
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis, biological evaluation and biosynthetic re-evaluation of Illicium-derived neolignans†
Robert E.
Arnold
a,
Jan
Saska
a,
Raquel
Mesquita-Ribeiro
b,
Federico
Dajas-Bailador
b,
Laurence
Taylor
 a,
William
Lewis
a,
Stephen
Argent
a,
Huiling
Shao
c,
Kendall N.
Houk
*c and
Ross M.
Denton
*a
a,
William
Lewis
a,
Stephen
Argent
a,
Huiling
Shao
c,
Kendall N.
Houk
*c and
Ross M.
Denton
*a
aThe GlaxoSmithKline Carbon Neutral Laboratories for Sustainable Chemistry, University of Nottingham, Jubilee Campus Triumph Road, Nottingham, NG7 2TU, UK. E-mail: ross.denton@nottingham.ac.uk
bSchool of Life Sciences, University of Nottingham, NG7 2UH, UK
cUniversity of California, Department of Chemistry and Biochemistry, 607 Charles E. Young Drive East, Box 951569, Los Angeles, CA 90095-1569, UK
First published on 19th June 2024
Abstract
We report the first total syntheses of simonsol F (3), simonsinol (5), fargenin (4), and macranthol (6) in addition to syntheses of simonsol C (2), simonsol G (1), and honokiol (14). The syntheses are based upon a phosphonium ylide-mediated cascade reaction and upon natural product isomerization reactions which proceed through Cope rearrangements of putative biosynthetic dienone intermediates. As a corollary of the natural product isomerization reactions, we propose an alternative biosynthesis of honokiol (14), simonsinol (5), and macranthol (6) which unites the natural products in this family under a single common precursor, chavicol (7). Finally, we demonstrate that simonsol C (2) and simonsol F (3) promote axonal growth in primary mouse cortical neurons.
Introduction
Plants of the genus Illicium have long been a rich source of natural products. For example, the seed pod of Illicium verum is star anise, a common flavoring, while the fruit of the same plant is a traditional Chinese medicine used for the treatment of abdominal pain. More recently Illicium-derived natural products have attracted considerable attention because of their potent neurotrophic properties.1,2 Depicted in Fig. 1 are two groups of neolignans 3 that are of interest as a result of their molecular structures, biosynthetic origin and potential biological activity. The first group (Fig. 1A) contains the skeletally distinct metabolites simonsol G (1),4 simonsol C (2),5 simonsol F (3)6 and fargenin (4),7 which share a cis-fused tetrahydrodibenzofuran ring system as their central structural element. The second group (Fig. 1B) contains the constitutionally isomeric aromatic sesquineolignans simonsinol (5)8 and macranthol (6).9 The first group is of particular interest because of their structural resemblance to the galantamine alkaloids10 and synthetic analogs such as narwedine (8, Fig. 1C), which is used in the clinic for the symptomatic treatment of Alzheimer's disease.11 This similarity was noted by Banwell and co-workers who reported the first synthesis of racemic simonsol C (2) in 2016.12 This was followed by a second total synthesis which was very recently disclosed by Qin and co-workers.13 Banwell's observation, coupled with the fact that several structurally related neolignans have been shown to exhibit neuroprotective effects,14–19 stimulated our interest in this family, and analogs thereof, as a possible source of new neuroprotective compounds.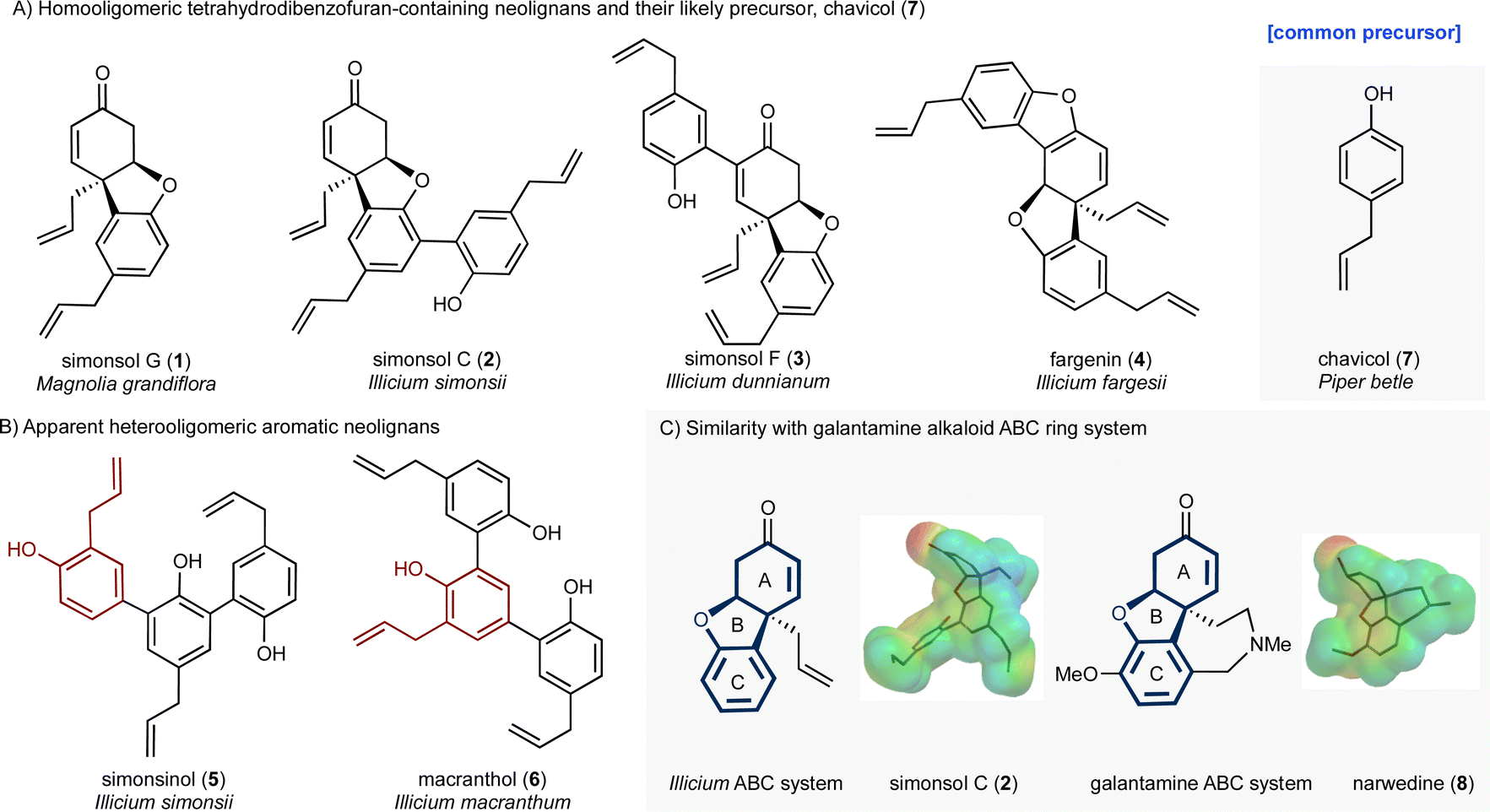 | ||
| Fig. 1 Structures of Illicium-derived neolignan natural products. | ||
Biosynthetic analysis
Alongside potential biological activity, we were intrigued by the biosynthetic origins of these natural products. For example, the compounds depicted in Fig. 1A are homooligomers likely derived from the natural product chavicol (7) and a plausible biosynthesis (Scheme 1A) involves oxidation and deprotonation of 7 (ref. 20 and 21) to generate radical 9 followed by ortho–ortho dimerization giving magnolol (10). From this branch point the trimeric natural products can be reached as follows. Oxidative coupling of magnolol (10) and chavicol (7) according to Path A gives dienone 13 and, following conjugate addition, simonsol C (2). An alternative oxidative coupling (Path B, Scheme 1) generates dienone 11, a second branch point, from which simonsol F (3) and fargenin (4) are derived through conjugate addition and addition/dehydration reactions respectively. Experimental support for these pathways comes from Brown and co-workers who have demonstrated that a variety of natural products including magnolol (10) and simsonol G (1) were formed non-selectively and in low yield when chavicol (7) was treated with iron trichloride.22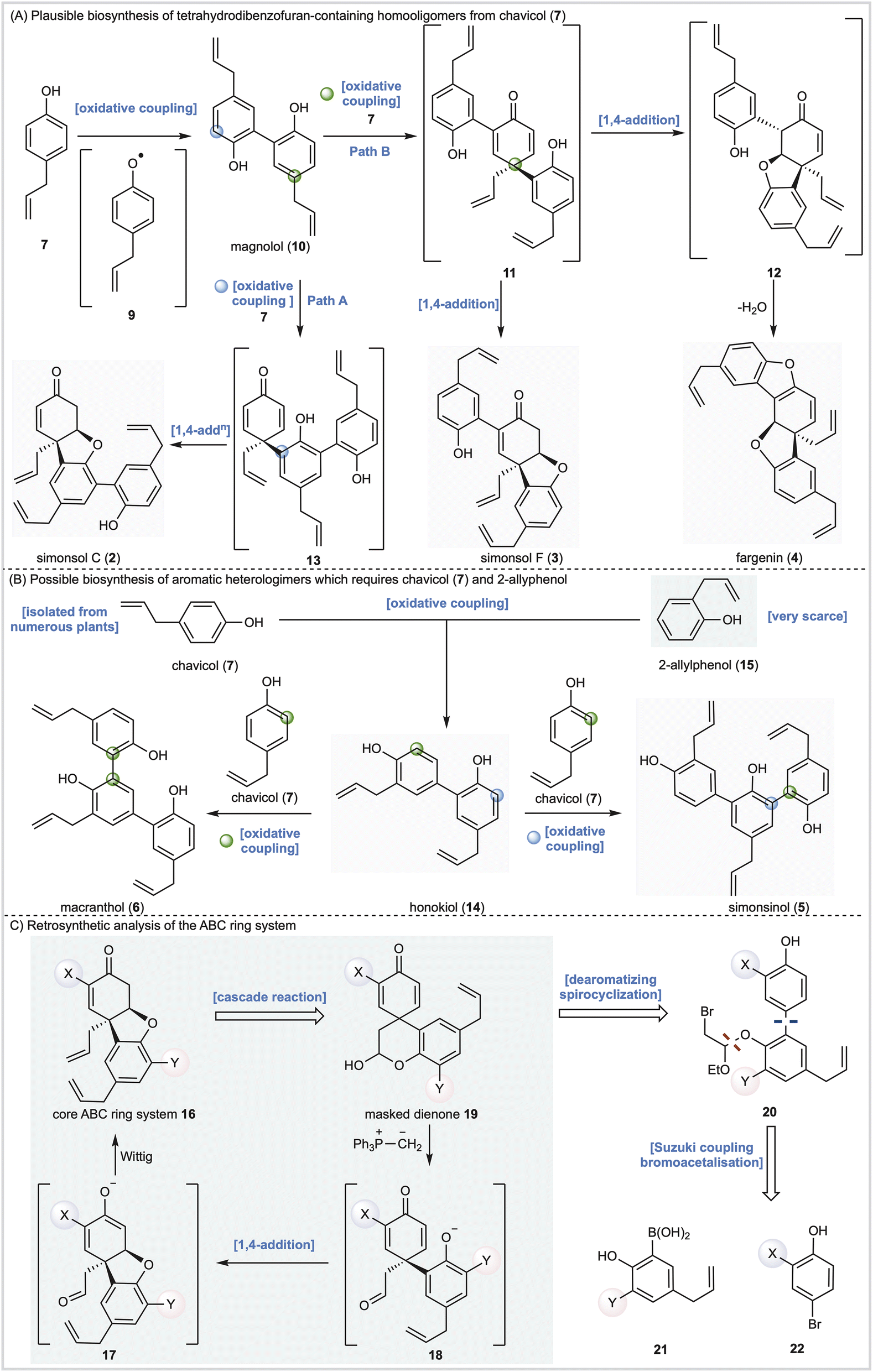 | ||
| Scheme 1 Proposed biosynthesis and retrosynthetic analysis of Illicium neolignans. | ||
In contrast simonsinol (5) and macranthol (6) (Scheme 1B) are prima facie heterooligomers derived from chavicol (7) and the additional phenolic precursor (15) through a presumed oxidative coupling which generates the natural product honokiol (14).23–28 From this point macranthol (6) and simonsinol (5) can be reached following further oxidative coupling with chavicol (7) as indicated (Scheme 1B).
However, while chavicol (7) and its methylated counterpart estragole have been isolated from numerous plants, including basil, fennel, and star anise (Illicum verum),29 phenol 15 is very scarce indeed.30 Furthermore, honokiol (14) is typically isolated with the homodimer magnolol (10) suggesting a common origin from chavicol (7) which undermines the presumed oxidative coupling of 7 and 15 depicted in Scheme 1B.31
Synthesis plan
In order to explore the biological activity and possible biosynthesis of these Illicium natural products we designed a method for the construction of the core [4.3.0] tetrahydrodibenzofuran ring system (Scheme 1C) featuring intermediate 16, with appropriate substituents X and Y, as a key building block. In principle 16 should be accessible from a cascade reaction in which a phosphonium ylide mediates a ring-opening, conjugate addition and methylenation sequence (19 →18→17→16, Scheme 1C). The cascade precursor 19 should be available through a dearomatizing spirocyclization reaction32 of biaryl 20, itself derived from cross-coupling of 21 and 22. Any such cross-coupling is potentially complicated by thermodynamic isomerization of the terminal alkenes. However, we have previously completed the total synthesis of simple aromatic neolignans33–35 and shown that, with care, cross-coupling and deprotection reactions could be carried out efficiently. Furthermore, we also investigated the proposed cascade reaction and demonstrated its feasibility on a simple model system, albeit with limited success.36Herein we report total syntheses of simonsol C (2), simonsol F (3) and simonsol G (1) based upon the successful implementation of this general approach. Additional syntheses of simonsinol (5), fargenin (4), macranthol (6), and honokiol (14) through isomerization processes provide evidence for a new biosynthesis proposal which unites the neolignans in Fig. 1 under a single common precursor. Finally, using neuronal models, the potential connection between neolignans and galantamine alkaloids is explored and tetrahydrodibenzofuran-containing neolignans are shown to promote axonal growth.
Results and discussion
Total synthesis of simonsol C (2) and simonsol F (3)
The synthesis of simonsol C (2) (Scheme 2) began with the preparation of boronic acid 24 through ortho-lithiation of 23 followed by quenching with trimethylborate and transesterification with pinacol. Subsequent treatment with boron trichloride dimethylsulfide complex effected cleavage of the methyl ether and pinacol ester and gave the boronic acid 24 and dimer 25 in a combined yield of 47% over three steps.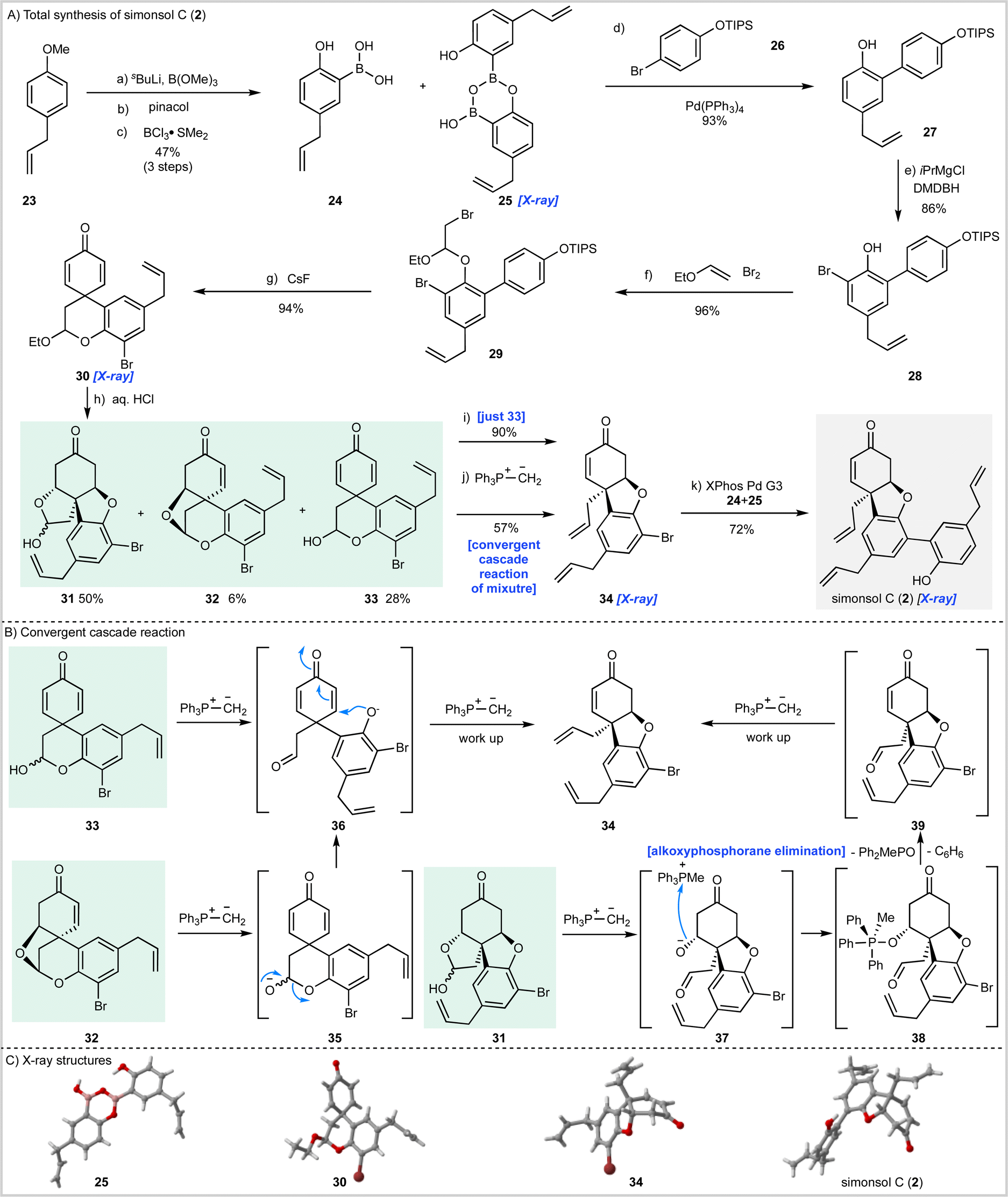 | ||
| Scheme 2 Total synthesis of simonsol C (2). (A) Reagents and conditions: (a) sBuLi, B(OMe)3, TMEDA, THF, −78 °C to r.t., 1 h; (b) pinacol, methanol, r.t., 16 h; (c) BCl3·SMe2, 1,2-DCE, 80 °C, 16 h, 47% (3 steps); (d) Pd(PPh3)4 (8 mol%), aq. Na2CO3, THF, r.t. 1.5 h, 93%; (e) DBDMH, iPrMgCl, THF, − 78 °C to r.t., 1.5 h, 86%; (f) ethyl vinyl ether, Br2, Hünig's base, DCM, 0 °C to r.t., 2.5 h, 96%; (g) CsF, Na2SO4, DMF, 130 °C, 1 h, 94%; (h) HCl aq., 1,4-dioxane, 80 °C, 10.5 h, 33 28%, 32 6%, 31 50%; (i) MePPh3 Br, KHMDS, THF, 0 °C, 1 h, 90% (just 33); (j) MePPh3Br, KHMDS, THF, 0 °C, 1 h, 57% (convergent cascade reaction of mixture); (k) XPhos Pd G3 10 mol%, aq. K3PO4, THF, 50 °C, 1.3 h, 72%. (B) Convergent cascade reaction pathways. (C) X-ray structures. THF = tetrahydrofuran, TMEDA = N,N,N′,N′-tetramethylethylenediame, DBDMH = dibromo dimethyl hydantoin, DMF = dimethylformamide, KHMDS = potassium hexamethyldisilazide. | ||
Suzuki–Miyaura cross-coupling of this mixture with bromide 26 then gave biaryl 27 which was brominated to afford intermediate 28 from which acetal 29 was prepared by reaction with bromine and ethyl vinyl ether. A dilute solution of 29 was then treated with cesium fluoride, which resulted in silyl ether cleavage and spirocyclization to afford dienone 30, the structure of which was confirmed by X-ray analysis.
Having reached this stage we sought hydrolysis conditions that would convert 30 into the corresponding hemi-acetal 33 required for the cascade reaction (Scheme 1C). Following some experimentation, we found that treatment of 30 with aqueous hydrochloric acid gave 28% of the desired hemiacetal 33 along with enone 32 (6%) and inseparable ketone diastereomers 31 (50%) which had arisen through post hydrolysis oxa-1,4-addition reactions. While hemiacetal 33 was not obtained as the major product it was available following chromatography and so we proceeded to examine the planned cascade reaction. To this end 33 was treated with methylenetriphenylphosphorane and the desired tricyclic intermediate 34 was obtained in an excellent 90% isolated yield and the structure of this material was confirmed by X-ray crystallography (Scheme 2C).
Although our synthesis plan had now been validated through the successful conversion of 33 into 34, we had to face the problem that hemiacetal 33 was only available as the minor product from the hydrolysis of acetal 30. To develop a viable synthesis of simonsol C (2) a means of recycling or, ideally, progressing the other hydrolysis products 31 and 32 was required. Since these by-products had arisen through conjugate addition, we reasoned it might be possible to effect β-elimination reactions under the basic conditions of the cascade reaction and thereby develop a process under which the hydrolysis products converge. Fortunately, this proved to be the case, and when a mixture of 31, 32 and 33 were treated with the phosphonium ylide we were delighted to isolate product 34 in 57% yield. We believe that this convergent process occurs as shown in Scheme 1B. It is likely that dienone 33 and enone 32 converge to give aldehyde 36 through deprotonation and ring-opening in the case of 33 and β-elimination/ring-opening in the case of 32. Subsequent conjugate addition of 36 and methylenation then gives product 34 after work up. In the case of the inseparable ketone diastereoisomers 31, deprotonation results in ring-opening to give alkoxide 37 which then forms alkoxyphosphorane intermediate 38 (ref. 37) after alkoxide addition to the phosphonium salt. Elimination of this intermediate to generate 39 may then proceed by an E1cB mechanism resulting in the generation of methyldiphenylphosphine oxide and benzene.
With a viable cascade sequence now developed we examined the Suzuki–Miyaura coupling reaction to access simonsol C (2). As expected, this proved challenging and was complicated by: (a) the free hydroxyl group present within the boronic acid; (b) isomerization of the allyl groups; and (c) additional reactions of the natural product which occurred post cross-coupling (vide infra). After numerous experiments, in which a wide range of palladium pre-catalysts were examined, an efficient cross-coupling was realized using Buchwald's third generation palladacycle XPhos Pd G3 (ref. 38) which gave simonsol C (2) in a 72% isolated yield. The structure of our synthetic material was confirmed by X-ray crystallography (Scheme 2C).
Having now fully validated our synthesis strategy we next turned our attention to simonsol F (3) which, prior to our work, had not been synthesized (Scheme 3A). To this end biaryl 40 was prepared by Suzuki–Miyaura coupling of the appropriate aryl halide and a mixture of boronic acids 24 and 25 (see ESI†). The biaryl then underwent bromoacetalization to give 41 followed by desilylation and spirocyclization to give an inconsequential mixture of bromodienone diastereoisomers 42, whose structures were confirmed by X-ray crystallography (Scheme 3C). Unfortunately, progressing either the single dienone isomers or the mixture through the required acetal hydrolysis proved to be extremely challenging. A range of conditions were examined, all of which gave intractable mixtures of debrominated material. As a result, we were forced to explore Suzuki–Miyaura cross-coupling prior to hydrolysis of the cyclic acetal. In our initial experiments we established that separate diastereoisomers of 42 underwent efficient coupling with boronic acid ester 43 (not depicted) before developing a more efficient method for cross-coupling of the mixture (Scheme 3A) which gave 44 as a mixture of diastereoisomers in excellent yield.
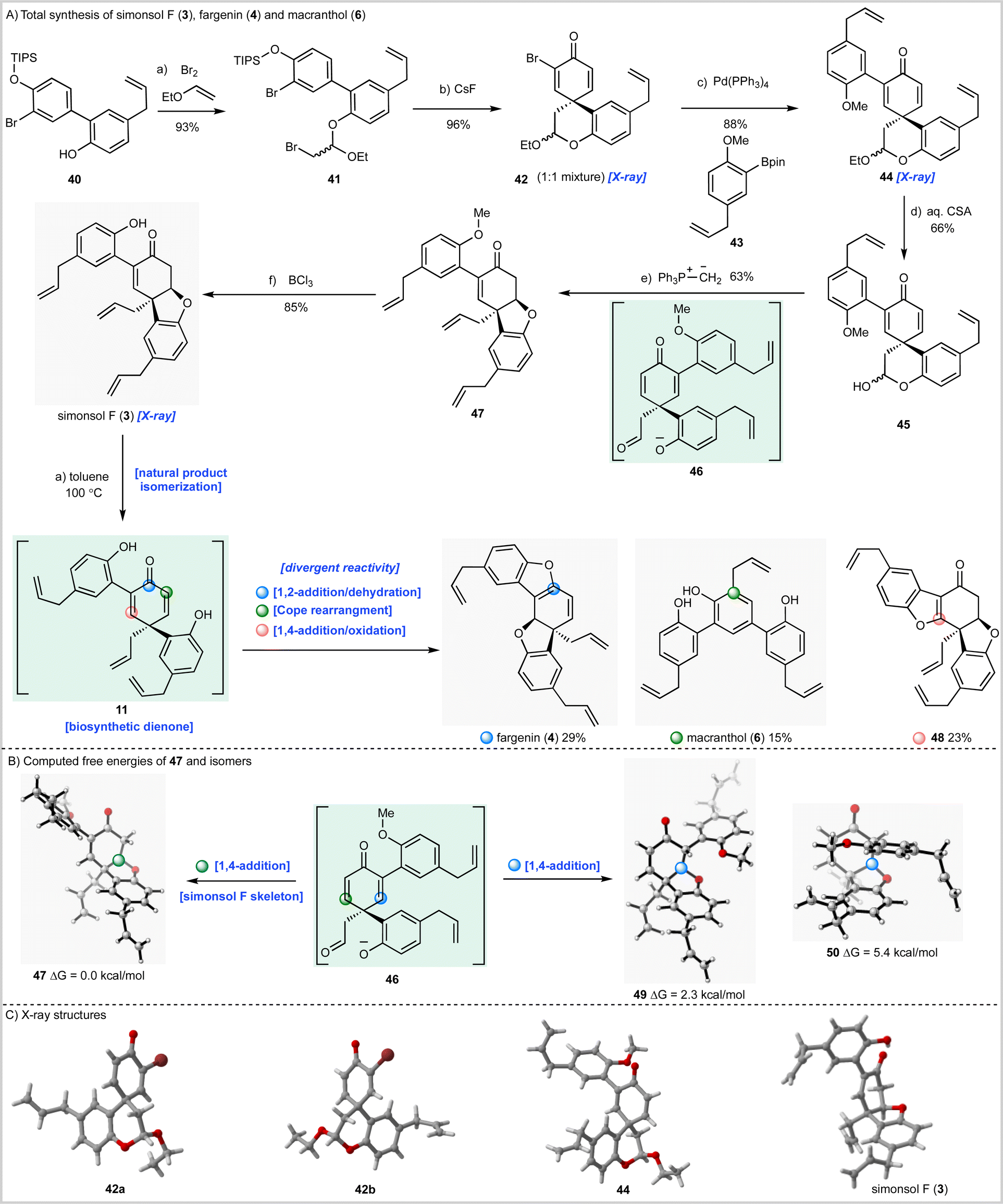 | ||
| Scheme 3 Total synthesis of simonsol F (3) and its possible biomimetic conversion into fargenin (4) and macranthol (6). (A) Reagents and conditions: (a) ethyl vinyl ether, Br2, Hünig's base, DCM, 0 °C to r.t., 2.5 h, 93%; (b) CsF, Na2SO4, DMF, 130 °C, 1 h, 96%; (c) Pd(PPh3)4, aq. Na2CO3, toluene°:°ethanol, 85 °C, 4 h, 88%; (d) CSA, H2O, 1,4-dioxane, 85 °C, 10.5 h, 66%; (e) MePPh3Br, KHMDS, THF, 0 °C, 0.5 h, 63%; (f) BCl3, DCM, 0 °C, 5.5 h, 85%; (a) toluene-d8, 100 °C, 228 h, macranthol (6) 15%, fargenin (4) 29%, 58 23%. (B) Computed free energies of 47 and isomers. (C) X-ray structures. DCM = dichloromethane, CSA = camphorsulfonic acid. | ||
We now had to develop hydrolysis conditions to obtain hemiacetal 45 for deployment in the cascade reaction. In this instance, when mixture 45 was heated with aqueous hydrochloric acid under the conditions used for the synthesis of simonsol C (2) a complex mixture of products was obtained. However, acid-catalyzed hydrolysis using camphorsulfonic acid gave hemiacetal 45 as the major product in an acceptable yield of 66%. Treatment of this mixture with methylenetriphenylphosphorane again proceeded smoothly to give methyl-protected simonsol F (47) establishing this sequence as a general method for the construction of functionalized tetrahydrodibenzofurans. In this iteration of the cascade reaction, the presumed dienone phenolate intermediate 46, or its methylenated equivalent, can undergo conjugate 1,4-addition at two sites (Scheme 3B). Experimentally, formation of simonsol F (3) via intermediate 47 was observed and QM calculations suggested that the site selectivity is thermodynamically controlled. The alternative conjugate addition products 49 and 50 are 2.3 and 5.4 kcal mol−1 higher in energy respectively than 47. This is consistent with nucleophilic attack on the more electron-deficient enone, not substituted by the electron-rich aromatic substituent. This reaction also preserves the conjugation of this substituent to the double bond, stabilizing the product.
As with simonsol C (2) the final step of the synthesis proved to be challenging as a result of the sensitive allyl groups and the reactivity of the natural product post deprotection. After considerable experimentation we identified demethylation conditions that involved treatment of 48 with BCl3 at 0 °C for 5.5 hours which gave simonsol F (3) in an excellent isolated yield of 85% (Scheme 3A). The structure of the natural product was confirmed by X-ray crystallography (Scheme 3C).
Synthesis of fargenin (4), macranthol (6), simonsinol (5), and honokiol (14) through natural product isomerization
During the syntheses of simonsol F (3) and simonsol C (2), we had observed that both natural products were thermally unstable to the extent that cross-coupling and demethylation conditions had to be carefully developed to avoid generating additional products. Therefore, we next explored the innate reactivity of these natural products and our notion that dienone intermediates e.g.11 (Scheme 1A), serve as biosynthetic branch points. If such intermediates could be accessed from the natural products themselves through retro conjugate addition, it would provide evidence for our biosynthesis, as well as opening isomerization pathways between natural products. Gratifyingly this reasoning proved to be correct, and we discovered that when simonsol F (3) was heated in toluene at 100 °C, it was converted into the pentacyclic natural product fargenin (4, 29%), the triaryl natural product macranthol (6, 15%) and benzofuran 48 (23%) (Scheme 3A). This remarkable series of transformations provides compelling evidence for intermediate 11 and the divergent reactivity associated with the cross-conjugated dienone moiety.As depicted in Scheme 3A the formation of these products can be rationalized by invoking retro conjugate addition to generate dienone 11. Fargenin (4) is then obtained through 1,2-addition of the phenolic hydroxyl group followed by dehydration, as indicated by the blue label on intermediate 11. An alternative oxidative 1,4-addition reaction (pink-labelled carbon) gives rise to benzofuran 48, and a Cope rearrangement (green label) is responsible for the generation of macranthol (6). The Cope rearrangement is of potential biosynthetic significance as it provides a hitherto unknown pathway to macranthol (6) which we had previously assumed to be a heterooligomer originating from the oxidative coupling of chavicol (7) and 2-allylphenol (15) (Scheme 1B).
Given these observations we next sought to effect isomerization of other homooligomeric natural products into their presumed heterooligomeric aromatic counterparts (Scheme 4A). In agreement with the results described above we discovered that simonsol G (1), which was prepared using our cascade strategy (see ESI† for the synthesis), could be converted into honokiol (18) under mild basic conditions. Similarly, simonsol C (2) and simonsol F (3) were converted into simonsinol (5) and macranthol (6) respectively. In the case of simoninsol (5), permethylation to give ether 52 was carried out for comparison with the literature compound derived from natural simonsinol (5).
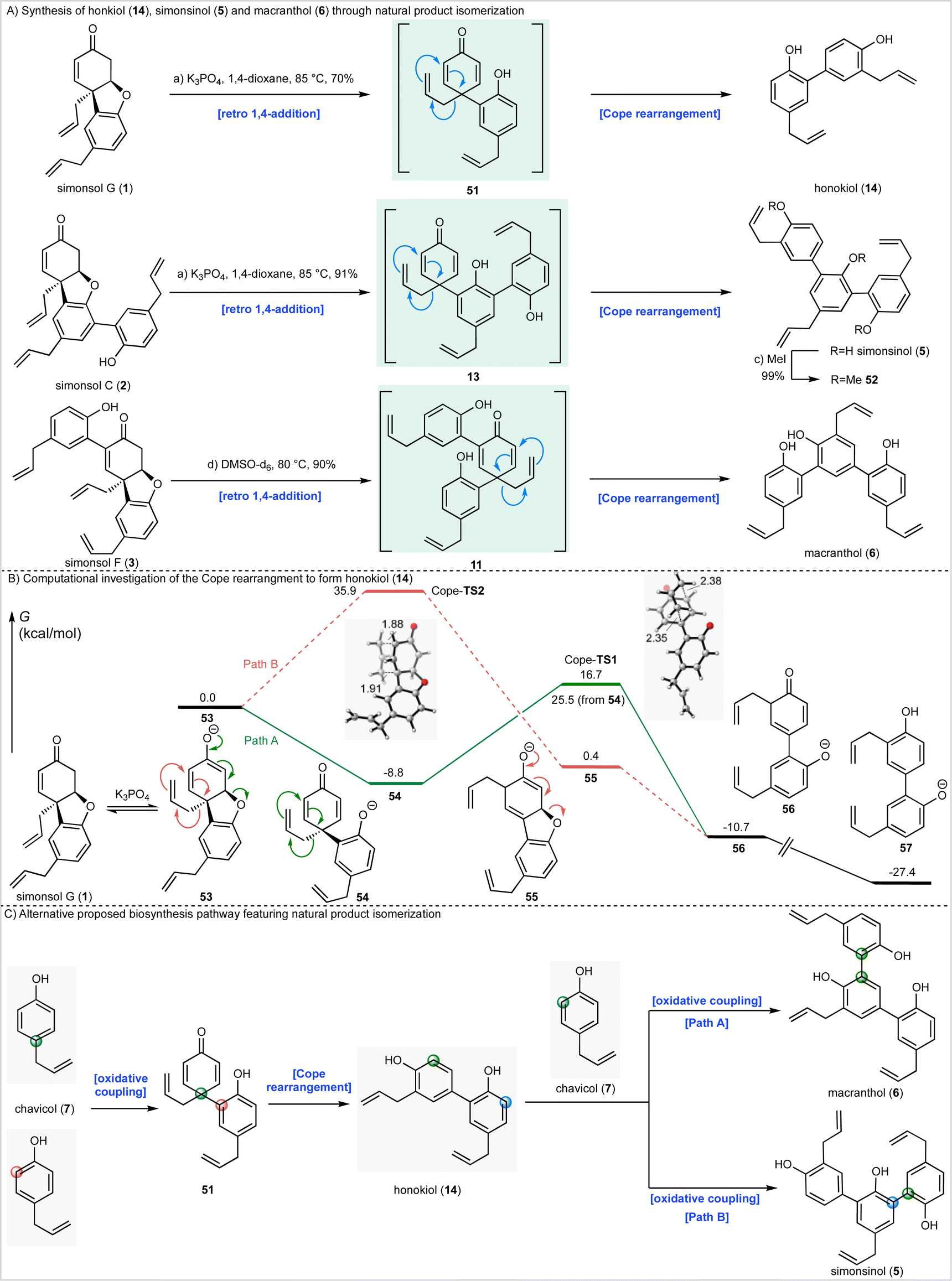 | ||
| Scheme 4 Synthesis of honokiol (14), simonsinol (5) and macranthol (6) through natural product isomerization. (A) Reagents and conditions: (a) K3PO4, 1,4-dioxane, 85 °C, 40 h, 70% (b) K3PO4 aq., THF, 80 °C, 7 h, 91%; (c) MeI, K2CO3, acetone, 55 °C, 24 h, 99%; (d) DMSO-d6, 85 °C, 16 h, 90%. (B) Computational analysis of potential Cope rearrangements. (C) Alternative biosynthesis proposal. DMSO = dimethylsulfoxide. | ||
To gain further insight into the Cope rearrangements we conducted DFT calculations to study the isomerization process that converts simonsol G (1) into honokiol (14). In the presence of base, this likely occurs through the generation of an equilibrium concentration of enolate 53 which could proceed through either Path A or Path B (Scheme 4B), depending on the order of the retro conjugate addition and Cope rearrangement. Path A, as invoked in Scheme 4A, involves a retro-conjugate addition of simonsol G enolate 53 to form the dienone intermediate 54, which undergoes Cope rearrangement and tautomerization to form anionic honokiol 57. Alternatively, along Path B, Cope rearrangement of 53 forms the dienolate intermediate 55, which then undergoes retro-conjugate addition and tautomerization to form 56. For completeness, we also investigated the corresponding neutral Cope rearrangements leading to honokiol (14) and macranthol (6) (see ESI†). Our computed reaction coordinate profile of Path A suggests that the formation of the dienone intermediate 54 from the simonsol G enolate 53 is exergonic by 8.8 kcal mol−1 (Scheme 4B). The subsequent Cope rearrangement transition state (Cope-TS1) has a relatively low free energy barrier of 25.5 kcal mol−1, and formation of the rearrangement product 56 is exergonic by 1.9 kcal mol−1. Subsequent tautomerization forms the honokinol phenolate 57, which is exogonic by 18.6 kcal mol−1.
Alternatively, Path B (Scheme 4B) via Cope-TS2 has a high free energy barrier of 35.9 kcal mol−1, which is kinetically unlikely at the reaction temperature (85 °C). Both Cope transition states adopt the favored chair-like conformation (Scheme 4B). As a result of the cross conjugated nature of dienone 54 the Cope-TS1 is favored over Cope-TS2 by 19.2 kcal mol−1. Based on these calculations we conclude that the natural product conversions take place through the proposed biosynthetic dienones 51, 13 and 11, or corresponding anions depending on the reaction conditions (Scheme 4A).
Biosynthetic ramifications
The isomerization processes that we have discovered and elucidated above have important consequences for the likely biosynthetic origin of honokiol (14), simonsinol (5), and macranthol (6). Specifically, it is no longer necessary to view these natural products as heterooligomers. Instead, we can draw a revised biosynthesis with chavicol (7) as the sole precursor (Scheme 4C). We suggest that honokiol arises from an ortho-para oxidative coupling of chavicol (7) to generate dienone 51 which can undergo a Cope rearrangement to generate honokiol (14) (Scheme 4C). Subsequent oxidative coupling with chavicol (7) can now generate macranthol (6) (Path A) or simonsinol (5) (Path B).Alternatively, honokiol (14), simonsinol (5) and macranthol (6) could arise from the isomerization processes that we have demonstrated experimentally (Scheme 4A) with the formation of an additional aromatic ring as the thermodynamic driving force. Although the calculated barrier of the Cope rearrangement of 54 is relatively high (t1/2 6 days at 25 °C), it is possible that this could be accelerated through enzyme catalysis. Examples of enzymes catalyzing pericyclic reactions are rare;39 however, there is evidence to support biosynthetic Cope rearrangements in alkaloid biosynthesis40–42 and there are biogenetically inspired syntheses of other Illicium natural products which feature Claisen and Cope rearrangements.43
Biological investigations: axonal growth assay
Having completed our total synthesis studies, we sought to investigate the potential neuronal functional properties of tetrahydrodibenzofuran-containing Illicium neolignans. When simonsol C (2) was isolated and characterized by Wang and co-workers, no anti-acetylcholinesterase activity was observed in vitro (no inhibition below 100 μM) nor was any anti-butyrylcholinesterase activity observed (no inhibition below 50 μM). However, Banwell suggested that the similarity between the ABC rings of simonsol C (2) and galantamine alkaloids (Fig. 1C) was such that biological investigation for the former was warranted.12The fact that both magnolol (10), which is contained within simonsol C (2), and honokiol (14) have been shown to possess neuroprotective and restorative effects44–46 lends credence to this notion. As does the additional observations that a variety of structurally related neolignans and sesquiterpene neoligans exhibit neurite outgrowth properties.14–19,47,48 Given that axonal functional loss and degeneration is known to occur at the early stages of neurodegenerative diseases which precedes cell death and the onset of clinical symptoms49 we opted to screen compounds in an axonal growth assay. We began by examining magnolol (10), simonsinol (5), simsonol C (2), and simonsol F (3) along with selected synthetic intermediates 58, 59 and 25 which contained phenolic, phenylpropenoid and ether moieties (Scheme 5A). The compounds were applied as DMSO solutions to primary embryonic mouse cortical neurons that had been cultured for two days on poly-L-ornithine coated glass coverslips. Following a forty-eight-hour incubation, neurons were fixed and immunostained for the axonally-enriched acetylated tubulin to reveal overall neuron morphology. Each coverslip was imaged at 20 random points and the axonal length of neurons was measured computationally (Scheme 5B).
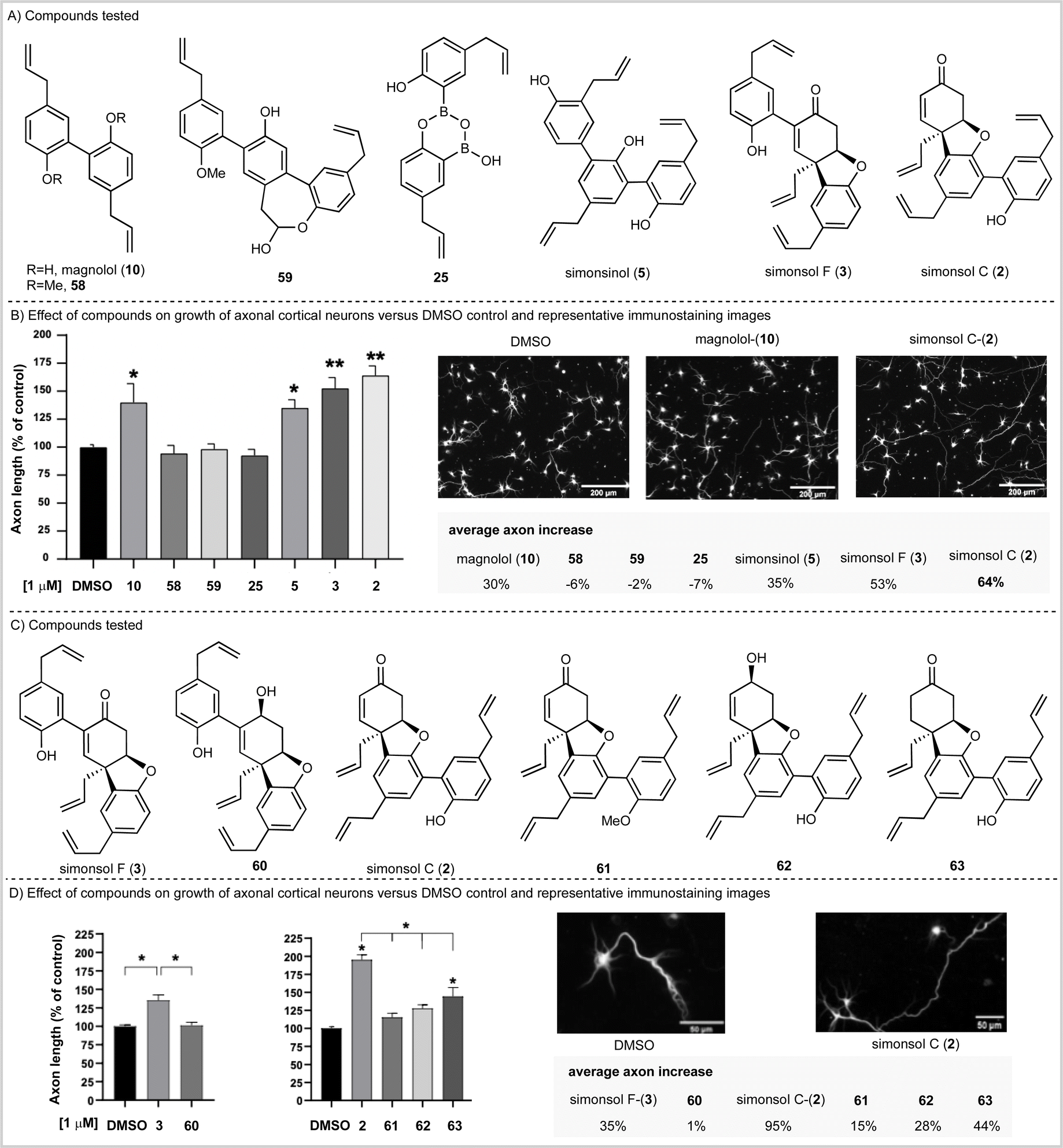 | ||
| Scheme 5 Effect of Illicium natural products and analogs on axonal growth. (A and C) Compounds tested. (B and D) Analysis of the effect of Illicium-derived neolignans and intermediate compounds on the growth of the axon of primary cortical neurons 48 hours after incubation in culture, as compared to control DMSO. Data obtained from 3–5 independent experiments, ∼160–200 axons measured per condition in each; *p < 0.05, **p < 0.01, one-way ANOVA with posthoc Dunnet's multiple comparison test. | ||
This study established that magnolol (10), simonsinol (5), simonsol F (3) and simonsol C (2) increased the length of mouse primary cortical axons at 1 μM. Furthermore, simonsol F (3) and simonsol C (2) promoted axonal growth at a level above that of the dimeric and trimeric aromatic natural products magnolol (10) and simonsinol (5) (Scheme 5B). Therefore, the galantamine alkaloid-like tetrahydrodibenzofuran core is biologically relevant in the promotion of axonal growth.
To gain further insight, we prepared a range of novel analogs (see SI for synthesis) in order to further probe the role of the tetrahydrodibenzofuran core (Scheme 5C). We began by examining simonsol F (3) and its reduced counterpart 60 at a concentration of 1 μM (Scheme 5D). The allylic alcohol 60 did not promote axonal growth, which illustrates the requirement for the enone within the tetrahydrodibenzofuran ring system. A second comparison between simonsol C-(2) and derivatives 61, 62 and 63 was then made (Scheme 5C and D). These results show that, in the case of simonsol C (2), a free phenol and the enone are required for the significant axonal growth which is observed. Taken together these data support Banwell's suggestion that simonsol C (2), and analogs thereof, may be promising starting points for the development of novel small molecules of therapeutic relevance in neurodevelopmental and neurodegenerative diseases.
Conclusion
In this Article we have documented the first total synthesis of simonsol F (3), simonsinol (5), fargenin-(4), macranthol (6) and reported additional total syntheses of simonsol C (2), simonsol G (1), and honokiol (14). The syntheses were based upon a phosphonium ylide-mediated cascade reaction which allowed for the efficient construction of the characteristic [4.3.0] core ring system, and upon natural product isomerization reactions which feature unusual dienone Cope rearrangements. As a corollary of the rearrangement processes, we proposed an alternative biosynthetic origin of honokiol (14), simonsinol (5) and macranthol (6), from a single common precursor chavicol (7). Finally, we have shown that both simonsol C (2) and simonsol (F) promote axonal growth and that the enone-containing tetrahydrodibenzofuran ring system is required for this activity.Data availability
The experimental and computational data supporting this article have been included as part of the ESI.†Author contributions
R. M. D. conceived the work. R. M. D. and R. E. A. prepared the manuscript. The synthesis work was carried out by R. E. A. and J. S. and R. E. A., J. S., and R. M. D. analysed the data. The computational chemistry was carried out by H. S. and K. N. H. The X-ray crystallography was carried out by S. A., L. T., and W. L. The axonal growth assays and subsequent data analysis was carried out by R. M.-B. and F. D.-J.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Mice (C57/BL6) for primary neuron cultures were housed, bred and sacrificed (Schedule 1) in compliance with the ethics and animal welfare in accordance with the Animal (Scientific Procedures) Act 1986, which is in place at the University of Nottingham, UK. We acknowledge financial support from the School of Chemistry, University of Nottingham (PhD Studentship to R. E. A. and J. S.).Notes and references
- J. Xu, M. H. Lacoske and E. A. Theodorakis, Angew. Chem., Int. Ed., 2014, 53, 956–987 CrossRef CAS.
- J. Richers, A. Pöthig, E. Herdtweck, C. Sippel, F. Hausch and K. Tiefenbacher, Chem.–Eur. J , 2017, 23, 3178–3183 CrossRef CAS.
- Y. Yang, Z. Wang, J. Wu and Y. Chen, Chem. Biodiversity, 2016, 13, 269–274 CrossRef CAS.
- A. Latif, Y. Du, S. R. Dalal, M. L. Fernández-Murga, E. F. Merino, M. B. Cassera, M. Goetz and D. G. I. Kingston, Chem. Biodiversity, 2017, 14, e1700209 CrossRef.
- C. Dong, L. Liu, X. Li, Z. Guan, H. Luo and Y. Wang, Planta Med., 2013, 79, 338–347 CAS.
- L. K. Sy and G. D. Brown, Phytochemistry, 1996, 43, 1417–1419 CrossRef CAS.
- M. Moriyama, J.-M. Huang, C.-S. Yang, H. Hioki, M. Kubo, K. Harada and Y. Fukuyama, Tetrahedron, 2007, 63, 4243–4249 CrossRef CAS.
- I. Kuono, T. Morisaki, Y. Hara and C.-S. Yang, Chem. Pharm. Bull., 1997, 117, 415–434 Search PubMed.
- I. Kuono, A. Hashimoto, K. Nobusuke and C.-S. Yang, Chem. Pharm. Bull., 1989, 1291–1292 CrossRef.
- J. Marco-Contelles, M. do Carmo Carreiras, C. Rodríguez, M. Villarroya and A. G. García, Chem. Rev., 2006, 106, 116–133 CrossRef CAS PubMed.
- S. Majumder, A. Yadav, S. Pal, A. Khatua and A. Bisai, J. Org. Chem., 2022, 87, 7786–7797 CrossRef CAS.
- J. Nugent, M. G. Banwell and B. D. Schwartz, Org. Lett., 2016, 18, 3798–3801 CrossRef CAS PubMed.
- J. J. Sui, K. Wang, S. J. Luo, K. Guo, M. W. Yuan and H. B. Qin, J. Org. Chem., 2024, 7821–7827 CrossRef CAS.
- X. Cheng, N. L. Harzdorf, T. Shaw and D. Siegel, Org. Lett., 2010, 12, 1304–1307 CrossRef CAS PubMed.
- Z. Khaing, D. Kang, A. M. Camelio, C. E. Schmidt and D. Siegel, Bioorg. Med. Chem. Lett., 2011, 21, 4808–4812 CrossRef CAS PubMed.
- Y. Fukuyama, K. Nakade, Y. Minoshima, R. Yokoyama, H. Zhai and Y. Mitsumoto, Bioorg. Med. Chem. Lett., 2002, 12, 1163–1166 CrossRef CAS.
- T. Esumi, G. Makado, H. Zhai, Y. Shimizu, Y. Mitsumoto and Y. Fukuyama, Bioorg. Med. Chem. Lett., 2004, 14, 2621–2625 CrossRef CAS.
- Y. Fukuyama, Y. Otoshi, K. Miyoshi, K. Nakamura, M. Kodama, M. Nagasawa, T. Hasegawa, H. Okazaki and M. Sugawara, Tetrahedron, 1992, 48, 377–392 CrossRef CAS.
- K. Zlotkowski, J. Pierce-Shimomura and D. Siegel, ChemBioChem, 2013, 14, 307–310 CrossRef CAS PubMed.
- G. Brunow, I. Kilpeläinen, J. Sipilä, K. Syrjänen, P. Karhunen, H. Setälä and P. Rummakko, ACS Symp. Ser., 1998, 697, 131–147 CrossRef CAS.
- S. C. Tzeng and Y. C. Liu, J. Mol. Catal. B: Enzym., 2004, 32, 7–13 CrossRef CAS.
- L. K. Sy and G. D. Brown, J. Chem. Res., 1998, 476–477 RSC.
- R. M. Denton, J. T. Scragg, A. M. Galofré, X. Gui and W. Lewis, Tetrahedron, 2010, 66, 8029–8035 CrossRef CAS.
- P. R. Khan, T. Mujawar, P. Shekhar, G. Shankar, B. V. Subba Reddy and R. Subramanyam, Tetrahedron Lett., 2020, 61, 152229–152230 CrossRef CAS.
- A. M. Wright and G. W. O'Neil, Tetrahedron Lett., 2016, 57, 3441–3443 CrossRef CAS.
- K. Harada, C. Arioka, A. Miyakita, M. Kubo and Y. Fukuyama, Tetrahedron Lett., 2014, 55, 6001–6003 CrossRef CAS.
- C. M. Chen and Y. C. Liu, Tetrahedron Lett., 2009, 50, 1151–1152 CrossRef CAS.
- T. Esumi, G. Makado, H. Zhai, Y. Shimizu, Y. Mitsumoto and Y. Fukuyama, Bioorg. Med. Chem. Lett., 2004, 14, 2621–2625 CrossRef CAS PubMed.
- J. K. Patra, G. Das, S. Bose, S. Banerjee, C. N. Vishnuprasad, M. del Pilar Rodriguez-Torres and H. S. Shin, Phytother Res., 2020, 34, 1248–1267 CrossRef CAS.
- M. Arumugam, A. Mitra, P. Jaisankar, S. Dasgupta, T. Sen, R. Gachhui, U. Kumar Mukhopadhyay and J. Mukherjee, Appl. Microbiol. Biotechnol., 2010, 86, 109–117 CrossRef CAS.
- F. Amblard, D. Delinsky, J. L. Arbiser and R. F. Schinazi, J. Med. Chem., 2006, 49, 3426–3427 CrossRef CAS PubMed.
- P. Magnus, N. Sane, B. P. Fauber and V. Lynch, J. Am. Chem. Soc., 2009, 131, 16045–16047 CrossRef CAS PubMed.
- R. M. Denton and J. T. Scragg, Synlett, 2010, 4, 633–635 CrossRef.
- R. M. Denton, J. T. Scragg and J. Saska, Tetrahedron Lett., 2011, 52, 2554–2556 CrossRef CAS.
- R. M. Denton, J. T. Scragg, A. M. Galofré, X. Gui and W. Lewis, Tetrahedron, 2010, 66, 8029–8035 CrossRef CAS.
- R. M. Denton and J. T. Scragg, Org. Biomol. Chem., 2012, 10, 5629–5635 RSC.
- P. A. Byrne and D. G. Gilheany, Chem.–Eur. J., 2016, 22, 9140–9154 CrossRef CAS PubMed.
- N. C. Bruno, M. T. Tudge and S. L. Buchwald, Chem. Sci., 2013, 4, 916 RSC.
- K. N. Houk, X.-S. Xue, F. Liu, Y. Chen, X. Chen and C. Jamieson, Isr. J. Chem., 2022, 62, e202100071 CrossRef CAS.
- H. P. Chen and I. Abe, Synth. Syst. Biotechnol., 2021, 6, 51–62 CrossRef CAS.
- S. A. Newmister, S. Li, M. Garcia-Borràs, J. N. Sanders, S. Yang, A. N. Lowell, F. Yu, J. L. Smith, R. M. Williams, K. N. Houk and D. H. Sherman, Nat. Chem. Biol., 2018, 14, 345–351 CrossRef CAS.
- L. Y. P. Luk, Q. Qian and M. E. Tanner, J. Am. Chem. Soc., 2011, 133, 12342–12345 CrossRef CAS.
- J. A. Homer, I. De Silvestro, E. J. Matheson, J. T. Stuart and A. L. Lawrence, Org. Lett., 2021, 23, 3248–3252 CrossRef CAS PubMed.
- C. P. Hoi, Y. P. Ho, L. Baum and A. H. L. Chow, Phytother Res., 2010, 24, 1538–1542 CrossRef CAS PubMed.
- H. H. Chen, S. C. Lin and M. H. Chan, Neurodegener. Dis., 2011, 8, 364–374 CrossRef CAS PubMed.
- N. Matsui, K. Takahashi, M. Takeichi, T. Kuroshita, K. Noguchi, K. Yamazaki, H. Tagashira, K. Tsutsui, H. Okada, Y. Kido, Y. Yasui, N. Fukuishi, Y. Fukuyama and M. Akagi, Brain Res., 2009, 1305, 108–117 CrossRef CAS PubMed.
- Z. Khaing, D. Kang, A. M. Camelio, C. E. Schmidt and D. Siegel, Bioorg. Med. Chem. Lett., 2011, 21, 4808–4812 CrossRef CAS PubMed.
- Y. Fukuyama, M. Kubo and K. Harada, J. Nat. Med., 2020, 74, 648–671 CrossRef CAS PubMed.
- N. Salvadores, M. Sanhueza, P. Manque and F. A. Court, Front. Neurosci., 2017, 11, 451 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2327688–2327695. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03232b |
| This journal is © The Royal Society of Chemistry 2024 |