
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coherent spin-control of S = 1 vanadium and molybdenum complexes†
Daniel W.
Laorenza
 a,
Kathleen R.
Mullin
b,
Leah R.
Weiss
cd,
Sam L.
Bayliss
ce,
Pratiti
Deb
cf,
David D.
Awschalom
*cfg,
James M.
Rondinelli
*b and
Danna E.
Freedman
*a
a,
Kathleen R.
Mullin
b,
Leah R.
Weiss
cd,
Sam L.
Bayliss
ce,
Pratiti
Deb
cf,
David D.
Awschalom
*cfg,
James M.
Rondinelli
*b and
Danna E.
Freedman
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. E-mail: danna@mit.edu
bDepartment of Materials Science and Engineering, Northwestern University, Evanston, Illinois 60208, USA. E-mail: jrondinelli@northwestern.edu
cPritzker School of Molecular Engineering, University of Chicago, Chicago, Illinois 60637, USA. E-mail: awsch@uchicago.edu
dAdvanced Institute for Materials Research (AIMR-WPI), Tohoku University, Sendai 980-8577, Japan
eJames Watt School of Engineering, University of Glasgow, Glasgow G12 8QQ, UK
fDepartment of Physics, University of Chicago, Chicago, Illinois 60637, USA
gCenter for Molecular Engineering and Materials Science Division, Argonne National Laboratory, Lemont, Illinois 60439, USA
First published on 5th August 2024
Abstract
The burgeoning field of quantum sensing hinges on the creation and control of quantum bits. To date, the most well-studied quantum sensors are optically active, paramagnetic defects residing in crystalline hosts. We previously developed analogous optically addressable molecules featuring a ground-state spin-triplet centered on a Cr4+ ion with an optical-spin interface. In this work, we evaluate isovalent V3+ and Mo4+ congeners, which offer unique advantages, such as an intrinsic nuclear spin for V3+ or larger spin–orbit coupling for Mo4+, as optically addressable spin systems. We assess the ground-state spin structure and dynamics for each complex, illustrating that all of these spin-triplet species can be coherently controlled. However, unlike the Cr4+ derivatives, these pseudo-tetrahedral V3+ and Mo4+ complexes exhibit no measurable emission. Coupling absorption spectroscopy with computational predictions, we investigate why these complexes exhibit no detectable photoluminescence. These cumulative results suggest that design of future V3+ complexes should target pseudo-tetrahedral symmetries using bidentate or tridentate ligand scaffolds, ideally with deuterated or fluorinated ligand environments. We also suggest that spin-triplet Mo4+, and by extension W4+, complexes may not be suitable candidate optically addressable qubit systems due to their low energy spin-singlet states. By understanding the failures and successes of these systems, we outline additional design features for optically addressable V- or Mo-based molecules to expand the library of tailor-made quantum sensors.
Introduction
The second quantum revolution is driven by the design, creation, and control of quantum bits, or qubits, the fundamental units of quantum information processing.1–10 Harnessing the power of quantum control, quantum sensors have provided unprecedented visualizations of nanoscale magnetic and electric fields in diverse physical environments.11–14 In many impressive demonstrations – such as magnetic resonance of individual proteins,12 detection of action potentials of individual neurons,15 or mapping electron flow in two-dimensional materials16,17 – optically active, paramagnetic defects embedded in a crystalline host serve as the qubits.3,7,18–22 The electronic structure of these color centers provides a valuable platform for quantum control. First, the ground state spin sublevels of these defects provide a two-level quantum system to act as the qubit. Crucially, the spin sublevel populations may be optically prepared into a non-thermal equilibrium state, i.e. initialized, while the spin state information may be read out using spin-dependent optical emission.18,23 These combined features enable remote optical control and single qubit addressability, both of which are valuable features for quantum sensing technologies.24,25Although solid-state color centers offer incredible coherence properties and quantum control, deterministic spin localization and defect tunability remain major challenges.7 To complement existing top-down creation of these solid-state systems, molecular synthesis provides a bottom-up approach, offering a modular and scalable route to develop emerging quantum technologies.5,7,26–31 To create bespoke quantum sensors, we sought to develop molecular analogues of solid-state color centers that host a similar optical-spin interface, but with their molecular nature facilitating bottom-up design and solution processibility towards specific sensor–analyte interactions.32,33 In recapitulating this optical-spin interface, we needed (i) a ground state spin that can be coherently controlled, (ii) an electronic excited state that is ‘connected’ to this ground-state spin through a spin-selective optical process (e.g., excitation or relaxation, see ESI† for further discussion), (iii) radiative decay from the excited state to the ground state for optical readout, and (iv) a ground-state spin lifetime, or spin-lattice relaxation time (T1), that is longer than the optical lifetime (τopt).34 These criteria initially led us to spin-triplet (S = 1) transition metal complexes that share a similar electronic structure with optically addressable solid-state spins such as the anionic nitrogen vacancy center in diamond, divacancy defects in silicon carbide, or more similarly, tetravalent chromium (Cr4+) dopants in silicon carbide.35–38
While these generic features provide a framework to build molecular color centers, S = 1 molecular systems pose two key challenges for spin-dependent optical readout. First, to achieve a spin-selective optical process and emission in S = 1 complexes, a ligand field that ensures that the first excited state is a spin-singlet excited state (1ES) is desirable.39 For resonant optical control (off-resonant optical control protocol addressed in the ESI†), the spin-flip transition from the 1ES to the spin-triplet ground state (3GS) should exhibit limited vibronic coupling, allowing for both narrow optical linewidths for spin-selective optical excitation and radiative decay to the ground state.34,40 This desired electronic structure requires a sufficiently strong ligand field around the spin-bearing metal center such that the first excited state is a 1ES, eliminating lower energy 3ES's that provide non-radiative decay pathways.39 Second, the ground-state spin must be capable of coherent control. However, the spin of S = 1 transition metal systems often cannot be coherently controlled with readily accessible microwave frequencies (i.e., 1–40 GHz). The inability to coherent drive the ground-state spin of these systems arises from both the integer spin and large zero-field splitting (ZFS) values, requiring high microwave frequencies (>95 GHz) to probe ground-state spin transitions.41,42 This challenge may be overcome by designing orbitally non-degenerate, S = 1 ground states, wherein the spin-bearing ion resides in a (near) cubic ligand field.
Considering these design parameters, we previously demonstrated that pseudo-tetrahedral (Td) Cr4+ molecules in a strong ligand field exhibit small ground-state ZFS values of 1.8–4.1 GHz for microwave control at X-band frequency (9–10 GHz), and an optical-spin interface.34,40 Yet, the air-sensitivity, small spin–orbit coupling, and low percentage of nuclear spin-bearing isotopes of Cr4+-based systems may not be optimal for every sensing task. Herein, we aim to translate this combination of features from Td Cr4+ systems into molecular hosts that offer (i) intrinsic nuclear spins, I, to serve as quantum memories for prolonged information storage,7,43,44 and (ii) larger spin–orbit coupling for improved sensitivity to electric fields.35,45 To that end, we turned to Td trivalent vanadium (V3+) and tetravalent molybdenum (Mo4+) derivatives in the same ligand fields as their Cr4+ congeners. The vanadium-51 (i.e., 51V, I = 7/2, 99.8% abundance) center provides a potential nuclear spin memory that is intrinsically coupled to the electronic spin through hyperfine interactions,42,46 while the Mo4+ center increases the spin–orbit coupling experienced by the electron spin, offering enhanced sensitivity of the spin Hamiltonian parameters to certain external fields through the Zeeman splitting term (e.g., strain or electric fields).47,48 However, the metal ion substitution should substantially change both the electronic structure and spin dynamics that dictate the quantum sensor performance. To initiate our studies with these ions, we synthesized and evaluated three spin-triplet systems with V3+ and Mo4+ to compare with previously studied Cr4+ congeners. Coupling spectroscopic analysis with computational predictions, we evaluate how to design these systems, addressing their advantages and limitations. From these results, we suggest that reducing nearby high energy oscillators and using ligand scaffolds could enable both emission and low frequency microwave control for V3+ systems while Mo4+ systems may be fundamentally limited by their intrinsically low lying 1ES.
Results and discussion
We synthesized [Li(THF)4][V(o-tolyl)4] (1, THF = tetrahydrofuran), Mo(o-tolyl)4 (2),49 and [Li(12-crown-4)2][V(trimethylsilylmethyl)4] (3) to directly compare with previous candidate color centers,34,40 Cr(o-tolyl)4 (4)50 and Cr(trimethylsilylmethyl)4 (5)51 (Fig. 1). Detailed synthetic procedures for 1 and 3 are outlined in the ESI† while 2, 4, and 5 were synthesized according to previously reported methods.49–51 We also note that previous efforts to synthesize the molybdenum analogue of Cr(trimethylsilylmethyl)4 resulted in the hexakis(trimethylsilylmethyl)dimolybdenum species, so we could not investigate the Mo4+ analogue of 5.52,53We first characterized the solid-state structure of 1 and 3 through single crystal X-ray diffraction, finding that their inner MC4 coordination sphere remains close to ideal tetrahedral symmetry based on both the τ4 and τ4′ metrics (0.95–1.01).54,552 had been previously characterized at room temperature and shows only a slight contraction in Mo–C bond lengths upon cooling to 100 K.49 As such, these systems should exhibit relatively small, but non-zero, ZFS values. The ground-state structure may be approximated as a descent in symmetry from ideal Td to C2v, as discussed for [Li(THF)4][V(mesityl)4] and [Li(THF)4] [V(pentachlorophenyl)4].56
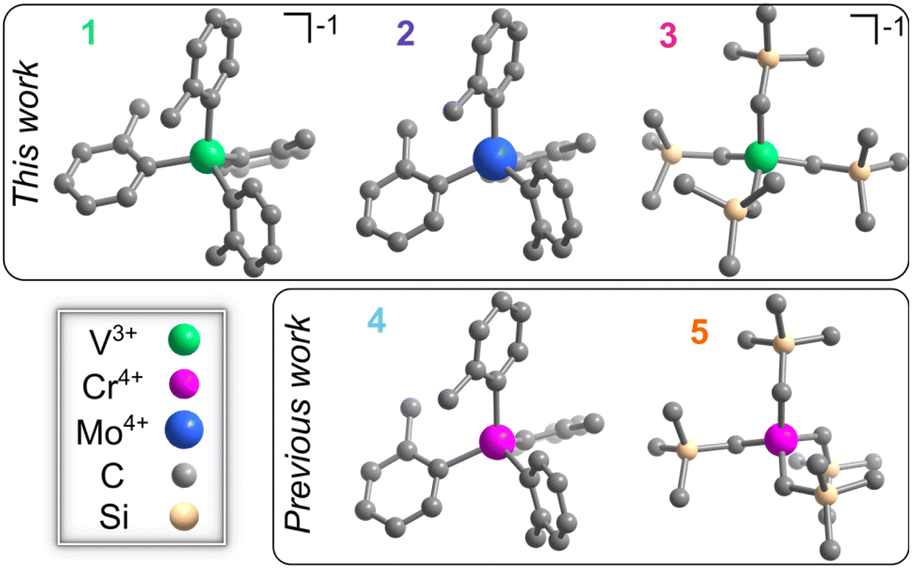 | ||
| Fig. 1 Molecular structures of 1, 2, 3, 4 (ref. 34), and 5 (ref. 40) determined at 100 K. Carbon, silicon, vanadium, chromium, and molybdenum given in gray, peach, green, pink, and blue, respectively. Hydrogen atoms and counter ions omitted for clarity. | ||
Microwave addressability remains a challenging parameter to design and tune in S = 1 compounds.41,42 Thus, we initiated our investigation by probing the ground-state spin structure of these systems to determine if they are capable of ground-state spin control. We characterized 1–3 with both continuous-wave (cw) and pulsed electron paramagnetic resonance (EPR) to understand their ground-state spin structure and dynamics. To reduce decoherence from electron spin-electron spin interactions, we cocrystallized 1–3 in their corresponding isostructural, diamagnetic analogues, [Li(THF)4][Al(o-tolyl)4] (1-Al), Sn(o-tolyl)4 (2-Sn),57 and [Li(12-crown-4)2][Al(trimethylsilylmethyl)4] (3-Al), respectively (see ESI† for details). We denote the cocrystallized samples as 1′–3′ for all subsequent experiments with relative electron spin concentrations of 0.36–3% (Table S8†). From the X-band cw-EPR spectra in Fig. 2a–c, we find that the axial ZFS values, D, are larger for the V3+ and Mo4+ derivatives than the corresponding Cr4+ congeners.401′ and 2′ exhibit |D| values of 5.62 GHz and 7.3 GHz, respectively, while |D| for 4′ at 5 K is 3.63 GHz.34 The largest contribution of the two-fold increase in |D| for 2′ likely arises from the increased spin–orbit coupling of the heavier Mo4+ ion, where the free ion spin–orbit coupling parameter, λ, is ∼425 and 167.5 cm−1 for Mo4+ and Cr4+.58 Given the similarity in λ for V3+ and Cr4+, the increase in |D| for 1′ more likely results from symmetry lowering around the V3+ center due to crystal packing with the [Li(THF)4]+ cation. For example, |D| = 5.62 GHz for 1′ is similar to the |D| value of 5.55 GHz for Cr(o-tolyl)4 (4) diluted in a lower symmetry (orthorhombic) host matrix.59
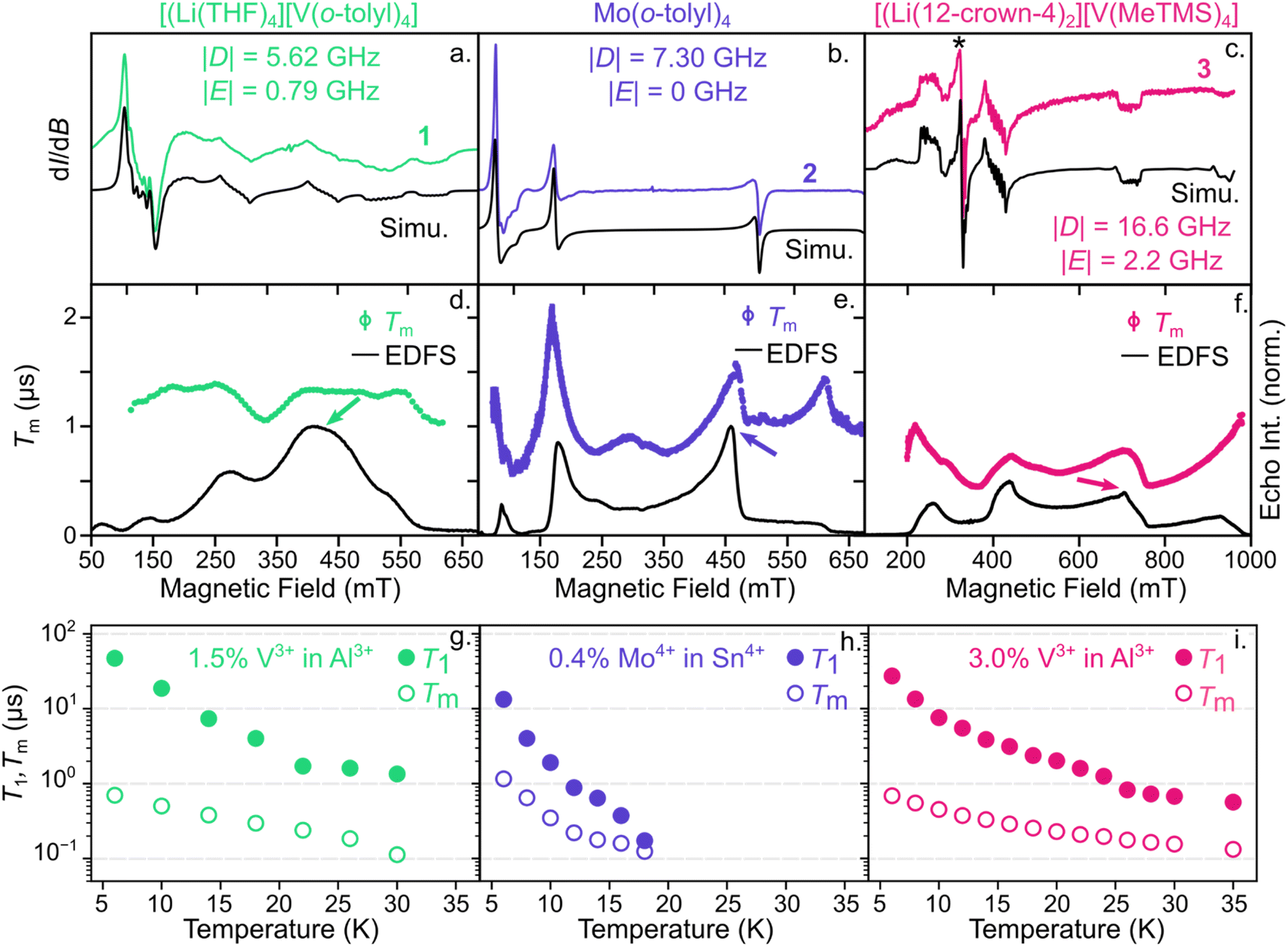 | ||
| Fig. 2 (top) cw-EPR spectra of (a) 1′, (b) 2′, and (c) 3′ at 5 K and 9.37–9.39 GHz with resulting D and E values. * denotes V4+ impurity in 3′, representing 1% of spin density in the sample. (middle) Magnetic field dependence of Tm (hollow symbols) at 6 K for (d) 1′, (e) 2′, and (f) 3 overlayed on the echo-detected field swept spectrum. Error bars are within the data points. (bottom) Temperature dependence of T1 (filled circles) and Tm (hollow circles) for (g) 1′, (h) 2′, and (i) 3′ at X-band frequency and measured at the magnetic field indicated by the arrow in the (d–f). | ||
While 1′ and 2′ show modest increases in |D| relative to their Cr4+ analogue (4′), 3′ exhibits a dramatic increase to 16.6 GHz, which is ∼13 times greater than its Cr4+ analogue (5′) where |D| = 1.23 GHz.40 To understand this order-of-magnitude increase in |D| for 3′, we quantified the deviation from ideal Td symmetry using the τ4 and τ4′ parameters54,55 for 3′ and 5′. Between 3′ and 5′, the τ4 and τ4′ values vary by only 0.004 and 0.015, respectively. While these geometric deviations are small, the absolute spin splitting energies of 3′ and 5′ vary by only 63 μeV, suggesting that subtle structural changes may result in significant variations in |D|. However, variations in |D| may also arise from a convolution of competing effects, such as changes in SOC, ligand field strength, symmetry-driven coupling to excited states, and electron delocalization.60,61 Thus, to maintain microwave addressability across diverse host media for sensing, it will be key to accurately predict and measure how growth on different surfaces or substrates affects local structure around the spin center and, consequently, |D|.62
The two V3+ complexes, 1′ and 3′, also exhibit two advantageous features. First, the crystal symmetries of 1′ and 3′ result in non-zero transverse ZFS, |E|, values of 0.8 and 2.2 GHz, respectively. Non-zero |E| should result in clock-like transitions around zero magnetic field at frequencies of |D − E|, |D + E| and 2|E|, resulting from mixing of the MS = +1 and −1 states (Fig. S1†).63 Clock-like transitions show little variation in their transition frequency with small changes in applied magnetic field.64,65 As a result, the clock transitions at zero magnetic field should exhibit longer coherence times, Tm, than typical Zeeman transitions, which exhibit a linear dependence on the external magnetic field.30,59,64–67 Second, the cw-EPR spectra in Fig. 2a and c exhibit hyperfine coupling to the 51V nucleus of 155 and 165 MHz for 1′ and 3′, respectively. Thus, the built-in 51V nuclear spin and relatively large hyperfine coupling to the electron spin provides a potential nuclear spin memory to prolong the storage time of quantum information.44,68,69 For example, previous work with S = 1/2 V4+ as well as 173Yb3+ molecular spins suggested that the coherence of strongly coupled nuclear spin may be prolonged beyond electronic spin coherence time by increasing the magnetic field strength.46,70 As a result, both the clock-like transitions around zero field and the intrinsic nuclear spin of the V3+-based systems provide potential routes to lengthen coherence lifetimes.
To then determine the spin dynamics of these Mo4+ and V3+ systems in their isostructural matrices, we examined the temperature and magnetic field dependence on spin-lattice relaxation (T1) and coherence (Tm) times for 1′–3′ at X-band frequency (9–10 GHz). In each case, these complexes exhibit shorter T1 times (10–15 μs) at low temperature than 4′ or 5′, where we previously measured T1 times of 2–3 ms at 5 K.40 However, the concentration of both 1′ and 3′ were 1.5 and 3% in the spin-diluted lattice, which we have previously shown leads to a reduction in T1 at low temperatures for 4′.40 Thus, T1 for 1′ and 3′ may likely be improved with further dilution. Notably, 1′ and 3′ exhibit T1 times of >0.5 μs up to 30 K while 2′ shows a steep decline in T1 < 0.5 μs by 18 K (Fig. 2g–i). These contrasting temperature dependencies likely arise from the larger spin–orbit coupling of the Mo4+-based spin centers, as well as lower energy phonon modes arising from the heavier metal center.71,72
Turning to the Tm times, a key metric to evaluate the performance of qubit candidates, we find relatively similar temperature dependencies for 1′–3′. In general, each compound exhibits Tm times of approximately 1 μs at 6 K, likely limited by the high density of 1H nuclei on the ligands.23,73,74 To verify that the nuclear spin environment inhibits Tm, we also performed power-dependent Hahn-echo experiments to mitigate the influence of instantaneous diffusion from nearby paramagnetic spin centers in relatively spin concentrated samples.64,75 We find that the Tm times of 1′ and 3′ show no significant change with decreasing power (Fig. S2†), suggesting that the nuclear spin environment of the matrix limits Tm in these samples. In contrast, the Tm time of 2′ approaches ∼2 μs at decreasing small microwave powers, which is similar to the Tm time previously measured for 4′ at similar concentrations40 (see ESI† for further details). This analysis does not result in large increases in Tm times, indicating that coherence times reported here are likely limited by the 1H nuclear spin environment. The Tm times then decrease with increasing temperature until the Tm times are limited by T1. The larger spin–orbit coupling in 2′ likely leads to T1-limited Tm times by 18 K while the V3+-based systems are measurable up to at least 30 K. However, both the T1 and Tm times in 1′ and 3′ are shorter than their Cr4+ congeners, 4′ and 5′.40 We posit that in these low field measurements, we may be either simultaneously driving multiple overlapping spin transitions (Fig. S1†) or driving a highly mixed spin transition, which has previously resulted in decreased spin-lattice relaxation times for an S = 5/2 Fe3+ complex.76 However, this assertion requires a multifrequency EPR study for validation, which will be the subject of future work. Additionally, we note that the T1 and Tm times reported here for 1′–3′ will vary substantially when measured in other matrices as the matrix tends to most strongly influence the spin dynamics of molecular qubits.77
We further evaluate the magnetic field dependence of these Tm times. Similar to the behavior of 4′,40 we find local maxima in Tm for 2′ at 170, 460, and 620 mT, where the applied field is parallel or perpendicular to the principal axis of the ZFS tensor (Fig. 2e). Across this field range, Tm changes by a factor of ∼4. For 1′ and 3′ where E > 0 and I = 7/2, the field dependence of Tm is less exaggerated since these systems exhibit are more transitions over a similar field range (Fig. S1†). As a result, Tm changes by less than a factor of 2 for 1′ over a similar field range to 2′. While the variation is less significant for 1′ and 3′, these data still highlight that no single value of Tm provides a complete picture of the spin relaxation, especially for anisotropic metal complexes. We similarly expect T1 to show a strong magnetic field dependence for 1′–3′, as demonstrated with Cr(o-tolyl)4.78
Most importantly, even the maxima in Tm times measured here still fall well short of coherence times for state-of-the-art quantum sensors, such as anionic nitrogen vacancy centers in diamond (e.g., from 10 s to 100 s of microseconds).79 In fact, low temperature (≤10 K) Tm times across most transition metal-based systems to date are ≤15 μs, regardless of spin state,30,73,77,80,81 ligand nuclear spin environment,77,82 or magnetic field,40,83 illustrating how the surrounding matrix promotes decoherence. Thus, introducing clock-like transitions (e.g., Fig. S1†) into molecular sensors while be critical to improve their coherence properties, and consequently their sensitivity,84 in magnetically noisy matrices.23,63
Turning to the excited state structure, we initially measured optical absorption spectra of these systems in solution at room temperature. The solution-phase UV-visible near-infrared spectra in Fig. 3a illustrate that the Mo4+ ion in 2 leads to both higher energy and more intense transitions than either of the first row congeners, 1 and 4. The UV-vis-NIR spectrum of 2 also appears qualitatively similar to 4, but the transitions in 2 are shifted to higher energy (Fig. S6†). This spectrum suggests that, similar to 4, the 1ES of 2 is the lowest energy excited state, which should yield the correct energy level structure for optical-spin control.34 Conversely, the V3+ ion in the pseudo-Tdo-tolyl ligand field of 1 leads to lower energy transitions than both 2 or 4. In fact, 1 exhibits absorption extending well into the NIR, leading to substantial spectral overlap with aromatic C–H overtones (see green and blue shaded regions in Fig. 3a), which may provide a multi-phonon mediated non-radiative decay pathway.85
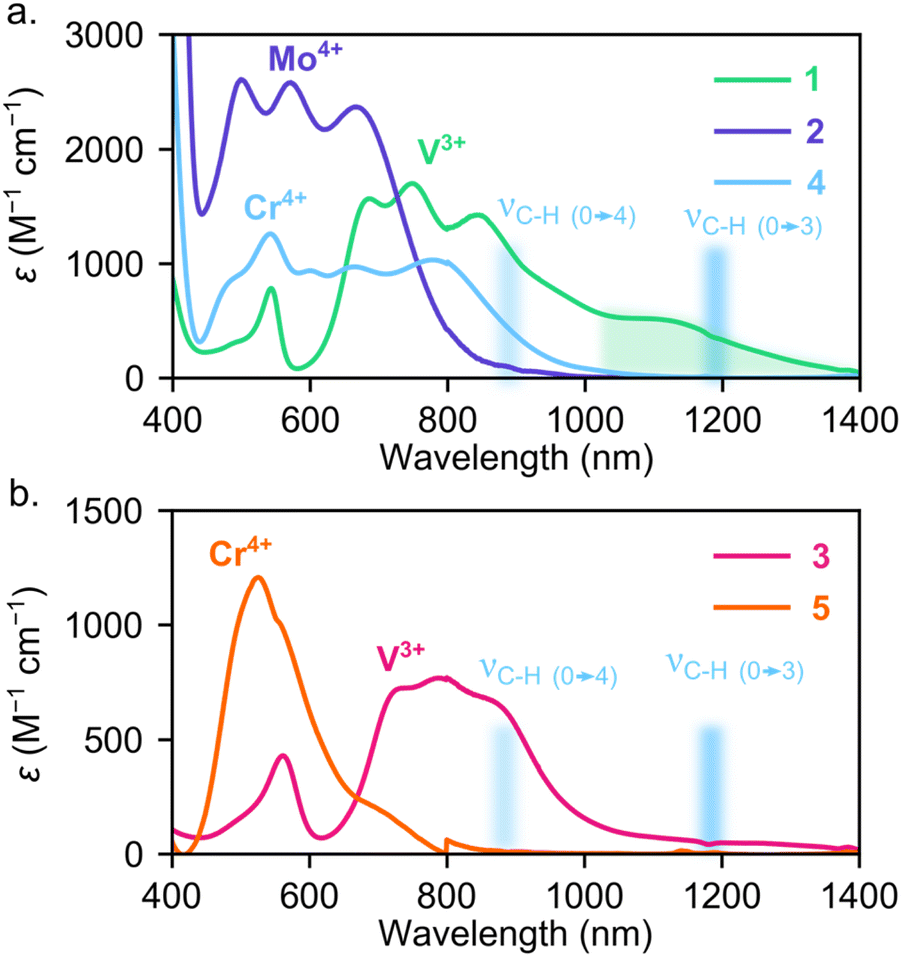 | ||
| Fig. 3 (a) Solution-state electronic absorption spectra of 1 and 2 compared with Cr4+ analogue, 4 (ref. 40). Green highlighted region indicates low energy absorption present for [V(aryl)4]− anions. (b) Solution-state electronic absorption spectra of 3 compared with Cr4+ analogue, 5 (ref. 40). Blue shaded region highlights energy of C–H stretching overtones that can mediate non-radiative decay. | ||
When replacing the aryl ligands in 1 with (trimethylsilyl)methyl ligands in 3, we find that the absorption features from 1100–1400 nm are suppressed while the shorter wavelength transitions from 400–1000 nm appear similar between 1 and 3. The reduction in absorption between 1100–1400 nm for 3 suggests that either the stronger alkyl ligand field increases the transition energy of the 3ES manifold or the σ-only alkyl ligands do not mix with the lowest energy 3ES metal-centered transitions, resulting in very low oscillator strengths of the d–d transitions. We observed similar behavior in tetraaryl- and tetralkyl-Cr4+ systems. For example, 4 exhibits intense absorption between 600 and 800 nm while those transitions are present, but much weaker, for 5.40
To determine if 1–3 exhibited emission from a 1ES, we then performed steady-state photoluminescence measurements. When exciting pure, single-crystalline samples of 1–3 at 4 K with 660 or 785 nm excitation, we observe no emission in the range of 900–1700 nm. Even performing photoluminescence experiments on dilute single-crystalline samples of 1′–3′ or films of 5–10% (w/w) of 1–3 in polystyrene at 4 K yielded no emission. The lack of emission prevents optical readout of the ground-state spin, the essential component for molecular color centers. We sought to understand why some systems emit and other similar ones did not, to see if failure might serve as a guide for future systems.
We performed spin polarized density functional theory calculations using VASP (Vienna Ab initio Simulation Package) 6.3.2 (ref. 86–89) with projector augmented wave pseudopotentials90,91 and the PBE exchange correlation functional92,93 (see ESI† for further details). For calculations of the charged molecules 1 and 3, we explicitly included counterions but, for clarity, we only highlight spin-bearing orbitals here. Fig. 4 shows the electronic structures of 1–3, highlighting the d character of the molecular orbitals. We visualized the highest occupied molecular orbital (HOMO) and spin up and down lowest unoccupied molecular orbital (LUMO) orbitals. Similar to 4 and 5,40 all molecules have a HOMO and LUMO with significant d character. Thus, the qualitative picture of all five compounds is similar, yet only 4 and 5 exhibit measurable emission.
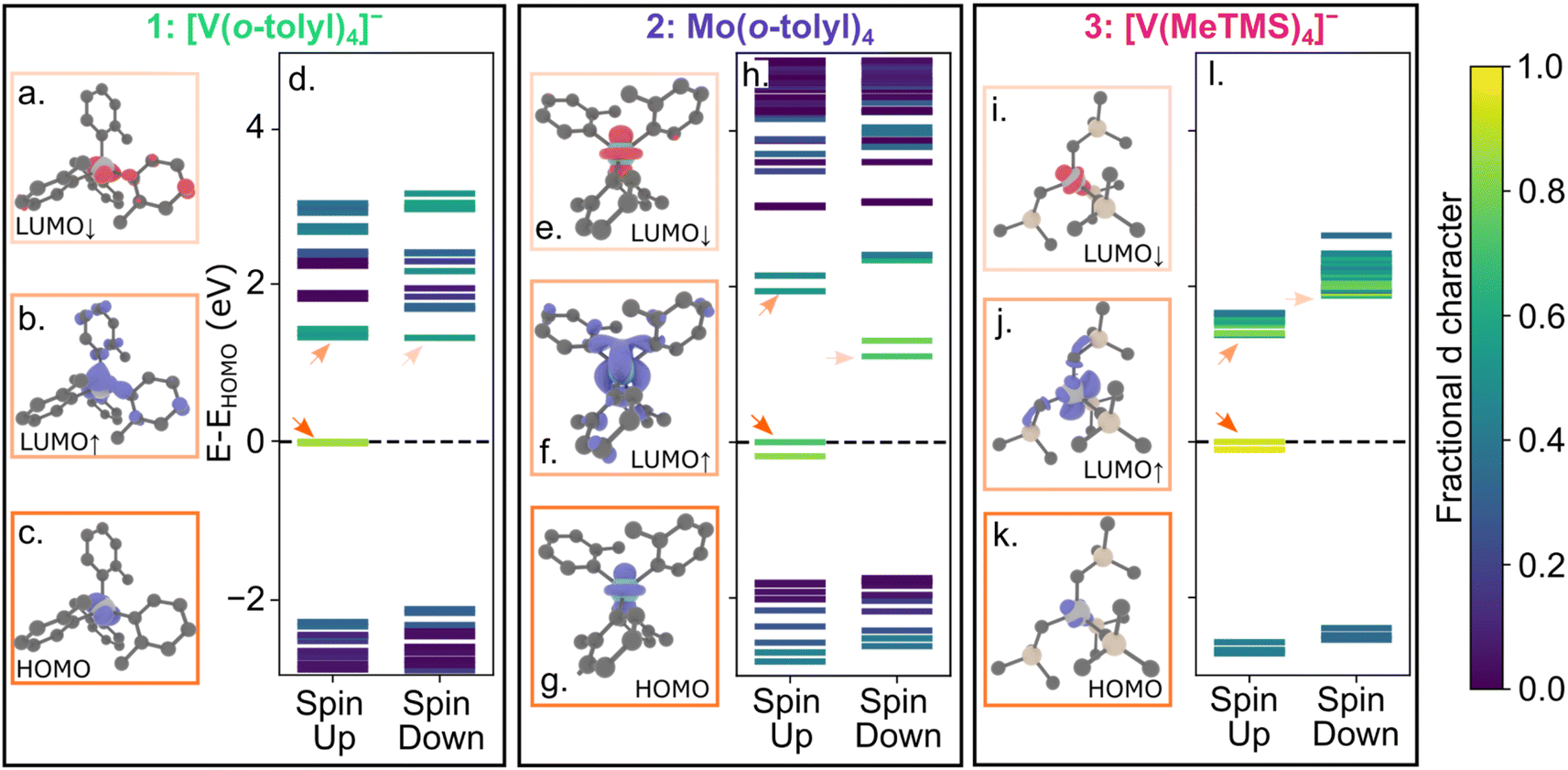 | ||
| Fig. 4 Visualizations of selected molecular orbitals for 1 (a–c), 2 (e–g), and 3 (i–k). The color of the box around each molecular orbital visualization corresponds to the quantitative molecular orbital diagram for 1 (d), 2 (h), and 3 (l). The LUMO↑ and LUMO↓ orbitals represent the first spin-triplet (3ES) and first spin-singlet (1ES) excited states, respectively. | ||
To evaluate where the 1ES lies relative to the 3GS manifold, we performed excited state calculations using the ΔSCF (delta self consistent field) method.94,95 We calculated the 1ES-3GS gap by subtracting the total energy of a constrained occupancy calculation, where the electron in the spin-up HOMO is promoted to the spin-down LUMO, from a standard ground-state DFT calculation (see ESI† for further details). We calculate 1ES-3GS gaps of 0.355 eV, 0.319 eV, and 0.407 eV, eV for 1, 2, and 3, respectively. Previously, we estimated the 1ES-3GS gap is 0.537 eV for 4 using the same methodology.62 Thus, the calculated 1ES energies for 1, 2, and 3 are only 66, 59, and 76% of 4. Although these values have limited quantitative accuracy, the trend suggests that the 1ES energy of 1 and 2 is significantly lower than 4. These results align with the expectation that the spin-pairing energetic penalty decreases with increasing ionic radii, such that the 1ES energies should be Mo4+ < V3+ < Cr4+ in the same ligand field. We further find that 3 also exhibits a lower calculated 1ES value than 4, despite the stronger alkyl ligand field of 3.
If we estimate hypothetical emission wavelengths by assuming the ratio between the calculated 1ES and experimental emission energies is similar to 4, where calculated 1ES/emission energy = 0.537/1.209 eV,34,62 we find that 1, 2, and 3 would emit at roughly 1550, 1800, and 1400 nm. Considering this wavelength range, the lack of measurable emission may therefore be rationalized by four possible effects. First, any photons emitted at longer wavelengths than 1700 nm are outside of the range of commercially available InGaAs NIR detectors that are optimal for typical NIR emission measurements. Second, if any emission occurs in the measurement window, the higher strain sensitivity of 1–3 with lower symmetry or larger spin orbit couplings could result in broader emission than Cr4 derivatives, resulting in fewer photons counted per pixel in the CCD measurements, rendering the measurement less sensitive. Third, the posited lower energy 1ES (≫1200 nm) may exhibit significant spectral overlap with high energy C–H stretching overtones (e.g., Fig. 3) that provide highly favorable non-radiative decay through multi-phonon mediated relaxation pathways. This potential explanation for the lack of emission could be ameliorated through selective deuteration or fluorination of the ligands.96 Further experimental and theoretical studies are required to investigate this pathway. Fourth, the energy-gap law suggests that as the emissive state becomes lower in energy, non-radiative decay rates will increase.97,98 In each compound, a combination of these effects may be operative, leading to no detectable emission. Based on the literature and our studies of similar compounds, we hypothesize that the C–H overtones are critical for quenching emission in this energy region.96,99–101 These results suggest a pathway forward for the design of molecular color centers featuring depleted C–H modes for compounds that emit in the near-IR or telecom region.
Designing emissive V3+ systems with a microwave addressable ground-state spin
The aggregate of these results provides us with a series of new S = 1 molecules featuring coherent control over their ground-state spin and a set of design principles for next generation quantum sensors. Previous data illustrate that emissive V3+ systems may be achieved using trigonal bipyramidal or octahedral geometries.102,103 However, trigonal bipyramidal or octahedral geometries with d2 metal ions result in either a non-cubic (e.g., C3v, D3h) symmetry or an orbitally degenerate (3T2) ground state, respectively. These features result in |D| values that well exceed frequencies of conventional microwave sources, making their ground-state spin control more challenging. Thus, we attempted to unify the emissive nature of five and six coordinate V3+ with the low |D| values of pseudo-Td by introducing the V3+ ion into sufficiently strong ligand fields. However, we never observed emission for V3+ systems, even with the strong-field alkyl donors of 3. These results suggest that a stronger ligand field should be enforced through a rigid ligand coordination with, for example, C3 symmetric ligand scaffolds (e.g. tris(pyridyl)methane, tris(N-heterocyclic carbene)borate) or C2-symmetric bidentate ligand scaffolds (e.g. 1,1′-binaphthyl-2,2′-diamine).104 Notably, increased rigidity is often ascribed to improved radiative efficiency of Cr3+ systems.96 However, the hard nature and high charge of early transition metals likely necessitates using suitably hard, anionic ligand donors, precluding the use of softer strong field ligands such as phosphines. Additionally, the lower energy transitions in V3+ systems likely undergo rapid non-radiative decay mediated by nearby C–H groups. Thus, the spectral overlap of these modes with excited states may be mitigated through deuteration or fluorination, also demonstrated with Cr3+ systems to greatly enhance emission.96,99,100 A similar approach may be employed with future systems to realize emission from these V3+ systems that feature generally small (e.g., <30 GHz) |D| values.Designing emissive Mo/W systems with microwave addressable ground-state spin
In contrast to the V3+ analogues, Mo4+-based systems pose a unique challenge. The energy gap between the 1ES and 3GS is generally quite small given the large size of the Mo4+ center. Additionally, to unlock the full potential of large spin–orbit coupling from the Mo4+ center to achieve enhanced sensitivities of the spin to electric field perturbations, the ion should sit in a polar molecular symmetry, such as C3v.45 For Mo4+ systems, these symmetries may simply yield an S = 0 complexes or prohibitively large |D| values. Thus, for Mo- and even W-based quantum sensors, an alternative spin-state may be better suited to enable strong spin-electric field coupling.47,105 For example, Mo5+S = 1/2 defects in silicon carbide have demonstrated NIR emission106 and may be capable of coherent optical control, similar to anionic tin vacancies in diamond.107 Thus, future investigations with S = 1/2 Mo5+/W5+ systems may provide a more suitable framework to achieve a ground-state spin capable of coherent control connected to an emissive optical state than the S = 1 derivatives with second or third row metal ions.Conclusions
In this work, we examined the electronic structure and spin dynamics of three novel V3+ and Mo4+ molecular color center candidates that could feature valuable spectroscopic handles for sensing. Notably, each system showed appropriately small ground-state anisotropy for coherent spin control. We demonstrated this control over a series of molecules, including a rare example of spin control in a second row transition metal with a spin-triplet ground state. We also suggest that the transverse anisotropy (i.e., |E| > 0) and nuclear spin of the V3+ analogues may offer two avenues to extend quantum coherence. Despite these potential advantageous features, none of the systems investigated here exhibited measurable emission, likely resulting from a combination of a low energy 1ES and rapid non-radiative decay mediated by the C–H-rich ligand environment. Coupling experimental and computational insight, we suggest that the use of ligand scaffolds and deuteration/fluorination may result in emissive V3+ while maintaining ground-state spin control, while larger transition metal ions, such as Mo and W, in S = 1/2 states may be better suited for optical-spin control. Thus, we offer design considerations that will hopefully lead to the realization of V- or Mo-based molecular quantum sensors in future studies.Data availability
The data underlying this work are available at https://doi.org/10.5281/zenodo.12794616. CCDC codes: 1 (2334908), 2 at 100 K (2334909), 3 (2334912), 1-Al (2334910), and 3-Al (2334911).Author contributions
D. W. L., L. R. W., S. L. B., P. D. performed the experimental measurements. K. R. M. performed the computations. D. W. L. synthesized the compounds. D. D. A., J. M. R. and D. E. F. advised on all efforts. All authors contributed to the data analysis and manuscript preparation.Conflicts of interest
The authors declare the following competing financial interests: D. W. L., S. L. B., D. E. F., and D. D. A. are inventors on patent application no. 63008589 submitted by the University of Chicago that covers chemically tunable optically addressable molecular-spin qubits and associated methods. S. L. B., P. D., D. W. L., D. E. F., and D. D. A. are inventors on patent application No. 22-T121 by the University of Chicago that covers enhancing quantum properties of optically addressable molecular qubits through host-matrix engineering.Acknowledgements
We thank Dr Peter Mintun and Dr Berk Kovos for scientific discussion and comments on the manuscript. We thank Dr Yifan Quan and Rebecca Sponenburg for experimental assistance with pulsed EPR experiments and ICP-OES analysis, respectively. D. W. L. and D. E. F gratefully acknowledge support from the US Army Research Office under award number W911NF2010088 for the synthesis and design of new molecular color centers. This work used resources at the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 using NERSC Award No. BES-ERCAP0020980. S. L. B. acknowledges support from UK Research and Innovation [grant number MR/W006928/1]. L. R. W. acknowledges the support from the University of Chicago/Advanced Institute for Materials Research Joint Research Center. Creating these molecules supports the work of Q-NEXT, one of the U.S. Department of Energy Office of Science National Quantum Information Science Research Centers, and will enable integration of these molecules into a larger quantum ecosystem. Work on magnetic dilution of these molecules, as a pathway for thin layer deposition was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under Award No. DE-SC0019356. This work made use of the IMSERC Crystallography facility at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633), and Northwestern University. For the purpose of open access, the authors have applied a Creative Commons Attribution (CC BY) license to any Author Accepted Manuscript version arising from this submission.References
- L. M. K. Vandersypen, H. Bluhm, J. S. Clarke, A. S. Dzurak, R. Ishihara, A. Morello, D. J. Reilly, L. R. Schreiber and M. Veldhorst, Interfacing Spin Qubits in Quantum Dots and Donors—Hot, Dense, and Coherent, Npj Quantum Inf., 2017, 3, 1–10 CrossRef.
- R. Li, L. Petit, D. P. Franke, J. P. Dehollain, J. Helsen, M. Steudtner, N. K. Thomas, Z. R. Yoscovits, K. J. Singh, S. Wehner, L. M. K. Vandersypen, J. S. Clarke and M. Veldhorst, A Crossbar Network for Silicon Quantum Dot Qubits, Sci. Adv., 2018, 4, eaar3960 CrossRef PubMed.
- B. C. Rose, D. Huang, Z. H. Zhang, A. M. Tyryshkin, S. Sangtawesin, S. Srinivasan, L. Loudin, M. L. Markham, A. M. Edmonds, D. J. Twitchen, S. A. Lyon and N. P. de Leon, Observation of an Environmentally Insensitive Solid State Spin Defect in Diamond, Science, 2018, 361, 60–63 CrossRef CAS PubMed.
- E. J. Davis, G. Bentsen, L. Homeier, T. Li and M. H. Schleier-Smith, Photon-Mediated Spin-Exchange Dynamics of Spin-1 Atoms, Phys. Rev. Lett., 2019, 122, 10405 CrossRef CAS PubMed.
- M. Atzori and R. Sessoli, The Second Quantum Revolution: Role and Challenges of Molecular Chemistry, J. Am. Chem. Soc., 2019, 141, 11339–11352 CrossRef CAS PubMed.
- M. Kjaergaard, M. E. Schwartz, J. Braumüller, P. Krantz, J. I. J. Wang, S. Gustavsson and W. D. Oliver, Superconducting Qubits: Current State of Play, Annu. Rev. Condens. Matter Phys., 2020, 11, 369–395 CrossRef.
- D. D. Awschalom, R. Hanson, J. Wrachtrup and B. B. Zhou, Quantum Technologies with Optically Interfaced Solid-State Spins, Nat. Photonics, 2018, 12, 516–527 CrossRef CAS.
- A. M. Ferrenti, N. P. de Leon, J. D. Thompson and R. J. Cava, Identifying Candidate Hosts for Quantum Defects via Data Mining, npj Comput. Mater., 2020, 6, 2–7 CrossRef.
- I. Harris, C. J. Ciccarino, J. Flick, D. R. Englund and P. Narang, Group-III Quantum Defects in Diamond Are Stable Spin-1 Color Centers, Phys. Rev. B, 2020, 102, 195206 CrossRef CAS.
- H. S. Zhong, H. Wang, Y. H. Deng, M. C. Chen, L. C. Peng, Y. H. Luo, J. Qin, D. Wu, X. Ding, Y. Hu, P. Hu, X. Y. Yang, W. J. Zhang, H. Li, Y. Li, X. Jiang, L. Gan, G. Yang, L. You, Z. Wang, L. Li, N. L. Liu, C. Y. Lu and J. W. Pan, Quantum Computational Advantage Using Photons, Science, 2020, 370, 1460–1463 CrossRef CAS PubMed.
- F. Shi, Q. Zhang, P. Wang, H. Sun, J. J. Wang, X. Rong, M. Chen, C. Ju, F. Reinhard, H. Chen, J. Wrachtrup, J. J. Wang and J. Du, Single-Protein Spin Resonance Spectroscopy under Ambient Conditions, Science, 2015, 347, 1135–1138 CrossRef CAS PubMed.
- I. Lovchinsky, A. O. Sushkov, E. Urbach, N. P. de Leon, S. Choi, K. De Greve, R. Evans, R. Gertner, E. Bersin, C. Müller, L. McGuinness, F. Jelezko, R. L. Walsworth, H. Park and M. D. Lukin, Nuclear Magnetic Resonance Detection and Spectroscopy of Single Proteins Using Quantum Logic, Science, 2016, 351, 836–841 CrossRef CAS PubMed.
- A. A. Clerk, K. W. Lehnert, P. Bertet, J. R. Petta and Y. Nakamura, Hybrid Quantum Systems with Circuit Quantum Electrodynamics, Nat. Phys., 2020, 16, 257–267 Search PubMed.
- D. D. Awschalom, C. R. Du, R. He, F. Joseph Heremans, A. Hoffmann, J. Hou, H. Kurebayashi, Y. Li, L. Liu, V. Novosad, J. Sklenar, S. E. Sullivan, D. Sun, H. Tang, V. Tyberkevych, C. Trevillian, A. W. Tsen, L. R. Weiss, W. Zhang, X. Zhang, L. Zhao and C. H. W. Zollitsch, Quantum Engineering With Hybrid Magnonic Systems and Materials, IEEE Trans. Quantum Eng., 2021, 2, 5500836 Search PubMed.
- J. F. Barry, M. J. Turner, J. M. Schloss, D. R. Glenn, Y. Song, M. D. Lukin, H. Park and R. L. Walsworth, Optical Magnetic Detection of Single-Neuron Action Potentials Using Quantum Defects in Diamond, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14133–14138 CrossRef CAS PubMed.
- M. J. H. Ku, T. X. Zhou, Q. Li, Y. J. Shin, J. K. Shi, C. Burch, L. E. Anderson, A. T. Pierce, Y. Xie, A. Hamo, U. Vool, H. Zhang, F. Casola, T. Taniguchi, K. Watanabe, M. M. Fogler, P. Kim, A. Yacoby and R. L. Walsworth, Imaging Viscous Flow of the Dirac Fluid in Graphene, Nature, 2020, 583, 537–541 CrossRef CAS PubMed.
- U. Vool, A. Hamo, G. Varnavides, Y. Wang, T. X. Zhou, N. Kumar, Y. Dovzhenko, Z. Qiu, C. A. C. Garcia, A. T. Pierce, J. Gooth, P. Anikeeva, C. Felser, P. Narang and A. Yacoby, Imaging Phonon-Mediated Hydrodynamic Flow in WTe2, Nat. Phys., 2021, 17, 1216–1220 Search PubMed.
- F. J. Heremans, C. G. Yale and D. D. Awschalom, Control of Spin Defects in Wide-Bandgap Semiconductors for Quantum Technologies, Proc. IEEE, 2016, 104, 2009–2023 CAS.
- M. W. Doherty, N. B. Manson, P. Delaney, F. Jelezko, J. Wrachtrup and L. C. L. Hollenberg, The Nitrogen-Vacancy Colour Centre in Diamond, Phys. Rep., 2013, 528, 1–45 CrossRef CAS.
- D. J. Christle, A. L. Falk, P. Andrich, P. V. Klimov, J. U. Hassan, N. T. Son, E. Janzén, T. Ohshima and D. D. Awschalom, Isolated Electron Spins in Silicon Carbide with Millisecond Coherence Times, Nat. Mater., 2015, 14, 160–163 CrossRef CAS PubMed.
- S. Chen, M. Raha, C. M. Phenicie, S. Ourari and J. D. Thompson, Parallel Single-Shot Measurement and Coherent Control of Solid-State Spins below the Diffraction Limit, Science, 2020, 370, 592–595 CrossRef CAS PubMed.
- P. Siyushev, K. Xia, R. Reuter, M. Jamali, N. Zhao, N. Yang, C. Duan, N. Kukharchyk, A. D. Wieck, R. Kolesov and J. Wrachtrup, Coherent Properties of Single Rare-Earth Spin Qubits, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- G. Wolfowicz, F. J. Heremans, C. P. Anderson, S. Kanai, H. Seo, A. Gali, G. Galli and D. D. Awschalom, Quantum Guidelines for Solid-State Spin Defects, Nat. Rev. Mater., 2021, 6, 906–925 CrossRef CAS.
- D. P. DiVincenzo, The Physical Implementation of Quantum Computation, Fortschr. Phys., 2000, 48, 771–783 CrossRef.
- C. L. Degen, F. Reinhard and P. Cappellaro, Quantum Sensing, Rev. Mod. Phys., 2017, 89, 1–39 CrossRef.
- T. Yamabayashi, M. Atzori, L. Tesi, G. Cosquer, F. Santanni, M. E. Boulon, E. Morra, S. Benci, R. Torre, M. Chiesa, L. Sorace, R. Sessoli and M. Yamashita, Scaling Up Electronic Spin Qubits into a Three-Dimensional Metal-Organic Framework, J. Am. Chem. Soc., 2018, 140, 12090–12101 CrossRef CAS PubMed.
- A. M. Ariciu, D. H. Woen, D. N. Huh, L. E. Nodaraki, A. K. Kostopoulos, C. A. P. Goodwin, N. F. Chilton, E. J. L. McInnes, R. E. P. Winpenny, W. J. Evans and F. Tuna, Engineering Electronic Structure to Prolong Relaxation Times in Molecular Qubits by Minimising Orbital Angular Momentum, Nat. Commun., 2019, 10, 1–8 CrossRef CAS PubMed.
- I. Cimatti, L. Bondì, G. Serrano, L. Malavolti, B. Cortigiani, E. Velez-Fort, D. Betto, A. Ouerghi, N. B. Brookes, S. Loth, M. Mannini, F. Totti and R. Sessoli, Vanadyl Phthalocyanines on Graphene/SiC(0001): Toward a Hybrid Architecture for Molecular Spin Qubits, Nanoscale Horiz., 2019, 4, 1202–1210 RSC.
- S. Thiele, F. Balestro, R. Ballou, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Electrically Driven Nuclear Spin Resonance in Single-Molecule Magnets, Science, 2014, 344, 1135–1138 CrossRef CAS PubMed.
- J. M. Zadrozny, A. T. Gallagher, T. D. Harris and D. E. Freedman, A Porous Array of Clock Qubits, J. Am. Chem. Soc., 2017, 139, 7089–7094 CrossRef CAS PubMed.
- C. J. Yu, M. D. Krzyaniak, M. S. Fataftah, M. R. Wasielewski and D. E. Freedman, A Concentrated Array of Copper Porphyrin Candidate Qubits, Chem. Sci., 2019, 10, 1702–1708 RSC.
- M. S. Fataftah and D. E. Freedman, Progress towards Creating Optically Addressable Molecular Qubits, Chem. Commun., 2018, 54, 13773–13781 RSC.
- C. J. Yu, S. Von Kugelgen, D. W. Laorenza and D. E. Freedman, A Molecular Approach to Quantum Sensing, ACS Cent. Sci., 2021, 7, 712–723 CrossRef CAS PubMed.
- S. L. Bayliss, D. W. Laorenza, P. J. Mintun, B. Diler, D. E. Freedman and D. D. Awschalom, Optically Addressable Molecular Spins for Quantum Information Processing, Science, 2020, 370, 1309–1312 CrossRef CAS PubMed.
- R. Schirhagl, K. Chang, M. Loretz and C. L. Degen, Nitrogen-Vacancy Centers in Diamond: Nanoscale Sensors for Physics and Biology, Annu. Rev. Phys. Chem., 2014, 65, 83–105 CrossRef CAS PubMed.
- A. L. Falk, B. B. Buckley, G. Calusine, W. F. Koehl, V. V. Dobrovitski, A. Politi, C. A. Zorman, P. X. L. Feng and D. D. Awschalom, Polytype Control of Spin Qubits in Silicon Carbide, Nat. Commun., 2013, 4, 1–7 Search PubMed.
- W. F. Koehl, B. Diler, S. J. Whiteley, A. Bourassa, N. T. Son, E. Janzén and D. D. Awschalom, Resonant Optical Spectroscopy and Coherent Control of Cr4+, Phys. Rev. B, 2017, 95, 035207 CrossRef.
- B. Diler, S. J. Whiteley, C. P. Anderson, G. Wolfowicz, M. E. Wesson, E. S. Bielejec, F. Joseph Heremans and D. D. Awschalom, Coherent Control and High-Fidelity Readout of Chromium Ions in Commercial Silicon Carbide, Npj Quantum Inf., 2020, 6, 11 CrossRef.
- N. B. Figgis and A. Hitchman, M. Tanabe-Sugano Diagrams, in Ligand Field Theory and Its Applications, Wiley-VCH, New York, 2000, pp 131–141 Search PubMed.
- D. W. Laorenza, A. Kairalapova, S. L. Bayliss, T. Goldzak, S. M. Greene, L. R. Weiss, P. Deb, P. J. Mintun, K. A. Collins, D. D. Awschalom, T. C. Berkelbach and D. E. Freedman, Tunable Cr4+ Molecular Color Centers, J. Am. Chem. Soc., 2021, 143, 21350–21363 CrossRef CAS PubMed.
- J. Krzystek, J. H. Park, M. W. Meisel, M. A. Hitchma, H. Stratemeier, L. C. Brunel and J. Telser, EPR Spectra from “EPR-Silent” Species: High-Frequency and High-Field EPR Spectroscopy of Pseudotetrahedral Complexes of Nickel(II), Inorg. Chem., 2002, 41, 4478–4487 CrossRef CAS PubMed.
- J. Krzystek, A. Ozarowski, J. Telser and D. C. Crans, High-Frequency and -Field Electron Paramagnetic Resonance of Vanadium(IV, III, and II) Complexes, Coord. Chem. Rev., 2015, 301–302, 123–133 CrossRef CAS.
- P. Neumann, R. Kolesov, B. Naydenov, J. Beck, F. Rempp, M. Steiner, V. Jacques, G. Balasubramanian, M. L. Markham, D. J. Twitchen, S. Pezzagna, J. Meijer, J. Twamley, F. Jelezko and J. Wrachtrup, Quantum Register Based on Coupled Electron Spins in a Room-Temperature Solid, Nat. Phys., 2010, 6, 249–253 Search PubMed.
- A. Ruskuc, C.-J. Wu, J. Rochman, J. Choi and A. Faraon, Nuclear Spin-Wave Quantum Register for a Solid-State Qubit, Nature, 2022, 602, 408–413 CrossRef CAS PubMed.
- F. Dolde, H. Fedder, M. W. Doherty, T. Nöbauer, F. Rempp, G. Balasubramanian, T. Wolf, F. Reinhard, L. C. L. Hollenberg, F. Jelezko and J. Wrachtrup, Electric-Field Sensing Using Single Diamond Spins, Nat. Phys., 2011, 7, 459–463 Search PubMed.
- S. Chicco, A. Chiesa, G. Allodi, E. Garlatti, M. Atzori, L. Sorace, R. De Renzi and S. Carretta, Controlled Coherent Dynamics of [VO(TPP)], a Prototype Molecular Nuclear Qudit with an Electronic Ancilla, Chem. Sci., 2021, 12, 12046–12055 RSC.
- J. Liu, J. Mrozek, W. K. Myers, G. A. Timco, R. E. P. Winpenny, B. Kintzel, W. Plass and A. Ardavan, Electric Field Control of Spins in Molecular Magnets, Phys. Rev. Lett., 2019, 122, 37202 CrossRef CAS PubMed.
- Z. Liu, Y. Wang, Y. Fang, S. Qin, Z. Wang, S. Jiang and S. Gao, Electric Field Manipulation Enhanced by Strong Spin-Orbit Coupling: Promoting Rare-Earth Ions as Qubits, Natl. Sci. Rev., 2020, 7, 1557–1563 CrossRef CAS PubMed.
- J. Arnold, G. Wilkinson, B. Hussain and M. B. Hursthouse, Synthesis and X-Ray Crystal Structure of Tetra (2- Methylphenyl)Molybdenum(IV), Mo(2-MeC6H4)4. Redox Chemistry of M(2-MeC6H4)4 Compounds of Molybdenum Rhenium, Ruthenium, and Osmium, J. Chem. Soc., Dalton Trans., 1989, 2149–2153 RSC.
- S. U. Koschmieder, B. S. McGilligan, G. McDermott, J. Arnold, G. Wilkinson, B. Hussain-Bates and M. B. Hursthouse, Aryl and Aryne Complexes of Chromium, Molybdenum, and Tungsten. X-Ray Crystal Structures of [Cr(2-MeC6H4)(μ-2-MeC6H4)(PMe3)]2, Mo(η2-2-MeC6H3)(2-MeC6H4)2(PMe2Ph)2, and W(η2-2,5-Me2C6H2)(2,5-Me2C6H3)2-(PMe3)2, J. Chem. Soc., Dalton Trans., 1990,(11), 3427–3433 RSC.
- C. Schulzke, D. Enright, H. Sugiyama, G. LeBlanc, S. Gambarotta, G. P. A. Yap, L. K. Thompson, D. R. Wilson and R. Duchateau, The Unusual Stability of Homoleptic Di- and Tetravalent Chromium Alkyls, Organometallics, 2002, 21, 3810–3816 CrossRef CAS.
- B. F. Huq, W. Mowat, A. Shortland, A. C. Skapski and G. Wilkinson, Crystal Structure of Hexakis (Trimethylsilylmethyl) Dimolybdenum, J. Chem. Soc. D, 1971, 1079–1080 RSC.
- W. Mowat, A. Shortland, G. Yagupsky, N. J. Hill, M. Yagupsky and G. Wilkinson, Elimination Stabilized Alkyls. Part I. Chromium, Molybdenum, Tungsten, and Vanadium, J. Chem. Soc., Dalton Trans., 1972, 533–542 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Structural Variation in Copper(i) Complexes with Pyridylmethylamide Ligands: Structural Analysis with a New Four-Coordinate Geometry Index, T4, J. Chem. Soc., Dalton Trans., 2007,(9), 955–964 RSC.
- A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Coordination Polymers and Molecular Structures among Complexes of Mercury(II) Halides with Selected 1-Benzoylthioureas, Polyhedron, 2015, 90, 47–57 CrossRef CAS.
- P. J. Alonso, J. Forniés, M. A. García-Monforte, A. Martín and B. Menjón, New Homoleptic Organometallic Derivatives of Vanadium(III) and Vanadium(IV): Synthesis, Characterization, and Study of Their Electrochemical Behaviour, Chem.–Eur. J., 2005, 11, 4713–4724 CrossRef CAS PubMed.
- C. Schneider-Koglin, B. Mathiasch and M. Dräger, Über Tetraaryl-Methan-Analoga in Der Gruppe 14. III. Ar4Sn/Pb (Ar Ph, p-, m-, o-Tol, 2,4-Xyl Und 2,5-Xyl): Gegenüberstellung von Bindungslängen Und Winkeln, von NMR Chemischen Verschiebungen Und Kopplungskonstanten Und von Schwingungsdaten, J. Organomet. Chem., 1994, 469, 25 CrossRef CAS.
- B. N. Figgis and M. A. Hitchman, Spin-Orbit Coupling Constants, Ligand Field Theory and Its Applications, Wiley-VCH, New York, 2000, pp 108–110 Search PubMed.
- S. L. Bayliss, P. Deb, D. W. Laorenza, M. Onizhuk, G. Galli, D. E. Freedman and D. D. Awschalom, Enhancing Spin Coherence in Optically Addressable Molecular Qubits through Host-Matrix Control, Phys. Rev. X, 2022, 12, 031028 CAS.
- S. Ye, F. Neese, A. Ozarowsk, D. Smirnov, J. Krzystek, J. Telser, J. H. Liao, C. H. Hung, W. C. Chu, Y. F. Tsai, R. C. Wang, K. Y. Chen and H. F. Hsu, Family of V(III)-Tristhiolato Complexes Relevant to Functional Models of Vanadium Nitrogenase: Synthesis and Electronic Structure Investigations by Means of High-Frequency and -Field Electron Paramagnetic Resonance Coupled to Quantum Chemical Computations, Inorg. Chem., 2010, 49, 977–988 CrossRef CAS PubMed.
- C. V. Stappen, D. Maganas, S. Debeer, E. Bill and F. Neese, Investigations of the Magnetic and Spectroscopic Properties of V (III) and V (IV) Complexes, Inorg. Chem., 2018, 57, 6421–6438 CrossRef PubMed.
- K. R. Mullin, D. W. Laorenza, D. E. Freedman and J. M. Rondinelli, Quantum Sensing of Magnetic Fields with Molecular Color Centers, Phys. Rev. Res., 2023, 5, 1–9 Search PubMed.
- M. Onizhuk, K. C. Miao, J. P. Blanton, H. Ma, C. P. Anderson, A. Bourassa, D. D. Awschalom and G. Galli, Probing the Coherence of Solid-State Qubits at Avoided Crossings, PRX Quantum, 2021, 2, 010311 CrossRef.
- G. Wolfowicz, A. M. Tyryshkin, R. E. George, H. Riemann, N. V. Abrosimov, P. Becker, H. J. Pohl, M. L. W. Thewalt, S. A. Lyon and J. J. L. Morton, Atomic Clock Transitions in Silicon-Based Spin Qubits, Nat. Nanotechnol., 2013, 8, 561–564 CrossRef CAS PubMed.
- K. C. Miao, J. P. Blanton, C. P. Anderson, A. Bourassa, A. L. Crook, G. Wolfowicz, H. Abe, T. Ohshima and D. D. Awschalom, Universal Coherence Protection in a Solid-State Spin Qubit, Science, 2020, 369, 1493–1497 CrossRef CAS PubMed.
- M. Shiddiq, D. Komijani, Y. Duan, A. Gaita-Ariño, E. Coronado and S. Hill, Enhancing Coherence in Molecular Spin Qubits via Atomic Clock Transitions, Nature, 2016, 531, 348–351 CrossRef CAS PubMed.
- K. Kundu, J. R. K. White, S. A. Moehring, J. M. Yu, J. W. Ziller, F. Furche, W. J. Evans and S. Hill, A 9.2-GHz Clock Transition in a Lu(II) Molecular Spin Qubit Arising from a 3,467-MHz Hyperfine Interaction, Nat. Chem., 2022, 14, 392–397 CrossRef CAS PubMed.
- G. Wolfowicz, C. P. Anderson, B. Diler, O. G. Poluektov, F. J. Heremans and D. D. Awschalom, Vanadium Spin Qubits as Telecom Quantum Emitters in Silicon Carbide, Sci. Adv., 2020, 6, eaaz1192 CrossRef CAS PubMed.
- C. E. Bradley, J. Randall, M. H. Abobeih, R. C. Berrevoets, M. J. Degen, M. A. Bakker, M. Markham, D. J. Twitchen and T. H. Taminiau, A Ten-Qubit Solid-State Spin Register with Quantum Memory up to One Minute, Phys. Rev. X, 2019, 9, 031045 CAS.
- R. Hussain, G. Allodi, A. Chiesa, E. Garlatti, D. Mitcov, A. Konstantatos, K. S. Pedersen, R. De Renzi, S. Piligkos and S. Carretta, Coherent Manipulation of a Molecular Ln-Based Nuclear Qudit Coupled to an Electron Qubit, J. Am. Chem. Soc., 2018, 140, 9814–9818 CrossRef CAS PubMed.
- P. L. Scott and C. D. Jeffries, Spin-Lattice Relaxation in Some Rare-Earth Salts at Helium Temperatures; Observation of the Phonon Bottleneck, Phys. Rev., 1962, 127, 32–51 CrossRef CAS.
- M. Briganti, F. Santanni, L. Tesi, F. Totti, R. Sessoli and A. Lunghi, A Complete Ab Initio View of Orbach and Raman Spin-Lattice Relaxation in a Dysprosium Coordination Compound, J. Am. Chem. Soc., 2021, 143, 13633–13645 CrossRef CAS PubMed.
- A. Ardavan, O. Rival, J. J. L. Morton, S. J. Blundell, A. M. Tyryshkin, G. A. Timco and R. E. P. Winpenny, Will Spin-Relaxation Times in Molecular Magnets Permit Quantum Information Processing?, Phys. Rev. Lett., 2007, 98, 057201 CrossRef PubMed.
- A. Bourassa, C. P. Anderson, K. C. Miao, M. Onizhuk, H. Ma, A. L. Crook, H. Abe, J. Ul-Hassan, T. Ohshima, N. T. Son, G. Galli and D. D. Awschalom, Entanglement and Control of Single Nuclear Spins in Isotopically Engineered Silicon Carbide, Nat. Mater., 2020, 19, 1319–1325 CrossRef CAS PubMed.
- S. Von Kugelgen, M. D. Krzyaniak, M. Gu, D. Puggioni, J. M. Rondinelli, M. R. Wasielewski and D. E. Freedman, Spectral Addressability in a Modular Two Qubit System, J. Am. Chem. Soc., 2021, 143, 8069–8077 CrossRef PubMed.
- J. M. Zadrozny, M. J. Graham, M. D. Krzyaniak, M. R. Wasielewski and D. E. Freedman, Unexpected Suppression of Spin-Lattice Relaxation via High Magnetic Field in a High-Spin Iron(III) Complex, Chem. Commun., 2016, 52, 10175–10178 RSC.
- J. M. Zadrozny, J. Niklas, O. G. Poluektov and D. E. Freedman, Millisecond Coherence Time in a Tunable Molecular Electronic Spin Qubit, ACS Cent. Sci., 2015, 1, 488–492 CrossRef CAS PubMed.
- N. P. Kazmierczak, K. M. Luedecke, E. T. Gallmeier and R. G. Hadt, T1 Anisotropy Elucidates Spin Relaxation Mechanisms in an S = 1 Cr(IV) Optically Addressable Molecular Qubit, J. Phys. Chem. Lett., 2023, 14, 7658–7664 CrossRef CAS PubMed.
- E. Bauch, S. Singh, J. Lee, C. A. Hart, J. M. Schloss, M. J. Turner, J. F. Barry, L. M. Pham, N. Bar-Gill, S. F. Yelin and R. L. Walsworth, Decoherence of Ensembles of Nitrogen-Vacancy Centers in Diamond, Phys. Rev. B, 2020, 102, 134210 CrossRef CAS.
- M. J. Amdur, K. R. Mullin, M. J. Waters, D. Puggioni, M. K. Wojnar, M. Gu, L. Sun, P. H. Oyala, J. M. Rondinelli and D. E. Freedman, Chemical Control of Spin-Lattice Relaxation to Discover a Room Temperature Molecular Qubit, Chem. Sci., 2022, 13, 7034–7045 RSC.
- S. Lenz, H. Bamberger, P. P. Hallmen, Y. Thiebes, S. Otto, K. Heinze and J. Van Slageren, Chromium(III)-Based Potential Molecular Quantum Bits with Long Coherence Times, Phys. Chem. Chem. Phys., 2019, 21, 6976–6983 RSC.
- C. E. Jackson, C. Y. Lin, S. H. Johnson, J. Van Tol and J. M. Zadrozny, Nuclear-Spin-Pattern Control of Electron-Spin Dynamics in a Series of V(IV) Complexes, Chem. Sci., 2019, 10, 8447–8454 RSC.
- C. E. Jackson, C.-Y. Lin, J. van Tol and J. M. Zadrozny, Orientation Dependence of Phase Memory Relaxation in the V(IV) Ion at High Frequencies, Chem. Phys. Lett., 2020, 739, 137034 CrossRef CAS.
- J. F. Barry, J. M. Schloss, E. Bauch, M. J. Turner, C. A. Hart, L. M. Pham and R. L. Walsworth, Sensitivity Optimization for NV-Diamond Magnetometry, Rev. Mod. Phys., 2020, 92, 15004 CrossRef CAS.
- W. Robert, K. Johannes and M. Katja, Spin-flip Luminescence, Photochem. Photobiol. Sci., 2022, 21, 1309–1331 CrossRef PubMed.
- G. Kresse and J. Hafner, Ab Initio Molecular Dynamics for Liquid Metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal-Amorphous-Semiconductor Transition in Germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- J. Gavnholt, T. Olsen, M. Engelund and J. Schiøtz, Δ Self-Consistent Field Method to Obtain Potential Energy Surfaces of Excited Molecules on Surfaces, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 75441 CrossRef.
- A. Hellman, B. Razaznejad and B. I. Lundqvist, Potential-Energy Surfaces for Excited States in Extended Systems, J. Chem. Phys., 2004, 120, 4593–4602 CrossRef CAS PubMed.
- S. Otto, M. Dorn, C. Förster, M. Bauer, M. Seitz and K. Heinze, Understanding and Exploiting Long-Lived near-Infrared Emission of a Molecular Ruby, Coord. Chem. Rev., 2018, 359, 102–111 CrossRef CAS.
- R. Englman and J. Jortner, The Energy Gap Law for Radiationless Transitions in Large Molecules, Mol. Phys., 1970, 18, 285–287 CrossRef.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Spin-Vibronic Mechanism for Intersystem Crossing, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- C. Wang, S. Otto, M. Dorn, E. Kreidt, J. Lebon, L. Sršan, P. Di Martino-Fumo, M. Gerhards, U. Resch-Genger, M. Seitz and K. Heinze, Deuterated Molecular Ruby with Record Luminescence Quantum Yield, Angew. Chem., Int. Ed., 2018, 57, 1112–1116 CrossRef CAS PubMed.
- C. Wegeberg and O. S. Wenger, Luminescent First-Row Transition Metal Complexes, JACS Au, 2021, 1, 1860–1876 CrossRef CAS PubMed.
- O. S. Wenger, Photoactive Complexes with Earth-Abundant Metals, J. Am. Chem. Soc., 2018, 140, 13522–13533 CrossRef CAS PubMed.
- M. S. Fataftah, S. L. Bayliss, D. W. Laorenza, X. Wang, B. T. Phelan, C. B. Wilson, P. J. Mintun, B. D. Kovos, M. R. Wasielewski, S. Han, M. S. Sherwin, D. D. Awschalom and D. E. Freedman, Trigonal Bipyramidal V3+ Complex as an Optically Addressable Molecular Qubit Candidate, J. Am. Chem. Soc., 2020, 142, 20400–20408 CrossRef CAS PubMed.
- M. Dorn, J. Kalmbach, P. Boden, A. Päpcke, S. Gómez, C. Förster, F. Kuczelinis, L. M. Carrella, L. A. Büldt, N. H. Bings, E. Rentschler, S. Lochbrunner, L. González, M. Gerhards, M. Seitz and K. Heinze, A Vanadium(III) Complex with Blue and NIR-II Spin-Flip Luminescence in Solution, J. Am. Chem. Soc., 2020, 142, 7947–7955 CrossRef CAS PubMed.
- D. Riegert, J. Collin, J. C. Daran, T. Fillebeen, E. Schulz, D. Lyubov, G. Fukin and A. Trifonov, Neutral Yttrium Tris(Amide) and Ate Complexes Coordinated by an (R)-N,N′-Diisopropyl-1,1′-Binaphthyl-2,2′-Diamido Ligand as Enantioselective Catalysts for Intramolecular Hydroamination, Eur. J. Inorg. Chem., 2007,(8), 1159–1168 CrossRef CAS.
- V. N. Golovach, M. Borhani and D. Loss, Electric-Dipole-Induced Spin Resonance in Quantum Dots, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 1–10 Search PubMed.
- T. Bosma, G. J. J. Lof, C. M. Gilardoni, O. V. Zwier, F. Hendriks, B. Magnusson, A. Ellison, A. Gällström, I. G. Ivanov, N. T. Son, R. W. A. Havenith and C. H. van der Wal, Identification and Tunable Optical Coherent Control of Transition-Metal Spins in Silicon Carbide, Npj Quantum Inf., 2018, 4, 48 CrossRef.
- R. Debroux, C. P. Michaels, C. M. Purser, N. Wan, M. E. Trusheim, J. Arjona Martínez, R. A. Parker, A. M. Stramma, K. C. Chen, L. De Santis, E. M. Alexeev, A. C. Ferrari, D. Englund, D. A. Gangloff and M. Atatüre, Quantum Control of the Tin-Vacancy Spin Qubit in Diamond, Phys. Rev. X, 2021, 11, 041041 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, crystallographic data for 1-3, 1-Al, and 3-Al, additional EPR, optical, electrochemistry, and computational data and discussion. CCDC 2334908–2334912. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03107e |
| This journal is © The Royal Society of Chemistry 2024 |