52, 8106–8125 RSC; (h) Y. Li, S. Dana and L. Ackermann, Curr. Opin. Electrochem., 2023, 40, 101312 CrossRef CAS.
- (a) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC; (b) M. Rafiee, B. Karimi and S. Alizadeh, ChemElectroChem, 2014, 1, 455–462 CrossRef; (c) M. Rafiee, K. C. Miles and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 14751–14757 CrossRef CAS PubMed; (d) K. Liu, C. Song and A. Lei, Org. Biomol. Chem., 2018, 16, 2375–2387 RSC; (e) J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed; (f) P. Xiong, H.-H. Xu, J. Song and H.-C. Xu, J. Am. Chem. Soc., 2018, 140, 2460–2464 CrossRef CAS PubMed; (g) P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339–3350 CrossRef CAS PubMed; (h) C. M. Galvin and R. M. Waymouth, J. Am. Chem. Soc., 2020, 142, 19368–19378 CrossRef CAS PubMed; (i) F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53, 561–574 CrossRef CAS PubMed.
- (a) Y. Liu, B. Shi, Z. Liu, R. Gao, C. Huang, H. Alhumade, S. Wang, X. Qi and A. Lei, J. Am. Chem. Soc., 2021, 143, 20863–20872 CrossRef CAS PubMed; (b) T. Feng, S. Wang, Y. Liu, S. Liu and Y. Qiu, Angew. Chem., Int. Ed., 2022, 61, e202115178 CrossRef CAS PubMed; (c) S. Hosseini, J. N. Janusz, M. Tanwar, A. D. Pendergast, M. Neurock and H. S. White, J. Am. Chem. Soc., 2022, 144, 21103–21115 CrossRef CAS PubMed; (d) A. D. Stergiou and M. D. Symes, Cell Rep. Phys. Sci., 2022, 3, 100914 CrossRef CAS; (e) J. Zhang, B. Das, O. Verho and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2022, 61, e202212131 CrossRef CAS PubMed; (f) A. G. Reid and C. W. Machan, J. Am. Chem. Soc., 2023, 145, 2013–2027 CrossRef CAS PubMed.
- (a) A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406–410 CrossRef CAS PubMed; (b) Z.-H. Wang, P.-S. Gao, X. Wang, J.-Q. Gao, X.-T. Xu, Z. He, C. Ma and T.-S. Mei, J. Am. Chem. Soc., 2021, 143, 15599–15605 CrossRef CAS PubMed; (c) M. A. Cribari, M. J. Unger and J. D. Martell, ACS Catal., 2022, 12, 12246–12252 CrossRef CAS PubMed; (d) M. A. Hoque, J. Twilton, J. Zhu, M. D. Graaf, K. C. Harper, E. Tuca, G. A. DiLabio and S. S. Stahl, J. Am. Chem. Soc., 2022, 144, 15295–15302 CrossRef CAS PubMed; (e) H. Park, M. Kim, J. Kang, H. Song and H. Kim, Chem. Commun., 2023, 59, 5447–5450 RSC; (f) S. Sharma, S. Shaheeda, K. Shaw, A. Bisai and A. Paul, ACS Catal., 2023, 13, 2118–2134 CrossRef CAS; (g) X. Zhao, J.-D. Yang and J.-P. Cheng, J. Org. Chem., 2023, 88, 540–547 CrossRef CAS PubMed.
- (a) B. Wang, P. Peng, W. Ma, Z. Liu, C. Huang, Y. Cao, P. Hu, X. Qi and Q. Lu, J. Am. Chem. Soc., 2021, 143, 12985–12991 CrossRef CAS PubMed; (b) A. A. Folgueiras-Amador, A. E. Teuten, M. Salam-Perez, J. E. Pearce, G. Denuault, D. Pletcher, P. J. Parsons, D. C. Harrowven and R. C. D. Brown, Angew. Chem., Int. Ed., 2022, 61, e202203694 CrossRef CAS PubMed; (c) Y. Wang, Z. Zhao, D. Pan, S. Wang, K. Jia, D. Ma, G. Yang, X. Xue and Y. Qiu, Angew. Chem., Int. Ed., 2022, 61, e202210201 CrossRef CAS PubMed; (d) X. Kong, Y. Chen, X. Chen, C. Ma, M. Chen, W. Wang, Y.-Q. Xu, S.-F. Ni and Z.-Y. Cao, Nat. Commun., 2023, 14, 6933 CrossRef CAS PubMed; (e) V. K. Rawat, H. Hayashi, H. Katsuyama, S. R. Mangaonkar and T. Mita, Org. Lett., 2023, 25, 4231–4235 CrossRef CAS PubMed; (f) W. Yu, S. Wang, M. He, Z. Jiang, Y. Yu, J. Lan, J. Luo, P. Wang, X. Qi, T. Wang and A. Lei, Angew. Chem., Int. Ed., 2023, 62, e202219166 CrossRef CAS PubMed; (g) K. Yang, T. Feng and Y. Qiu, Angew. Chem., Int. Ed., 2023, 62, e202312803 CrossRef CAS PubMed.
- (a) A. Kurimoto, R. S. Sherbo, Y. Cao, N. W. X. Loo and C. P. Berlinguette, Nat. Catal., 2020, 3, 719–726 CrossRef CAS; (b) P. Li, C. Guo, S. Wang, D. Ma, T. Feng, Y. Wang and Y. Qiu, Nat. Commun., 2022, 13, 3774 CrossRef CAS PubMed; (c) P. L. Norcott, Chem. Commun., 2022, 58, 2944–2953 RSC; (d) D. Wood and S. Lin, Angew. Chem., Int. Ed., 2023, 62, e202218858 CrossRef CAS PubMed; (e) W. Deng, Y. Hu, J. Hu, X. Li, Y. Li and Y. Huang, Chem. Commun., 2022, 58, 12094–12097 RSC; (f) R. Li, Y. Wu, C. Wang, M. He, C. Liu and B. Zhang, Nat. Commun., 2022, 13, 5951 CrossRef PubMed; (g) C. Liu, S. Han, M. Li, X. Chong and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18527–18531 CrossRef CAS PubMed; (h) S. Kolb and D. B. Werz, Chem.–Eur. J., 2023, 29, e202300849 CrossRef CAS PubMed.
- (a) R. Alvarado De La Torre and J. W. Sease, J. Am. Chem. Soc., 1979, 101, 1687–1690 CrossRef CAS; (b) L. Lu, H. Li, Y. Zheng, F. Bu and A. Lei, CCS Chem., 2021, 3, 2669–2675 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03049d |
| This journal is © The Royal Society of Chemistry 2024 |