
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed regiodivergent sulfonylarylation of 1,3-enynes to access allenes and dienes†
Zhuomin
Chi
,
Yongchao
Zhou
,
Bingbing
Liu
,
Xiaojing
Xu
,
Xueyuan
Liu
 and
Yongmin
Liang
*
and
Yongmin
Liang
*
State Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: liangym@lzu.edu.cn
First published on 23rd July 2024
Abstract
The radical-mediated difunctionalization of 1,3-enynes facilitates rapid access to structurally diverse allenes and dienes. Whereas, owing to the existence of multiple active sites in conjugated 1,3-enynes, regulating selectivity in difunctionalized addition via a single transition-metal-catalyzed radical tandem process remains elusive. Herein, we disclose an intriguing protocol of substrate-controlled nickel-catalyzed regiodivergent sulfonylarylation of 1,3-enynes with the assistance of sulfonyl chlorides and arylboronic acids. This valuable synthetic utility respectively delivers a series of highly functionalized and synthetically challenging allenyl sulfones and dienyl sulfones from fine-tuned 1,3-enynes by one step, which provides a facile approach for complex sulfone-containing drug molecules synthesis.
Introduction
Molecular scaffolds containing sulfones are important structures in many areas of chemical research from agrochemicals to pharmaceuticals (Fig. 1a).1 Hence considerable efforts have been devoted to the efficient incorporation of sulfonyl motifs into organic molecules.2 Among the different kinds of sulfonyl sources, commercially available sulfonyl chlorides have emerged as a particularly attractive candidate due to their abundance, low toxicity, stable physiochemical properties and cost efficiency.2e In this context, sulfonyl chloride has continually been employed as a radical precursor in photo-catalytic sulfonylation3 and transition-metal-catalyzed C–H activated sulfonylation4 during the past decades (Fig. 1b, left). In contrast, only limited progress has been made in transition-metal-catalyzed multicomponent radical sulfonation of unsaturated C–C bonds with sulfonyl chlorides. For example, nickel-catalyzed intermolecular sulfonylarylation of alkynes with sulfonyl chlorides and arylboronic acids has been established by the group of Nevado in 2017.5 Recently, Li and co-workers have discovered a sulfonylarylation of alkenes or 1,3-dienes employing sulfonyl chlorides and organozinc pivalates as reaction partners through a cobalt-catalyzed radical relay pathway (Fig. 1b, right).6 To date, reported advances have mainly focused on the sulfonylarylations of conventional alkynes or alkenes frameworks, thus the related multicomponent sulfonylarylation7 for introducing a sulfonyl group into unactivated 1,3-enynes8 with sulfonyl chloride by a single transition-metal-catalyzed radical procedure still deserves further investigation.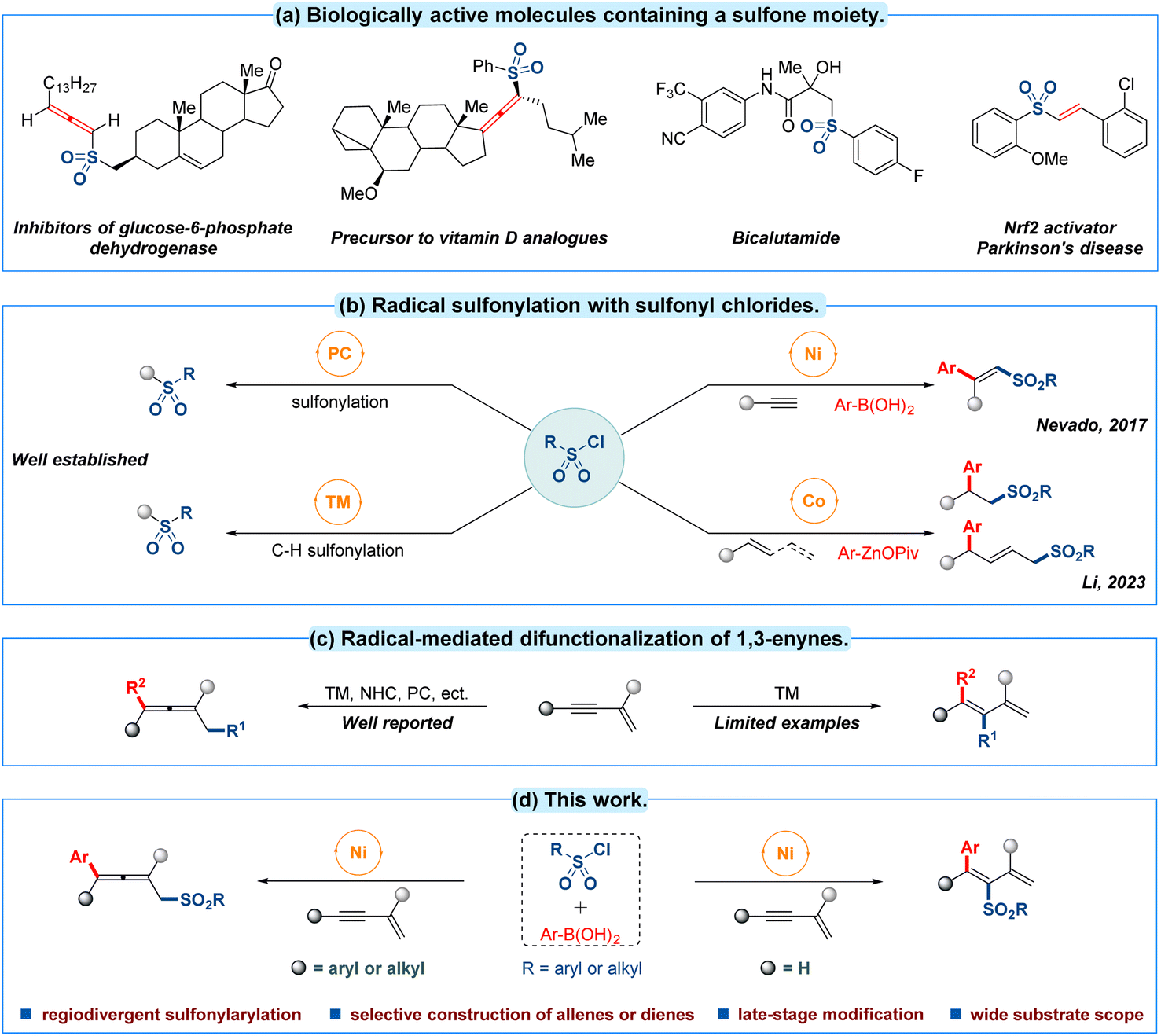 | ||
| Fig. 1 (a) Biologically active molecules containing a sulfone moiety. (b) Radical sulfonylation with sulfonyl chlorides. (c) Radical-mediated difunctionalization of 1,3-enynes. (d) This work: nickel-catalyzed regiodivergent sulfonylarylation of 1,3-enynes with sulfonyl chlorides. | ||
All the time, the radical-mediated 1,4-difunctionalization of 1,3-enynes is recognized as one of most straightforward strategy to introduce two functional groups simultaneously towards the synthesis of polysubstituted allenes.9 Several impressive approaches for the 1,4-difunctionalization of conjugated 1,3-enynes have well been developed through a radical pathway in recent years (Fig. 1c, left).10 Compared with 1,4-difunctionalization, radical 3,4-addition research of 1,3-enynes is still in its infancy, and there are only a handful of significant reports for generating multiple functionalized 1,3-dienes (Fig. 1c, right).11 In 2021, dual photoredox/nickel-catalyzed regiodivergent sulfonylarylation of 1,3-enynes has been reported by the group of Lu.12 Despite such breakthroughs, it is undeniable that the selective construction of allenes or dienes from fine-tuned 1,3-enynes13 through a 1,4-addition or 3,4-addition process remains a formidable challenge under a simpler catalytic system.
Inspired by these pioneering works and our interest in 1,3-enynes radical addition,14 we validated the possibility of regiodivergent difunctionalization of 1,3-enynes by switching the positions of the triple bonds in the substrates to realize the selective preparation of allenyl sulfones and dienyl sulfones using sulfonyl chloride as careful selection of radical precursor with the help of the unique catalytic reactivity of the nickel complex.15 The following crucial challenges need to be overcome: (i) controlling addition site in difunctionalization of such more intricate π-system substrate; (ii) compatibility of radical and reaction partners; (iii) direct competitive sulfonylarylation between sulfonyl source and arlynucleophile. With these clues in mind, we describe a novel strategy to achieve a substrate-controlled regiodivergent radical sulfonylarylation of 1,3-enynes utilizing sulfonyl chlorides as sulfonyl source and arylboronic acids as nucleophile within the same nickel-catalyzed mild conditions, affording the desired sulfonylarylated allenes and dienes respectively (Fig. 1d).
Results and discussion
To commence our investigation, the easily prepared 1,3-enyne 1a was employed as the template substrate, which reacted with p-toluenesulfonyl chloride 2a and phenyl boronic acid 3a in the presence of commercially available Ni-catalyst, ligand and base at oil bath (Table 1). Initially, a series of bipyridine-based ligands were tested in this preliminary catalytic system using the Ni(PPh3)2Cl2 as the catalyst and the K3PO4 as the base in toluene at 80 °C for 24 h (entries 1–5). Of particular note, goal product 4 was obtained with a yield of 25% when bidentate ligand L5 was chosen (entry 3). To further improve the yield, several organic solvents were also tested, and the 1,4-dioxane was proved to be the most suitable one (entries 6–10). Afterwards screening of different nickel catalysts showed that the NiBr2·DME was also indispensable in promoting reaction efficiency, which was superior to other Ni-catalysts examined (entries 12–14). To our delight, a slight increase in reaction outcome was detected respectively when the ratio of 1a/2a/3a was changed to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5, and the levels of Ni-catalyst and bipyridine-based ligand were all added to 12% (entries 11 and 15). Most notably, the yield of 4 was improved to 82% when the solvent concentration was 0.2 mol L−1 (entry 16). Furthermore, it was found that the K3PO4 has always been the best base compared with other bases. Finally, the reaction temperature was also briefly evaluated. Either lower or higher temperature decreased the yields to some extent. Control reactions revealed that the catalyst, ligand and base played a crucial role in this reaction (see ESI† for more detailed conditions exploring).
:2.5:2.5, and the levels of Ni-catalyst and bipyridine-based ligand were all added to 12% (entries 11 and 15). Most notably, the yield of 4 was improved to 82% when the solvent concentration was 0.2 mol L−1 (entry 16). Furthermore, it was found that the K3PO4 has always been the best base compared with other bases. Finally, the reaction temperature was also briefly evaluated. Either lower or higher temperature decreased the yields to some extent. Control reactions revealed that the catalyst, ligand and base played a crucial role in this reaction (see ESI† for more detailed conditions exploring).
|
|
||||
|---|---|---|---|---|
| Entry | Ni-catalyst | Ligand | Solvent | Yield of 4 |
| a Reaction conditions: 1,3-enyne (0.1 mmol), arylboronic acids (0.2 mmol), sulfonyl chlorides (0.2 mmol), Ni-catalyst (10 mol%), ligand (10 mol%), base (0.3 mmol) and solvent (1.0 mL), Ar, 80 °C, 24 h. Yield was determined by 1H NMR with CH2Br2 as internal standard. b Arylboronic acids (0.25 mmol), sulfonyl chlorides (0.25 mmol). c Ni-catalyst (12 mol%), ligand (12 mol%). d Solvent (0.5 mL). e Isolated yield. N.D. = desired product not detected. | ||||
| 1 | Ni(PPh3)2Cl2 | L1 | Toluene | 4% |
| 2 | Ni(PPh3)2Cl2 | L2–4 | Toluene | 4–21% |
| 3 | Ni(PPh3)2Cl2 | L5 | Toluene | 25% |
| 4 | Ni(PPh3)2Cl2 | L6 | Toluene | N.D. |
| 5 | Ni(PPh3)2Cl2 | L7–8 | Toluene | Trace |
| 6 | NiCl2·dppe | L5 | Toluene | 38% |
| 7 | NiCl2·dppe | L5 | Xylene | 35% |
| 8 | NiCl2·dppe | L5 | DCE | 35% |
| 9 | NiCl2·dppe | L5 | THF | N.D. |
| 10 | NiCl2·dppe | L5 | 1,4-Dioxane | 46% |
| 11b | NiCl2·dppe | L5 | 1,4-Dioxane | 52% |
| 12b | Ni(COD)2 | L5 | 1,4-Dioxane | 51% |
| 13b | NiBr2·DME | L5 | 1,4-Dioxane | 62% |
| 14b | NiBr2·diglyme | L5 | 1,4-Dioxane | 45% |
| 15b,c | NiBr2·DME | L5 | 1,4-Dioxane | 69% |
| 16b,c,d | NiBr2·DME | L5 | 1,4-Dioxane | 82%/73%e |

Having determined the optimal reaction conditions, we first explored the substrate scope of different 1,3-enynes for the 1,4-addition research. As shown in Fig. 2, the 1,4-sulfonylarylation occurred smoothly with a variety of aryl-substituted 1,3-enynes under standard conditions. Notably, substrates bearing electron-donating groups (t-Bu, OAc and Oi-Pr) or electron-withdrawing groups (CN, CO2Me and CF3) were compatible with this reaction, affording moderate to good yields of the expected products (5–12). Various halogen atoms (F, Cl and Br) at the ortho, meta or para position of the phenyl ring were all tolerated with this procedure in generally decent yields (7, 10 and 13). Multisubstituted 1,3-enynes featuring different functional groups gave the desired product in 74%, 40% and 53% yields, respectively (14–16). Then, a sequence of heterocyclic substrates, such as thiophene, pyridine and quinoline, were viable with the protocol to produce corresponding products (17–19), and target products were also detected with acceptable yields when R2 was altered to trifluoromethyl or other long-chain alkyl in substrates (20–22). Regretfully, alkyl-substituted 1,3-enynes were subjected for further testing, and the yields all decreased dramatically, which were more residual in system (23 and 24). Of note, about 10% direct sulfonylarylation side product from the competitive reaction could be observed for most of the low yield examples.
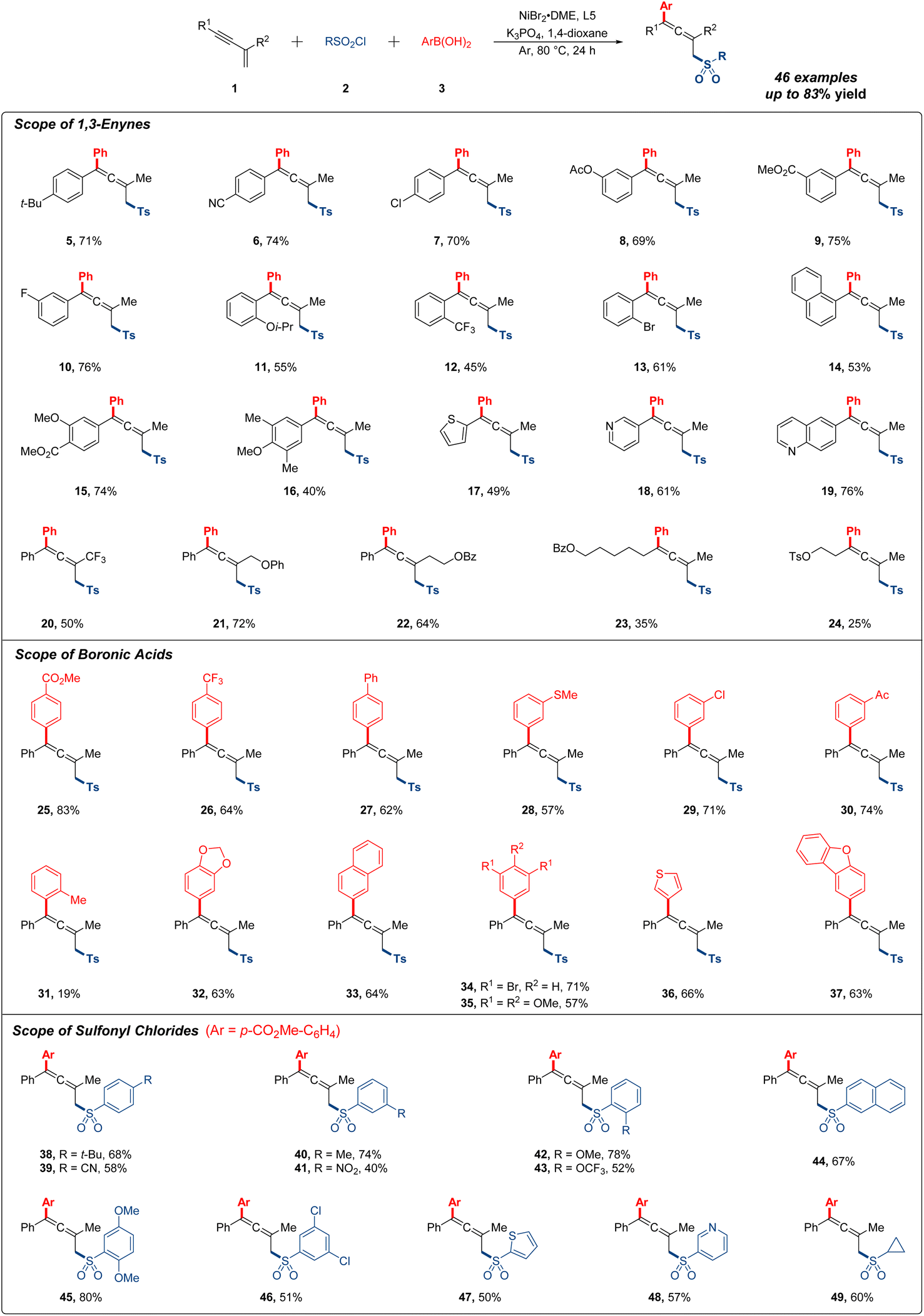 | ||
| Fig. 2 Scope of 1,3-enynes, boronic acids and sulfonyl chlorides. Reaction conditions: 1,3-enynes (0.1 mmol), arylboronic acids (0.25 mmol), sulfonyl chlorides (0.25 mmol), NiBr2·DME (12 mol%), 5,5′-bis(trifluoromethyl)-2,2′-bipyridine (12 mol%), K3PO4 (0.3 mmol) and 1,4-dioxane (0.5 mL), Ar, 80 °C, 24 h, isolated yield. | ||
Next, we turned our attention to the research on generality of organoboronic acids (Fig. 2). It was found that readily accessible arylboronic acids possessing all kinds of useful functional groups (CO2Me, CF3, Ph, SMe, Cl, and Ac) at the meta or para position of the phenyl ring were all applicable in this transformation, providing the target products in excellent yields (25–30). However, when arylboronic acid bearing a methyl at the ortho-position was used, the yield of 1,4-sulfonylarylation was only 19% probably due to the steric hindrance effect (31). In the case of this reaction, the substrate 1a was recovered in 65% yield, and no other side products were monitored. Additionally, multisubstituted arylboronic acids also participated in this system with satisfactory yields (32–35), and the heteroarylboronic acids containing thiophene and dibenzofuran could be well applied in this reaction, giving the allenyl sulfones in 66% and 63% yields, independently (36 and 37).
Subsequently, we moved on to investigate the effect of sulfonyl chlorides (Fig. 2). A range of arylsulfonyl chlorides with different substituent groups (t-Bu, CN, Me, OMe and OCF3) were all accommodated with this procedure delivering moderate to good reaction outcomes (38–43). Generally, electron-rich arylsulfonyl chlorides gave better yields than the electron-deficient ones. It was worth mentioning that utilizing arylsulfonyl chloride bearing a nitryl at the meta-position, expected product was observed in 40% yield (41). The employment of disubstituted arylsulfonyl chlorides and heteroarylsulfonyl chlorides were also compatible with current conditions in generally satisfied yields (44–48). Of particular note, cyclopropane sulfonyl chloride could also be well exploited in this reaction (49).
During our study, we surprisingly observed an unexpected reactivity with commercially available 2-methylbut-1-en-3-yne as the model substrate. Interestingly, this 1,3-enyne substrate unpredictably underwent 3,4-sulfonylarylation process instead of 1,4-sulfonylarylation under the same catalytic system, resulting in the formation of sulfonylarylated 1,3-diene. After screening the several bipyridine-based ligands (see ESI†), we further verified the possibility of 3,4-addition research (Fig. 3). A few terminal 1,3-enynes were briefly examined to generate corresponding products in 39–51% yields (50–52). Unambiguous proof of the structure of 50 was achieved by single-crystal X-ray analysis. Conspicuously, expected product was generated into the E configuration. In addition, a series of arylboronic acids and sulfonyl chlorides were tested under current conditions with generally acceptable yields (53–63). It should be mentioned that 1,4-addition products were not detected for these substrates, and 3,4-sulfonylarylation was less effective than 1,4-sulfonylarylation as a result of the effumability and instability of these terminal 1,3-enynes in system.
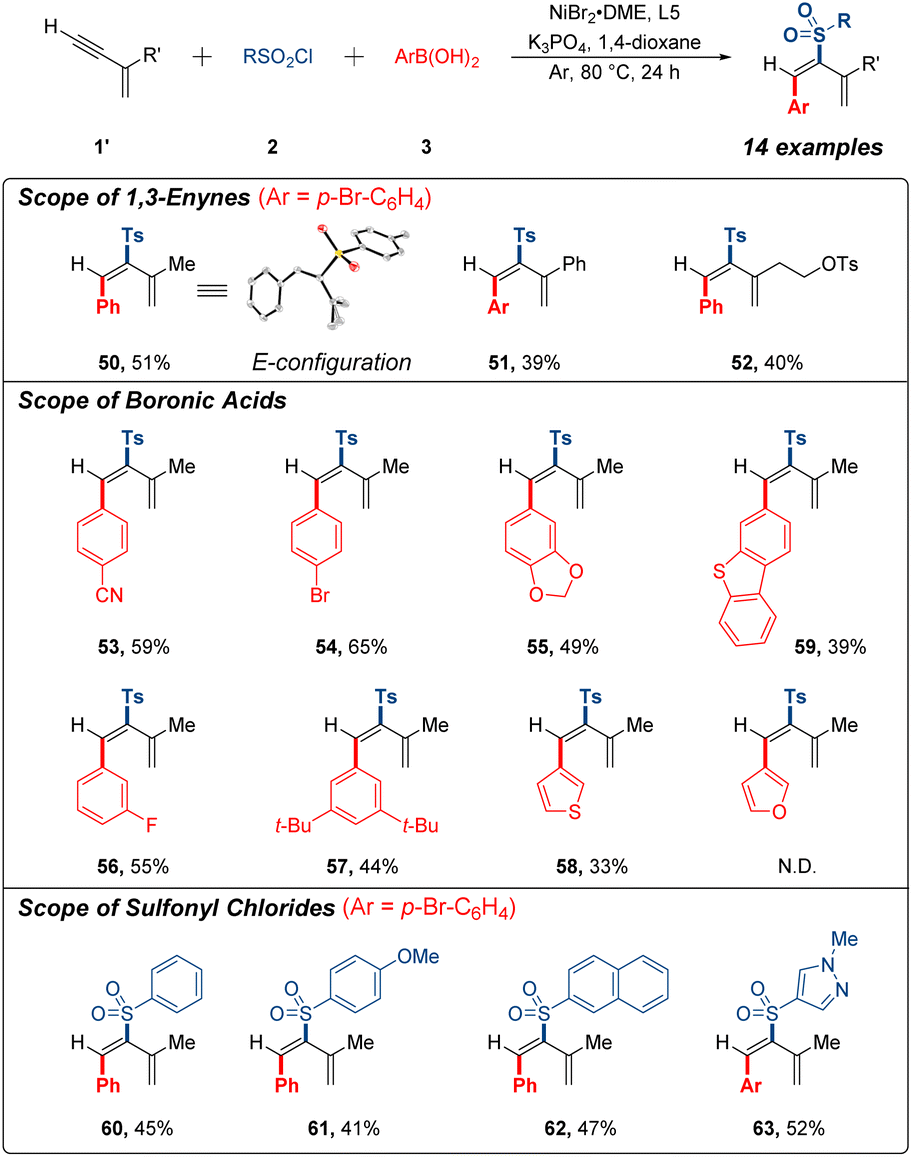 | ||
| Fig. 3 Scope of 1,3-enynes, boronic acids and sulfonyl chlorides. Reaction conditions: 1,3-enynes (0.1 mmol), arylboronic acids (0.25 mmol), sulfonyl chlorides (0.25 mmol), NiBr2·DME (12 mol%), 5,5′-bis(trifluoromethyl)-2,2′-bipyridine (12 mol%), K3PO4 (0.3 mmol) and 1,4-dioxane (0.5 mL), Ar, 80 °C, 24 h, isolated yield. N.D. = desired product not detected. | ||
To further demonstrate the comprehensive versatility of this procedure, a series of extended experiments were conducted as depicted in Fig. 4. First of all, late-stage functionalization of bioactive natural products and pharmaceutical drugs proceeded easily. Gratifyingly, a number of structurally complex 1,3-enyne substrates derived from (pre)drug compounds and natural products, such as probenecid, ibuprofen, estrone, L-borneol and the precursor of canagliflozin, dapagliflozin and tazarotene, and the sulfonyl chloride modified from valdecoxib, were employed as the reaction partners, which successfully underwent the 1,4-sulfonylarylation cross-coupling process to furnish the highly functionalized allenyl sulfones in 25–73% yields (64–71). Beyond that, we also examined the feasibility of regiodivergent sulfonylarylation of 1,3-enynes with the arylboronic acid that was derivatized from glycine, leading to the corresponding products in 74% and 20% yields, separately (72 and 73). The successful applications of this facile approach to these molecules will have significant implications for synthesizing complex sulfone-containing compounds. Next, two gram-scale syntheses were executed effortlessly on 3.0 mmol and 5.0 mmol scale without much decrease in the product yield. The post-transformations of sulfonylarylated allene and diene were also briefly carried out to produce corresponding products in high yields (74 and 75). Finally, the interesting protocol could extend to arylsulfonylalkylation with other radical precursor in 46% yield (76), and the four-component 1,4-sulfonylacylation of CF3-containing 1,3-enyne also occurred in 21% yield under 1 atm CO gas (77).
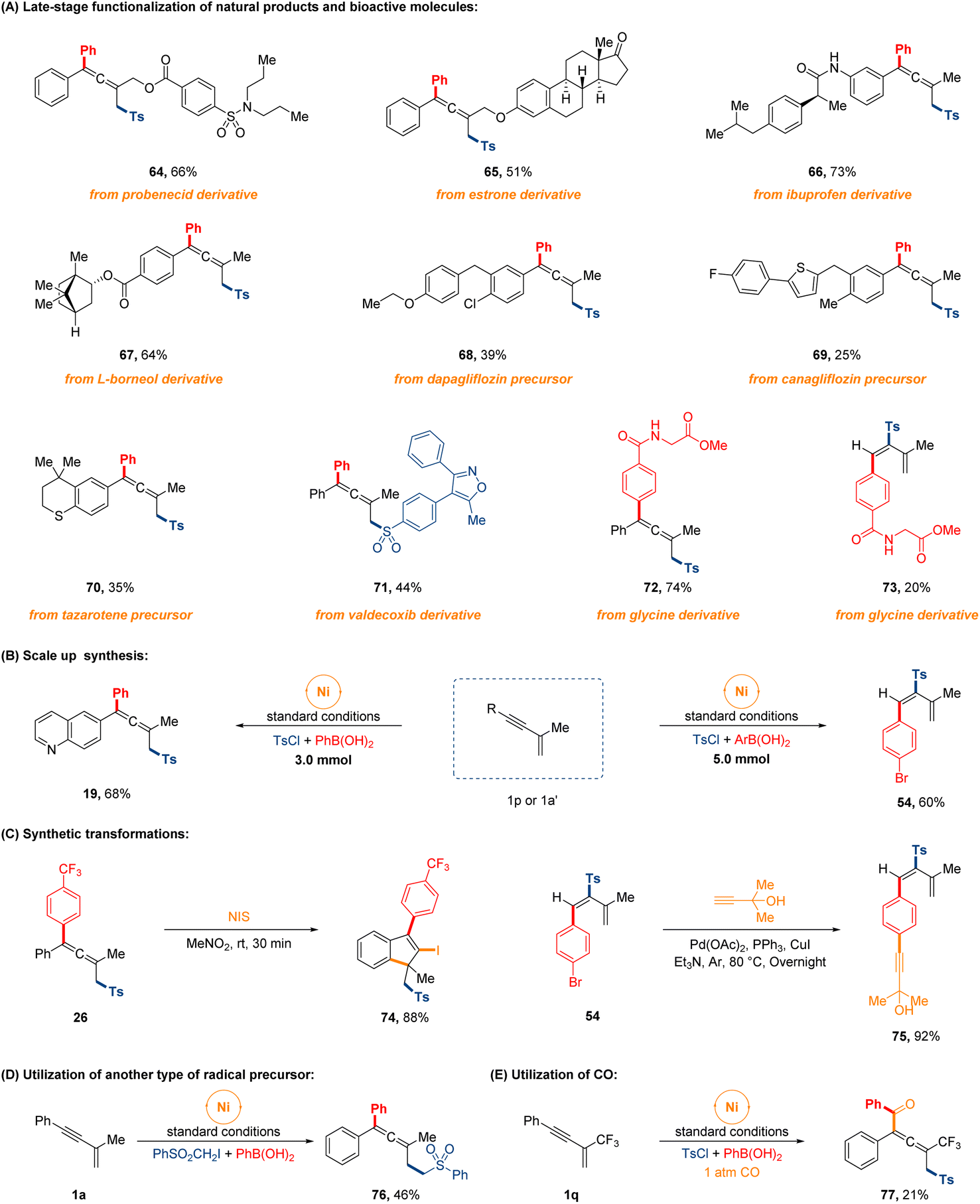 | ||
| Fig. 4 Synthetic utilizations. | ||
Preliminary studies were conducted to provide some insight into the mechanism of the reaction (Fig. 5). Initially, we set out to conduct radical inhibition experiments using radical scavengers. The addition of either 1,1-diphenyl ethylene or 2,2,6,6-tetramethylpiperidinooxy (TEMPO) under standard conditions significantly suppressed this sulfonylarylation process and product (78) was isolated in 30% yield fortunately. When the transformation was performed under air atmosphere, no target product was observed. These observations suggested that a sulfonyl radical might be involved along the reaction course. Furthermore, the radical nature of the reaction was further supported by the radical clock experiment with α-cyclopropyl styrene affording the formation of cyclized product (79) through sequential ring-opening of the cyclopropylmethyl radical intermediate and intramolecular cyclization, while the ring-opened sulfonylarylated product was not detected. On the other hand, the reaction was performed with a catalytic amount of phenyl boronic acid, giving the trace allenyl sulfones. While none of propargyl chloride or allenyl chloride was monitored in system. Besides, in order to eliminate the influence of light-mediated pathway of radical generation, the reactions were conducted in the dark under standard conditions, and the isolated yields were almost unchanged whether 1,4-sulfonylarylation or 3,4-sulfonylarylation. Finally, no expected products were observed when other terminal alkynes substrates were tested under our conditions, which might indicate the formation of intermediate π-allylnickel species in our catalytic system.
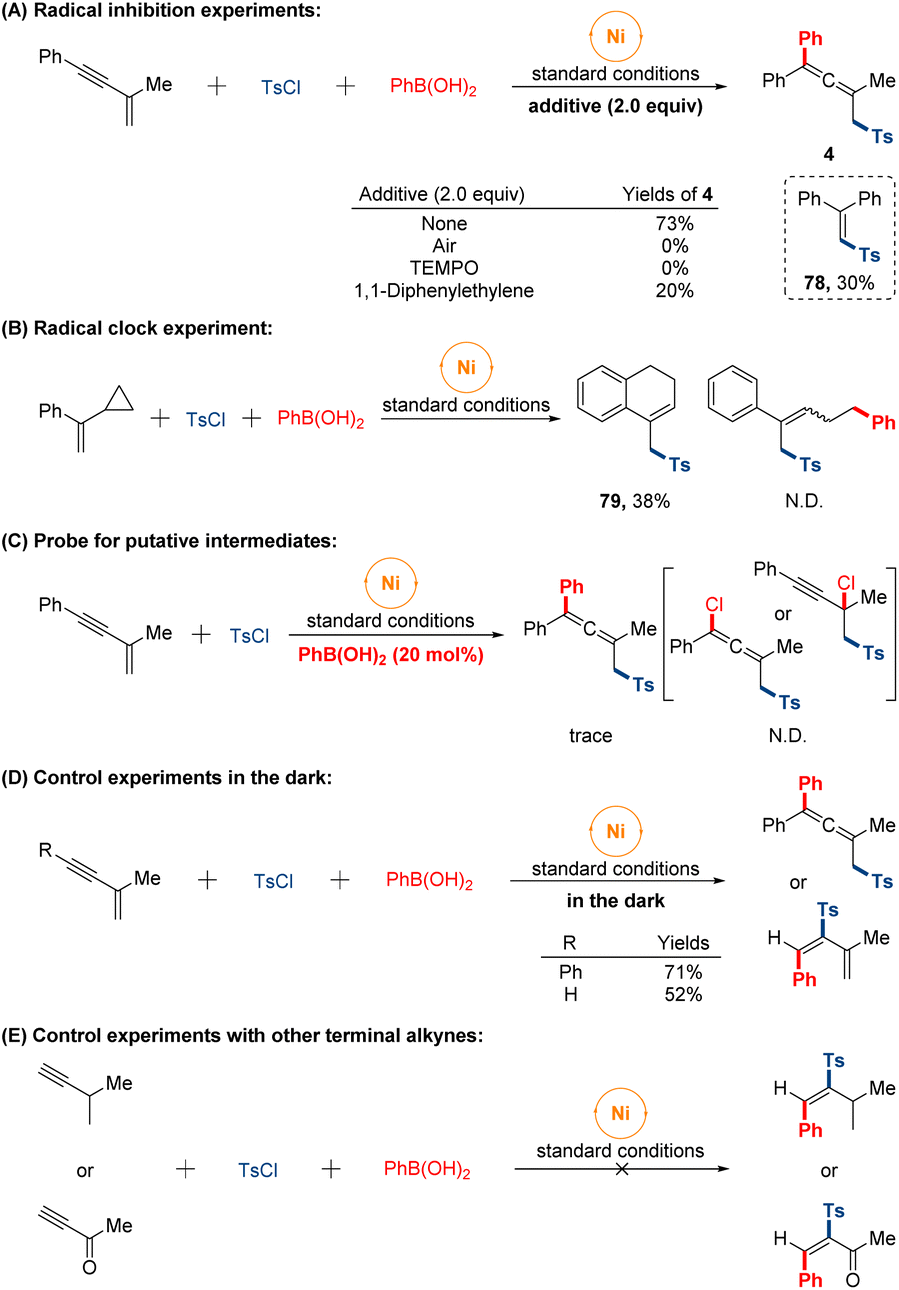 | ||
| Fig. 5 Mechanistic studies. | ||
A mechanistic scenario is proposed for this transformation based on the above experimental studies and pervious literature reports (Fig. 6).5,12,16 Initially, the transmetalation of the Ni(I) species (A), which might be generated by in situ reduction from Ni(II), with arylboronic acid would deliver the Ni(I) species (B). Then, the Ni(I) species (B) could react with sulfonyl chloride through a single electron transfer pathway, leading to the generation of the Ni(II) species (C) and the corresponding radical. Subsequently, the sulfonyl radical might add to the double bond in 1,3-enyne 1a to afford a propargyl radical, and the tautomerized allenyl radical might be direct trapped by the Ni(II) species (C). Moreover, this pathway of 1,3-Ni shift process could not be excluded. When terminal 1,3-enyne 1a′ was used, the Ni(II) species (C) might preferentially undergo migratory insertion into the less substituted terminal triple bond in the substrates probably due to the steric hindrance effect. The intermediate Ni(II) species (D) might undergo isomerization to generate the π-allylnickel(II) species (D′). Then, the sulfonyl radical might be trapped by the Ni(II) species (D′′), which might undergo π-allylnickel-assisted syn/anti isomerization from the Ni(II) species (D′). Finally, the reductive elimination of the produced intermediate Ni(III) species (E and F) could release the desired products and regenerate the Ni(I) species (A) for the next catalytic cycle.
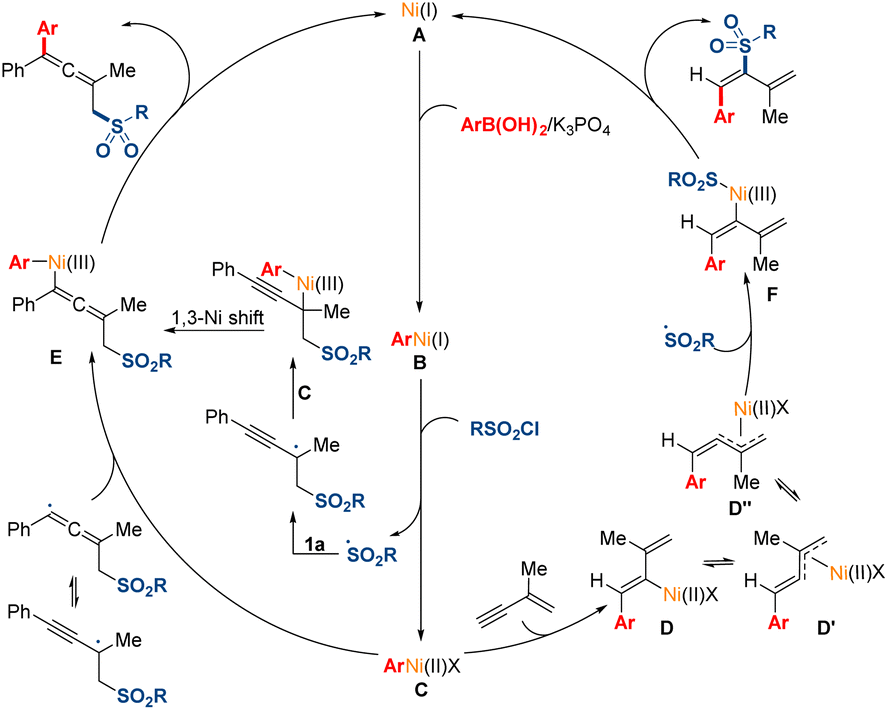 | ||
| Fig. 6 Proposed mechanism. | ||
Conclusions
In summary, a novel methodology for nickel-catalyzed substrate-controlled regiodivergent sulfonylarylation of 1,3-enynes is presented. The versatile protocol rapidly delivers the selective construction of valuable sulfonylarylated allenes or dienes from fine-tuned 1,3-enynes with wide substrate scope and acceptable functional group tolerance under the same catalytic system. Preliminary control experiments provide evidence supporting the involvement of sulfonyl radicals. This facile approach also provides a valuable strategy for the late-stage modification of complex sulfone-containing molecules.Data availability
All data associated with this study are available in the article and ESI.†Author contributions
Z. Chi performed the experiments and wrote the manuscript and ESI. Y. Zhou and B. Liu prepared some of the substrates and checked the ESI. X. Xu and X. Liu checked the manuscript. Y. Liang directed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant 22171114). The authors thank Dr P. Cheng and Dr Z. Wen from College of Chemistry and Chemical Engineering, Xiamen University, and Dr S. Li from Fujian Institute of Research on the Structure of Matter, University of Chinese Academy of Sciences, and Dr J. Yang from College of Chemistry and Chemical Engineering, Lanzhou University.Notes and references
- (a) H. Tucker and G. J. Chesterson, J. Med. Chem., 1988, 31, 885–887 CrossRef CAS PubMed; (b) A. El-Awa, M. N. Noshi, X. M. du Jourdin and P. L. Fuchs, Chem. Rev., 2009, 109, 2315–2349 CrossRef CAS PubMed; (c) M. Feng, B. Tang, H. S. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed; (d) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed.
- (a) Y. Fang, Z. Luo and X. Xu, RSC Adv., 2016, 6, 59661–59676 RSC; (b) N.-W. Liu, S. Liang and G. Manolikakes, Synthesis, 2016, 48, 1939–1973 CrossRef CAS; (c) G. Qiu, K. Zhou, L. Gao and J. Wu, Org. Chem. Front., 2018, 5, 691–705 RSC; (d) D. Zeng, M. Wang, W.-P. Deng and X. Jiang, Org. Chem. Front., 2020, 7, 3956–3966 RSC; (e) D. Joseph, M. A. Idris, J. Chen and S. Lee, ACS Catal., 2021, 11, 4169–4204 CrossRef CAS; (f) S. Huang, M. Wang and X. Jiang, Chem. Soc. Rev., 2022, 51, 8351–8377 RSC; (g) T. Yu, Z.-Z. Shi, M.-Z. Zhang, S. You and C. Deng, Tetrahedron, 2023, 138, 133409 CrossRef CAS.
- (a) S. M. Hell, C. F. Meyer, A. Misale, J. B. I. Sap, K. E. Christensen, M. C. Willis, A. A. Trabanco and V. Gouverneur, Angew. Chem., Int. Ed., 2020, 59, 11620–11626 CrossRef CAS PubMed; (b) S. Cao, D. Kim, W. Lee and S. Hong, Angew. Chem., Int. Ed., 2023, 62, e202312780 CrossRef CAS PubMed; (c) X. Chen, D. Zheng, L. Jiang, Z. Wang, X. Duan, D. Cui, S. Liu, Y. Zhang, X. Yu, J. Ge and J. Xu, Angew. Chem., Int. Ed., 2023, 62, e202218140 CrossRef CAS PubMed; (d) C.-S. Dong, W.-Y. Tong, P. Ye, N. Cheng, S. Qu and B. Zhang, ACS Catal., 2023, 13, 6983–6993 CrossRef CAS; (e) C.-M. Li, X.-X. Dong, Z. Wang and B. Zhang, Green Chem., 2023, 25, 4122–4128 RSC; (f) J. Liu, H. Liu, X. Guo, Z. Wang, X. Wu, J. Li and C. Zhu, Green Chem., 2023, 25, 3847–3851 RSC; (g) K. Muralirajan, R. Kancherla, B. Maity, S. Karuthedath, F. Laquai, L. Cavallo and M. Rueping, Nat. Commun., 2023, 14, 6622 CrossRef CAS PubMed; (h) A. Sookezian and G. A. Molander, Org. Lett., 2023, 25, 1014–1019 CrossRef CAS PubMed; (i) J. D. Lasso, D. J. Castillo-Pazos, J. M. Salgado, C. Ruchlin, L. Lefebvre, D. Farajat, D. F. Perepichka and C.-J. Li, J. Am. Chem. Soc., 2024, 146, 2583–2592 CrossRef CAS PubMed; (j) W. Li, Z. Huang, D. Zhong and H. Li, Org. Lett., 2024, 26, 2062–2067 CrossRef CAS PubMed; (k) F. Tang, Y.-S. Feng, W. Yang and H.-J. Xu, Org. Lett., 2024, 26, 236–240 CrossRef CAS PubMed; (l) L. Tang, G. Lv, F. Jia, R. Zhao, X. Wang and Q. Zhou, Adv. Synth. Catal., 2024, 366, 70–76 CrossRef CAS.
- J. Liu and L. Zheng, Adv. Synth. Catal., 2019, 361, 1710–1732 CrossRef CAS.
- A. García-Domínguez, S. Müller and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 9949–9952 CrossRef PubMed.
- X. Liu, H. Chen, D. Yang, B. Hu, Y. Hu, S. Wang, Y. Lan, A. Lei and J. Li, ACS Catal., 2023, 13, 9254–9263 CrossRef CAS.
- (a) T. Xu, S. Wu, Q.-N. Zhang, Y. Wu, M. Hu and J.-H. Li, Org. Lett., 2021, 23, 8455–8459 CrossRef CAS PubMed; (b) D.-D. Hu, Q. Gao, J.-C. Dai, R. Cui, Y.-B. Li, Y.-M. Li, X.-G. Zhou, K.-J. Bian, B.-B. Wu, K.-F. Zhang, X.-S. Wang and Y. Li, Sci. China: Chem., 2022, 65, 753–761 CrossRef CAS; (c) C. Li, D.-D. Hu, R.-X. Jin, B.-B. Wu, C.-Y. Wang, Z. Ke and X.-S. Wang, Org. Chem. Front., 2022, 9, 788–794 RSC.
- (a) Y. Song, S. Song, X. Duan, X. Wu, F. Jiang, Y. Zhang, J. Fan, X. Huang, C. Fu and S. Ma, Chem. Commun., 2019, 55, 11774–11777 RSC; (b) C.-Y. Zhang, J. Zhu, S.-H. Cui, X.-Y. Xie, X.-D. Wang and L. Wu, Org. Lett., 2021, 23, 3530–3535 CrossRef CAS PubMed; (c) Y. Lv, W. Han, W. Pu, J. Xie, A. Wang, M. Zhang, J. Wang and J. Lai, Org. Chem. Front., 2022, 9, 3775–3780 RSC; (d) Y. Lv, J. Lai, W. Pu, J. Wang, W. Han, A. Wang, M. Zhang and X. Wang, J. Org. Chem., 2023, 88, 2034–2045 CrossRef CAS PubMed.
- (a) Q. Dherbassy, S. Manna, F. J. T. Talbot, W. Prasitwatcharakorn, G. J. P. Perry and D. J. Procter, Chem. Sci., 2020, 11, 11380–11393 RSC; (b) L. Fu, S. Greßies, P. Chen and G. Liu, Chin. J. Chem., 2020, 38, 91–100 CrossRef CAS; (c) Y. Li and H. Bao, Chem. Sci., 2022, 13, 8491–8506 RSC; (d) Y. Yu and J. Zhang, Eur. J. Org. Chem., 2022, 2022, e202201017 CrossRef; (e) X. Liu, C. Jiao, S. Cui, Q. Liu, X. Zhang and G. Zhang, ChemCatChem, 2023, 15, e202300601 CrossRef CAS.
- (a) F. Wang, D. Wang, Y. Zhou, L. Liang, R. Lu, P. Chen, Z. Lin and G. Liu, Angew. Chem., Int. Ed., 2018, 57, 7140–7145 CrossRef CAS PubMed; (b) K.-F. Zhang, K.-J. Bian, C. Li, J. Sheng, Y. Li and X.-S. Wang, Angew. Chem., Int. Ed., 2019, 58, 5069–5074 CrossRef CAS PubMed; (c) X. Zhu, W. Deng, M.-F. Chiou, C. Ye, W. Jian, Y. Zeng, Y. Jiao, L. Ge, Y. Li, X. Zhang and H. Bao, J. Am. Chem. Soc., 2019, 141, 548–559 CrossRef CAS PubMed; (d) Y. Zeng, M.-F. Chiou, X. Zhu, J. Cao, D. Lv, W. Jian, Y. Li, X. Zhang and H. Bao, J. Am. Chem. Soc., 2020, 142, 18014–18021 CrossRef CAS PubMed; (e) L. Chen, C. Lin, S. Zhang, X. Zhang, J. Zhang, L. Xing, Y. Guo, J. Feng, J. Gao and D. Du, ACS Catal., 2021, 11, 13363–13373 CrossRef CAS; (f) X.-Y. Dong, T.-Y. Zhan, S.-P. Jiang, X.-D. Liu, L. Ye, Z.-L. Li, Q.-S. Gu and X.-Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 2160–2164 CrossRef CAS PubMed; (g) F.-S. He, P. Bao, F. Yu, L.-H. Zeng, W.-P. Deng and J. Wu, Org. Lett., 2021, 23, 7472–7476 CrossRef CAS PubMed; (h) W. Liu, C. Liu, M. Wang and W. Kong, ACS Catal., 2022, 12, 10207–10221 CrossRef CAS; (i) L. Wang, R. Ma, J. Sun, G. Zheng and Q. Zhang, Chem. Sci., 2022, 13, 3169–3175 RSC; (j) F.-H. Zhang, X. Guo, X. Zeng and Z. Wang, Nat. Commun., 2022, 13, 5036 CrossRef CAS PubMed; (k) H. Chen, C. Zhu, H. Yue and M. Rueping, Angew. Chem., Int. Ed., 2023, 62, e202306498 CrossRef CAS PubMed; (l) C. Cheng, G.-F. Lv, S. Wu, Y. Li and J.-H. Li, Org. Lett., 2023, 25, 4236–4240 CrossRef CAS PubMed; (m) S. Dong, Z. Tian, J. Wang, L. He and J. Li, J. Catal., 2023, 425, 350–358 CrossRef CAS; (n) S. Li, W. Yang, J. Shi, T. Dan, Y. Han, Z.-C. Cao and M. Yang, ACS Catal., 2023, 13, 2142–2148 CrossRef CAS; (o) X. Li, N. Li, L. Yang, R. Jiang, Y. He, J. Shi, Z.-X. Jiang and Z. Yang, ACS Catal., 2023, 13, 12755–12765 CrossRef CAS; (p) H.-J. Miao, J.-H. Zhang, S. Liu, W.-H. Wang, X. Yang, X.-H. Duan and L.-N. Guo, Org. Lett., 2023, 25, 5563–5568 CrossRef CAS PubMed; (q) H. Song, X. Zhang, G. Chen, X. He and Z. Lian, Org. Lett., 2023, 25, 5916–5921 CrossRef CAS PubMed; (r) C.-Y. Tan, M. Kim and S. Hong, Angew. Chem., Int. Ed., 2023, 62, e202306191 CrossRef CAS PubMed; (s) Q. Wang, Y. Chen, K. Peng, Y. Li, L. Cheng and G.-J. Deng, Org. Lett., 2023, 25, 8889–8894 CrossRef CAS PubMed; (t) Z. Zhang, Y. Tian, X. Li, Z. Wang, R. Liu, P. Chen, X. Li, J. Dai and D. Shi, Green Chem., 2023, 25, 2723–2729 RSC; (u) C. Zhu, H. Chen, H. Yue and M. Rueping, Nat. Synth., 2023, 2, 1068–1081 CrossRef; (v) C.-S. Kuai, B.-H. Teng and X.-F. Wu, Angew. Chem., Int. Ed., 2024, 63, e202318257 CrossRef CAS PubMed; (w) Y. Pu, S. Ding, H. Zhao, Q. Xue, H. Zhang, X. Xie, Y. Shang and J. Wang, Org. Lett., 2024, 26, 1834–1839 CrossRef CAS PubMed; (x) Q.-C. Shan, Y. Zhao, S.-T. Wang, H.-F. Liu, X.-H. Duan and L.-N. Guo, ACS Catal., 2024, 14, 2144–2150 CrossRef CAS; (y) B.-H. Teng, C.-S. Kuai, Y. Zhao and X.-F. Wu, Tetrahedron, 2024, 157, 133965 CrossRef CAS; (z) G. Zhang, W. Tan, D. Zhang, K. Wang, P. Gao, S. Wang, S.-L. Liu and F. Chen, Org. Lett., 2024, 26, 536–541 CrossRef CAS PubMed.
- (a) H. Hou, B. Zhou, J. Wang, D. Zhao, D. Sun, X. Chen, Y. Han, C. Yan, Y. Shi and S. Zhu, Org. Lett., 2021, 23, 2981–2987 CrossRef CAS PubMed; (b) Y. Li, D. Liu, L. Wan, J.-Y. Zhang, X. Lu and Y. Fu, J. Am. Chem. Soc., 2022, 144, 13961–13972 CrossRef CAS PubMed.
- Y. Chen, K. Zhu, Q. Huang and Y. Lu, Chem. Sci., 2021, 12, 13564–13571 RSC.
- S.-N. Yang, S.-H. Sun, C.-H. Liu, X.-T. Min, B. Wan, D.-W. Ji and Q.-A. Chen, Chin. Chem. Lett., 2023, 34, 107914 Search PubMed.
- (a) Y. Zhao, J.-L. Wang, Z. Zhang, X.-S. Li, Z.-J. Niu and X.-Y. Liu, J. Org. Chem., 2021, 86, 18056–18066 CrossRef CAS PubMed; (b) M. Li, G.-Q. Sun, Y.-Y. Liu, S.-X. Li, H.-C. Liu, Y.-F. Qiu, D.-P. Chen, X.-C. Wang, Y.-M. Liang and Z.-J. Quan, J. Org. Chem., 2023, 88, 1403–1410 CrossRef CAS PubMed.
- (a) Y.-L. Xiao, W.-H. Guo, G.-Z. He, Q. Pan and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 9909–9913 CrossRef CAS PubMed; (b) Z. Feng, Y.-L. Xiao and X. Zhang, Acc. Chem. Res., 2018, 51, 2264–2278 CrossRef CAS PubMed; (c) M. Zhou, H.-Y. Zhao, S. Zhang, Y. Zhang and X. Zhang, J. Am. Chem. Soc., 2020, 142, 18191–18199 CrossRef CAS PubMed; (d) S. Ma, F. Li, G. Zhang, L. Shi and X. Wang, ACS Catal., 2021, 11, 14848–14853 CrossRef CAS; (e) N. Rao, Y.-Z. Li, Y.-C. Luo, Y. Zhang and X. Zhang, ACS Catal., 2023, 13, 4111–4119 Search PubMed.
- (a) S. Tobisch, Chem.–Eur. J., 2002, 8, 4756–4766 CrossRef CAS PubMed; (b) S. Tobisch, Acc. Chem. Res., 2002, 35, 96–104 CrossRef CAS PubMed; (c) P. Fan, R. Wang and C. Wang, Org. Lett., 2021, 23, 7672–7677 CrossRef CAS PubMed; (d) P. Lv, A. Wang, X. Xie, Y. Chen and Y. Liu, Org. Lett., 2023, 25, 3319–3324 CrossRef CAS PubMed; (e) G. Zhang, C. Zhang, H. Jiao and F. Chen, J. Catal., 2023, 418, 312–319 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2344342. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03067b |
| This journal is © The Royal Society of Chemistry 2024 |