
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDefect engineering of air-stable Li5FeO4 towards an ultra-high capacity cathode prelithiation additive†
Bin
Zhu
 a,
Naifeng
Wen
a,
Jingyang
Wang
*b,
Qiyu
Wang
a,
Jingqiang
Zheng
a and
Zhian
Zhang
*a
a,
Naifeng
Wen
a,
Jingyang
Wang
*b,
Qiyu
Wang
a,
Jingqiang
Zheng
a and
Zhian
Zhang
*a
aSchool of Metallurgy and Environment, National Energy Metal Resources and New Materials Key Laboratory, Central South University, Changsha 410083, People's Republic of China. E-mail: zhangzhian@csu.edu.cn
bSchool of Sustainable Energy and Resources, Nanjing University, Suzhou, 215163, China. E-mail: jy_wang@nju.edu.cn
First published on 16th July 2024
Abstract
Antifluorite-type Li5FeO4 (LFO) belongs to a class of promising prelithiation materials for next-generation high-energy lithium-ion batteries. Unfortunately, the incomplete de-lithiation performance and inferior air stability hinder its application. In this work, ultra-high capacity is achieved by selective doping of Zr into the Fe sites (LFO-Zr) of LFO to form a large number of defects. The underlying defect formation mechanism is comprehensively investigated using density functional theory, revealing that such selective site doping not only enlarges the unit cell volume but also induces Li vacancies into the structure, both of which facilitate lithium-ion migration at a high-rate and promote the redox of oxygen anions. As a result, under 0.05 and 1C rates, the capacity of LFO-Zr reaches 805.7 and 624.5 mA h g−1, which are 69.0 and 262.0 mA h g−1 higher than those of LFO, translating to an increase of 9.4% and 73.3%, respectively. In addition, LFO-Zr exhibits excellent electrochemical performance in a humidity of 20%, with a high capacity of 577.6 mA h g−1 maintained. With the LFO-Zr additive, the full cell delivered 193.6 mA h g−1 for the initial cycle at 0.1C. The defect engineering strategy presented in this work delivers insights to promote ultra-high capacity and high-rate performance of air-stable LFO.
1. Introduction
Lithium-ion batteries (LIBs) have been widely used in our lives to power portable electronic devices and electric vehicles by virtue of their long life and high specific energy.1,2 However, upon the first cycle, up to 15% active lithium-ions will be inevitably consumed due to the in situ electrochemical reduction of lithium salt anions (e.g., PF6−) or organic solvents to produce a lithium-containing solid electrolyte interphase (SEI) layer, leading to a lower specific energy in the initial cycle.3 For instance, the initial coulombic efficiency (ICE) of the silicon oxide anode is very low, which reduces the energy density. Therefore, an efficient solution to improve the ICE becomes particularly important for high-energy-density LIBs. Prelithiation is a promising technology to generate extra active lithium ions to offset initial lithium losses and increase the energy density.4–6 There have been several prelithiation methods developed, such as electrochemical prelithiation,7,8 chemical prelithiation,9 and incorporation of prelithiation additives into the cathode and anode.10 Both electrochemical prelithiation and chemical prelithiation are constrained by the high reactivity of lithium metal and safety issues during processing, which leads to harsh production environments and restricts their large-scale application.8 In contrast, by adding high-specific-capacity and irreversible lithium-containing compounds with suitable oxidation potential to the cathode material, cathode prelithiation can be achieved.Until now, various cathode prelithiation additives have been proposed. Binary lithium-rich compounds (Li–N, Li–O, and Li–C, etc.)11–13 can donate a large amount of irreversible active Li+ ions. However, it is accompanied by the production of undesired N2, CO or CO2 gases, which will lead to increased polarization, dissolution of metal ions, and contact loss between the electrodes and current collectors. In contrast, ternary lithium-rich compounds (Li2CuO2, Li5FeO4 (LFO), Li2NiO2, and Li6CoO4, etc.)4,14–19 seem to be better candidates with inhibited gas release. Among them, Li5FeO4 holds great promise by virtue of its inexpensive raw materials, high theoretical specific capacity (867 mA h g−1), simple synthesis method, and process compatibility with existing cathode materials. Hence, as a cathode additive, Li5FeO4 serves as a Li+ donor to improve ICE and boost the overall energy density.20,21 Nevertheless, it still suffers from incomplete de-lithiation, poor electronic conductivity, and severe surface degradation in air.19,22 To illustrate, insufficient delithiation leads to the actual capacity of LFO being about 693.6 mA h g−1 (4 Li+ per chemical formula unit), which could be more severe under high rates, leading to prolonged prelithiation process time and higher production costs. In addition, LFO is prone to forming surface alkaline species (e.g., Li2O, LiOH, or Li2CO3) when exposed to air, resulting in high interfacial resistance, poor kinetics, and a significant increase in cathode slurry alkalinity and slurry gelation. In our previous work,19,22 we presented that the air stability of LFO can be effectively improved by a carbon encapsulation strategy. Nevertheless, it is still worth further research to improve the de-lithiation kinetics of LFO to achieve ultra-high capacity and high-rate performance.
Herein, we propose a defect engineering strategy to achieve ultra-high capacity and high-rate performance for air-stable LFO. A comprehensive study on the underlying mechanisms of defect formation and the effects on the structural and electrochemical performance of the material is performed. The introduction of Zr4+ at the tetrahedral Fe sites promotes the formation of Li+ vacancies and a decrease in the Li+ migration barrier, which favors rapid lithium-ion migration and improves the lithium compensation capability. In addition, we found that the introduction of Zr ions facilitates the shift of O 2p state energy to higher energies and promotes anion redox. Overall, LFO-Zr achieves an ultra-high initial irreversible capacity of 805.7 mA h g−1 at 0.05C. Under 1C rate, the capacity of LFO-Zr also reaches 624.5 mA h g−1, which is 262.0 mA h g−1 higher than that of pure LFO. In addition, LFO-Zr exhibits excellent stability in a humidity of 20%, with a capacity of 577.6 mA h g−1 maintained, as compared to 333.4 mA h g−1 of the pristine sample. This is attributed to the carbon layer of the material, which can effectively hinder airborne erosion. The LFO-Zr electrode exhibits excellent prelithiation performance in graphite‖NCM811 full cells with a high specific capacity of 193.6 mA h g−1 at 0.1C, which is 18.8 mA h g−1 higher than that of the pristine graphite‖NCM811 full cell. This simple strategy enables improved de-lithiation performance and air stability of LFO, contributing to the acceleration of large-scale applications of LFO as an effective cathode prelithiation additive.
2. Results and discussion
2.1 Theoretical calculation
The defect formation mechanism of Zr doping in LFO was first explored using Density Function Theory (DFT) calculations, considering Zr sitting either in the Fe or the Li site. To maintain the charge neutrality of the compound, the introduction of high-valence Zr4+ can be compensated by the reduction of Fe3+ to Fe2+, which involves the generation of O2 gas, or by the creation of Li+ vacancies without the participation of oxygen. Therefore, the defect formation energy is either in positive correlation, or independent of the oxygen chemical potential (μO2), respectively. By including the entropy contribution of gaseous O2 in μO2, the defect formation energies at different temperatures can be therefore estimated, as higher temperature indicates a more negative μO2 and vice versa.23,24 As displayed in Fig. 1a, within the temperature range of typical solid-state synthesis, Zr is more likely to occupy Fe sites accompanied by the formation of Li vacancies, which exhibit the lowest defect formation energy among all types of defects generated upon Zr doping. Furthermore, positron annihilation lifetime spectra (PALS) were employed to analyze the species of vacancy defects.25 The positron lifetime spectra of LFO and LFO-Zr exhibit similar shapes in Fig. 1b. Table S1† shows the three lifetime components of LFO and LFO-Zr. The longest lifetime τ3 (=1.5 ns) is ascribed to positron annihilation from large-size vacancies in the pre-lithiation. Larger vacancies have a lower average electron density leading to a lower annihilation rate facilitating the prolongation of the positron lifetime, whereas the existence of small defects reduces the surrounding electron density, which leads to an increase in the lifetime of τ1.26 Based on theoretical calculations of positron lifetime, the shorter lifetime component τ1 in LFO-Zr is attributed to Li vacancies.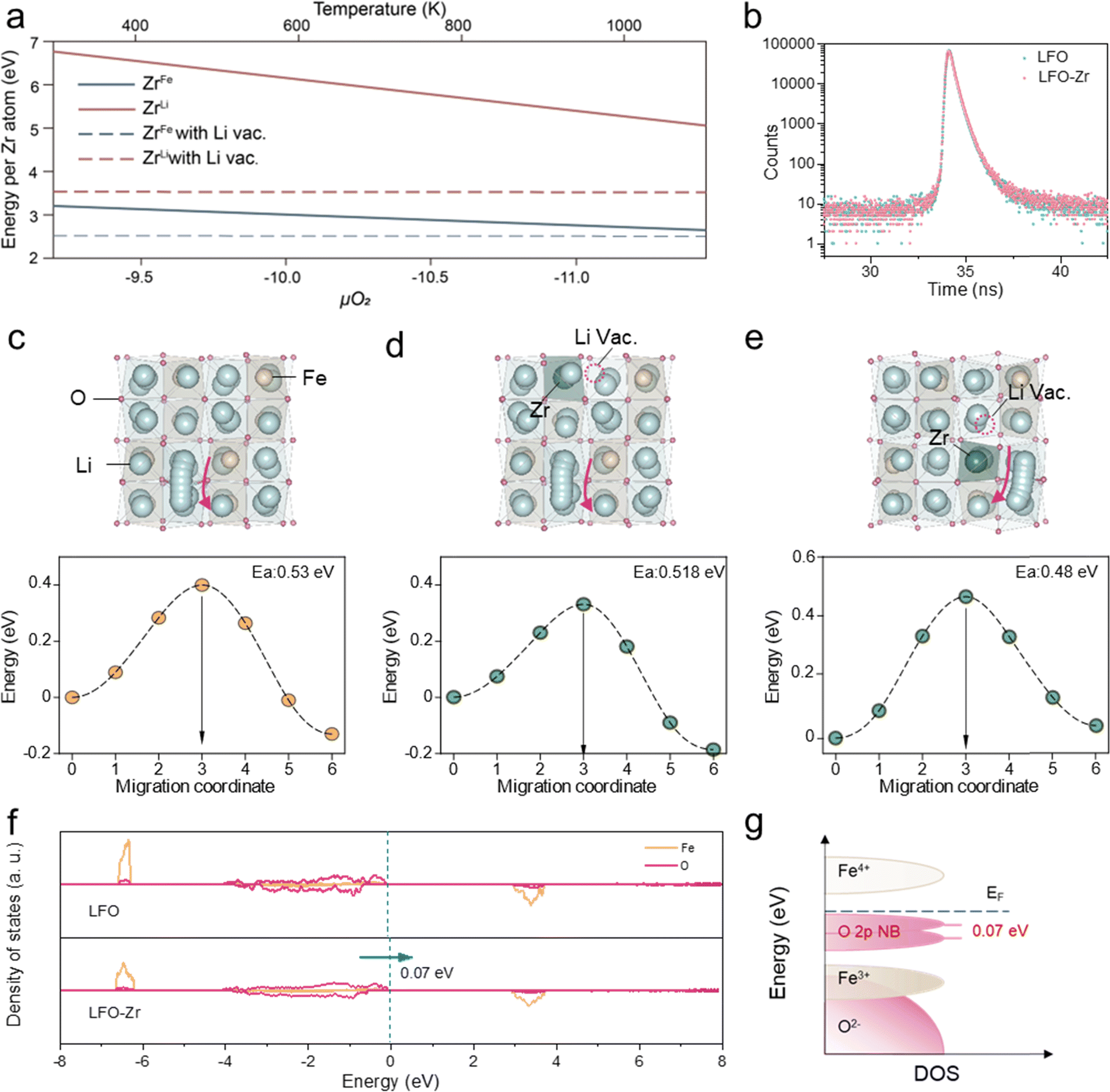 | ||
| Fig. 1 DFT calculations. (a) Defect formation energy as a function of oxygen chemical potential (temperature) for various types of defects introduced by Zr doping. Zr4+ may substitute either Fe3+ or Li+, and the charge can be compensated by the reduction of Fe3+ to Fe2+ (black and red lines), or the creation of Li vacancies (black and pink dashed lines). (b) Positron annihilation spectra. Migration pathway and corresponding energy barrier obtained from ci-NEB for pristine Li5FeO4 (c), and Zr-doped Li5FeO4 (d and e). Li, Fe, Zr and O ions are denoted by blue, yellow, green and pink spheres. Li vacancy is denoted by pink dashed circles. The red arrows demonstrate the migration path for Li ions. (f) The density of states of O 2p states and Fe 2p states in LFO and LFO-Zr. (g) Schematic diagrams of the electronic structure in LFO and LFO-Zr samples according to the above density of states. | ||
To gain insight into the effect of Zr-doping on the de-lithiation performance of LFO-Zr at high rates, the lithium migration pathway was compared for the pristine LFO (Fig. 1c) and LFO-Zr (Fig. 1d and e). As shown in Fig. 1c, for the pristine LFO, the migration of Li ions from the initial tetrahedral to the neighboring tetrahedral site going through a distorted tetrahedral transition state exhibits a migration barrier of 0.53 eV. For the LFO-Zr, two distinct tetrahedra-to-tetrahedra diffusion paths were considered, i.e., one with the initial Li site edge-sharing with FeO4 tetrahedra (Fig. 1d), and the other edge-sharing with ZrO4 (Fig. 1e), featuring low migration barriers of 0.518 and 0.48 eV, respectively. In addition, the percolation of Li+ depends heavily on the lithium-ion concentration27 and the formation of lithium vacancies upon Zr doping may further facilitate lithium-ion diffusion. The redox behaviors were also investigated via the density of states calculation. As shown in Fig. 1f and g, the O 2p state of LFO-Zr can be shifted to higher energy by 0.07 eV compared with that of LFO, indicating that the introduction of zirconium ions is beneficial to reduce the redox potential of the oxygen anion. This is because the redox voltage depends on the state energy of the O 2p electrons involved in the redox reaction, and the higher the state energy, the lower the redox potential. These results together show that defect engineering can indeed enhance the de-lithiation kinetics of LFO-Zr.
2.2 Material characterization
LFO-Zr samples were prepared through a feasible solid-state reaction with different Zr contents. As shown in Fig. 2a, for all samples there is no impurity detected, even with the addition of Zr content by 5%, indicating pure LFO-Zr phases were synthesized. As shown in Fig. S1,† the sample with 3% Zr shows the most excellent de-lithiation performance, and it is denoted as LFO-Zr0.03. The XRD peaks located at 20.5°–22.5° and 23.0°–24.0° shift to the left (Fig. 2b and c) with the a and c lattice parameters exhibiting evident expansion (Table S3†), demonstrating that Zr is successfully doped into the phase, which is consistent with the calculated results in Table S2.† The Rietveld XRD pattern of LFO-Zr0.03 is shown in Fig. 2d. The Rwp of LFO-Zr0.03 is 1.38% indicating that the material has good crystallinity and credibility. The intensity of LFO-Zr0.03 weakens compared to LFO as shown in Fig. S2a.† SEM and TEM were applied to characterize the morphology change of surface LFO-Zr0.03 and the pristine material. In sharp contrast to the smooth surface of LFO (Fig. 2e–h), there is an uneven coating layer on the surface of LFO-Zr0.03. | ||
| Fig. 2 Characterization of the as-prepared LFO-Zr material. (a) XRD patterns of LFO and the modified sample. (b and c) Detailed XRD patterns of 20.5°–22.5° and 23°–24°, showing the peak shift. (d) The Rietveld XRD pattern of LFO-Zr0.03. SEM images of LFO (e and f) and LFO-Zr0.03 (g and h). TEM image (i), HRTEM image (j), HADDF and EDS mapping (k) of LFO-Zr0.03. (l) FIB-SEM and Zr, Fe content as a function of the radial distance of LFO-Zr0.03. Zr 3d (m) of etched LFO-Zr0.03 at 0, 40, and 80 s. (n) The Raman spectra of LFO, C@LFO and LFO-Zr0.03. | ||
According to HRTEM analysis and the energy dispersive X-ray (EDS) mapping (Fig. 2i–k and S3†), the carbon layer is uniformly coated on the surface of LFO-Zr0.03 with a thickness of 6 nm and the even-distribution of Fe, C, O and Zr elements can be observed. Fig. S4† presents the HRTEM and the homogeneous distribution of each element of the pristine LFO. Moreover, the sulfur carbon analysis test showed that the modified sample contained 0.99% carbon content, as shown in Table S4.† Fig. S5† shows the line sweep along the direction of the arrow indicating that a large amount of Zr is present in the surface phase of the material. To further confirm whether zirconium ions enter the bulk phase of the material, a Focused Ion Beam (FIB) coupled with a scanning electron microscope (SEM) was used to observe the elemental distribution within the material. As displayed in Fig. 2l and S6,† the signals of Zr elements are still present within the bulk phase of the material indicating that Zr is successfully doped into LFO. In addition, X-ray photoelectron spectroscopy (XPS) was applied to analyze the surface composition information. The high spectra XPS (Fig. S7†) display that the C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O are located at 286.0 eV and 288.5 eV respectively, suggesting that the carbon layer exists on the surface of the sample. In order to prove that Zr doped into LFO, the material was etched at 0, 40, and 80 s. As shown in (Fig. 2m), the content of Zr still exists in the sample and the content of Zr gradually decreases in Fig. S8,† indicating that Zr is doped in the phase and decreases with depth. Combined with the conclusion from XRD, FIB-SEM and XPS analysis in Fig. 2, the Zr element was successfully incorporated into the bulk phase of the material. As shown in Fig. 2n, the Raman spectra of C@LFO and LFO-Zr0.03 show the D and G bands are located at 1360 cm−1 and 1585 cm−1, respectively.28 The ID/IG values of C@LFO and LFO-Zr0.03 are 0.96, which confirms that the graphitization of the C@LFO and LFO-Zr0.03 carbon cladding layers is consistent.
O are located at 286.0 eV and 288.5 eV respectively, suggesting that the carbon layer exists on the surface of the sample. In order to prove that Zr doped into LFO, the material was etched at 0, 40, and 80 s. As shown in (Fig. 2m), the content of Zr still exists in the sample and the content of Zr gradually decreases in Fig. S8,† indicating that Zr is doped in the phase and decreases with depth. Combined with the conclusion from XRD, FIB-SEM and XPS analysis in Fig. 2, the Zr element was successfully incorporated into the bulk phase of the material. As shown in Fig. 2n, the Raman spectra of C@LFO and LFO-Zr0.03 show the D and G bands are located at 1360 cm−1 and 1585 cm−1, respectively.28 The ID/IG values of C@LFO and LFO-Zr0.03 are 0.96, which confirms that the graphitization of the C@LFO and LFO-Zr0.03 carbon cladding layers is consistent.
To gain insight into the effect of Zr doping on the electrochemical performance, LFO and LFO-Zr0.03 electrodes were fabricated and measured by assembling coin cells with lithium metal foils. The initial charge capacities of LFO and LFO-Zr0.03 electrodes were characterized at different rates from 0.05C to 1C (1C = 867 mA h g−1). In Fig. 3a, there are two de-lithiation plateaus of LFO at 0.05C, which are consistent with the two-phase reaction mechanism, i.e., Li5FeO4 → Li3FeO3.5 and Li3FeO3.5 → LiFeO2. With increased current rate, both plateaus become less obvious indicating increased cell polarization at high rate, which can be attributed to the poor electronic conductivity of the material. Meanwhile, the capacity of LFO rapidly decays from 736.7 mA h g−1 at 0.05C to 362.5 mA h g−1 at 1C. Fig. S9† shows a lower de-lithiation platform and the charge capacity of C@LFO reaches 746.8 mA h g−1 at 0.1C, and it is still 528.1 mA h g−1 when the charge rate is up to 1C. However, compared with the electrochemical performance of LFO and C@LFO, in Fig. 3b, LFO-Zr0.03 exhibits excellent rate performance with the charge capacity reaching 805.7 mA h g−1, 790 mA h g−1, 739.4 mA h g−1, 690.9 mA h g−1 and 624.5 mA h g−1 at 0.05C, 0.1C, 0.2C, 0.5C and 1C, respectively. The de-lithiation plateaus of LFO-Zr0.03 can be distinguished at high current rates, providing more irreversible Li ions at 0.05 and 1C rates as compared to C@LFO. In Fig. 3c, LFO-Zr0.03 displays a lower attenuation rate than LFO along with the increase of current rates from 0.05 to 1C. The charge and discharge curves and cycle performance of LFO and LFO-Zr0.03 at 0.1C are illustrated in Fig. 3d and e. It is noteworthy that LFO-Zr0.03 exhibits lower charge voltage and higher discharge voltage, indicating less polarization. The initial charge and coulombic efficiency of LFO-Zr0.03 are 790 mA h g−1 and 8.23% at 0.1C, which are consistent with the characteristics of pre-lithiation additives. Fig. 3f shows cyclic voltammetry curves at different current densities. The polarization voltage of LFO increases with the increase of current density. When the sweep rate increases by 0.5 mV s−1, the second oxidation peak disappears and the first oxidation peak shifts to the right. The polarization voltage of the modified sample is less affected by the sweep rate, and the position of the oxidation peak does not change significantly under the condition of 0.5 mV s−1. Furthermore, the modified sample exhibits a smaller polarization voltage. It proves that the Zr doped in LFO reduces the redox voltage and promotes LFO further delithiation, which is consistent with the calculations in Fig. 1. In addition, LFO and LFO-Zr0.03 were exposed in 20% humidity air to explore the effects of air on materials. As displayed in Fig. 3g, a small amount of lithium carbonate heterogeneous phase has formed on LFO which is hardly detected on LFO-Zr0.03. SEM was applied to observe the morphological changes of LFO and LFO-Zr0.03 after exposure to air, as shown in Fig. S10.† The surface of LFO becomes very rough, while the surface of LFO-Zr0.03 hardly changes. Fig. S11.† shows the FT-IR spectra of LFO and LFO-Zr0.03 and their post-exposure products. After 8 h exposure in 20% humidity air, a peak in the range of 800–900 cm−1 for Li2CO3 and a peak in the range of 3600–3700 cm−1 for LiOH are observed on the surface of LFO, indicating that some Li2CO3 and LiOH form on the surface of LFO, while there are little Li2CO3 and LiOH on the surface of LFO-Zr0.03. Combined with the conclusion from XRD, SEM and FT-IR, LFO-Zr0.03 exhibits good air stability. Fig. 3h displays the initial charge–discharge curves of LFO and LFO-Zr0.03 after being exposed in air. The initial capacity of LFO-Zr0.03 still reaches 577.6 mA h g−1 higher than 244.2 mA h g−1 of LFO and Fig. S12† exhibits low initial coulombic efficiencies of LFO and LFO-Zr0.03. The initial CV plot of LFO-Zr0.03 (Fig. S13†) demonstrates that the first de-lithiation platform remains. The results of the above analyses show that LFO-Zr0.03 exhibits excellent de-lithiation properties and good air stability. We also compared LFO-Zr0.03 with other cathode prelithiation materials in terms of delithiation capacity, raw material price, air stability, and multiplicity performance.10,29–31 The radargrams of LFO-Zr0.03 (Fig. 3i) show its excellent multiplicity performance, and the price advantage is far beyond that of other cathode prelithiation materials.
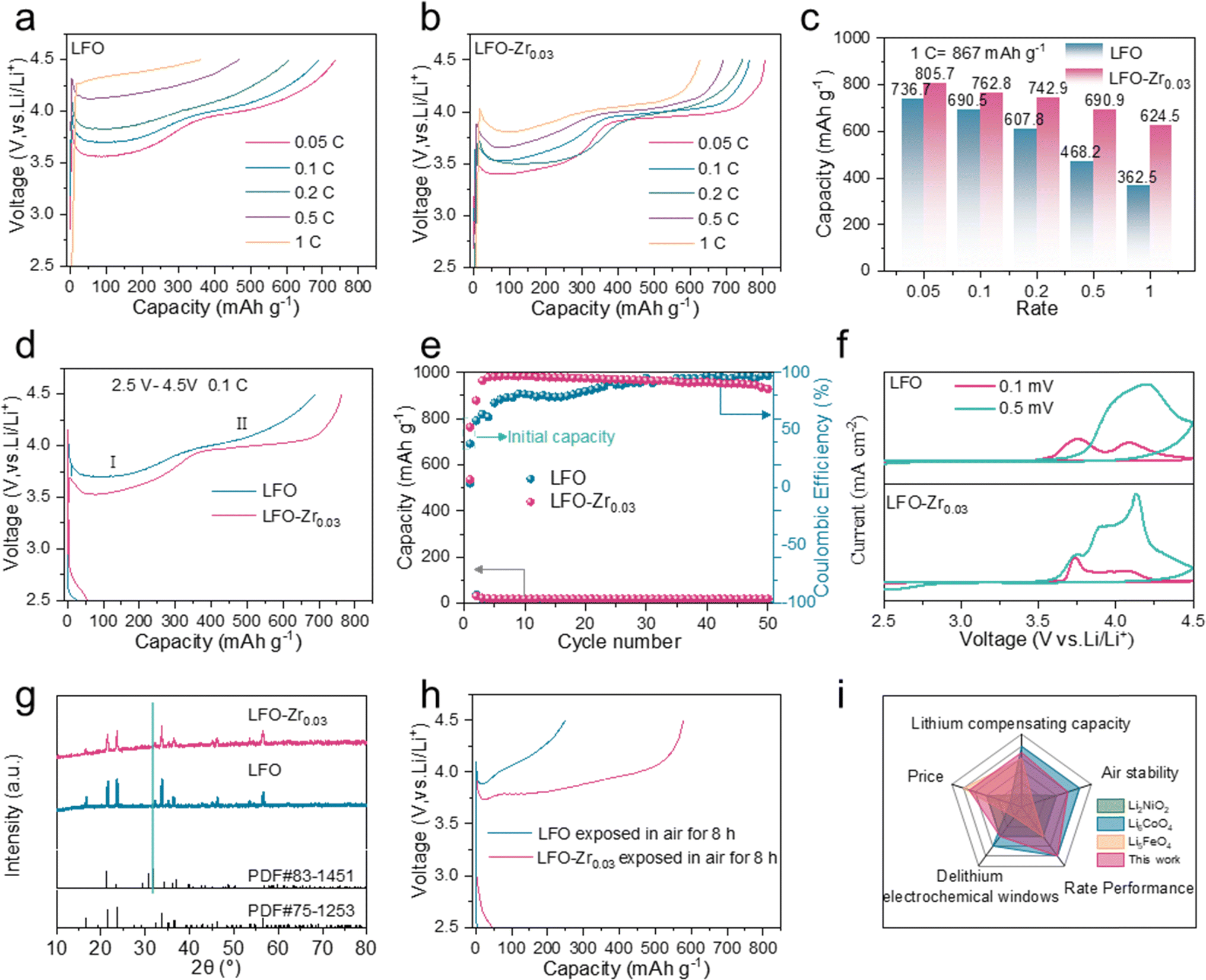 | ||
| Fig. 3 Electrochemical performance of LFO and LFO-Zr0.03 electrodes in the voltage region of 2.5–4.5 V at 25 °C. (a and b) Initial charge curves and (c) initial charge capacity at different rates of LFO and LFO-Zr0.03. (d) Initial charge/discharge curves and (e) cycle performance at 0.1C of LFO and LFO-Zr0.03. (f) CVs of LFO and LFO-Zr0.03 electrodes at various scan rates. The XRD patterns (g) and initial charge–discharge curves (h) of LFO and LFO-Zr0.03 after being exposed in 20% humidity air. (i) Radargrams. | ||
2.3 Electrochemical performance
To determine the origin of the excellent rate performance, the reaction kinetics of LFO-Zr0.03 was further studied. GITT was applied to measure the Li+ ion diffusion coefficients (DLi+) during the first cycle. As shown in Fig. 4a, the second de-lithiation platform of LFO-Zr0.03 is significantly longer than that of LFO. The DLi+ of LFO-Zr0.03 ranges from 10−12.5 to 10−9.5 cm2 s−1 in Fig. 4b, which is much higher than that of LFO. Such fast Li+ ion kinetics is attributed to the fact that the introduction of Zr ions not only creates a large number of lithium vacancies but also reduces the lithium-ion migration energy barrier. Moreover, lithium diffusion velocity at 3.5 V to 3.9 V is significantly higher than 3.9 V to 4.5 V, indicating that the kinetics of lithium diffusion velocity at 3.9 V to 4.5 V is relatively slow, which is consistent with the conclusion obtained in Fig. 3. What is more, the electrochemical impedance test in Fig. S14a† shows that the initial interface impedance of the modified sample is smaller than that of the original sample. After cycling, the impedance of the LFO-Zr0.03 electrode is still smaller than that of LFO. In addition, the diffusion coefficient of DLi+ can also be calculated using the following two equations:32| D = R2T2/2A2F4C2σw2 |
| Z′ = Re + Rct + σwω−1/2 |
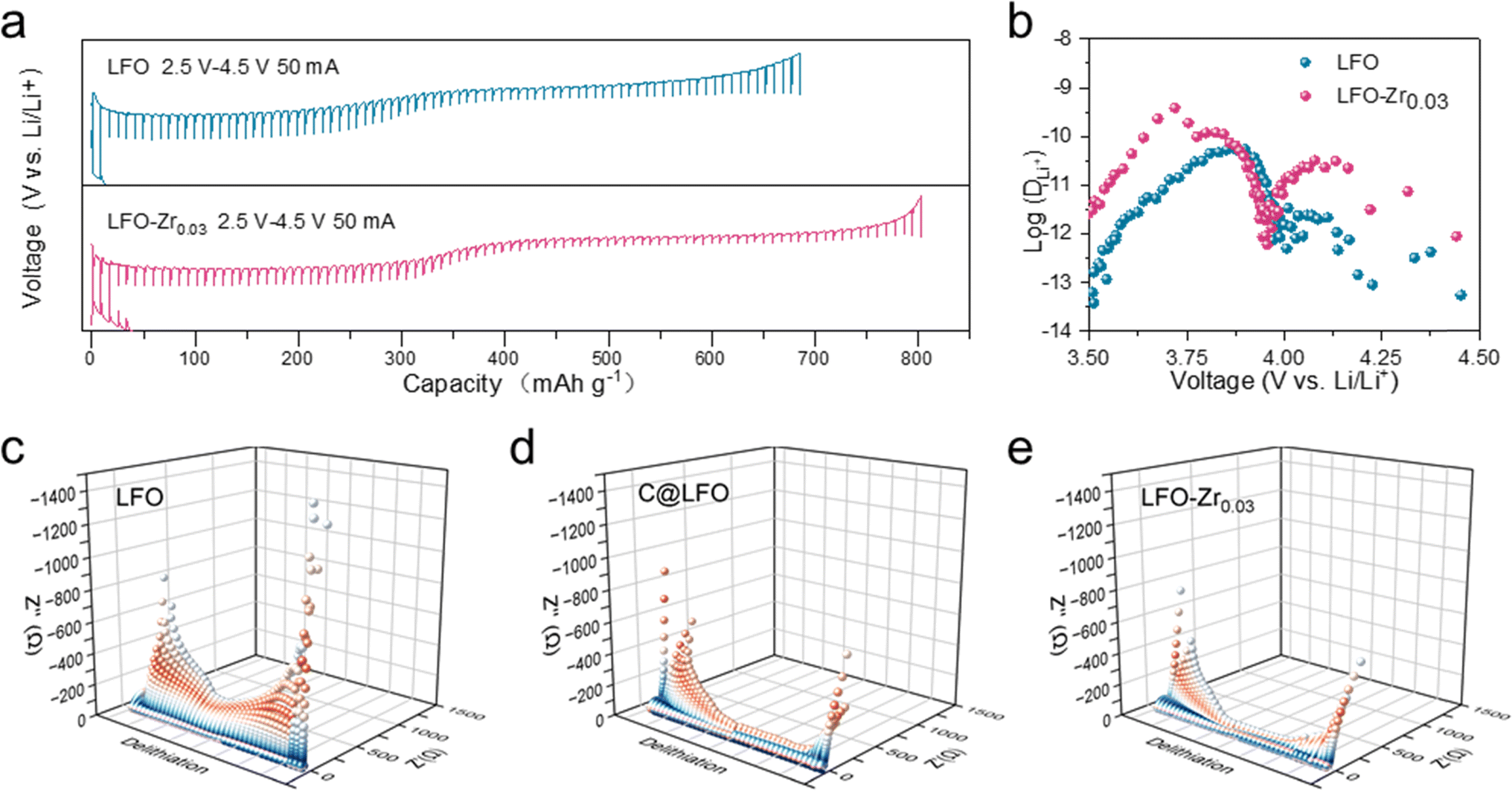 | ||
| Fig. 4 Kinetic properties of LFO and LFO-Zr0.03 electrodes. (a) Galvanostatic intermittent titration technique (GITT) profiles and (b) the log(DLi+)-potential plots during charge processes. The in situ EIS of LFO (c), C@LFO (d) and LFO-Zr0.03 (e) during the initial de-lithiation process. | ||
To further test the pre-lithiation effect of LFO and LFO-Zr0.03, NCM811, pre-lithiation reagents and the Li metal anode were assembled into 2025-coin type half cells for testing between 2.5 V and 4.5 V. As shown in Fig. 5a, the initial charge capacity and discharge capacity of NCM are 258.7 mA h g−1 and 231.7 mA h g−1 at 0.1C. When LFO and LFO-Zr0.03 pre-lithiation additives are added to the cathode material, the initial discharge capacity of the material becomes 223.9 mA h g−1 and 227.6 mA h g−1. After 100 cycles at 0.5C in Fig. 5b, the discharge capacity retention of the bare NCM811 was only 185.2 mA h g−1 and the discharge capacity of NCM with the LFO-Zr0.03 electrode reached 182.3 mA h g−1, with the capacity retention rate up to 86.0%. In addition, the NCM with the LFO-Zr0.03 electrode exhibits similar rate performance to the NCM with LFO in Fig. 5c, with reversible discharge capacities of 229.0, 212.8, 198.0, 184.7, 170.3 and 140.7 mA h g−1 at various rates of 0.1C, 0.2C, 0.5C, 1C, 2C and 5C, respectively. When back to 0.1C, a capacity of 216.4 mA h g−1 was restored indicating good reversibility. EIS was employed to understand the effects of LFO-Zr0.03 on the Li-ion kinetics properties of NCM811. The Nyquist plots of NCM811 with LFO and the NCM811 with the modified materials show a slightly larger resistance before and after cycling at 0.5C for 100 cycles. This observation can be ascribed to the carbon coating layer of LFO-Zr0.03 reducing the barrier of lithium-ion transfer at the cathode–electrolyte interface. In addition, both NCM811 with LFO and the NCM811 with LFO-Zr0.03 exhibit similar apparent diffusion coefficients in half cells in Fig. S15.† This indicates that a small amount of pre-lithiation additive in the half cell has little effect on the impedance and lithium-ion diffusion of NCM. Cyclic voltammetry (CV) was employed to analyze electrochemical reaction kinetics at a scan rate of 0.1 mV s−1 in the voltage range of 2.5–4.5 V in Fig. 5d. Compared to pure NCM, there is a small oxidation peak at 3.7 V, corresponding to the de-lithiation of Li5FeO4 into Li3FeO3.5. The other oxidation peaks observed in the CV plot are corresponding redox processes of Ni2+/Ni3+/Ni4+ and Co3+/Co4+ respectively. The peak strength of CV is significantly stronger than that of the pure phase, which is attributed to the lithium ions provided by the pre-lithiation material. The results suggest that LFO-Zr0.03 almost has no adverse effects on NCM811.
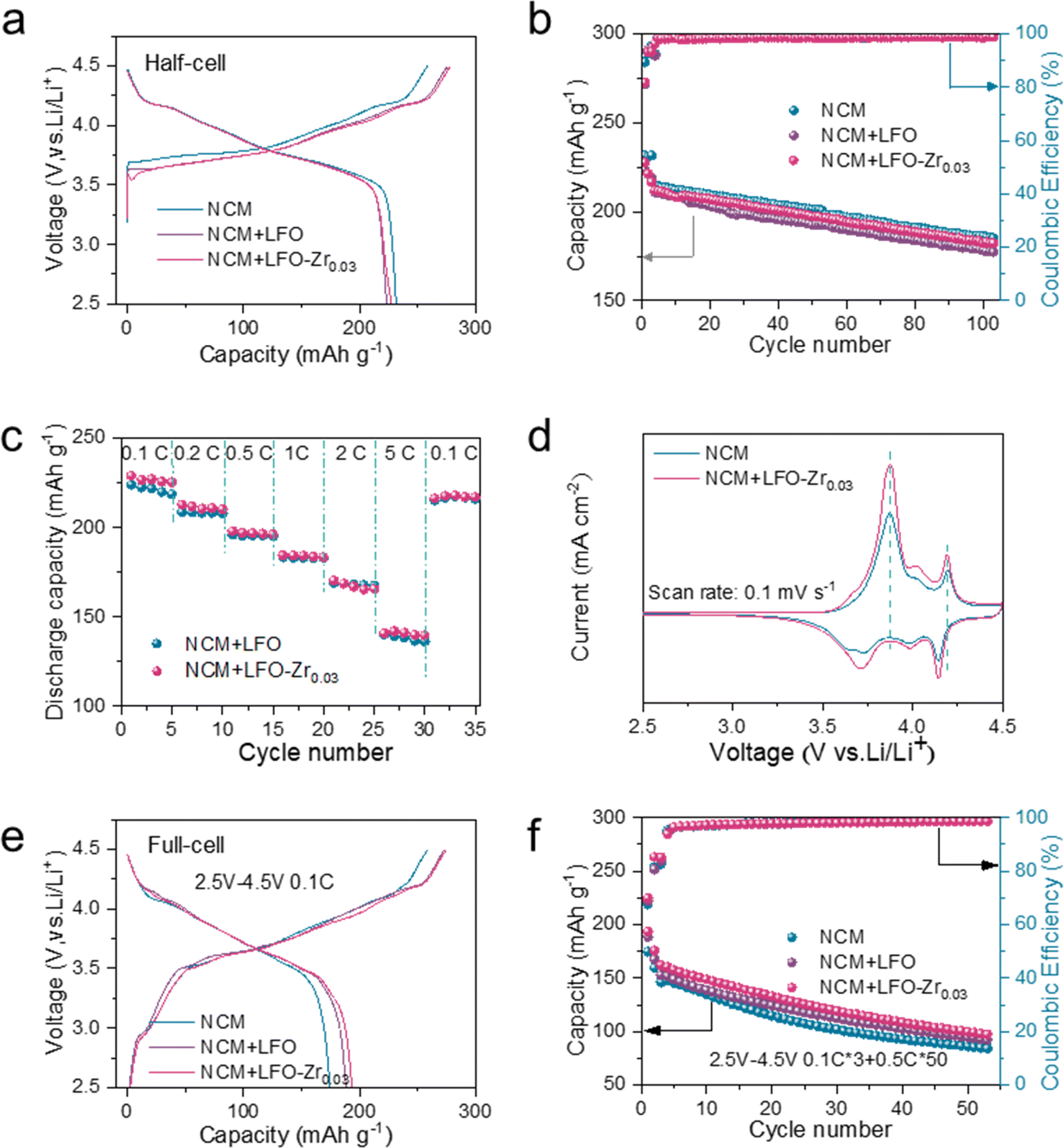 | ||
| Fig. 5 Electrochemical performances of half-cell and full-cell electrodes of NCM, NCM with LFO and LFO-Zr0.03. (a) The initial charge/discharge curves. (b) Cycle performance. (c) Rate performances. (d) CV curves at 0.1 mV s−1 scan rates. (e) The initial charge/discharge curve and (f) cycle performance of the full-cell. | ||
With more practical concerns, commercial NCM811, pre-lithiation reagent and hard carbon are assembled into full cells (graphite‖NCM811), as shown in Fig. 5e–f. The graphite‖NCM811 and graphite‖NCM811/LFO-Zr0.03 full cells were subjected to 0.5C to test the electrochemical performance of pre-lithiation effect within 2.5 V to 4.5 V. The pre-lithiation additive provides sufficient lithium ions to compensate for the lithium ions consumed by the SEI on the anode side. With the LFO-Zr0.03 additive, the full cell delivered 193.6 mA h g−1 for the initial cycle at 0.1C, while the pure graphite‖NCM811 full cell only delivered 174.8 mA h g−1. The capacity of NCM811 with LFO-Zr0.03 in a full cell maintains 97.3 mA h g−1 after 50 cycles, which is also higher than that of the graphite‖NCM811 full cell.
2.4 Analysis of the mechanism of delithiation
To investigate the detailed structural evolution of LFO and LFO-Zr0.03 during electrochemical de-lithiation, in situ XRD was carried out during the first charge/discharge cycle (Fig. 6). For LFO and LFO-Zr samples in Fig. 6a and b, upon charging, the (102), (112) and (222) peaks corresponding to the orthorhombic phase gradually attenuate with continuous extraction of Li-ions from LFO. During subsequent Li+ extraction, the orthorhombic phase gradually disappears, leaving only the characteristic signals of the disordered rock-salt phase (α-LiFeO2). In addition, in situ quantitative DEMS was applied to measure the gas production of LFO/Li and LFO-Zr/Li batteries during charging to 4.5 V on the first charge. As displayed in Fig. 6c and d, in LFO-Zr/Li batteries, a small amount of gas release is observed during the first charging plateau, followed by a large amount of gas release during the second plateau which is much more significant than that of the LFO/Li batteries, indicating a higher capacity with promoted reaction kinetics and oxygen redox. Furthermore, as more oxygen is generated in LFO-Zr/Li, the content of CO2 also increases, which can be attributed to more electrolyte decomposition with excess oxygen release.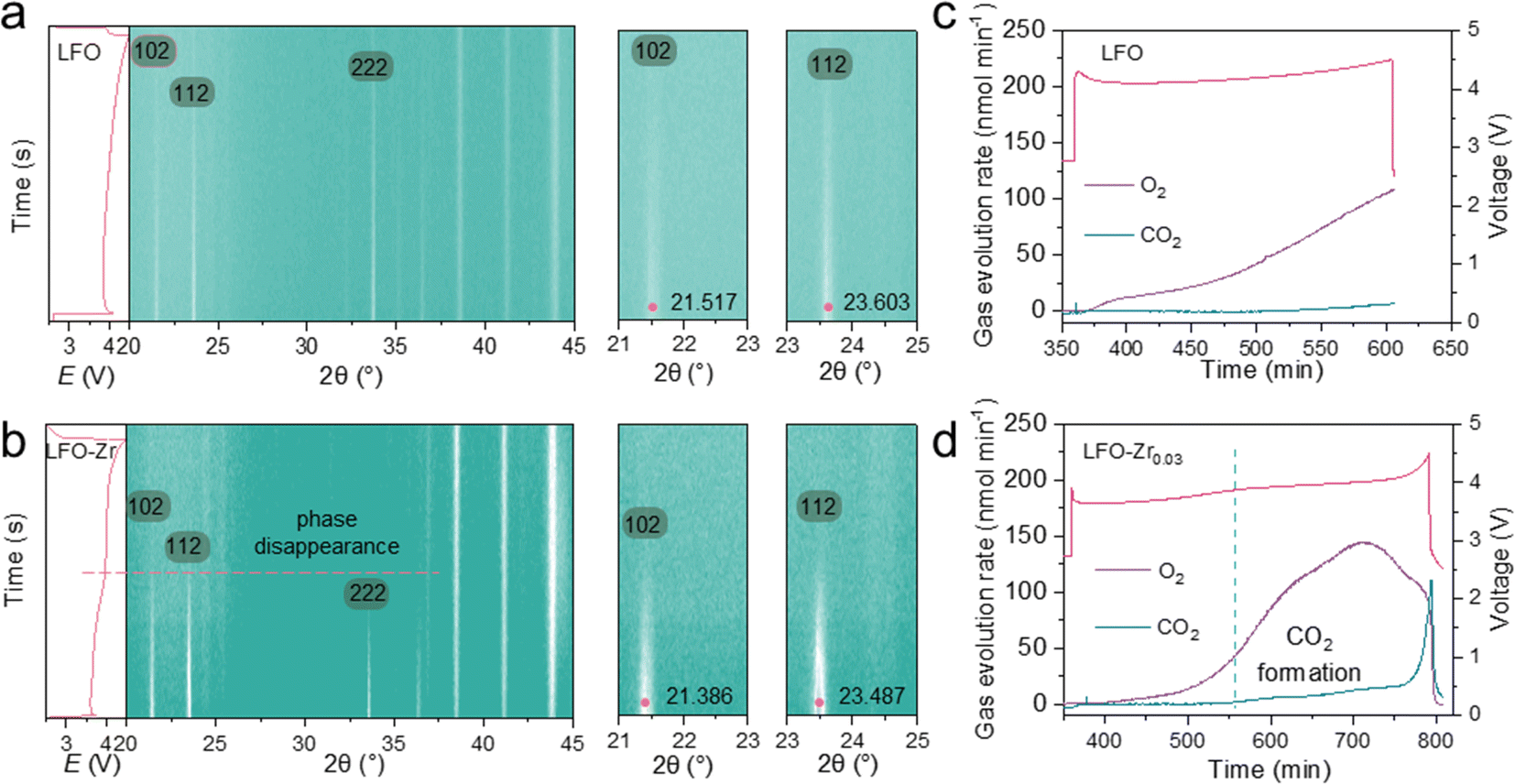 | ||
| Fig. 6 Phase conversion and evolution of oxygen in the initial charge of LFO and LFO-Zr0.03. The corresponding 2D contour plot and magnified details of the (102) and (112) peaks of LFO (a) and LFO-Zr0.03 (b), and quantitative DEMS data for O2 and CO2 released in the LFO (c) and LFO-Zr0.03 (d) during the first charge to 4.5 V. | ||
3. Conclusion
In summary, we theoretically investigated and experimentally achieved the production of lithium vacancies in the LFO structure by introducing Zr ions into the Fe site by defect engineering, which effectively reduces the migration energy barrier of lithium ions and facilitates the redox of oxygen anions, leading to lower interfacial impedance, lower redox potentials and higher lithium compensation capacity. It was proved that the delithiation capacity of LFO-Zr is up to 805.7 and 624.5 mA h g−1 under 0.05C and 1C rate, which are higher than those of LFO, respectively. In addition, the air stability of LFO-Zr is also well improved by a facile coating strategy. It turns out that after exposure to air for 8 h, the initial capacity of LFO-Zr remains at 577.6 mA h g−1, and is 244.2 mA h g−1 higher than that of the pristine LFO. Our work provides insights to promote the commercial use of the Li5FeO4 material as a sacrificial cathode prelithiation additive for high-energy Li-ion batteries.Data availability
The data that support these findings of this article are available on request from the corresponding authors.Author contributions
Bin Zhu: conceptualization; data curation; formal analysis; investigation; methodology; validation; writing – original draft; writing – review & editing, Naifeng Wen: formal analysis; investigation; software; validation; visualization; writing – original draft; writing – review & editing, Jingyang Wang: conceptualization; data curation; formal analysis; investigation; methodology; project administration; resources; supervision; validation; visualization; writing – original draft; writing – review & editing, Qiyu Wang: data curation; formal analysis; methodology; validation; writing – original draft; writing – review & editing, Jingqiang Zheng: investigation; methodology; software; visualization; writing – review & editing, Zhian Zhang: conceptualization; data curation; formal analysis; investigation; methodology; project administration; resources; supervision; visualization; writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the financial support from the National Natural Science Foundation of China (52304339).References
- G. Liang, Z. Wu, C. Didier, W. Zhang, J. Cuan, B. Li, K. Y. Ko, P. Y. Hung, C. Z. Lu, Y. Chen, G. Leniec, S. M. Kaczmarek, B. Johannessen, L. Thomsen, V. K. Peterson, W. K. Pang and Z. Guo, Angew. Chem., Int. Ed., 2020, 59, 10594–10602 CrossRef CAS.
- Z. Feng, R. Rajagopalan, S. Zhang, D. Sun, Y. Tang, Y. Ren and H. Wang, Advanced Science, 2020, 8, 2001809 CrossRef.
- B. Liu, T. Lv, A. Zhou, X. Zhu, Z. Lin, T. Lin and L. Suo, Nat. Commun., 2024, 15, 2922 CrossRef CAS.
- Q. Meng, M. Fan, X. Chang, H. Li, W. P. Wang, Y. H. Zhu, J. Wan, Y. Zhao, F. Wang, R. Wen, S. Xin and Y. G. Guo, Adv. Energy Mater., 2023, 13, 2300507 CrossRef CAS.
- H. Kobayashi, Y. Nakamura, M. Nakayama, S. Kodaki, R. Matsuo and I. Honma, Adv. Energy Mater., 2023, 13, 2203441 CrossRef CAS.
- S. Alvin, H. S. Cahyadi, J. Hwang, W. Chang, S. K. Kwak and J. Kim, Adv. Energy Mater., 2020, 10, 2000283 CrossRef CAS.
- C. Sun, X. Zhang, C. Li, K. Wang, X. Sun and Y. Ma, Energy Storage Mater., 2020, 32, 497–516 CrossRef.
- K. Zou, W. Deng, P. Cai, X. Deng, B. Wang, C. Liu, J. Li, H. Hou, G. Zou and X. Ji, Adv. Funct. Mater., 2020, 32, 2005581 Search PubMed.
- J. Jang, I. Kang, J. Choi, H. Jeong, K. W. Yi, J. Hong and M. Lee, Angew. Chem., Int. Ed., 2020, 14473–14480 CrossRef CAS.
- W. Lee, H. Lee, Y. Byeon, J. H. Kim, W. Choi, M. Choi, M. S. Park and W. S. Yoon, Adv. Energy Mater., 2023, 13, 2302316 CrossRef CAS.
- Y. Wang, J. Wang, X. Zhao, W. Qiu, E. Song, W. Zhang, X. Liu and J. Liu, Energy Environ. Sci., 2020, 13, 2540–2548 RSC.
- Z. Rao, J. Wu, B. He, W. Chen, H. Wang, Q. Fu and Y. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 38194–38201 CrossRef CAS PubMed.
- S.-Y. Yang, X.-Y. Yue, H.-Y. Xia, X.-L. Li, T. Wang, H. Li and Z.-W. Fu, J. Power Sources, 2020, 480, 229109 CrossRef CAS.
- Z. Chen, Z. Zhang and J. Li, Phys. Chem. Chem. Phys., 2018, 20, 20363–20370 RSC.
- Z. Hui, X. Fengxia, P. Ben and M. S. Whittingham, ACS Energy Lett., 2019, 1902–1906 Search PubMed.
- G. Vitins, E. A. Raekelboom, M. T. Weller and J. R. Owen, J. Power Sources, 2003, 119–121, 938–942 CrossRef CAS.
- C. S. Johnson, S. H. Kang, J. T. Vaughey, S. V. Pol, M. Balasubramanian and M. M. Thackeray, Chem. Mater., 2010, 22, 1263–1270 CrossRef CAS.
- X. Su, C. Lin, X. Wang, V. A. Maroni, Y. Ren, C. S. Johnson and W. Lu, J. Power Sources, 2016, 324, 150–157 CrossRef CAS.
- B. Zhu, W. Zhang, Q. Wang, Y. Lai, J. Zheng, N. Wen and Z. Zhang, Adv. Funct. Mater., 2024, 2315010 CrossRef CAS.
- M. D. Wesley, V. Cesar, H. Xiaobing, R. D. Alison, P.-M. Maria Jose, A. M. Victor, E. T. Stephen, B. Ira, D. Vinayak and S. J. Christopher, J. Electrochem. Soc., 2020, 167, 160543 CrossRef.
- L. Zhang, W. M. Dose, A. D. Vu, C. S. Johnson and W. Lu, J. Power Sources, 2018, 400, 549–555 CrossRef CAS.
- B. Zhu, W. Zhang, Z. Li, Q. Y. Wang, N. F. Wen and Z. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 45290–45299 CrossRef CAS.
- M. Bianchini, J. Wang, R. J. Clément, B. Ouyang, P. Xiao, D. Kitchaev, T. Shi, Y. Zhang, Y. Wang, H. Kim, M. Zhang, J. Bai, F. Wang, W. Sun and G. Ceder, Nat. Mater., 2020, 19, 1088–1095 CrossRef CAS PubMed.
- H. Li, J. Wang, S. Xu, A. Chen, H. Lu, Y. Jin, S. Guo and J. Zhu, Adv. Mater., 2024, 36, 2403073 CrossRef CAS PubMed.
- J. Sun, H. Xue, N. Guo, T. Song, Y. r. Hao, J. Sun, J. Zhang and Q. Wang, Angew. Chem., Int. Ed., 2021, 60, 19435–19441 CrossRef CAS.
- Z. Li, H. Cheng, Y. Lu, T. Wang, Y. Li, W. Zhang, G. He and Z. Tian, Adv. Energy Mater., 2023, 202203963 Search PubMed.
- F. Schipper, M. Dixit, D. Kovacheva, M. Talianker, O. Haik, J. Grinblat, E. M. Erickson, C. Ghanty, D. T. Major, B. Markovsky and D. Aurbach, J. Mater. Chem. A, 2016, 4, 16073–16084 RSC.
- H. Yang, R. Xu and Y. Yu, Energy Storage Mater., 2019, 22, 105–112 CrossRef.
- Y. Wu, W. Zhang, S. Li, N. Wen, J. Zheng, L. Zhang, Z. Zhang and Y. Lai, ACS Sustain. Chem. Eng., 2023, 11, 1044–1053 CrossRef CAS.
- M. G. Kim and J. Cho, J. Mater. Chem., 2008, 18, 5880–5887 RSC.
- J. Li, B. Zhu, S. Li, D. Wang, W. Zhang, Y. Xie, J. Fang, B. Hong, Y. Lai and Z. Zhang, J. Electrochem. Soc., 2021, 168, 080510 CrossRef CAS.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, e1800561 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03052d |
| This journal is © The Royal Society of Chemistry 2024 |