
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeted protein degradation in the mitochondrial matrix and its application to chemical control of mitochondrial morphology†
Wakana
Yamada
a,
Shusuke
Tomoshige
 *a,
Sho
Nakamura
a,
Shinichi
Sato
ab and
Minoru
Ishikawa
*a
*a,
Sho
Nakamura
a,
Shinichi
Sato
ab and
Minoru
Ishikawa
*a
aGraduate School of Life Sciences, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, Miyagi 980-8577, Japan. E-mail: stomohi@tohoku.ac.jp; minoru.ishikawa.e4@tohoku.ac.jp
bFrontier Research Institute for Interdisciplinary Sciences, Tohoku University, 6-3 Aramaki Aza-Aoba, Aoba-ku, Sendai-shi, Miyagi 980-8578, Japan
First published on 28th August 2024
Abstract
Dysfunction of mitochondria is implicated in various diseases, including cancer and neurodegenerative disorders, but drug discovery targeting mitochondria and mitochondrial proteins has so far made limited progress. Targeted protein degradation (TPD) technologies represented by proteolysis targeting chimeras (PROTACs) are potentially applicable for this purpose, but most existing TPD approaches leverage the ubiquitin-proteasome system or lysosomes, which are absent in mitochondria, and TPD in mitochondria (mitoTPD) remains little explored. Herein, we describe the design and synthesis of a bifunctional molecule comprising TR79, an activator of the mitochondrial protease complex caseinolytic protease P (ClpP), linked to desthiobiotin. This compound successfully induced the degradation of monomeric streptavidin (mSA) and its fusion proteins localized to the mitochondrial matrix. Furthermore, in cells overexpressing mSA fused to short transmembrane protein 1 (mSA-STMP1), which enhances mitochondrial fission, our mitochondrial mSA degrader restored the mitochondrial morphology by reducing the level of mSA-STMP1. A preliminary structure–activity relationship study indicated that a longer linker length enhances the degradation activity towards mSA. These findings highlight the potential of mitoTPD as a tool for drug discovery targeting mitochondria and for research in mitochondrial biology, as well as the utility of mSA as a degradation tag for mitochondrial protein.
Introduction
In recent years, targeted protein degradation (TPD) technology has attracted great attention as a new drug discovery modality. TPD is a method to degrade and remove target proteins in cells by using compounds called degraders that utilize the endogenous protein degradation machinery. Given their unique mode of action, degraders are expected to be effective even against classically undruggable targets, such as nonenzymatic proteins and aggregation-prone proteins.1–3 Ubiquitin-proteasome system (UPS)-harnessing degraders termed proteolysis targeting chimeras (PROTACs), which are chimeric molecules composed of a ubiquitin ligase ligand and a ligand for the protein of interest (POI), are the most actively studied degraders and many PROTACs have already entered Phase I to III clinical trials.4,5 PROTACs are also used as chemical protein knockdown tools as alternatives to RNA interference, and have contributed to biological research.6–8The emergence of PROTACs has already established that TPD technology is a promising approach, especially for hard-to-drug targets,9 and has led to the development of other TPD technologies such as hydrophobic tagging,10,11 which harnesses protein quality control machinery, and methods exploiting lysosomal degradation machineries,12 including AUTACs,13 ATTECs,14 AUTOTACs,15 and LYTACs.16 In 2022, Clausen et al. developed BacPROTAC, a degrader utilizing unique degradation machinery in Gram-positive bacteria, for TPD in bacteria.17 This study pioneered the use of protein degradation machineries other than UPS and lysosomes.
Mitochondria are important organelles that produce about 90% of the ATP metabolized in cells. In addition, they play multiple roles in processes such as ion homeostasis, cell growth, redox state maintenance, cell signaling, and stress response. Therefore, mitochondrial function is critical for the regulation of cell function, as well as being associated with the onset and progression of diseases such as cancer, neurodegenerative diseases, cardiovascular diseases, and metabolic diseases.18 For example, short transmembrane protein 1 (STMP1), a 47-mer transmembrane micropeptide localized to the mitochondrial inner membrane, is upregulated in various cancer cells and is associated with cancer metastasis and recurrence. It has been reported that overexpression of STMP1 promotes mitochondrial fission and enhances tumor cell migration. Further, knockdown of STMP1 suppresses mitochondrial fission, suggesting it may be a potential target for cancer therapy.19 For this reason, mitochondria and mitochondrial proteins that control mitochondrial function have attracted great attention as drug targets in recent years,20–22 but few drugs or therapies have yet been approved.22 The main reason for this is considered to be the complexity of mitochondrial biology.23 Consequently, the achievement of TPD in mitochondria (mitoTPD) is expected to contribute greatly to the elucidation of mitochondrial biology, as well as expanding drug discovery options. Indeed, quite recently, TPD strategies for degrading mitochondrial proteins have begun to be developed.24,25
Herein, we describe the design and synthesis of a bifunctional molecule comprising TR79, an activator of the mitochondrial protease complex caseinolytic protease P (ClpP), linked to desthiobiotin, and we show that it can rapidly induce the degradation of monomeric streptavidin (mSA) and an mSA-fused protein of interest overexpressed in mitochondria. As proof of concept, we demonstrate that in cells overexpressing mSA-STMP1 fusion protein, which enhances mitochondrial fission, our degrader restored the mitochondrial morphology by reducing the level of mSA-STMP1, thereby enabling control of mitochondrial morphology. We believe these results advance the prospects for employing mitoTPD as a modality to develop drugs targeting mitochondrial proteins, as well as validating the mSA-based degradation tag platform as a useful tool to study mitochondrial biology.
Results
Conceptual basis of this work
As mitochondria lack UPS and lysosomes, mitoTPD requires harnessing protein degradation machinery that is unique to mitochondria. Candidates include the following four ATP-dependent proteases: Lon, ClpXP (a complex of caseinolytic protease P (ClpP) and AAA + ATPase ClpX), the matrix-oriented AAA (m-AAA) protease, and the intermembrane space-oriented AAA (i-AAA) protease.26–28ClpP is a serine protease resident in the matrix and is well conserved across species, being found in mitochondria, chloroplasts, and most bacteria.26 We hypothesized that mitoTPD could be achieved by using a chimeric small molecule consisting of a ClpP activator and a ligand of the target protein to bring the target protein into close proximity with activated ClpP (Fig. 1a). Numerous activators of ClpP have already been reported and shown to degrade mitochondrial proteins non-specifically.29,30 Structural biological analyses of the well-studied ClpP activators, the acyldepsipeptide (ADEP) series, and their small molecule analogs, the ACP series, suggest that these ClpP activators induce conformational changes in ClpP and expansion of its pore, thereby activating it.31 We selected TR79,32 which has a primary amine in a pyrimidinedione scaffold and has analogs that bind to the pocket shared with ADEPs,33 as a ClpP activator. Since this amino group has already been used to conjugate TR79 to agarose resin for ClpP pull-down,32 we thought that it would be suitable as a linker site for chimeric compound design. As a POI, we selected monomeric streptavidin (mSA)34 for the following two reasons: (1) biotin and desthiobiotin are well known and commercially available as mSA ligands with high affinity, and (2) mSA has a sufficiently small molecular weight (12 kDa) and has a track record as a fusion protein.35 We selected desthiobiotin as a ligand for mSA because it is thought to be bioorthogonal in mammals that lack avidin, as discussed later. Accordingly, we designed and synthesized WY165 (1), which connects TR79 and desthiobiotin via a tetraethylene glycol linker (Fig. 1b and Scheme S1, S2†).
 | ||
| Fig. 1 Conceptual basis of TPD using mitochondrial ClpP. (a) Schematic illustration of mitoTPD by a chimeric compound consisting of a ClpP activator conjugated to an mSA ligand via a linker. (b) Structures of desthiobiotin (an mSA ligand), TR79 (a ClpP activator), and WY165 (1). The primary amino group of TR79 serves as the conjugation site for the PEG linker. | ||
In vitro mSA degradation by WY165 (1)
First, we conducted in vitro ClpP activation assay and pull-down assay using SA-conjugated beads to confirm that WY165 (1) is capable of ClpP activation and can form a [SA-1-ClpP] ternary complex (Fig. S1 and S2a†). The results validated our molecular design. Next, in vitro mSA degradation assay revealed that WY165 (1) decreased mSA in a dose-dependent manner (DC50,24h = 197 nM under the conditions of 50 nM ClpP, 2.5 μM mSA) (Fig. 2a). Excess desthiobiotin inhibited the mSA degradation activity of 1, suggesting that the formation of the [mSA-1-ClpP] ternary complex is required for the activity of 1 (Fig. 2b).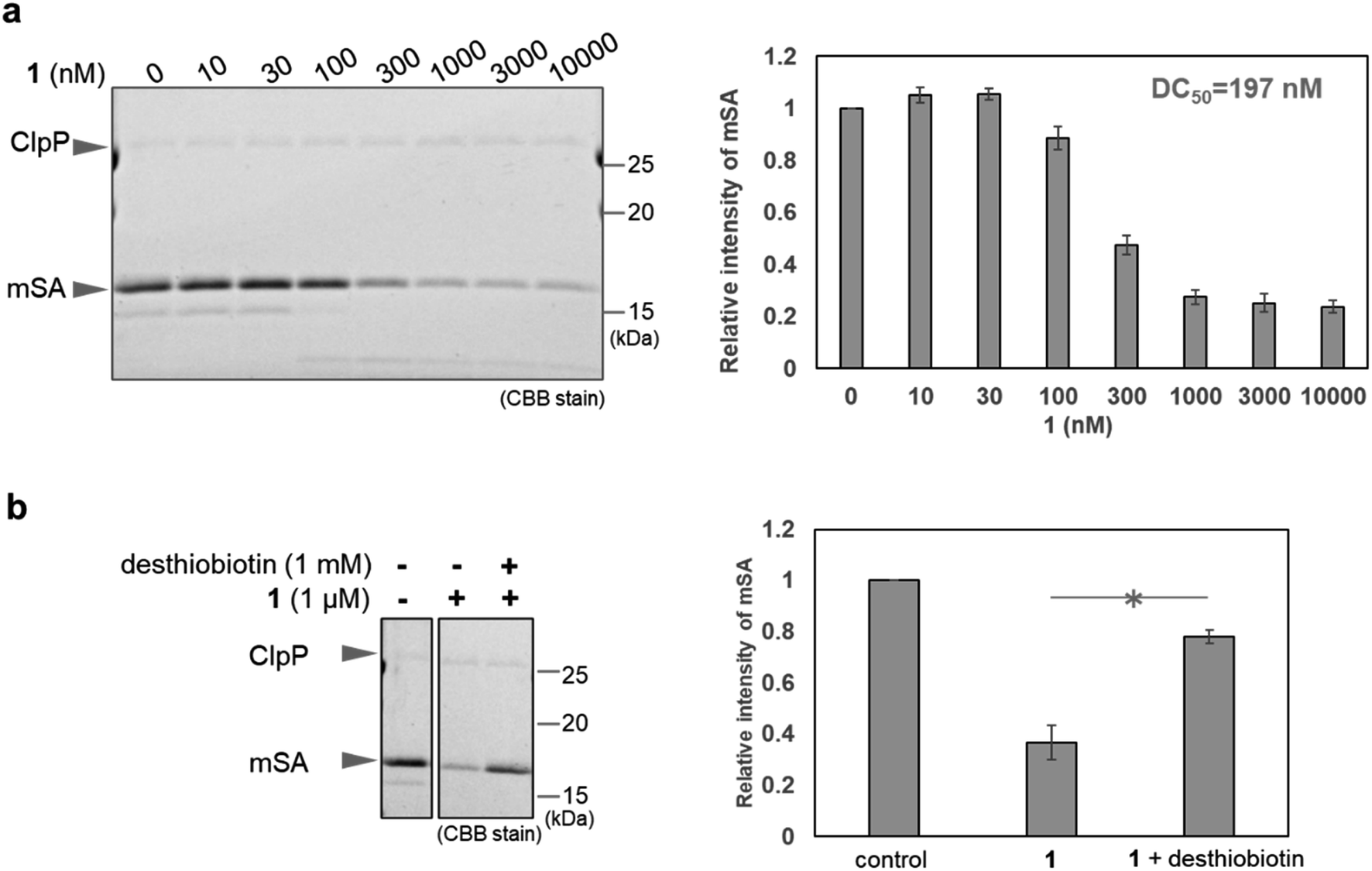 | ||
| Fig. 2 In vitro mSA degradation by WY165 (1). (a) Degradation of mSA in vitro by treatment with various concentrations of 1 (SDS-PAGE/CBB stain). The quantitative results are shown in the bar graph on the right. The intensity is normalized to that of the control sample (indicated as 0 nM), taken as 1. (b) Effect of desthiobiotin on mSA degradation mediated by 1. The quantitative results are shown in the bar graph on the right. The intensity is normalized to the control sample as above. Error bars indicate the SEM (n = 3 measurements). *: p < 0.05, Student's t-test. | ||
Whereas ClpP activators promiscuously enhance protein degradation, WY165 (1) is expected to mediate selective degradation of the target protein. Indeed, in cell lysates supplemented with mSA and ClpP, i.e. in mixture of mSA, ClpP, and whole cytoplasmic components including cytosolic, nuclear, and mitochondrial proteins, WY165 (1) led to a decrease of mSA, while other proteins appeared to be unaffected as indicated by CBB staining (Fig. S2b†).
To further evaluate the selectivity, we used mass spectrometry-based proteomics analysis. Analysis of the sample derived from the above cell lysates supplemented with mSA and ClpP detected 1410 proteins. In the presence of WY165 (1), mSA and 33 other proteins (2.4%) were decreased (Fig. 3a and Table S1†). Furthermore, in the presence of biotin as a competitor with 1, the abundances of 4 proteins, including mSA, were increased compared to the sample prepared in the absence of biotin. These results indicate that only 3 proteins other than mSA are recruited to ClpP by WY165 (1) as off-targets, and the 30 other proteins are degraded non-specifically by 1-activated ClpP (Fig. 3b), suggesting that the degradation-inducing activity of 1 is highly selective for mSA.
 | ||
| Fig. 3 Impact of WY165 (1) on other cellular proteins. Results of proteomics analysis. The vertical axis represents −log10 of the p-value (n = 3 measurements) (dotted lines: p = 0.01), and the horizontal axis represents the log2 fold change of protein abundance (dotted lines: 0.5-fold and 2-fold changes, respectively). (a) The 1-treated sample was compared to the control (DMSO-treated) sample. (b) The 1-treated sample was compared to the 1 + biotin-treated sample. | ||
Intracellular mSA degradation-inducing activity of WY165 (1)
To evaluate the cellular activity of WY165 (1), we next prepared HeLa cells transiently expressing mSA (cox8 (mitochondrial targeting signal sequence: MTS)36-His6-mSA-FLAG, hereafter cox8-mSA-FLAG or mSA) localized to the mitochondrial matrix (Fig. S3a†). Selective expression of cox8-mSA-FLAG in mitochondria was confirmed by isolation of the mitochondrial fraction followed by western blotting, which revealed three anti-FLAG-positive bands at 15–20 kDa. It is known that MTS is generally cleaved by mitochondrial processing peptidase at the mitochondrial matrix when the protein translocates from the cytosol to the mitochondria.37 We speculated that the largest protein (about 20 kDa) was intact cox8-mSA-FLAG remaining on the outer membrane or in the intermembrane space of the mitochondria. The lowest-molecular-weight band remained mainly in the cytoplasmic fraction, and may consist of defective or misfolded molecules. We considered that the middle band was the desired mitochondrial mSA, and focused on this band in the following experiments (Fig. S3b†).When HeLa cells transiently expressing mitochondrial cox8-mSA-FLAG were treated for 12 h with 1 (0–30 μM), we found that 10 μM and 30 μM 1 successfully reduced the mSA level with a DC50 value of 4.8 μM (Fig. 4a). A significant reduction of mSA was also observed upon 4 h treatment with 10 μM 1 (Fig. 4b). These results indicate that 1 reduces mSA in mitochondria in a dose- and time-dependent manner. We also performed similar experiments using MCF7 cells transiently expressing mitochondrial cox8-mSA-FLAG. After12 h treatment with 1 μM 1, we observed a significant decrease of mSA abundance, and the DC50 value was 0.96 μM. In the case of treatment with 3 μM 1, the mSA level was significantly reduced within 2 h (Fig. 4b). The reason for the difference in potency of WY165 (1) towards HeLa cells and MCF7 cells might be a difference in the expression levels of mSA (Fig. S4†).
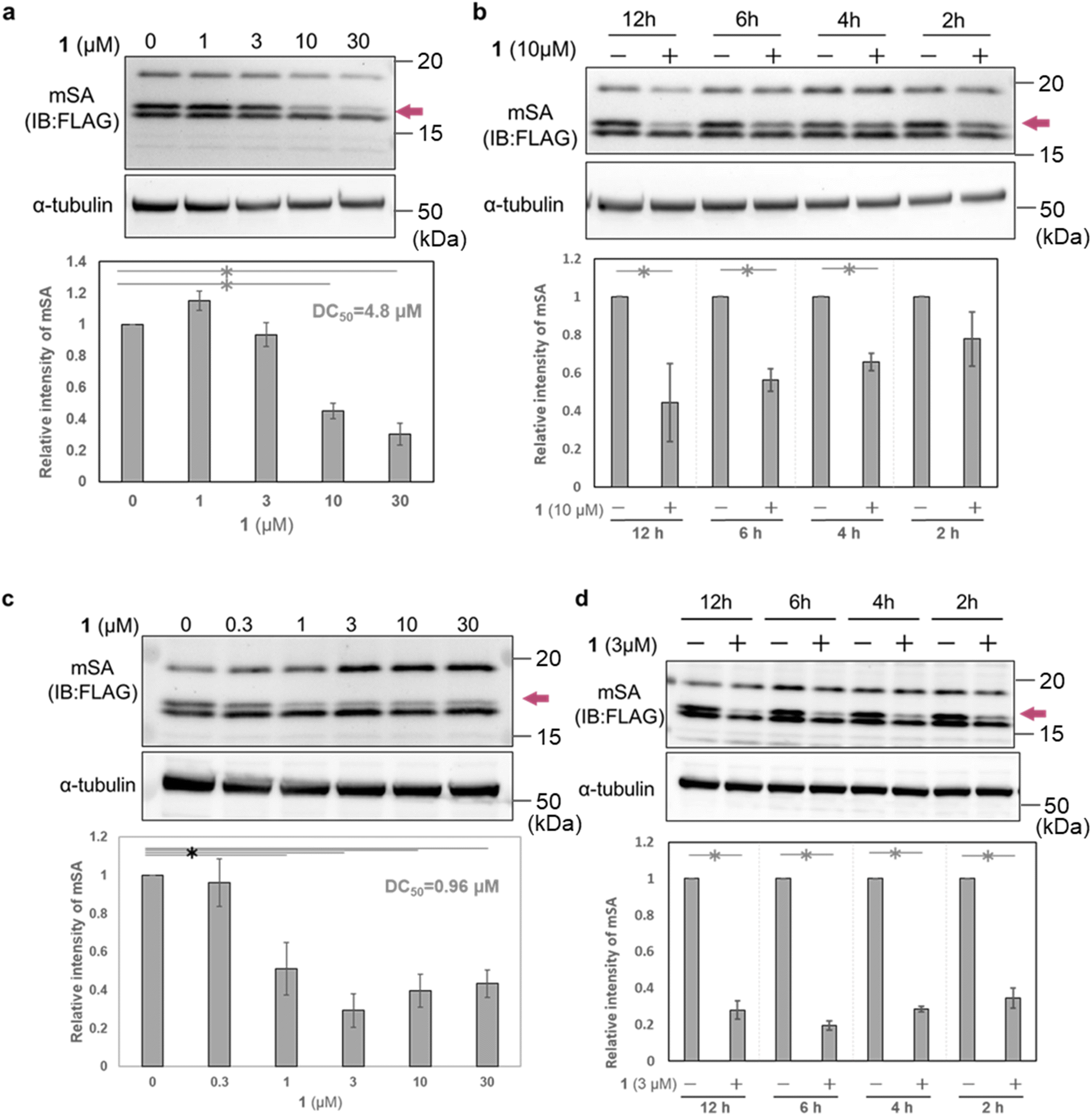 | ||
| Fig. 4 Degradation of mSA in mitochondria by treatment with WY165 (1). (a) Western blots and quantification of mSA in HeLa cells treated with various concentrations of 1 for 12 h. (b) Time-course of mSA level in HeLa cells treated with 10 μM 1. Western blots and quantification results are shown. (c) Western blots and quantification of mSA in MCF7 cells treated with various concentrations of 1 for 12 h. (d) Time-course of mSA level in MCF7 cells treated with 3 μM 1. Western blots and quantification results are shown. Brown arrows on western blot images indicate the putative mitochondrial mSA. Band intensities in the plots were normalized to the control band intensity, taken as 1. Error bars indicate SEM (n = 3 measurements). *: p < 0.05, Student's t-test. | ||
To confirm the involvement of ClpP in the mSA-degradation-inducing activity of 1, we carried out ClpP RNAi experiments in HeLa cells. The cells were first transfected with siRNA and incubated for 12 h, then transfected with mSA and treated with 1. Western blot analysis showed that ClpP RNAi suppressed the 1-induced degradation of mSA (Fig. 5a and S5†). We also examined whether proteasomes and lysosomes are involved in 1-induced mSA degradation. Pharmacological inhibition of proteasome or lysosome did not affect the mSA degradation induced by 1 (Fig. 5b). These results suggest that the 1-induced degradation of mSA depends on ClpP. Under conditions of proteasome inhibition, the intensity of the lowest-molecular-weight band related to mSA was increased, and this result is consistent with the idea that this band is due to mis-localized and/or mis-cleaved cox8-mSA-FLAG at least partially located in the cytosol.
 | ||
| Fig. 5 Analyses of mSA degradation pathway. (a) Degradation of mSA induced by 48 h treatment with WY165 (1) in HeLa cells with ClpP knockdown by transfection of siClpP. TF stands for transfection. (b) Degradation of mSA induced by 48 h treatment with 1 in HeLa cells in the presence of an inhibitor of proteasome or lysosome. Proteasome inhibition was performed by treatment with 300 nM bortezomib. Lysosome inhibition was performed by co-treatment with 10 μM leupeptin and 30 mM NH4Cl. Western blots and quantification results are shown. The data were normalized to the control value, taken as 1. Error bars indicate SEM (n = 3 measurements). *: p < 0.05, Student's t-test. | ||
To investigate the impact of WY165 (1) on total cellular protein abundance, proteomics analysis was conducted on cells treated with WY165 (1) (Fig. S6†). LC-MS/MS analysis detected 3105 proteins, among which 26 proteins (0.8%) were significantly decreased. Of these, 77% (20 proteins) were mitochondria-localized proteins, supporting the idea that WY165 (1) functions within mitochondria (Table S2†). Mitochondrial ClpP activator TR57, which shares the same pharmacophore as TR79, has been reported to decrease approximately 8% of detected proteins (686/∼8000) in SUM159 cells.38 Though these findings are not directly comparable, they suggest that WY165 (1) may reduce target proteins more selectively than ClpP activators.
Chemical control of mitochondrial morphology by mitoTPD
We next focused on STMP1, a mitochondrial short peptide enhancing mitochondrial fission, as a target protein to investigate whether mitochondrial morphology could be controlled. Since a ligand and ligand binding site of STMP1 were not available, we utilized mSA as a degradation tag and examined the degradation of mSA-STMP1 fusion protein. Although STMP1 is a trans-inner membrane peptide, we anticipated that mSA fused to the matrix-oriented N-terminus of STMP1 would be exposed to the matrix and would be targetable to our mitoTPD. First, we confirmed that cox8-His6-mSA-STMP1-FLAG (hereafter mSA-STMP1) is expressed in mitochondria (Fig. S7†) and that treatment with WY165 (1) decreased mSA-STMP1 in a dose-dependent manner (Fig. 6a). Next, we evaluated changes in mitochondrial morphology caused by the expression of mSA-STMP1 and treatment with 1, using fluorescence-microscopic images and an ImageJ/Fiji macro tool, Mitochondria Analyzer.39 Analysis of the number of mitochondrial branches, branch length, number of branch junctions, and form factor, which indicates mitochondrial shape, showed that expression of mSA-STMP1 leads to mitochondrial fission, resulting in a smaller, less branched structure, in accordance with previous work.19 Treatment of HeLa cells expressing mSA-STMP1 with WY165 successfully restored the changes in mitochondrial morphology, as judged according to all the evaluated factors (Fig. 6c). In contrast, WY165 did not change mitochondrial morphology in HeLa cells not expressing mSA-STMP1 (Fig. S8†). These results suggest that mitoTPD is able to control mitochondrial morphology. | ||
| Fig. 6 The impact of expression of mSA-STMP1 and its degradation by treatment with WY165 (1) on mitochondrial morphology. (a) Degradation of mSA-STMP1. Western blot and quantification results are shown. The data was normalized to the control, taken as 1. Error bars indicate SEM (n = 3 measurements). *: p < 0.05, Student's t-test. The two anti-FLAG antibody-positive bands are considered to be due to mSA-STMP1 (lower band) and its processing variant (upper band). (b) Representative microscopic images of mitochondria in HeLa cells expressing mSA-STMP1 after MitoTracker staining (top scale bars: 10 μm, bottom: 3 μm) with or without 1 μM 1 treatment. (c) Box-and-whisker plot of mitochondrial morphology in microscopic images. Mean branch length: total branch length divided by number of branches. Branches/mitochondria: total number of branches in the image, normalized to mitochondria count. Branch junctions/mitochondria: number of junctions within all skeletons in the image, normalized to mitochondria count. Junctions are points where 2 or more branches meet. Mean form factor: a measure of shape, where the value 1 indicates a round object and increases with elongation. Control: 98 cells, mSA-STMP1: 92 cells, mSA-STMP1 + compound 1: 128 cells. Steel–Dwass test (○, p < 0.05). | ||
Effect of linker structure of WY165 (1)
It is well established that the linker length and structure of PROTACs significantly affect their POI-degradation activity. To evaluate the structure–activity relationship of the linker in WY165 (1), we synthesized two other compounds, SN026 (2) and SN046 (3), with different PEG linker lengths. Their mSA degradation-inducing activity was assessed using HeLa cells expressing mitochondrial mSA. Western blotting results showed that the longer the linker length, the more effectively these compounds induce mSA degradation (Fig. 7). In particular, 10 μM 3 reduced the mSA level to approx. 20%, suggesting that the longer linker improves not only the half-maximal degradation concentration (DC50), but also the maximum degradation efficacy (Dmax).40 We observed no degradation of cox8-lacking mSA by these degraders (Fig. S9†).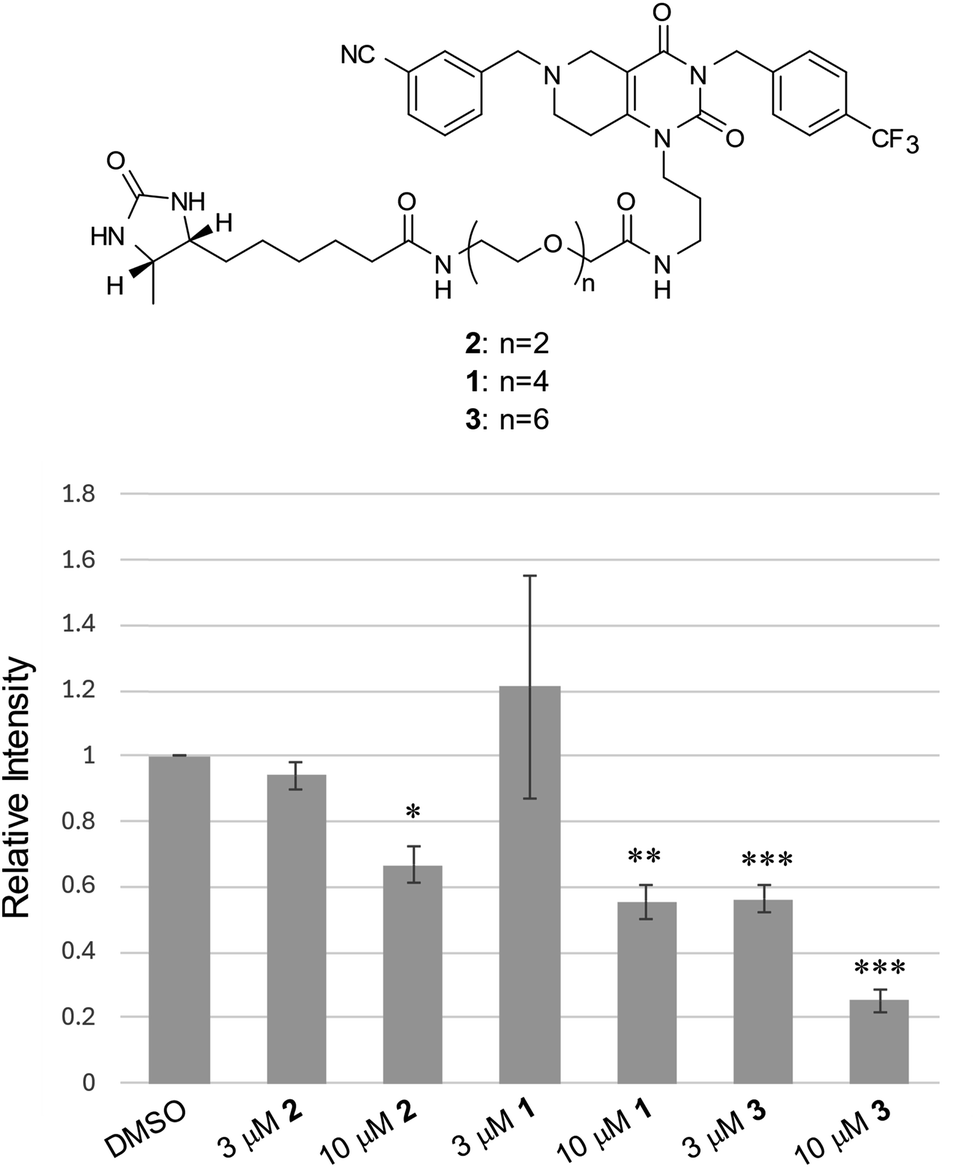 | ||
| Fig. 7 Structure–activity relationships for linker length of degraders. Quantitative western blot results are shown as a bar graph. Values are normalized to that of the control DMSO sample, taken as 1, and are the mean ± SEM (n = 4 measurements). *, p < 0.005; **, p < 0.0005; ***, p < 0.00005; Student's t-test. | ||
Discussion and conclusion
In this study, we designed degraders for mitoTPD technology, comprised of a mitochondrial protease activator linked to a ligand for the POI. By applying this conceptual framework, we successfully induced the degradation of mSA localized within mitochondria. Furthermore, by targeting STMP1, which is involved in mitochondrial fission, we demonstrated the feasibility of chemical control of mitochondrial morphology. Given the involvement of mitochondria and mitochondrial proteins in diseases such as cancer and neurodegenerative disorders, the TPD technology developed in this study has the potential to open up new options for drug discovery targeting mitochondria. Notably, our mitochondrial mSA degrader WY165 (1) exhibited degradation activity within 2–4 hours. Additionally, the degrader's potency could be adjusted by changing the linker length.A limitation of our study is that we targeted artificially expressed mSA and its fusion protein within the mitochondria, but not endogenous mitochondrial proteins. Nevertheless, it is noteworthy that we achieved the degradation of the fusion protein of STMP1, a transmembrane protein localized to the mitochondrial inner membrane, suggesting the applicability of mitoTPD technology not only to the mitochondrial matrix, but also to inner membrane-localized proteins whose ligand binding site is exposed to the matrix. Various PROTACs targeting protein tags such as HaloTag and FKBP12F36V have been reported and have been employed as rapid knockdown tools for biological research.7,41–47 Our mitochondrial mSA-tag degraders may therefore prove useful as research tools for mitochondrial biology. The use of biotin and its analogs in cells raises concerns about bioorthogonality, given that biotin functions as a coenzyme in cells. Indeed, to our knowledge, there has been no report of utilizing biotin-binding proteins, including mSA, as PROTAC degradation tags. However, in the case of bacteria, it has been reported that the affinity of desthiobiotin for protein biotin ligases is very low or undetectable.48 This implies that desthiobiotin would be a bioorthogonal molecule in mammals that lack avidin, and based on this, we employed the combination of mSA and desthiobiotin in this study, demonstrating for the first time its value as a cellular degradation tag.
Independently of our research, Wang et al. recently reported a TPD technology for mitochondrial proteins.24 They employed the known compound ONC201 (ref. 49) as a ClpP activator and developed a degrader targeting human mitochondrial RNA polymerase (POLRMT), an intramitochondrial protein. This degrader, 3B-1, consisted of ONC201 linked to a POLRMT inhibitor. However, 3B-1 required 16 hours at a concentration of 40 μM to induce degradation of POLRMT, whereas our degraders worked within only 2–4 hours, and the DC50 values were 4.8 μM (HeLa) and 0.96 μM (MCF7), respectively. This may be attributed to the weaker ClpP-activation potency of ONC201 as compared with TR79,32 though a difference in stability between POLRMT and mSA could also have contributed to the outcome.
Interestingly, Wang et al. found that increasing the length of the ethylene glycol linker in 3B-1 resulted in loss of activity, whereas we found that a longer linker increased the degradation activity. Two possible explanations can be considered: (1) the potency of ClpP activators affects the potency of ClpP-based degraders, and (2) the optimal linker length varies depending on the target protein. The technology reported by Wang et al. and us differs from PROTACs in using degraders that directly recruit the protease complex. Therefore, various features, such as characteristics arising from ternary complex formation and degradation time course, are expected to differ from those of PROTACs. Further work will be needed to fully elucidate the characteristics of this technology.
Data availability
All the data supporting the key findings in the paper have been included as part of ESI.†Author contributions
ST, SS, and MI conceived and directed the study. WY synthesized WY165 (1) and assessed its biological activities. SN synthesized compounds SN026 (2) and SN046 (3) and assessed their biological activities. WY and ST wrote and edited the manuscript with the contributions from all authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was funded by JSPS KAKENHI (JP22H00436, M.I. and JP22K15242, S.T.), JST ACT-X (JPMJAX2018, S.T.), AMED-CREST (JP21gm1410007, M.I.), and The Uehara Memorial Foundation (S.T.). We thank Dr Eiji Hishinuma and Naomi Matsukawa (Tohoku University) for their kind help in data collection of nanoLC-MS/MS analysis. A part of this study was supported by the research equipment sharing system, Tohoku University (042 Triple TOF5600, 373·Orbitrap Fusion). HEK293FT cells were kindly provided by Dr Shinichiro Kojima, Keio University. HeLa and MCF-7 cells were provided by RIKEN BRC.References
- P. M. Cromm and C. M. Crews, Targeted Protein Degradation: from Chemical Biology to Drug Discovery, Cell Chem. Biol., 2017, 24, 1181–1190 CrossRef CAS.
- Y. Itoh, M. Ishikawa, M. Naito and Y. Hashimoto, Protein Knockdown Using Methyl Bestatin−Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins, J. Am. Chem. Soc., 2010, 132, 5820–5826 CrossRef CAS.
- S. Tomoshige, S. Nomura, K. Ohgane, Y. Hashimoto and M. Ishikawa, Discovery of Small Molecules that Induce the Degradation of Huntingtin, Angew. Chem., Int. Ed., 2017, 56, 11530–11533 CrossRef CAS.
- M. Békés, D. R. Langley and C. M. Crews, PROTAC targeted protein degraders: the past is prologue, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef.
- D. Chirnomas, K. R. Hornberger and C. M. Crews, Protein degraders enter the clinic—a new approach to cancer therapy, Nat. Rev. Clin. Oncol., 2023, 20, 265–278 CrossRef CAS PubMed.
- M. A. Erb, T. G. Scott, B. E. Li, H. Xie, J. Paulk, H.-S. Seo, A. Souza, J. M. Roberts, S. Dastjerdi, D. L. Buckley, N. E. Sanjana, O. Shalem, B. Nabet, R. Zeid, N. K. Offei-Addo, S. Dhe-Paganon, F. Zhang, S. H. Orkin, G. E. Winter and J. E. Bradner, Transcription control by the ENL YEATS domain in acute leukaemia, Nature, 2017, 543, 270–274 CrossRef CAS.
- S. Mehta, A. Buyanbat, Y. Kai, O. Karayel, S. R. Goldman, D. Seruggia, K. Zhang, Y. Fujiwara, K. A. Donovan, Q. Zhu, H. Yang, B. Nabet, N. S. Gray, M. Mann, E. S. Fischer, K. Adelman and S. H. Orkin, Temporal resolution of gene derepression and proteome changes upon PROTAC-mediated degradation of BCL11A protein in erythroid cells, Cell Chem. Biol., 2022, 29, 1273–1287 CrossRef CAS.
- K. Yamano, R. Kikuchi, W. Kojima, R. Hayashida, F. Koyano, J. Kawawaki, T. Shoda, Y. Demizu, M. Naito, K. Tanaka and N. Matsuda, Critical role of mitochondrial ubiquitination and the OPTN–ATG9A axis in mitophagy, J. Cell Biol., 2020, 219, e201912144 CrossRef CAS.
- S. Tomoshige and M. Ishikawa, PROTACs and Other Chemical Protein Degradation Technologies for the Treatment of Neurodegenerative Disorders, Angew. Chem., Int. Ed., 2021, 60, 3346–3354 CrossRef CAS.
- M. Toure and C. M. Crews, Small-Molecule PROTACS: New Approaches to Protein Degradation, Angew. Chem., Int. Ed., 2016, 55, 1966–1973 CrossRef CAS.
- T. K. Neklesa, H. S. Tae, A. R. Schneekloth, M. J. Stulberg, T. W. Corson, T. B. Sundberg, K. Raina, S. A. Holley and C. M. Crews, Small-molecule hydrophobic tagging–induced degradation of HaloTag fusion proteins, Nat. Chem. Biol., 2011, 7, 538–543 CrossRef CAS.
- X. Li, Q. Liu, X. Xie, C. Peng, Q. Pang, B. Liu and B. Han, Application of Novel Degraders Employing Autophagy for Expediting Medicinal Research, J. Med. Chem., 2023, 66, 1700–1711 CrossRef CAS PubMed.
- D. Takahashi, J. Moriyama, T. Nakamura, E. Miki, E. Takahashi, A. Sato, T. Akaike, K. Itto-Nakama and H. Arimoto, AUTACs: Cargo-Specific Degraders Using Selective Autophagy, Mol. Cell, 2019, 76, 797–810 CrossRef CAS PubMed.
- Z. Li, C. Zhu, Y. Ding, Y. Fei and B. Lu, ATTEC: a potential new approach to target proteinopathies, Autophagy, 2020, 16, 185–187 CrossRef CAS PubMed.
- C. H. Ji, H. Y. Kim, M. J. Lee, A. J. Heo, D. Y. Park, S. Lim, S. Shin, S. Ganipisetti, W. S. Yang, C. A. Jung, K. Y. Kim, E. H. Jeong, S. H. Park, S. Bin Kim, S. J. Lee, J. E. Na, J. I. Kang, H. M. Chi, H. T. Kim, Y. K. Kim, B. Y. Kim and Y. T. Kwon, The AUTOTAC chemical biology platform for targeted protein degradation via the autophagy-lysosome system, Nat. Commun., 2022, 13, 904 CrossRef CAS PubMed.
- S. M. Banik, K. Pedram, S. Wisnovsky, G. Ahn, N. M. Riley and C. R. Bertozzi, Lysosome-targeting chimaeras for degradation of extracellular proteins, Nature, 2020, 584, 291–297 CrossRef CAS.
- F. E. Morreale, S. Kleine, J. Leodolter, S. Junker, D. M. Hoi, S. Ovchinnikov, A. Okun, J. Kley, R. Kurzbauer, L. Junk, S. Guha, D. Podlesainski, U. Kazmaier, G. Boehmelt, H. Weinstabl, K. Rumpel, V. M. Schmiedel, M. Hartl, D. Haselbach, A. Meinhart, M. Kaiser and T. Clausen, BacPROTACs mediate targeted protein degradation in bacteria, Cell, 2022, 185, 2338–2353 CrossRef CAS.
- P. A. Andreux, R. H. Houtkooper and J. Auwerx, Pharmacological approaches to restore mitochondrial function, Nat. Rev. Drug Discovery, 2013, 12, 465–483 CrossRef CAS.
- C. Xie, F.-Y. Wang, Y. Sang, B. Chen, J.-H. Huang, F.-J. He, H. Li, Y. Zhu, X. Liu, S.-M. Zhuang and J.-H. Fang, Mitochondrial Micropeptide STMP1 Enhances Mitochondrial Fission to Promote Tumor Metastasis, Cancer Res., 2022, 82, 2431–2443 CrossRef CAS PubMed.
- S. Javadov, A. V. Kozlov and A. K. S. Camara, Mitochondria in Health and Diseases, Cells, 2020, 9, 1177 CrossRef.
- R. H. Malty, M. Jessulat, K. Jin, G. Musso, J. Vlasblom, S. Phanse, Z. Zhang and M. Babu, Mitochondrial Targets for Pharmacological Intervention in Human Disease, J. Proteome Res., 2015, 14, 5–21 CrossRef CAS PubMed.
- A. Singh, D. Faccenda and M. Campanella, Pharmacological advances in mitochondrial therapy, eBioMedicine, 2024, 65, 103244 CrossRef.
- H. C. Delgado de la Herran, Y. Cheng and F. Perocchi, Towards a systems-level understanding of mitochondrial biology, Cell Calcium, 2021, 95, 102364 CrossRef CAS PubMed.
- D. Wang, W. Wang, L. Fang, L. Qi, Y. Zhang, J. Liu, Y. Liang, H. Yang, M. Wang, X. Wei, R. Jiang, Y. Liu, W. Zhou and X. Fang, Mitochondrial Protease Targeting Chimeras for Mitochondrial Matrix Protein Degradation, J. Am. Chem. Soc., 2023, 145, 12861–12869 CrossRef CAS PubMed.
- S. Sanyal, A. Kouznetsova, L. Ström and C. Björkegren, A system for inducible mitochondria-specific protein degradation in vivo, Nat. Commun., 2024, 15, 1454 CrossRef CAS.
- K. Nouri, Y. Feng and A. D. Schimmer, Mitochondrial ClpP serine protease-biological function and emerging target for cancer therapy, Cell Death Dis., 2020, 11, 1–12 CrossRef PubMed.
- C. A. Goard and A. D. Schimmer, Mitochondrial matrix proteases as novel therapeutic targets in malignancy, Oncogene, 2014, 33, 2690–2699 CrossRef CAS PubMed.
- R. Wedam, Y. E. Greer, D. J. Wisniewski, S. Weltz, M. Kundu, D. Voeller and S. Lipkowitz, Targeting Mitochondria with ClpP Agonists as a Novel Therapeutic Opportunity in Breast Cancer, Cancers, 2023, 15, 1936 CrossRef CAS.
- V. Bhandari, K. S. Wong, J. L. Zhou, M. F. Mabanglo, R. A. Batey and W. A. Houry, The Role of ClpP Protease in Bacterial Pathogenesis and Human Diseases, ACS Chem. Biol., 2018, 13, 1413–1425 CrossRef CAS PubMed.
- M. F. Mabanglo and W. A. Houry, Recent structural insights into the mechanism of ClpP protease regulation by AAA+ chaperones and small molecules, J. Biol. Chem., 2022, 298, 101781 CrossRef CAS PubMed.
- M. F. Mabanglo, E. Leung, S. Vahidi, T. V. Seraphim, B. T. Eger, S. Bryson, V. Bhandari, J. L. Zhou, Y. Q. Mao, K. Rizzolo, M. M. Barghash, J. D. Goodreid, S. Phanse, M. Babu, L. R. S. Barbosa, C. H. I. Ramos, R. A. Batey, L. E. Kay, E. F. Pai and W. A. Houry, ClpP protease activation results from the reorganization of the electrostatic interaction networks at the entrance pores, Commun. Biol., 2019, 2, 410 CrossRef CAS.
- P. R. Graves, L. J. Aponte-Collazo, E. M. J. Fennell, A. C. Graves, A. E. Hale, N. Dicheva, L. E. Herring, T. S. K. Gilbert, M. P. East, I. M. McDonald, M. R. Lockett, H. Ashamalla, N. J. Moorman, D. S. Karanewsky, E. J. Iwanowicz, E. Holmuhamedov and L. M. Graves, Mitochondrial Protease ClpP is a Target for the Anticancer Compounds ONC201 and Related Analogues, ACS Chem. Biol., 2019, 14, 1020–1029 CrossRef CAS.
- M. F. Mabanglo, K. S. Wong, M. M. Barghash, E. Leung, S. H. W. Chuang, A. Ardalan, E. M. Majaesic, C. J. Wong, S. Zhang, H. Lang, D. S. Karanewsky, A. A. Iwanowicz, L. M. Graves, E. J. Iwanowicz, A.-C. Gingras and W. A. Houry, Potent ClpP agonists with anticancer properties bind with improved structural complementarity and alter the mitochondrial N-terminome, Structure, 2023, 31, 185–200 CrossRef CAS PubMed.
- A. Kroetsch, B. Chin, V. Nguyen, J. Gao and S. Park, Functional expression of monomeric streptavidin and fusion proteins in Escherichia coli: applications in flow cytometry and ELISA, Appl. Microbiol. Biotechnol., 2018, 102, 10079–10089 CrossRef CAS PubMed.
- D. Demonte, C. M. Dundas and S. Park, Expression and purification of soluble monomeric streptavidin in Escherichia coli, Appl. Microbiol. Biotechnol., 2014, 98, 6285–6295 CrossRef CAS.
- H. Yu, R. D. Koilkonda, T.-H. Chou, V. Porciatti, S. S. Ozdemir, V. Chiodo, S. L. Boye, S. E. Boye, W. W. Hauswirth, A. S. Lewin and J. Guy, Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber's hereditary optic neuropathy in a mouse model, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, E1238–E1247 CAS.
- D. Stojanovski, M. Bohnert, N. Pfanner and M. van der Laan, Mechanisms of Protein Sorting in Mitochondria, Cold Spring Harb. Perspect. Biol., 2012, 4, a011320 Search PubMed.
- E. M. J. Fennell, L. J. Aponte-Collazo, W. Pathmasiri, B. R. Rushing, N. K. Barker, M. C. Partridge, Y.-Y. Li, C. A. White, Y. E. Greer, L. E. Herring, S. Lipkowitz, S. C. J. Sumner, E. J. Iwanowicz and L. M. Graves, Multi-omics analyses reveal ClpP activators disrupt essential mitochondrial pathways in triple-negative breast cancer, Front. Pharmacol., 2023, 14, 1136317 CrossRef CAS PubMed.
- A. Chaudhry, R. Shi and D. S. Luciani, A pipeline for multidimensional confocal analysis of mitochondrial morphology, function, and dynamics in pancreatic β-cells, Am. J. Physiol. Endocrinol. Metabol., 2020, 318, E87–E101 CrossRef CAS.
- D. P. Bondeson, A. Mares, I. E. D. Smith, E. Ko, S. Campos, A. H. Miah, K. E. Mulholland, N. Routly, D. L. Buckley, J. L. Gustafson, N. Zinn, P. Grandi, S. Shimamura, G. Bergamini, M. Faelth-Savitski, M. Bantscheff, C. Cox, D. A. Gordon, R. R. Willard, J. J. Flanagan, L. N. Casillas, B. J. Votta, W. den Besten, K. Famm, L. Kruidenier, P. S. Carter, J. D. Harling, I. Churcher and C. M. Crews, Catalytic in vivo protein knockdown by small-molecule PROTACs, Nat. Chem. Biol., 2015, 11, 611–617 CrossRef CAS PubMed.
- J. M. Etersque, I. K. Lee, N. Sharma, K. Xu, A. Ruff, J. D. Northrup, S. Sarkar, T. Nguyen, R. Lauman, G. M. Burslem and M. A. Sellmyer, Regulation of eDHFR-tagged proteins with trimethoprim PROTACs, Nat. Commun., 2023, 14, 7071 CrossRef CAS PubMed.
- C. Grohmann, C. M. Magtoto, J. R. Walker, N. K. Chua, A. Gabrielyan, M. Hall, S. A. Cobbold, S. Mieruszynski, M. Brzozowski, D. S. Simpson, H. Dong, B. Dorizzi, A. V. Jacobsen, E. Morrish, N. Silke, J. M. Murphy, J. K. Heath, A. Testa, C. Maniaci, A. Ciulli, G. Lessene, J. Silke and R. Feltham, Development of NanoLuc-targeting protein degraders and a universal reporter system to benchmark tag-targeted degradation platforms, Nat. Commun., 2022, 13, 2073 CrossRef CAS PubMed.
- A. G. Bond, C. Craigon, K.-H. Chan, A. Testa, A. Karapetsas, R. Fasimoye, T. Macartney, J. J. Blow, D. R. Alessi and A. Ciulli, Development of BromoTag: A “Bump-and-Hole”–PROTAC System to Induce Potent, Rapid, and Selective Degradation of Tagged Target Proteins, J. Med. Chem., 2021, 64, 15477–15502 CrossRef CAS PubMed.
- K. Okitsu, T. Hattori, T. Misawa, T. Shoda, M. Kurihara, M. Naito and Y. Demizu, Development of a Small Hybrid Molecule That Mediates Degradation of His-Tag Fused Proteins, J. Med. Chem., 2018, 61, 576–582 CrossRef CAS PubMed.
- B. Nabet, J. M. Roberts, D. L. Buckley, J. Paulk, S. Dastjerdi, A. Yang, A. L. Leggett, M. A. Erb, M. A. Lawlor, A. Souza, T. G. Scott, S. Vittori, J. A. Perry, J. Qi, G. E. Winter, K.-K. Wong, N. S. Gray and J. E. Bradner, The dTAG system for immediate and target-specific protein degradation, Nat. Chem. Biol., 2018, 14, 431–441 CrossRef CAS PubMed.
- S. Tomoshige, M. Naito, Y. Hashimoto and M. Ishikawa, Degradation of HaloTag-fused nuclear proteins using bestatin-HaloTag ligand hybrid molecules, Org. Biomol. Chem., 2015, 13, 9746–9750 RSC.
- D. L. Buckley, K. Raina, N. Darricarrere, J. Hines, J. L. Gustafson, I. E. Smith, A. H. Miah, J. D. Harling and C. M. Crews, HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins, ACS Chem. Biol., 2015, 10, 1831–1837 CrossRef CAS.
- S. Purushothaman, G. Gupta, R. Srivastava, V. G. Ramu and A. Surolia, Ligand Specificity of Group I Biotin Protein Ligase of Mycobacterium tuberculosis, PLoS One, 2008, 3, e2320 CrossRef.
- J. Ishizawa, S. F. Zarabi, R. E. Davis, O. Halgas, T. Nii, Y. Jitkova, R. Zhao, J. St-Germain, L. E. Heese, G. Egan, V. R. Ruvolo, S. H. Barghout, Y. Nishida, R. Hurren, W. Ma, M. Gronda, T. Link, K. Wong, M. Mabanglo, K. Kojima, G. Borthakur, N. MacLean, M. C. J. Ma, A. B. Leber, M. D. Minden, W. Houry, H. Kantarjian, M. Stogniew, B. Raught, E. F. Pai, A. D. Schimmer and M. Andreeff, Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality, Cancer Cell, 2019, 35, 721–737 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03145h |
| This journal is © The Royal Society of Chemistry 2024 |