
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulating Ni–S coordination in Ni3S2 to promote electrocatalytic oxidation of 5-hydroxymethylfurfural at ampere-level current density†
Lan
Chen‡
ab,
Zhaohui
Yang‡
c,
Chuanyu
Yan
ab,
Yijun
Yin
c,
Zhimin
Xue
 *ab,
Yiting
Yao
ab,
Shao
Wang
c,
Fanfei
Sun
*de and
Tiancheng
Mu
*c
*ab,
Yiting
Yao
ab,
Shao
Wang
c,
Fanfei
Sun
*de and
Tiancheng
Mu
*c
aBeijing Key Laboratory of Lignocellulosic Chemistry, Beijing Forestry University, Beijing 100083, China. E-mail: zmxue@bjfu.edu.cn
bState Key Laboratory of Efficient Production of Forest Resources, Beijing 100083, China
cSchool of Chemistry and Life Resources, Renmin University of China, Beijing 100872, China. E-mail: tcmu@ruc.edu.cn
dShanghai Synchrotron Radiation Facility, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201204, China. E-mail: sunff@sari.ac.cn
eShanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 201204, China
First published on 28th June 2024
Abstract
Electricity-driven oxidation of 5-hydroxymethylfurfural (HMF) to 2,5-furandicarboxylic acid (FDCA) is a highly attractive strategy for biomass transformation. However, achieving industrial-grade current densities remains a great challenge. Herein, by modulating the water content in a solvothermal system, Ni3S2/NF with stabilized and shorter Ni–S bonds as well as a tunable coordination environment of Ni sites was fabricated. The prepared Ni3S2/NF was highly efficient for electrocatalytic oxidation of HMF to produce FDCA, and the FDCA yield and Faraday efficiency could reach 98.8% and 97.6% at the HMF complete conversion. More importantly, an industrial-grade current density of 1000 mA cm−2 could be achieved at a potential of only 1.45 V vs. RHE for HMFOR and the current density could exceed 500 mA cm−2 with other bio-based compounds as the reactants. The excellent performance of Ni3S2/NF originated from the shorter Ni–S bonds and its better electrochemical properties, which significantly promoted the dehydrogenation step of oxidizing HMF. Besides, the gram-scale FDCA production could be realized on Ni3S2/NF in a MEA reactor. This work provides a robust electrocatalyst with high potential for practical applications for the electrocatalytic oxidation of biomass-derived compounds.
Introduction
In order to mitigate the environmental problems and energy crisis caused by the overconsumption of fossil resources, there is an urgent need to find alternative resources that should be renewable and abundantly available.1 Biomass, the world's most abundant renewable carbon-based resource, is a promising alternative to fossil resources. Generally, biomass can be converted into high-value chemicals,2,3 fuel products (e.g., bio-oils),4 and functional bio-based materials.5 In this regard, 5-hydroxymethylfurfural (HMF), a highly attractive platform molecule prepared from biomass-based carbohydrates (e.g., cellulose, and glucose),6 has great potential in the production of high value-added fine chemicals,7 such as 2,5-furandicarboxylic acid (FDCA), 2,5-furandicarboxaldehyde (DFF), 2,5-dimethyl furan (DMF), etc. Of these HMF-derived compounds, FDCA has been employed as the renewable monomer for the synthesis of polyethylene 2,5-furandicarboxylate (PEF), which has been considered an environmentally friendly alternative to petroleum-derived and widely-used polyethylene terephthalate (PET).8,9 Therefore, selective conversion of HMF to produce FDCA has become a hotspot in recent years.10–12Generally, FDCA could be synthesized from oxidation of HMF via the conventional thermal catalysis.13 However, thermal catalysis was usually carried out under harsh reaction conditions (i.e., high temperature, and/or high oxygen pressure).14,15 Recently, electrocatalytic oxidation of HMF (HMFOR) for the production of FDCA has been developed, which exhibits inherent advantages such as mild conditions, no need for precious metals and oxidants, as well as environmental friendliness. More importantly, HMFOR has favorable thermodynamic properties compared to the OER and can be coupled with different cathodic reduction reactions to obtain various high value-added products (e.g., H216,17 and CO18). Currently, the main challenge for HMFOR remains the development of robust catalysts for practical applications. In previous studies, transition metal materials, especially nickel-based catalysts, were the most used electrocatalysts19 because Ni sites (by the adsorption of hydroxide or being converted into hydroxide) could promote HMF oxidation by proton-coupled electron transfer processes. HMFOR generally requires a redox mediator as a sacrificial agent (e.g., TEMPO) for the conversion of HMF to FDCA under neutral or weakly acidic conditions.20,21 However, the Ni-based catalysts reported in recent articles can efficiently convert HMF to FDCA through an indirect oxidation mechanism without the need for a sacrificial agent.22 Although many Ni-based catalysts have been developed for HMFOR, the developed catalysts unfortunately underwent severe and uncontrollable surface reconstruction to form oxides or hydroxides under alkaline conditions, which might result in a significant decrease in catalytic activity.23 More challengingly, there were only a few catalytic systems that could conduct HMFOR in a high concentration of HMF (>50 mM) at industrial-scale current densities (>500 mA cm−2), and the systems reaching ampere-scale current densities were even less.24,25 Therefore, it is highly essential to construct robust and stable nickel-based electrocatalysts to realize HMFOR at high current densities.
The electro-catalytic performance of Ni-based catalysts was significantly affected by the electronic environment of Ni sites. Generally, the orbital hybridization of Ni sites could be modulated by changing the type and ratio of coordination bonds of the active sites, resulting in a delicate regulation of the electronic structure.26,27 Thus, modifying the coordination structure of Ni sites was a promising strategy to improve the activity of Ni-based catalysts for HMFOR. Besides, the electrochemical performance of the electrodes could also be improved by introducing heteroatoms (e.g., S, N and P) to optimize the electrode surface properties.28 Poor catalytic efficiency was often obtained due to excessive adsorption of substrates on the electrode surface with a very strong positive electric field.29 It has been reported that the S2− anion of good electronegativity could reduce the strength of the positive electric field on the electrode surface, thereby promoting the desorption of the product as well as the adsorption and desorption of the probable intermediates.30 Additionally, according to the Lewis acid–base theory, there was a M–X coordination (M and X represent the metal and the heteroatoms, respectively.) formed between the metal and the heteroatom.31 In this way, the Lewis acid–base electron pair could boost the activation of the catalyst and reduce the reaction potential barrier on the catalytic electrode effectively.32 Based on the discussions above, robust and stable Ni-based electrocatalytic materials to generate high current densities for HMFOR could be potentially constructed by finely tuning the coordination structure of Ni sites.
In this work, Ni3S2 nanosheets on the surface of nickel foam (NF) were synthesized by a one-step solvothermal method. The synthesized Ni3S2/NF with shorter Ni–S coordination offered the possibility of sustained and rapid electron transfer during the reaction as well as the deprotonation of HMF and showed high catalytic efficiency for HMFOR, in which the HMF conversion, the FDCA yield, and the Faraday efficiency (FE) of FDCA could reach close to 100%, 98.8% and 97.6%, respectively. More importantly, an ultra-high current density (1000 mA cm−2) could be achieved at a potential of only 1.45 V vs. RHE. The fact of achieving industrial current density and the high stability represented the major breakthrough, thus making the developed catalyst have great potential in practical applications.
Results and discussion
The desired Ni3S2/NF was prepared by a one-step solvothermal route as illustrated in Fig. 1a, where thioacetamide (TAA) was employed as the sulfur source. The morphology of the prepared Ni3S2/NF was characterized using field emission scanning electron microscopy (FE-SEM). As shown in Fig. S1a and S1b,† after the solvothermal process, the NF framework was well maintained, and Ni3S2 nanosheets were arranged neatly on the NF surface to form three-dimensional porous nanospheres (Fig. 1b–f). In comparison, the morphology of the Ni3S2/NF changed from nanospheres to nanodendrites when water was introduced into the synthetic system, and the size of dendrites became larger with the increase of the introduced water (Fig. S2–S5†). The transmission electron microscopy (TEM) image showed that the prepared Ni3S2/NF was composed of nanosheets (Fig. 1e). Importantly, the lattice crystal for Ni3S2 could be clearly observed (Fig. 1f and g), where 0.208 nm belonged to the (202) plane of Ni3S2, indicating the successful synthesis of Ni3S2 on NF.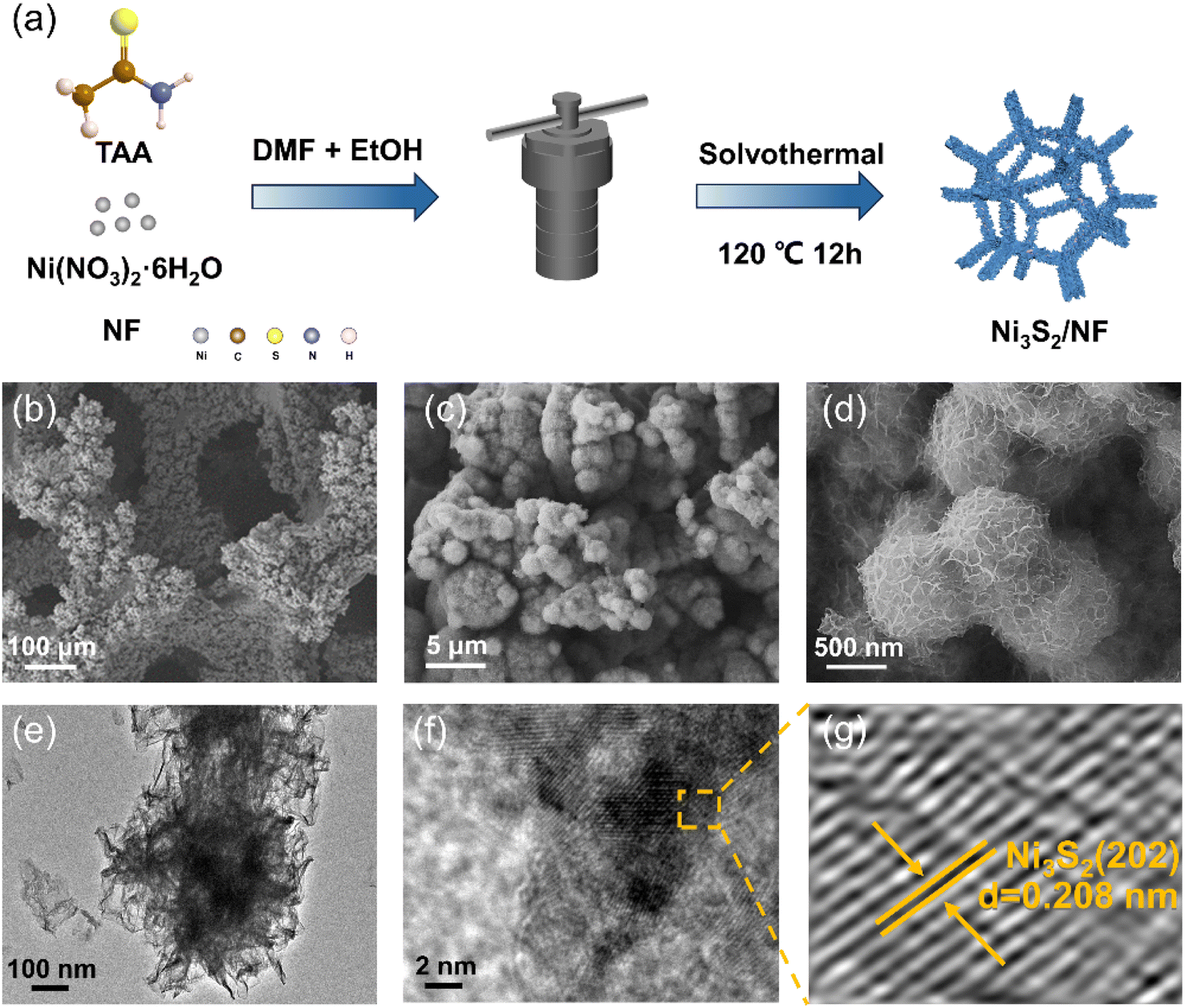 | ||
| Fig. 1 (a) The process of preparing the desired Ni3S2/NF, (b–d) SEM images of Ni3S2/NF, (e) TEM image of Ni3S2/NF, and (f and g) HRTEM images of Ni3S2/NF. | ||
The fine structure of the prepared Ni3S2/NF was further characterized by X-ray diffraction (XRD), Raman spectroscopy, and X-ray photoelectron spectroscopy (XPS). As presented in XRD patterns (Fig. 2a), both Ni3S2/NF and Ni3S2/NF-1 showed the characteristic peaks of Ni3S2 (PDF # 44-1418) and Ni (PDF # 04-0850), while Ni(OH)2/NF possessed the characteristic peaks of Ni(OH)2 (PDF # 38-0715) and Ni (PDF # 04-0850). More importantly, the peak intensity for Ni3S2 in Ni3S2/NF was greater than that in Ni3S2/NF-1, suggesting that Ni3S2/NF was more fully vulcanized and crystallized, which endowed Ni3S2/NF with a higher electronic conductivity. Moreover, the Raman spectrum was used to identify the molecular vibrational information of functional groups on the electrode surface. In Raman spectra of the prepared electrodes (Fig. 2b), the characteristic Ni–S vibrational modes were observed in the range of 100–800 cm−1 for each electrode, further confirming the formation of Ni3S2 during the solvothermal process. Notably, the intensity of the Ni–S bond decreased gradually as the water ratio increased in the reaction solvent, suggesting that the content of the Ni–S bond could be modulated by adjusting the water ratio in the solvothermal system. Considering the important role of surface species and their electronic state in electrocatalysis, the electronic states and the surface chemical compositions of the prepared materials were determined by XPS. XPS survey spectra (Fig. S6†) confirmed the coexistence of Ni, S, and O elements in the obtained materials. In the high-resolution XPS spectra of Ni 2p (Fig. 2c), characteristic peaks of Ni2+ at 855.7 (Ni2+ 2p3/2) and 873.7 eV (Ni2+ 2p1/2) accompanied by oscillating satellite peaks were observed. Besides, there was an extra peak assigned to the Ni–S bond at 852.3 eV.30,33 More importantly, the binding energy of Ni 2p increased with the increase of the water ratio in the solvothermal system, suggesting that the Ni sites in the Ni3S2/NF were more negatively charged. In the XPS spectra of S 2p, two peaks at 162.1 eV and 163.5 eV, which were assigned to S2− 2p3/2 and S2− 2p1/2, respectively, could be observed. Notably, the peak of S 2p in Ni3S2/NF moved towards lower binding energy with the increase of the water ratio in the solvothermal system (Fig. 2d), implying that S2− in Ni3S2/NF was more negatively charged. Based on the XPS results of Ni 2p and S 2p, the electronic interaction between Ni species and S species in the prepared materials could be tuned by the water ratio in the solvothermal system, and this interaction in Ni3S2/NF was the strongest. Additionally, the ratio of S–O/Ni–S bonds increased from 0 to 1.180 when the water ratio in the solvothermal system increased from 0 to 100% (Table S1†), indicating the controllable modification of the material structure by the water ratio in the solvothermal system. With the increase of water proportion in the solvothermal system, water molecules would occupy more Ni-ligand sites, resulting in the generation of S–O bonds on the surface of the prepared materials.34 Actually, the intensity of the Ni–S bonds would significantly increase when the surface of each material was etched by Ar+ ions for 100 s (Fig. S7 and S8†), further confirming that the surface structure of the obtained materials was tunable. Comparing the O 1s spectra of the samples (Fig. S9†), three characteristic peaks appear near 531.7, 530.6 eV and 529.4 eV attributed to the adsorbed water and oxides on the catalyst surface, O–H bonds and Ni–O bonds. Owing to the fact that the sample has been in contact with air, this leads to the oxidation of the surface metal to form Ni–O bonds. Ni3S2/NF has the lowest intensity of the characteristic peak belonging to the Ni–O bond, meaning that oxygen content is limited compared to the other prepared catalysts. The above results indicate that the Ni3S2/NF has been prepared successfully. Additionally, the electronic and coordination structures of Ni were further analyzed by the X-ray absorption fine structure (XAFS) of Ni3S2/NF and Ni3S2/NF-1. The Ni K-edge X-ray absorption near-edge structure (XANES) spectra (Fig. 2e) of the materials showed that the near-edge energy of Ni3S2/NF was between those of Ni foil and Ni(OH)2, implying that the valence state of Ni in Ni3S2/NF was higher than that in Ni foil while lower than that in Ni(OH)2. The Fourier-transformed (FT) k3-weighted extended X-ray absorption fine structure (EXAFS) showed that there were Ni–S coordination peaks in Ni3S2/NF and Ni3S2/NF-1 at 2.28 and 2.35 Å in R-space (Fig. 2f), respectively, which were in good agreement with the Ni–S bond reported in the literature.35,36 Meanwhile, the EXAFS fitting curves in R-space matched well with the experimental spectra of Ni3S2/NF and Ni3S2/NF-1 (Fig. S10 and Table S2†). Notably, the bond length of Ni–S in Ni3S2/NF was shorter than that in Ni3S2/NF-1, implying the higher bond dissociation energy of Ni–S and the more robust structure of Ni3S2 in Ni3S2/NF.
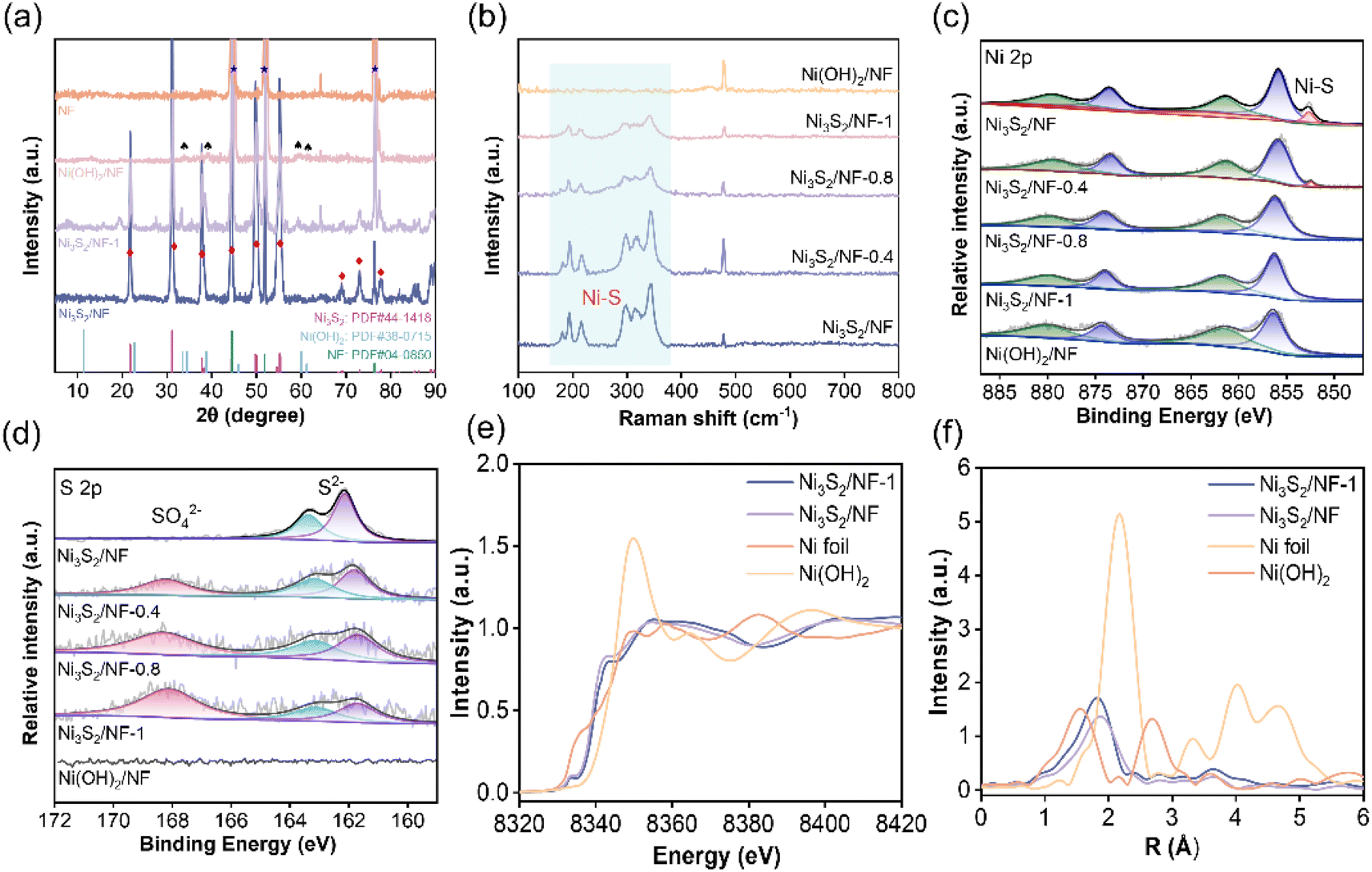 | ||
| Fig. 2 (a) XRD patterns, (b) Raman spectra, (c) XPS spectra of Ni 2p, (d) XPS spectra of S 2p, (e) Ni K-edge XANES, and (f) Ni K-edge Fourier transformed EXAFS spectra in the R space. | ||
The electrochemical performance of the prepared electrodes for the OER and HMFOR was investigated using an H-type electrolytic cell in 1.0 M KOH solution with and without 50 mM HMF. Before electrochemical testing, the electrodes were activated by cyclic voltammetry (CV) at a scan rate of 50 mV s−1 (Fig. S11†). In the LSV curves (Fig. 3a), a small peak appeared at the potential of 1.38 V vs. RHE for the OER, representing the oxidation of Ni2+ to Ni3+ as well as the adsorption of OH−. In comparison, the onset potential for HMFOR was lower than that of the OER, indicating that HMFOR was thermodynamically more favorable than the OER. Subsequently, the catalytic performance of the prepared electrodes for HMFOR was systematically investigated. It was observed that the current densities of Ni3S2/NF, Ni3S2/NF-0.4, Ni3S2/NF-0.8, Ni3S2/NF-1 and Ni(OH)2/NF decrease continuously (Fig. 3b and c), and the tendency was consistent with the content of S–O bond in the electrodes. Especially, Ni3S2/NF could achieve a current density of 1000 mA cm−2 at a potential of only 1.45 V vs. RHE. To the best of our knowledge, this current density was the maximum that could be achieved with non-precious metal electrodes in alkaline electrolyte (Fig. 3d and Table S3†). To analyze different catalytic performance, Tafel plots for different electrodes were collected (Fig. 3e). As expected, Ni3S2/NF had the lowest Tafel slope (63.5 mV dec−1) compared with Ni3S2/NF-0.4 (91.43 mV dec−1), Ni3S2/NF-0.8 (98.49 mV dec−1), Ni3S2/NF-1 (103.64 mV dec−1) and Ni(OH)2/NF (156.56 mV dec−1), indicating the superior catalytic kinetics for HMFOR over Ni3S2/NF. The lowest Tafel slope of Ni3S2/NF was probably caused by the stronger interaction between Ni and S species, which led to an optimized electronic configuration, thus enhancing the corresponding charge transfer process. Furthermore, the double layer capacitance (Cdl) was calculated from CV curves in the non-faradaic region (Fig. S12†) to estimate the electrochemically active surface area (ECSA) of the electrodes. Ni3S2/NF exhibited the highest ECSA (Fig. 3f), which could provide more electrochemically active sites for the HMFOR. Meanwhile, the interfacial charge transfer resistance was investigated based on electrochemical impedance spectroscopy (EIS). After being fitted with an equivalent circuit model (Fig. S13†), the Nyquist plot of Ni3S2/NF in 1 M KOH without HMF was more than five times larger than that after the addition of 50 mM HMF (Fig. S14†), suggesting that the OER had much larger charge transfer resistance (Rct) than HMFOR. More importantly, Ni3S2/NF exhibited a smaller Rct value in comparison with Ni3S2/NF-0.4, Ni3S2/NF-0.8, Ni3S2/NF-1 and Ni(OH)2/NF (Fig. 3g), indicating a faster charge transfer at the interface of Ni3S2/NF. Generally, most of the reported electrode materials tended to have a larger surface area than the geometry of the substrate electrode, resulting in a bias in the evaluation of apparent versus intrinsic activity.37–39 To reveal the inherent correlation between structural features and the catalytic activity of different electrodes, the turnover frequency (TOF) was evaluated (Fig. 3h), and the number of active sites was estimated by integration of the redox peak because the oxidation process of HMF involved a redox cycle between Ni2+ and Ni3+ (Fig. S15†).40 Based on the results in Fig. 3h and S13†, Ni3S2/NF not only had more active sites but also possessed the highest TOF. Based on the discussions above, we could deduce that the good performance of Ni3S2/NF probably originated from its high content of Ni–S bonds, which contributed to the exposure of the active sites, thereby enhancing the intrinsic activity of Ni3S2/NF for HMFOR. Encouraged by the excellent electrocatalytic performance of Ni3S2/NF for HMFOR, the electrooxidation of other bio-based compounds containing aldehyde or hydroxyl groups was evaluated. Based on the results in Fig. 3i and S16,† after adding 50 mM benzaldehyde (BZH), 5-hydroxymethyl furoic acid (HMFCA), 2,5-diformylfuran (DFF), ethylene glycol (EG), glycerol (Gly) and furfural (FF) into 1 M KOH solution, the onset potentials were all lower than that for the OER, indicating that the oxidation of these compounds was thermodynamically more favorable than the OER. More importantly, for the electrooxidation of all the mentioned compounds, Ni3S2/NF could achieve a current density of 500 mA cm−2 below the potential of 1.6 V vs. RHE. These results above not only confirmed the outstanding catalytic activity of Ni3S2/NF but also demonstrated its versatility for the oxidation of diverse substrates.
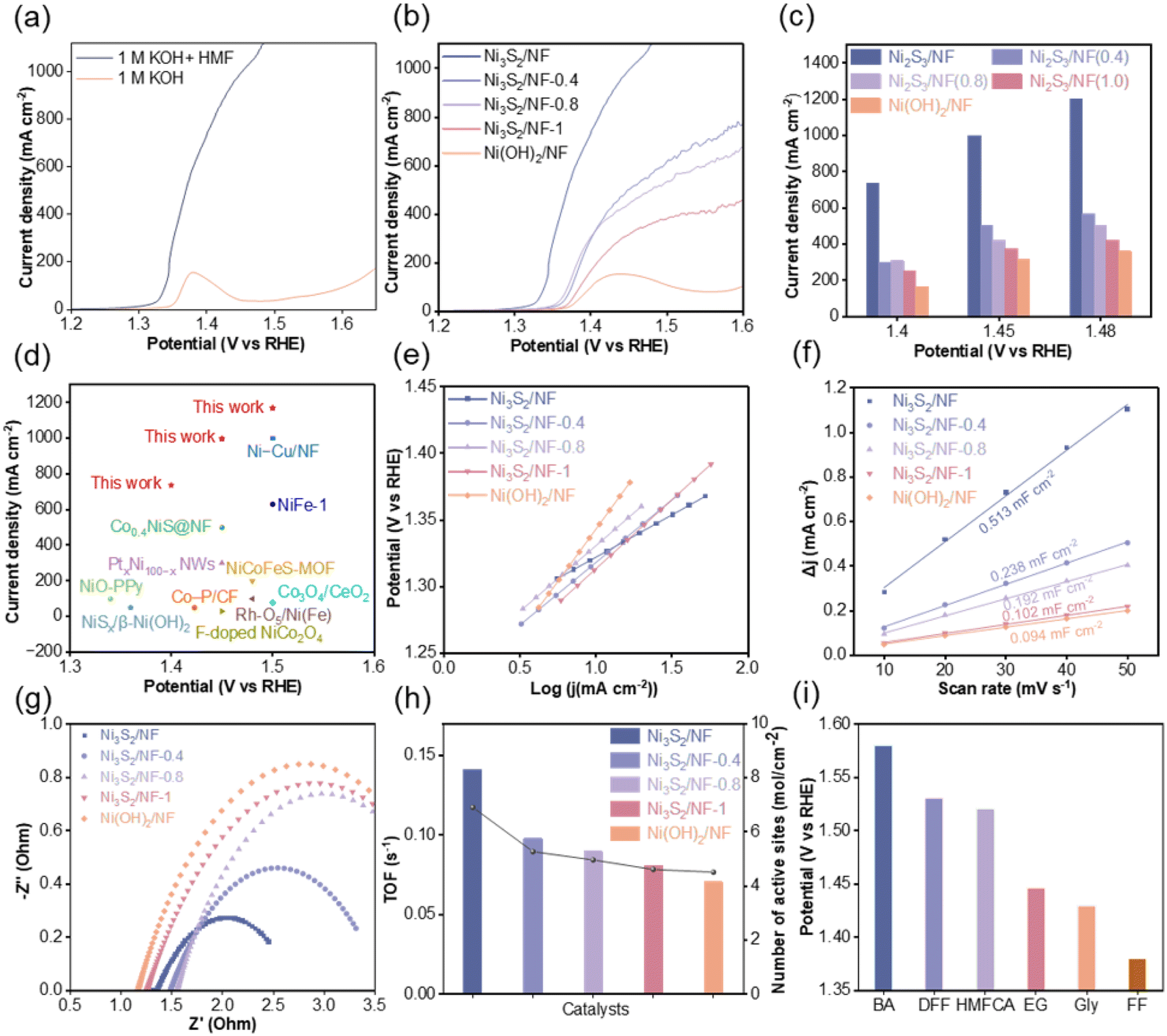 | ||
| Fig. 3 Electrochemical performance of the prepared materials. (a) Linear sweep voltammetry (LSV) curves of Ni3S2/NF in 1 M KOH with and without 50 mM HMF, (b) LSV curves of Ni3S2/NF, Ni3S2/NF-0.4, Ni3S2/NF-0.8, Ni3S2/NF-1 and Ni(OH)2/NF for HMFOR, (c) comparison of the current densities of Ni3S2/NF, Ni3S2/NF-0.4, Ni3S2/NF-0.8, Ni3S2/NF-1 and Ni(OH)2/NF at different potentials, (d) comparison of HMFOR performance with previously reported electrocatalysts, (e) Tafel plots, (f) change of current density plotted against the scan rate for Ni3S2/NF, Ni3S2/NF-0.4, Ni3S2/NF-0.8, Ni3S2/NF-1 and Ni(OH)2/NF, (g) Nyquist plots, (h) calculated TOFs, and (i) potentials for oxidation of different substrates containing hydroxyl and aldehyde groups at 500 mA cm−2. | ||
The reason for the excellent performance of Ni3S2/NF in HMFOR was systematically analyzed. As well-accepted, the adsorption of HMF molecules on the electrode surface played a crucial role in determining the HMFOR performance.41 Generally, the open-circuit potentials (OCP) could reflect the capacity of HMF adsorption in the Helmholtz layer on the electrode surface. After adding 50 mM HMF in the reaction system, the OCP values of Ni3S2/NF, Ni3S2/NF-1, and Ni(OH)2/NF were 402, 341, and 330 mV, respectively, implying that Ni3S2/NF with robust Ni–S coordination had the strongest adsorption capacity for HMF. Besides, the surface structures of Ni3S2/NF were monitored using potential-dependent operando Raman spectra to reveal the variation of active sites during the reaction. In the control experiments, Ni3S2/NF was used as the working electrode and the surface changes of the electrode were monitored at different potentials in 1 M KOH without or with 50 mM HMF. In general, Ni3+–OH was considered to be the active species of both the OER and HMFOR.42 For the OER, the characteristic Raman peaks were observed at 476 and 556 cm−1 attributed to bending and stretching vibrations of Ni3+–OH when the potential increased to 1.40 V vs. RHE (Fig. 4b), matching perfectly with the appearance of an oxidation peak in the potential range of 1.35–1.4 V vs. RHE (Fig. 3a). In comparison, after adding 50 mM HMF into 1 M KOH solution, the two peaks were not observed until the potential exceeded 1.45 V vs. RHE, probably because the generated Ni3+–OH before 1.45 V vs. RHE was rapidly consumed. In other words, Ni2+ was electrooxidized to form Ni3+–OH sites, which would be rapidly consumed by the spontaneous HMFOR through the interaction between Ni3+–OH and HMF adsorbed on the electrode, which was independent of the applied potential (Fig. 4c). In order to gain more insight into the behavior of the catalyst–electrolyte interface, operando EIS was utilized to further analyze the differences between the OER and HMFOR.43 For HMFOR, the Rct of Ni3S2/NF decreased sharply after the potential exceeded 1.3 V vs. RHE and was smaller than that of Ni3S2/NF-1 and Ni(OH)2/NF (Fig. 4d), suggesting the faster charge transfer rate of HMFOR on Ni3S2/NF. However, the Rct increased slightly after the potential of 1.45 V vs. RHE, which was attributed to the competition between the HMFOR and OER. Besides, the semicircle radius of the Nyquist plot (Fig. S17†) showed that the impedance of the OER was consistently larger than that of HMFOR, implying the faster charge transfer rate of HMFOR. Based on the above discussions, Ni3S2/NF had the highest electrochemical surface area, lowest charge transfer resistance, and strongest adsorption capacity for HMF. These advantages could significantly improve the oxidation efficiency of HMF to generate FDCA. Thereby, much better performance in HMF electrooxidation could be achieved over Ni3S2/NF in comparison with other materials.
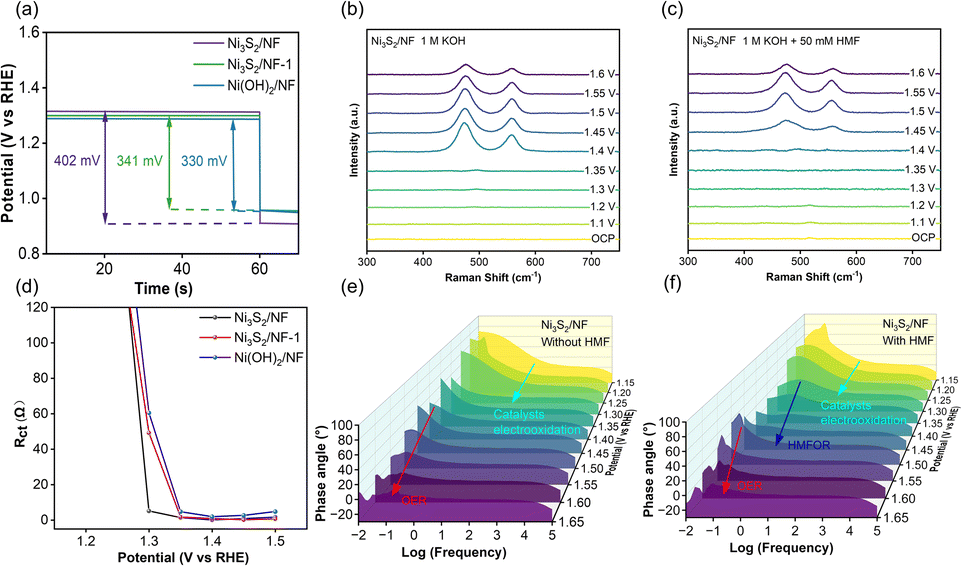 | ||
| Fig. 4 (a) OCP curves of Ni3S2/NF, Ni3S2/NF-1 and Ni(OH)2/NF in 1 M KOH and 50 mM HMF injected subsequently, (b) in situ Raman spectroscopy of Ni3S2/NF in KOH, (c) in situ Raman spectroscopy of Ni3S2/NF in 1 M KOH containing 50 mM HMF, (d) Rct values of Ni3S2/NF, Ni3S2/NF-1 and Ni(OH)2/NF during HMFOR, (e) Bode plots of Ni3S2/NF in 1 M KOH, and (f) Bode plots of Ni3S2/NF in 1 M KOH containing 50 mM HMF. | ||
According to previous reports, the signals in the middle and high frequency regions (101–105 Hz) corresponded to electron transfers between the interior and interface of the electrode, which were indicative of deprotonation/oxidation (i.e., HMFOR). The signals in the low-frequency region (10−2–100 Hz) were related to interfacial reactions involving non-uniform charge distribution (i.e., OER).44,45 In the absence of HMF (Fig. 4e), the intensity of peaks appearing in the high-frequency region decreased whereas it increased in the low-frequency region with the increase of the applied potentials, implying the structural transformation and occurrence of the OER.46 After adding HMF, new peaks, appearing at potentials of 1.3–1.5 V vs. RHE in the middle-frequency region, were attributed to the dehydrogenation of HMF (Fig. 4f). Compared with the peaks belonging to the OER only at the potential over 1.5 V vs. RHE, the peaks for HMFOR at lower potentials further confirmed the favorable HMFOR on Ni3S2/NF. In situ examinations verified the superiority of Ni3S2 with strong Ni–S bonds for charge transfer during HMFOR compared to that on Ni(OH)2.
Selectivity of the targeted products is a key aspect of biomass upgrading. As is well-known, there were two main pathways in the electrooxidation of HMF due to the co-existence of carbonyl and hydroxyl groups (Fig. 5a). In pathway 1, the aldehyde group on HMF was first oxidized to generate 5-hydroxymethyl 2-furan carboxylic acid (HMFCA) as the reaction intermediate. In pathway 2, 2,5-dicarboxyfuran (DFF) was generated as the reaction intermediate by the preferential oxidation of the hydroxymethyl group on HMF. Subsequently, both HMFCA and DFF were further oxidized to form FFCA, which was ultimately converted to FDCA. To confirm the predominant pathway, the concentrations of HMF and the products were monitored by high performance liquid chromatography (HPLC) and quantitatively calculated based on the standard curves (Fig. S18†). With the prolonging of the electrolysis time, the concentration of HMF gradually decreased and the concentration of FDCA gradually increased, indicating that HMF was successfully converted into FDCA (Fig. 5c and d). Simultaneously, HMFCA and FFCA were identified during the electrolysis process, but negligible amounts of DFF could be detected, indicating that the electrooxidation of HMF on Ni3S2/NF mainly proceeded through the pathway 1 (Fig. 5a). Finally, nearly complete conversion of HMF was achieved with a FDCA yield of 98.8% and a Faraday efficiency of 97.6% when the charge was accumulated up to 580C. Additionally, the current density continuously decreased during the electrocatalysis process until the charge reached 580C (Fig. S19†), resulting from the continuous consumption of HMF. More importantly, the cycling stability test revealed that the Ni3S2/NF electrode could be recycled for six consecutive electrocatalytic cycles, and the HMF conversion (99.4–99.8%), the FDCA selectivity (97.9–99.6%) and FDCA Faraday efficiency (95.6–98.1%) varied within a highly narrow range. Meanwhile, the used Ni3S2/NF was characterized by SEM, XPS and XRD techniques (Fig. S20–S22†), and the results showed that the properties of the Ni3S2/NF were very similar to those of the fresh electrode. Particularly, the XPS spectra of S 2p in the used Ni3S2/NF remained almost devoid of S–O bonds, demonstrating the solidity of Ni–S coordination. The above results confirmed the outstanding stability (both catalytic activity and its structure) of Ni3S2/NF, and its surface was not electrochemically reconfigured to hydroxide or oxyhydroxides as in previously reported work. To further reveal the possibility for the practical application of the Ni3S2/NF, a membrane electrode assembly (MEA) reactor of alkaline electrolysis with Ni3S2/NF and NF as the anode and cathode was fabricated for the HMFOR. When employing 1 M KOH with 50 mM HMF as the electrolyte, the current density of the HMFOR//HER was much higher than that of the OER//HER, indicating that substitution of the OER for HMFOR was favorable as a means to reduce energy consumption (Fig. 5f). Especially, industrial-grade current density of more than 500 mA cm−2 could be achieved at an input voltage of 1.55 V vs. RHE. More importantly, 2.002 g FDCA could be obtained from the electrolyte after acidification and the purity of the generated FDCA was more than 99%, which could be proved by HPLC (Fig. 5f and S23†).
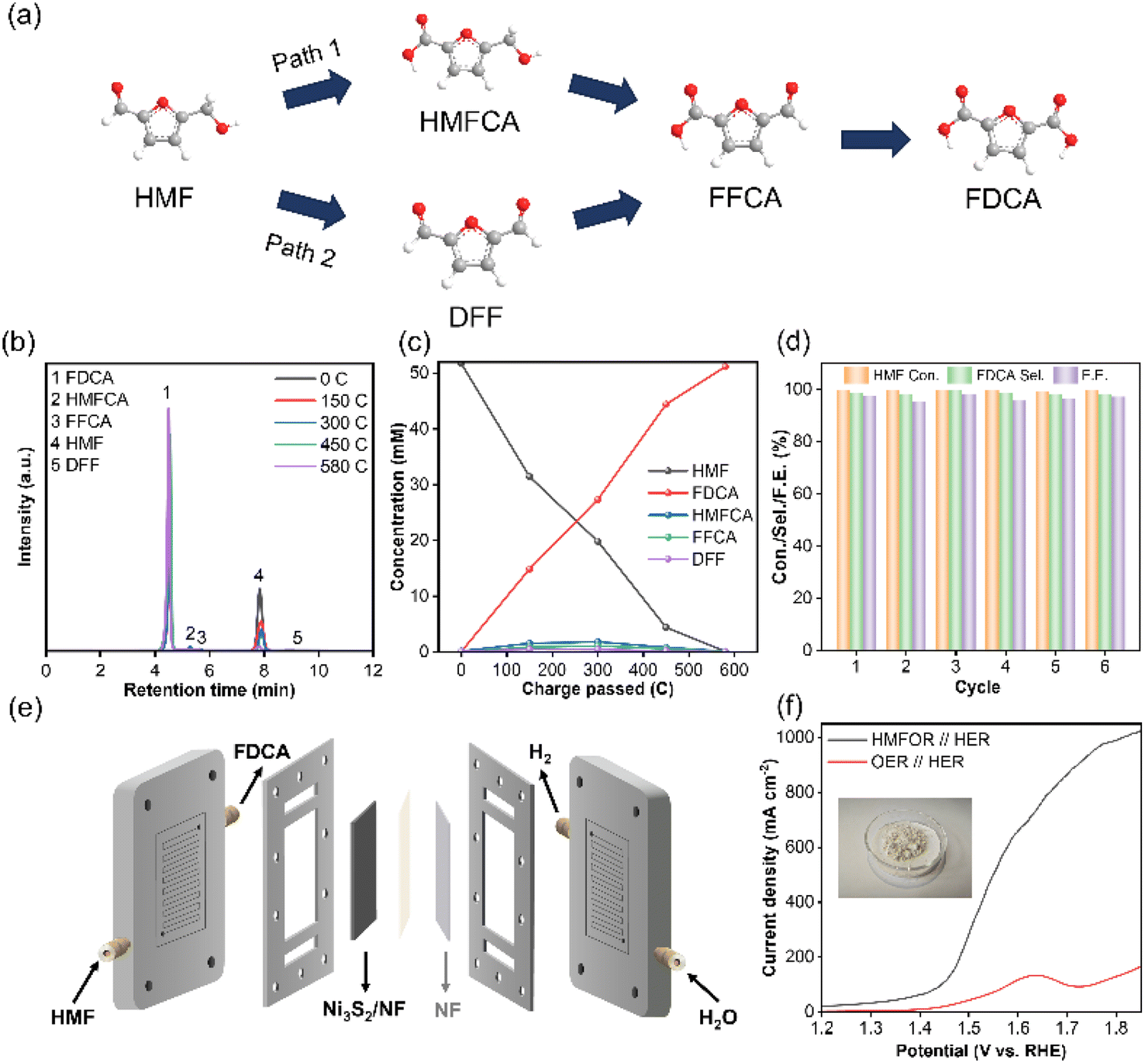 | ||
| Fig. 5 The chronoamperometric electrolysis measurements of HMFOR using Ni3S2/NF at 1.4 V vs. RHE. (a) The possible pathways of HMF oxidation to FDCA, (b) HPLC chromatogram traces of the electrolyte during the electrochemical oxidation of HMF, (c) the concentration changes of HMF and its oxidation products during HMF electrooxidation, (d) HMF conversion, selectivity and FE of FDCA during HMF electrooxidation at a potential of 1.4 V vs. RHE in 6 successive cycles, (e) schematic representation of the MEA reactor, and (f) LSV curves of Ni3S2/NF in 1 M KOH with and without 50 mM HMF in the MEA reactor. | ||
Density functional theory (DFT) calculations were finally used to theoretically uncover the differences in structure as well as reactivity regarding Ni3S2 with robust Ni–S coordination and Ni3S2-1 of being conventionally oxygen-embedded. Based on the above experimental results, the optimal theoretical models for Ni3S2/NF and Ni3S2/NF-1 were constructed (Fig. 6a and b), and the density of electronic states (DOS) of these two catalysts were analyzed to obtain the differences in the electronic structure (Fig. 6b). Compared with Ni3S2/NF-1, Ni3S2/NF had higher TDOS near the Fermi energy, suggesting an enhanced intrinsic conductivity of Ni3S2/NF and thus facilitating the electron transfer in the electrocatalytic process. In order to further demonstrate the bonding strength in both catalysts, the crystal orbital Hamilton population (COHP) was used to quantitatively analyze the Ni–S bond interactions.47 As shown in Fig. 5c, d, and S24†, the COHP value of Ni–S in Ni3S2/NF was −4.01, which was more negative than that in Ni3S2/NF-1 (−3.06). Besides DFT, the behavior of HMFOR on the catalysts was further stimulated by the calculations on the HMF dehydrogenation. HMFOR was a stepwise deprotonation process, in which the oxidation of the aldehyde group of HMF in alkaline electrolyte required hydration to form a diol group and then gradual dehydrogenation to generate a carboxyl group. The kinetic energy barriers for proton transfer from HMF to the Ni3S2/NF surface with adsorbed hydroxyl groups were calculated. As shown in Fig. 6f and g, HMF was adsorbed on the Ni sites while the absorbed OH acted as a proton transfer receptor, followed by detachment of H from HMF which combines with OH to generate water. The energy barrier of hydrogen transfer on Ni3S2/NF was 1.35 eV, which was lower than that of Ni3S2/NF-1 (1.59 eV), suggesting that Ni3S2/NF enhanced the deprotonation ability of HMF and thus facilitated the oxidation of HMF. All these calculation results were in agreement with the experimental observations for electrocatalytic oxidation of HMF.
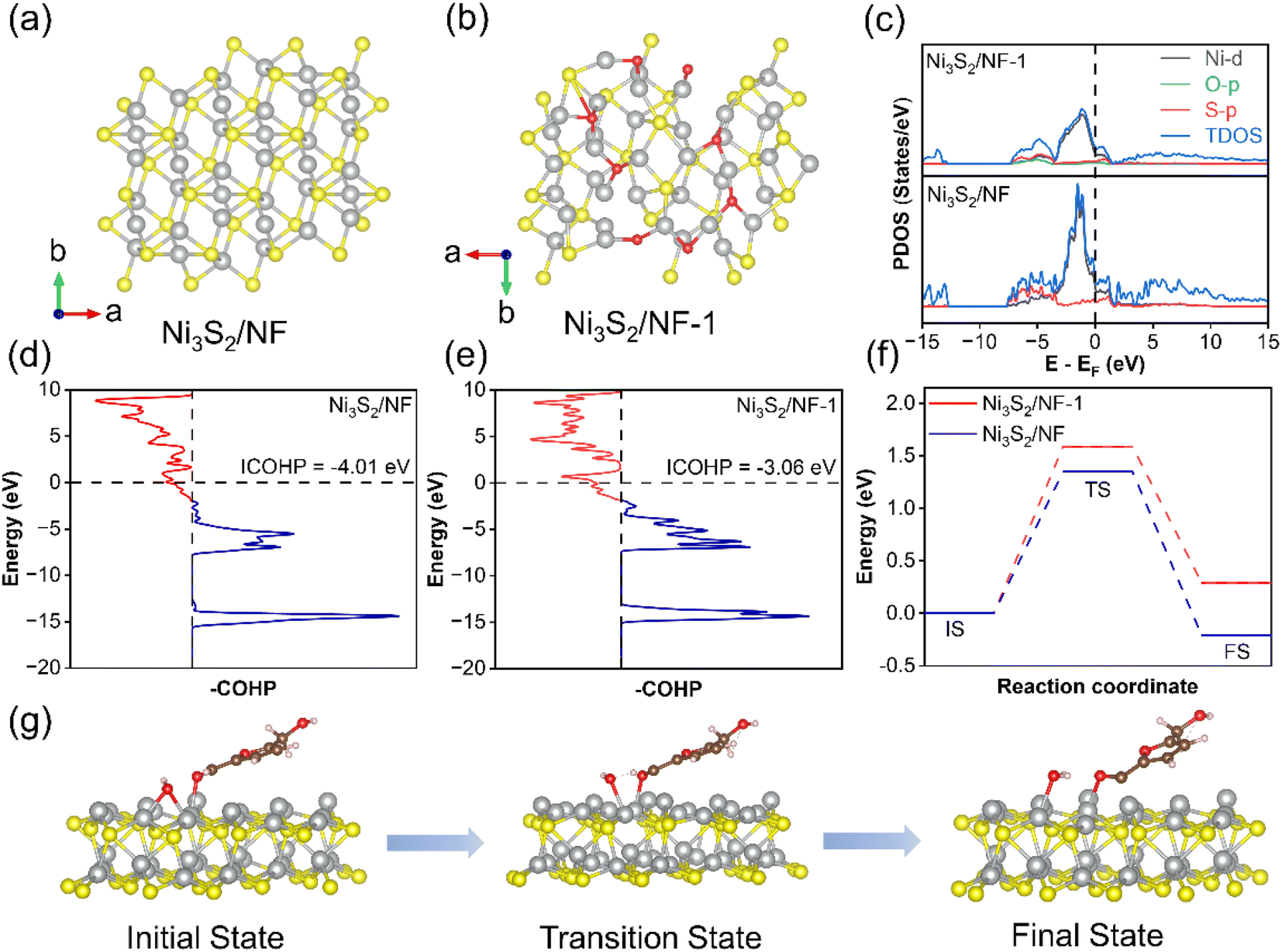 | ||
| Fig. 6 (a and b) Theoretical models of Ni3S2/NF and Ni3S2/NF-1, (c) the density of states (DOS) of Ni3S2/NF and Ni3S2/NF-1, (d and e) crystal orbital Hamilton population (–COHP) of Ni–S bonds of Ni3S2/NF and Ni3S2/NF-1 (the bonding and antibonding states of the Ni–S bonding interactions are shown in blue and red, respectively), (f) energy changes in the deprotonation process on Ni3S2/NF and Ni3S2/NF-1, and (g) the initial, transition and final states in the deprotonation process. | ||
Conclusions
In summary, Ni3S2 nanosheets supported on NF were fabricated directly by a one-step solvothermal method, and the Ni–S coordination in these materials could be controllably tuned by changing the water content in the solvothermal system. The as-prepared materials as electrodes could catalyze the electro-oxidation of HMF to produce FDCA. It was observed that the catalytic activity of the obtained electrodes increased with the decrease of the water content in the solvothermal system of preparing the catalysts, and Ni3S2/NF showed the best performance for the reaction with current densities up to 1000 mA cm−2 at a potential of only 1.45 V vs. RHE. The HMF conversion was almost 100% with a FDCA yield of 98.8% and a Faraday efficiency of 97.6% on Ni3S2/NF. Besides, industrial-scale current densities exceeding 500 mA cm−2 could also be achieved in the electrooxidation of other bio-based compounds, including benzaldehyde, 5-hydroxymethyl furoic acid, 2,5-diformylfuran, ethylene glycol, glycerol and furfural. Systematic investigations revealed that Ni3S2/NF had higher electronic conductivity, lower charge transfer resistance, and higher electrochemical surface area, which all could promote its catalytic performance on the HMFOR. More importantly, Ni3S2/NF could be applied in a MEA reactor, which allowed the gram-scale FDCA production with a purity of more than 99%. We believe that the constructed Ni3S2/NF has great potential for practical applications for the synthesis of FDCA from electrocatalytic oxidation of HMF.Data availability
The data supporting this article have been uploaded as part of the ESI.†Author contributions
Lan Chen: methodology, investigation, data curation, writing the original draft. Zhaohui Yang: methodology, investigation, writing the original draft. Chuanyu Yan: conceptualization, supervision. Yijun Yin: methodology, investigation. Zhimin Xue: conceptualization, writing – review & editing supervision. Yiting Yao: methodology, investigation, data curation. Shao Wang: methodology. Fanfei Sun: investigation, data curation. Tiancheng Mu: conceptualization, methodology, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support by Natural Science Foundation of Beijing (2242010), the Forestry and Grassland Science and Technology Innovation Youth Top Talent Project of China (No. 2020132609), and National Natural Science Foundation of China (22238011). We thank the staff at the BL20U1 beamline of the Shanghai Synchrotron Radiation Facilities (SSRF) for assistance with the EXAFS measurements.Notes and references
- J. Wu, Z. Zhai, S. Yin and S. Wang, General Formation of Interfacial Assembled Hierarchical Micro-Nano Arrays for Biomass Upgrading-Coupled Hydrogen Production, Adv. Funct. Mater., 2023, 34, 2308198, DOI:10.1002/adfm.202308198
.
- W. Deng, Y. Feng, J. Fu, H. Guo, Y. Guo, B. Han, Z. Jiang, L. Kong, C. Li, H. Liu, P. T. T. Nguyen, P. Ren, F. Wang, S. Wang, Y. Wang, Y. Wang, S. S. Wong, K. Yan, N. Yan, X. Yang, Y. Zhang, Z. Zhang, X. Zeng and H. Zhou, Catalytic conversion of lignocellulosic biomass into chemicals and fuels, Green Energy Environ., 2023, 8, 10–114, DOI:10.1016/j.gee.2022.07.003
- R. Gérardy, D. P. Debecker, J. Estager, P. Luis and J.-C. M. Monbaliu, Continuous Flow Upgrading of Selected C2–C6 Platform Chemicals Derived from Biomass, Chem. Rev., 2020, 120, 7219–7347, DOI:10.1021/acs.chemrev.9b00846
- R. Kumar, V. Strezov, H. Weldekidan, J. He, S. Singh, T. Kan and B. Dastjerdi, Lignocellulose biomass pyrolysis for bio-oil production: A review of biomass pre-treatment methods for production of drop-in fuels, Renewable Sustainable Energy Rev., 2020, 123, 109763, DOI:10.1016/j.rser.2020.109763
- J.-G. Rosenboom, R. Langer and G. Traverso, Bioplastics for a circular economy, Nat. Rev. Mater., 2022, 7, 117–137, DOI:10.1038/s41578-021-00407-8
- Y. Yang and T. Mu, Electrochemical oxidation of biomass derived 5-hydroxymethylfurfural (HMF): pathway, mechanism, catalysts and coupling reactions, Green Chem., 2021, 23, 4228–4254, 10.1039/D1GC00914A
- Y. Gao, L. Ge, H. Xu, K. Davey, Y. Zheng and S.-Z. Qiao, Electrocatalytic Refinery of Biomass-Based 5-Hydroxymethylfurfural to Fine Chemicals, ACS Catal., 2023, 13, 11204–11231, DOI:10.1021/acscatal.3c02272
-
T. Werpy and G. Petersen, Top value added chemicals from biomass: volume I--results of screening for potential candidates from sugars and synthesis gas, National Renewable Energy Lab.(NREL), Golden, CO (United States), 2004 Search PubMed
- G. Liu, T. Nie, Z. Song, X. Sun, T. Shen, S. Bai, L. Zheng and Y.-F. Song, Pd Loaded NiCo Hydroxides for Biomass Electrooxidation: Understanding the Synergistic Effect of Proton Deintercalation and Adsorption Kinetics, Angew. Chem., Int. Ed., 2023, 62, e202311696, DOI:10.1002/anie.202311696
- Y. Zhang, Z. Xue, X. Zhao, B. Zhang and T. Mu, Controllable and facile preparation of Co9S8–Ni3S2 heterostructures embedded with N,S,O-tri-doped carbon for electrocatalytic oxidation of 5-hydroxymethylfurfural, Green Chem., 2022, 24, 1721–1731, 10.1039/D1GC04499K
- M. Sajid, X. Zhao and D. Liu, Production of 2,5-furandicarboxylic acid (FDCA) from 5-hydroxymethylfurfural (HMF): recent progress focusing on the chemical-catalytic routes, Green Chem., 2018, 20, 5427–5453, 10.1039/C8GC02680G
- B. Zhang, Z. Li, Y. Zhou, Z. Yang, Z. Xue and T. Mu, Fluorine Induced In Situ Formation of High Valent Nickel Species for Ultra Low Potential Electrooxidation of 5-Hydroxymethylfurfural, Small, 2023, 20, 2306663, DOI:10.1002/smll.202306663
- M. G. Davidson, S. Elgie, S. Parsons and T. J. Young, Production of HMF, FDCA and their derived products: a review of life cycle assessment (LCA) and techno-economic analysis (TEA) studies, Green Chem., 2021, 23, 3154–3171, 10.1039/D1GC00721A
- M. Kim, Y. Su, A. Fukuoka, E. J. M. Hensen and K. Nakajima, Aerobic Oxidation of 5-(Hydroxymethyl)furfural Cyclic Acetal Enables Selective Furan-2,5-dicarboxylic Acid Formation with CeO2-Supported Gold Catalyst, Angew. Chem., Int.
Ed., 2018, 57, 8235–8239, DOI:10.1002/anie.201805457
- B. Tharat, L. Ngamwongwan, T. Seehamongkol, B. Rungtaweevoranit, J. Nonkumwong, S. Suthirakun, K. Faungnawakij, N. Chanlek, A. Plucksacholatarn, W. Nimsaila, C. Prommin and A. Junkaew, Hydroxy and surface oxygen effects on 5-hydroxymethylfurfural oxidation to 2,5-furandicarboxylic acid on β-MnO2: DFT, microkinetic and experiment studies, Nanoscale, 2024, 16, 678–690, 10.1039/D3NR03075J
- M. Park, M. Gu and B.-S. Kim, Tailorable Electrocatalytic 5-Hydroxymethylfurfural Oxidation and H2 Production: Architecture–Performance Relationship in Bifunctional Multilayer Electrodes, ACS Nano, 2020, 14, 6812–6822, DOI:10.1021/acsnano.0c00581
- C. Chen, Z. Zhou, J. Liu, B. Zhu, H. Hu, Y. Yang, G. Chen, M. Gao and J. Zhang, Sustainable biomass upgrading coupled with H2 generation over in-situ oxidized Co3O4 electrocatalysts, Appl. Catal., B, 2022, 307, 121209, DOI:10.1016/j.apcatb.2022.121209
- J. Bi, Q. Zhu, W. Guo, P. Li, S. Jia, J. Liu, J. Ma, J. Zhang, Z. Liu and B. Han, Simultaneous CO2 Reduction and 5-Hydroxymethylfurfural Oxidation to Value-Added Products by Electrocatalysis, ACS Sustainable Chem. Eng., 2022, 10, 8043–8050, DOI:10.1021/acssuschemeng.2c02117
- B. Zhang, Z. Yang, C. Yan, Z. Xue and T. Mu,
Operando Forming of Lattice Vacancy Defect in Ultrathin Crumpled NiVW-Layered Metal Hydroxides Nanosheets for Valorization of Biomass, Small, 2023, 19, 2207236, DOI:10.1002/smll.202207236
- H. G. Cha and K.-S. Choi, Combined biomass valorization and hydrogen production in a photoelectrochemical cell, Nat. Chem., 2015, 7, 328–333, DOI:10.1038/nchem.2194
- T. Wang, X. Cao and L. Jiao, Progress in Hydrogen Production Coupled with Electrochemical Oxidation of Small Molecules, Angew. Chem., Int. Ed., 2022, 61, e202213328, DOI:10.1002/anie.202213328
- N. Jiang, B. You, R. Boonstra, I. M. Terrero Rodriguez and Y. Sun, Integrating Electrocatalytic 5-Hydroxymethylfurfural Oxidation and Hydrogen Production via Co–P-Derived Electrocatalysts, ACS Energy Lett., 2016, 1, 386–390, DOI:10.1021/acsenergylett.6b00214
- Y. Song, W. Xie, Y. Song, H. Li, S. Li, S. Jiang, J. Y. Lee and M. Shao, Bifunctional integrated electrode for high-efficient hydrogen production coupled with 5-hydroxymethylfurfural oxidation, Appl. Catal., B, 2022, 312, 121400, DOI:10.1016/j.apcatb.2022.121400
- D. Chen, Y. Ding, X. Cao, L. Wang, H. Lee, G. Lin, W. Li, G. Ding and L. Sun, Highly Efficient Biomass Upgrading by a Ni−Cu Electrocatalyst Featuring Passivation of Water Oxidation Activity, Angew. Chem., Int. Ed., 2023, 62, e202309478, DOI:10.1002/anie.202309478
- Y. Sun, J. Wang, Y. Qi, W. Li and C. Wang, Efficient Electrooxidation of 5-Hydroxymethylfurfural Using Co-Doped Ni3S2 Catalyst: Promising for H2 Production under Industrial-Level Current Density, Adv. Sci., 2022, 9, 2200957, DOI:10.1002/advs.202200957
- T. Kwon, T. Kim, Y. Son and K. Lee, Dopants in the Design of Noble Metal Nanoparticle Electrocatalysts and their Effect on Surface Energy and Coordination Chemistry at the Nanocrystal Surface, Adv. Energy Mater., 2021, 11, 2100265, DOI:10.1002/aenm.202100265
- S. Kogularasu, Y.-Y. Lee, B. Sriram, S.-F. Wang, M. George, G.-P. Chang-Chien and J.-K. Sheu, Unlocking Catalytic Potential: Exploring the Impact of Thermal Treatment on Enhanced Electrocatalysis of Nanomaterials, Angew. Chem., Int. Ed., 2024, 63, e202311806, DOI:10.1002/anie.202311806
- J. Wang, T. Liao, Z. Wei, J. Sun, J. Guo and Z. Sun, Heteroatom-Doping of Non-Noble Metal-Based Catalysts for Electrocatalytic Hydrogen Evolution: An Electronic Structure Tuning Strategy, Small Methods, 2021, 5, 2000988, DOI:10.1002/smtd.202000988
- Y. Qi, L. Zhang, L. Sun, G. Chen, Q. Luo, H. Xin, J. Peng, Y. Li and F. Ma, Sulfur doping enhanced desorption of intermediates on NiCoP for efficient alkaline hydrogen evolution, Nanoscale, 2020, 12, 1985–1993, 10.1039/C9NR08583A
- W. Peng, Y.-R. Lu, H. Lin, M. Peng, T.-S. Chan, A. Pan and Y. Tan, Sulfur-Stabilizing Ultrafine High-Entropy Alloy Nanoparticles on MXene for Highly Efficient Ethanol Electrooxidation, ACS Nano, 2023, 17, 22691–22700, DOI:10.1021/acsnano.3c07110
- J. Becica and G. E. Dobereiner, The roles of Lewis acidic additives in organotransition metal catalysis, Org. Biomol. Chem., 2019, 17, 2055–2069, 10.1039/C8OB02856G
- B. Zhang, K. Jiang, H. Wang and S. Hu, Fluoride-Induced Dynamic Surface Self-Reconstruction Produces Unexpectedly Efficient Oxygen-Evolution Catalyst, Nano Lett., 2019, 19, 530–537, DOI:10.1021/acs.nanolett.8b04466
- P. Xu, Z. Bao, Y. Zhao, L. Zheng, Z. Lv, X. Shi, H.-E. Wang, X. Fang and H. Zheng, Anionic Regulation and Heteroatom Doping of Ni-Based Electrocatalysts to Boost Biomass Valorization Coupled with Hydrogen Production, Adv. Energy Mater., 2024, 14, 2303557, DOI:10.1002/aenm.202303557
- J. Sun, X. Yu, S. Zhao, H. Chen, K. Tao and L. Han, Solvent-Controlled Morphology of Amino-Functionalized Bimetal Metal–Organic Frameworks for Asymmetric Supercapacitors, Inorg. Chem., 2020, 59, 11385–11395, DOI:10.1021/acs.inorgchem.0c01157
- W. He, R. Zhang, D. Cao, Y. Li, J. Zhang, Q. Hao, H. Liu, J. Zhao and H. L. Xin, Super-Hydrophilic Microporous Ni(OH)x/Ni3S2 Heterostructure Electrocatalyst for Large-Current-Density Hydrogen Evolution, Small, 2023, 19, 2205719, DOI:10.1002/smll.202205719
- B. Zhao, J. Liu, R. Feng, L. Wang, J. Zhang, J.-L. Luo and X.-Z. Fu, Less-Energy Consumed Hydrogen Evolution Coupled with Electrocatalytic Removal of Ethanolamine Pollutant in Saline Water over Ni@Ni3S2/CNT Nano-Heterostructured Electrocatalysts, Small Methods, 2022, 6, 2101195, DOI:10.1002/smtd.202101195
- L. Yu, S. Sun, H. Li and Z. J. Xu, Effects of catalyst mass loading on electrocatalytic activity: An example of oxygen evolution reaction, Fundam. Res., 2021, 1, 448–452, DOI:10.1016/j.fmre.2021.06.006
- P. Kuang, Y. Wang, B. Zhu, F. Xia, C.-W. Tung, J. Wu, H. M. Chen and J. Yu, Pt Single Atoms Supported on N-Doped Mesoporous Hollow Carbon Spheres with Enhanced Electrocatalytic H2-Evolution Activity, Adv. Mater., 2021, 33, 2008599, DOI:10.1002/adma.202008599
- G. Piao, S. H. Yoon, H. G. Cha, D. S. Han and H. Park, Porous dendritic BiSn electrocatalysts for hydrogenation of 5-hydroxymethylfurfural, J. Mater. Chem. A, 2022, 10, 24006–24017, 10.1039/D2TA05969J
- S. Anantharaj, P. E. Karthik and S. Noda, The Significance of Properly Reporting Turnover Frequency in Electrocatalysis Research, Angew. Chem., Int. Ed., 2021, 60, 23051–23067, DOI:10.1002/anie.202110352
- D. Xiao, X. Bao, D. Dai, Y. Gao, S. Si, Z. Wang, Y. Liu, P. Wang, Z. Zheng, H. Cheng, Y. Dai and B. Huang, Boosting the Electrochemical 5-Hydroxymethylfurfural Oxidation by Balancing the Competitive Adsorption of Organic and OH− over Controllable Reconstructed Ni3S2/NiOx, Adv. Mater., 2023, 35, 2304133, DOI:10.1002/adma.202304133
- Z. Zhou, Y.-n. Xie, L. Sun, Z. Wang, W. Wang, L. Jiang, X. Tao, L. Li, X.-H. Li and G. Zhao, Strain-induced in situ formation of NiOOH species on CoCo bond for selective electrooxidation of 5-hydroxymethylfurfural and efficient hydrogen production, Appl. Catal., B, 2022, 305, 121072, DOI:10.1016/j.apcatb.2022.121072
- J. Wu, Z. Kong, Y. Li, Y. Lu, P. Zhou, H. Wang, L. Xu, S. Wang and Y. Zou, Unveiling the Adsorption Behavior and Redox Properties of PtNi Nanowire for Biomass-Derived Molecules Electrooxidation, ACS Nano, 2022, 16, 21518–21526, DOI:10.1021/acsnano.2c10327
- B. Zhou, Y. Li, Y. Zou, W. Chen, W. Zhou, M. Song, Y. Wu, Y. Lu, J. Liu, Y. Wang and S. Wang, Platinum Modulates Redox Properties and 5-Hydroxymethylfurfural Adsorption Kinetics of Ni(OH)2 for Biomass Upgrading, Angew. Chem., Int. Ed., 2021, 60, 22908–22914, DOI:10.1002/anie.202109211
- S. Li, S. Wang, Y. Wang, J. He, K. Li, Y. Xu, M. Wang, S. Zhao, X. Li, X. Zhong and J. Wang, Doped Mn Enhanced NiS Electrooxidation Performance of HMF into FDCA at Industrial-Level Current Density, Adv. Funct. Mater., 2023, 33, 2214488, DOI:10.1002/adfm.202214488
- Y. Yang, W. H. Lie, R. R. Unocic, J. A. Yuwono, M. Klingenhof, T. Merzdorf, P. W. Buchheister, M. Kroschel, A. Walker, L. C. Gallington, L. Thomsen, P. V. Kumar, P. Strasser, J. A. Scott and N. M. Bedford, Defect-Promoted Ni-Based Layer Double Hydroxides with Enhanced Deprotonation Capability for Efficient Biomass Electrooxidation, Adv. Mater., 2023, 35, 2305573, DOI:10.1002/adma.202305573
- Y. Wang, Y. Xu, C. Cheng, B. Zhang, B. Zhang and Y. Yu, Phase-Regulated Active Hydrogen Behavior on Molybdenum Disulfide for Electrochemical Nitrate-to-Ammonia Conversion, Angew. Chem., Int. Ed., 2024, 63, e202315109, DOI:10.1002/anie.202315109
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03470h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |