DOI:
10.1039/D4SC03571B
(Edge Article)
Chem. Sci., 2024,
15, 16612-16617
The H/F substitution strategy can achieve large spontaneous polarization in 1D hybrid perovskite ferroelectrics†
Received
31st May 2024
, Accepted 12th September 2024
First published on 13th September 2024
Abstract
Hybrid organic–inorganic perovskite (HOIP) ferroelectrics exhibit polarization reversibility and have a wide range of applications in the fields of smart switches, memorizers, sensors, etc. However, the inherent limitations of small spontaneous polarization (Ps) and large coercive field (Ec) in ferroelectrics have impeded their broader utilization in electronics and data storage. Molecular ferroelectrics, as a powerful supplement to inorganic ferroelectrics, have shown great potential in the new generation of flexible wearable electronic devices. The important research responsibility is to greatly improve progressiveness and overcome the above limitations. Here, a novel one-dimensional (1D) HOIP ferroelectric, (3-F-BTAB)PbBr3 (3-F-BTAB = 3-fluorobenzyltrimethylammonium), was successfully synthesized by employing the H/F substitution strategy to modify parent compound (BTAB)PbBr3 (BTAB = benzyltrimethylammonium), which undergoes a ferroelectric phase transition with Aizu notation 2/mF2 at 420 K. Notably, (3-F-BTAB)PbBr3 demonstrates exceptional ferroelectric properties with a large Ps of 7.18 μC cm−2 and a low Ec of 1.78 kV cm−1. As far as we know, (3-F-BTAB)PbBr3 features the largest Ps among those reported for 1D lead-based HOIP ferroelectrics. This work enriches the 1D lead-based ferroelectric family and provides guidance for applying ferroelectrics in low-voltage polar memories.
Introduction
Ferroelectrics with switchable spontaneous polarization under the applied electric field are widely used in the fields of data storage devices, transducers and sensors.1–9 The main research on ferroelectrics has always been inorganic oxides, such as BaTiO3 (BTO) and Pb(Zr, Ti)O3 (PZT) for their large Ps and longitudinal piezoelectric coefficient (d33) since the discovery of Rochelle salt (potassium sodium tartrate tetrahydrate).10–12 Molecular ferroelectrics, as a powerful supplement to inorganic ceramic ferroelectrics, have shown great potential in the new generation of flexible wearable electronic devices. With the development of molecular ferroelectrics in the past decades, ferroelectrochemistry has been a hot topic in the interdisciplinary fields of physics, chemistry, information, and electronics,13–17 which has led to the subsequent discovery of organic ferroelectrics such as diisopropylammonium bromide (DIPAB) with large Ps (23 μC cm−2) and high Curie temperature (Tc = 426 K). DIPAB is the first molecular ferroelectric to rival BTO, and its discovery is a notable achievement in the advancement of molecular ferroelectrics.18 The organic and inorganic components are able to self-assemble into HOIPs through weak intermolecular interactions, which have advantages of both organic cations and inorganic anions, exhibiting diverse structures, rich properties, and mechanical flexibility.19–30 This structure serves as an ideal template for constructing ferroelectrics and facilitates the optimization of their properties by chemical modification strategies.18,31–33 As we all know, Ps and Ec are two important parameters to characterize ferroelectricity. Optimizing the performance of ferroelectrics by increasing Ps and decreasing Ec has been a longstanding endeavor, aiming to apply them in the field of non-volatile memory and electronic devices with lower energy consumption and longer work lifespan.34–37 Despite the extensive research that has been undertaken, the improvement of ferroelectricity for HOIPs is still confronted with challenges.
Fortunately, HOIPs exhibit rich structural modifiability and compositional flexibility, allowing for chemical modification strategies such as homochirality, quasi-spherical strategy and H/F substitution strategy.38–43 Among them, the H/F substitution is regarded as an effective strategy to design ferroelectrics and optimize ferroelectric properties. Fluorine atoms exhibit higher electronegativity than hydrogen atoms, as well as they have analogous van der Waal's radii and steric hindrances, but substituting hydrogen atoms with fluorine atoms can regulate significantly the chemical and physical properties of materials. Compared to the isotope substitution only applicable to hydrogen-bonded molecular ferroelectrics, H/F substitution can increase the dipole moment, induce phase transitions, and improve the phase transition temperature, which has greater development space for the design and regulation of high-performance ferroelectrics.
Sha et al. synthesized the initial fluorinated 2D lead iodide perovskite ferroelectric (DFCHA)2PbI4 (DFCHA = 4, 4-difluorocyclohexylammonium) through the H/F substitution strategy, the Ps of which is estimated to be 4.5 μC cm−2 only according to the point charge model.44 Additionally, Shi et al. methodically incorporated fluorine substituents into the organic cations of [BA]2PbCl4 (BA = benzylammonium), and acquired a novel HOIP ferroelectric [2-FBA]2PbCl4 (2-FBA = 2-fluorobenzylammonium) with a Ps of 5.35 μC cm−2 and an Ec of 15 kV cm−1.45 Although extensive research has been conducted based on the H/F substitution strategy, the issue of small Ps and high Ec remains. We still need to further explore the research and development of HOIP ferroelectrics with large Ps and low Ec for better commercial applications.
Here, we synthesized a 1D HOIP ferroelectric (3-F-BTAB)PbBr3 through the H/F substitution strategy based on the parent (BTAB)PbBr3 (Scheme 1). Compared to (BTAB)PbBr3 crystallized in the centrosymmetric space group C2/c, the fluorinated (3-F-BTAB)PbBr3 crystallizes in the non-centrosymmetric space group C2 and undergoes a ferroelectric phase transition (the Aizu notation 2/mF2) at a high temperature of 420 K.46 The measurement results of the ferroelectric hysteresis loop indicate that (3-F-BTAB)PbBr3 can realize polarization reversal at room temperature when applied a small electric field (Ec = 1.78 kV cm−1). In fact, as far as we know, the Ps of (3-F-BTAB)PbBr3 is 7.18 μC cm−2 which is the highest among those reported for reported 1D organic–inorganic hybrid lead-based ferroelectrics.40,47–49 Notably, (3-F-BTAB)PbBr3 exhibits a high decomposition temperature and can also possess a long service life when below the Tc (Tc = 420 K), which is essential for the application of ferroelectric-related electronic components. In addition to ferroelectricity, (3-F-BTAB)PbBr3 reveals excellent nonlinear optical, photoluminescence and semiconductor properties. This work sheds light on exploring more high-performance 1D lead-based HOIP ferroelectrics for application in low-voltage memories.
 |
| | Scheme 1 Design strategy for obtaining organic–inorganic hybrid ferroelectrics. | |
Results and discussion
Thermal properties and dielectric properties
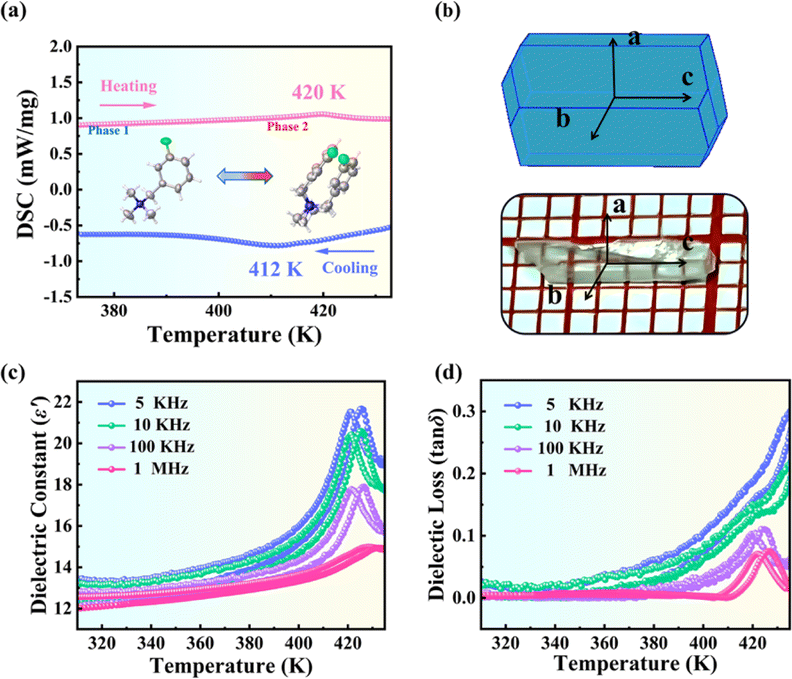
The colorless transparent single crystal of (3-F-BTAB)PbBr3 in a size of 2 × 2 × 7 mm3 was prepared by solvent volatilization (Fig. 1b), and powder X-ray diffraction (PXRD) was used to verify its phase purity (Fig. S1†). Differential scanning calorimetry (DSC) is a commonly employed thermal analysis method, which can determine the existence of phase transition behavior by studying the heat change of a substance with temperature. DSC curves of (BTAB)PbBr3 and (3-F-BTAB)PbBr3 are shown in Fig. S2† and 1a. For (BTAB)PbBr3, there are no obvious thermal anomalies within the temperature range of 260–420 K, suggesting that no phase transition behavior occurred in (BTAB)PbBr3 throughout the examined temperature range. However, for (3-F-BTAB)PbBr3, its DSC curves show a pair of significant endothermic and exothermic peaks at 420 and 412 K, respectively. According to the DSC curves of (3-F-BTAB)PbBr3, the entropy change ΔS is determined to be 2.08 J mol−1 K−1 by the formula ΔS = ΔH/T (ΔH = 873.6 J mol−1). For simplicity, the structure of (3-F-BTAB)PbBr3 when the temperature is above Tc is regarded as the high-temperature phase (HTP), and below Tc is regarded as the low-temperature phase (LTP). The results show that the H/F substitution strategy successfully induced the phase transition behavior of (3-F-BTAB)PbBr3, which possesses high phase transition temperature and small thermal hysteresis. For solid–solid phase transition materials, their dielectric constants usually exhibit significant anomalies at their phase transition temperatures and show temperature and frequency dependency. As shown in Fig. 1c and S3†, the dielectric constant slowly increases with the increase of temperature below the phase transition temperature and a λ-shaped dielectric peak appears near 420 K, which is in accordance with the DSC measurements. The dielectric loss (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) of (3-F-BTAB)PbBr3 as a function of temperature is shown in Fig. 1d. In addition, the Curie–Weiss law fits the dielectric response well, indicating the presence of a ferroelectric phase transition. Based on the formula ε′ = Cpara/(T − T0) for the paraelectric phase, or ε′ = Cferro/(T0′ − T) for the ferroelectric phase (Cpara and Cferro represent the Curie constants, T0′and T0 represent the Curie–Weiss temperatures for the ferroelectric and paraelectric phases, respectively). Cpara and Cferro are calculated to be approximately 10482 and 3676 K respectively at 1 MHz (Fig. S4†). The Cpara/Cferro ratio of (3-F-BTAB)PbBr3 is 2.85 at 1 MHz, which tends to be a second-order ferroelectric transition around 420 K.50
δ) of (3-F-BTAB)PbBr3 as a function of temperature is shown in Fig. 1d. In addition, the Curie–Weiss law fits the dielectric response well, indicating the presence of a ferroelectric phase transition. Based on the formula ε′ = Cpara/(T − T0) for the paraelectric phase, or ε′ = Cferro/(T0′ − T) for the ferroelectric phase (Cpara and Cferro represent the Curie constants, T0′and T0 represent the Curie–Weiss temperatures for the ferroelectric and paraelectric phases, respectively). Cpara and Cferro are calculated to be approximately 10482 and 3676 K respectively at 1 MHz (Fig. S4†). The Cpara/Cferro ratio of (3-F-BTAB)PbBr3 is 2.85 at 1 MHz, which tends to be a second-order ferroelectric transition around 420 K.50
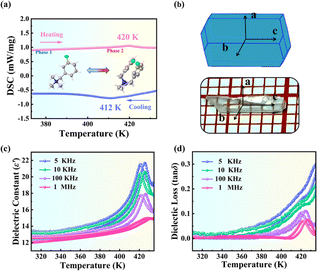 |
| | Fig. 1 (a) DSC curves of (3-F-BTAB)PbBr3 in the heating–cooling cycle. (b) The image and simulated crystal growth morphology of the crystal. (c) Temperature dependence of the real part (ε′) of (3-F-BTAB)PbBr3 at different frequencies. (d) Dielectric loss (tanδ) of (3-F-BTAB)PbBr3 as a function of temperature. | |
Crystallographic structure analysis
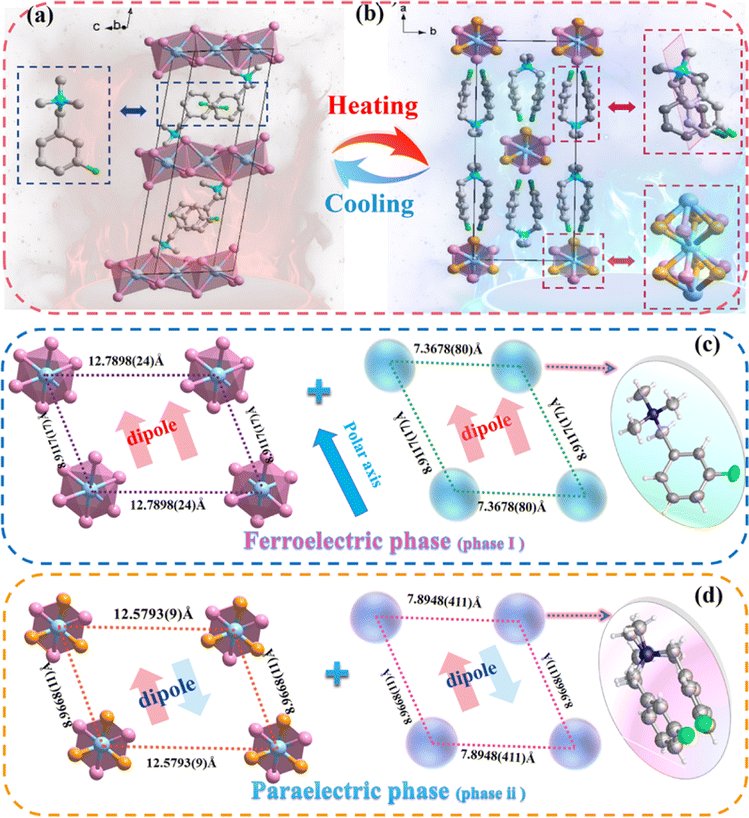
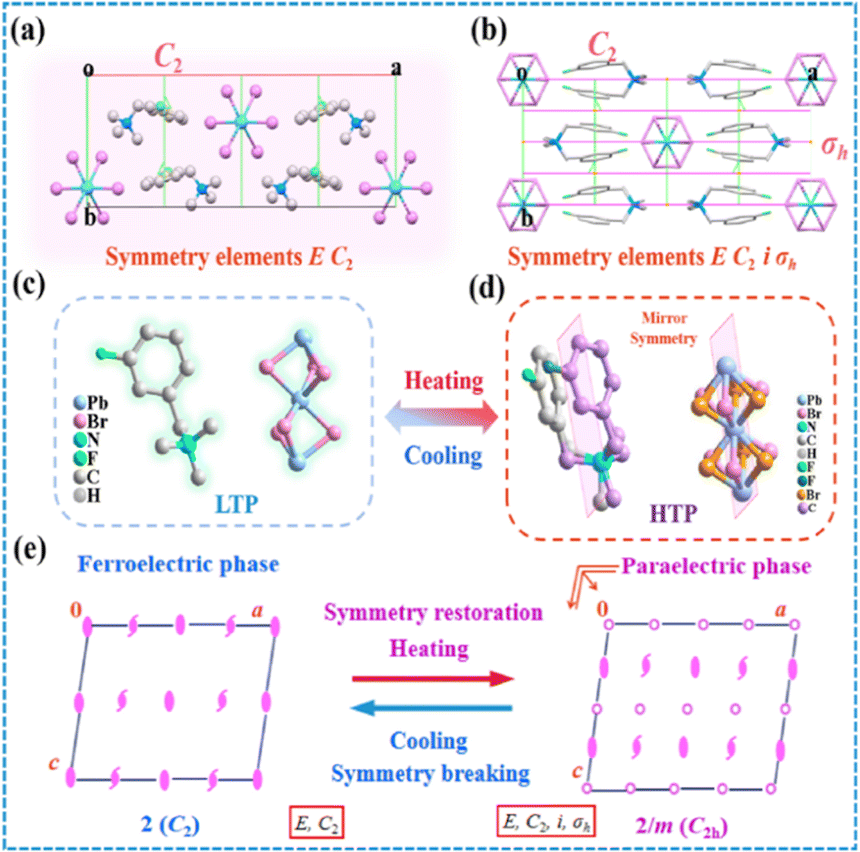
In order to elucidate the mechanism of the reversible phase transition, the crystal structures of (BTAB)PbBr3 and (3-F-BTAB)PbBr3 were determined using single crystal X-ray diffraction (SC-XRD). As shown in Table S1† the parent (BTAB)PbBr3 crystallizes at room temperature in the monoclinic space group C2/c (point group 2/m), with cell parameters a = 23.110(3) Å, b = 9.6090(13) Å, c = 26.851(4) Å, β = 106.990(14)°, V = 5702.4(13) Å3. Interestingly, the fluorinated (3-F-BTAB)PbBr3 crystallizes at LTP in the monoclinic non-centrosymmetric space group C2 (point group 2), with cell parameters aLTP = 23.977(5) Å, bLTP = 8.9117(16) Å, cLTP = 7.7036(15) Å, βLTP = 101.973(5)°, VLTP = 1610.3(5) Å3 (Table S1†). The organic cations and inorganic anions form the 1D ABX3 perovskite structure through intermolecular interactions (Fig. 2a and b). The inorganic components are composed of face-shared octahedral [PbBr3]− infinite chains (Fig. S5†), with Br–Pb–Br bond angles ranging from 81.59(2)° to 100.24(4)° and bond lengths ranging from 2.9080(10) Å to 3.1875(10) Å (Table S2†), respectively. To better illustrate the arrangement of the compound, the inorganic skeleton and the organic part are separately drawn. In LTP, the dipoles of the whole composed of inorganic and organic parts are along the b-axis. However, in HTP, the dipoles of inorganic and organic parts cancel out (Fig. 2c and d). As shown in Fig. 3a, from the polarization perspective, the centers of positive and negative charges do not coincide along the b direction, such an arrangement should lead to spontaneous polarization along the b-axis. In HTP, (3-F-BTAB)PbBr3 crystallizes in the centrosymmetric space group C2/c, with cell parameters aHTP = 23.5064(18) Å, bHTP = 8.9668(5) Å, cHTP = 6.8118(4) Å, βHTP = 98.299(7)°, VHTP = 1420.74(16) Å3. Due to the increase temperature, the vibration of the organic cation and inorganic anion frameworks becomes intense, accompanied by an ordered state to a disordered state (Fig. 3c and d). Therefore, the phase transition is driven by the order–disorder transition of both the organic cation and the inorganic anionic framework. The symmetric position of cations becomes a special position on the symmetry plane perpendicular to the b-axis, and the dipole moments along the b-axis cancel each other out, resulting in the disappearance of spontaneous polarization (Fig. 3b). According to the phase transition of compound from 2/m (HTP) to 2 (LTP) point group, the symmetry elements are reduced from four (E, C2, σh, i) to two (E, C2), belonging to the ferroelectric phase transition characterized by the Aizu notation of 2/mF2 (Fig. 3e). Through comparison, it was found that the distance between the inorganic skeletons of (BTAB)PbBr3 (15.9899(20) Å) is greater than that of (3-F-BTAB)PbBr3 (12.7898(24) Å), so the arrangement of organic cations and inorganic skeletons of (3-F-BTAB)PbBr3 is more compact. In addition, there are intermolecular interactions between the H and F atoms of adjacent cations, as well as between the H atom on the cation and the inorganic skeleton in (3-F-BTAB)PbBr3, compared to (BTAB)PbBr3 (Fig. S8 and S9†). A new polar substance was obtained by precise molecular modification and design of non-polar substances, and it is exciting that this work will be a greater space to enrich the molecular ferroelectric family with the H/F substitution strategy in the future.
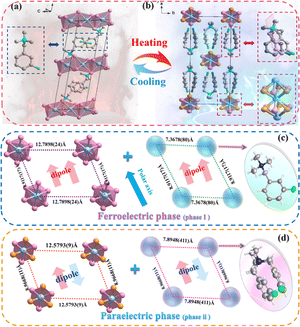 |
| | Fig. 2 Comparison of crystal structures of (3-F-BTAB)PbBr3 in the LTP and HTP. (a) Packing view in the LTP. The [3-F-BTAB]+ cations and [PbBr3]− frameworks are ordered. (b) Packing view in the HTP. Both the [3-F-BTAB]+ cations and [PbBr3]− frameworks become disordered. The distance between adjacent inorganic frameworks along the b-axis and the distance between adjacent cations in the LTP (c) and HTP (d). H atoms were omitted for clarity. | |
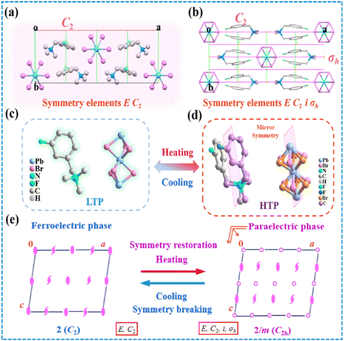 |
| | Fig. 3 Comparison of packing views of (3-F-BTAB)PbBr3 at (a) LTP and (b) HTP along the c-axis. The [3-F-BTAB]+ cations occupy a general symmetry position at LTP, yet a special position of the symmetry plane at HTP. (c) Molecular structure of (3-F-BTAB)PbBr3 crystallized as the low temperature phase. (d) Molecular structure of (3-F-BTAB)PbBr3 crystallized as the high temperature phase, the pink planes are mirrors only existing in the high temperature phase. (e) Symmetry breaking occurs with an Aizu notation of 2/mF2 in (3-F-BTAB)PbBr3. | |
Second harmonic generation (SHG) and ferroelectricity
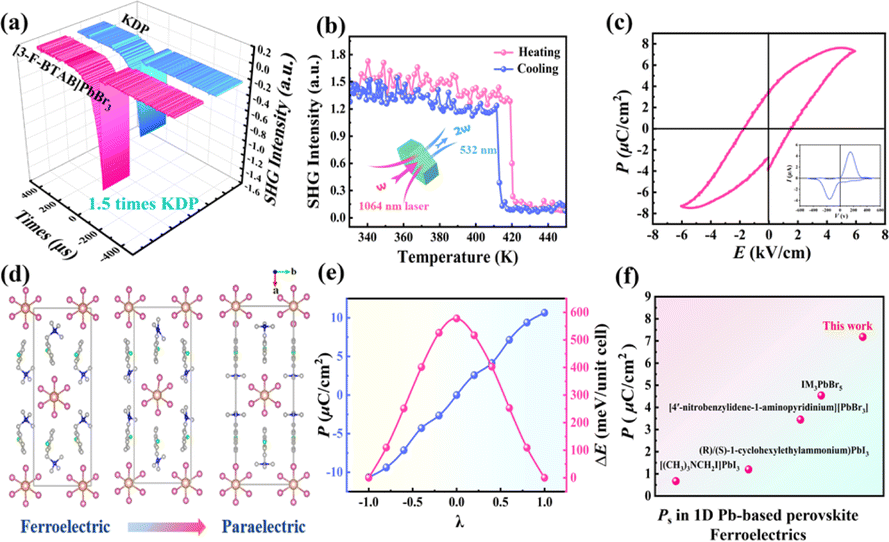
Generally, the second harmonic generation (SHG) response only exists in non-centrosymmetric crystals (except for point groups 422, 622 and 432) and is a common method for characterizing structural symmetry and verifying phase transition behavior. The intense SHG signal (1.5 KDP) of (3-F-BTAB)PbBr3 can be observed at room temperature, consistent with the non-centrosymmetric C2 space group (Fig. 4a). To further verify the phase transition of (3-F-BTAB)PbBr3 from non-centrosymmetric C2 to centrosymmetric C2/c, we measured the variation of SHG response with temperature (Fig. 4b). At 300–405 K, the SHG intensity is approximately 1.5 times that of KH2PO4 (KDP), presenting a high SHG signal response. The SHG intensity undergoes a sudden change near the phase transition temperature and the signal intensity sharply decreases to nearly 0, indicating a low SHG response. The reversible step-like response between LTP and HTP was observed during the heating–cooling cycle, in accordance with the symmetry requirements from the polar ferroelectric phase to the centrosymmetric paraelectric phase, indicating that (3-F-BTAB)PbBr3 is a potential nonlinear optical switching material. Combined with notable variations in dielectric anomalies, the ferroelectricity of (3-F-BTAB)PbBr3 necessitates additional validation through the measurement of the polarization–electric field (P–E) hysteresis loop.51 As shown in Fig. 4c, the P–E loop exhibits a characteristic rectangular shape, providing confirmation of the ferroelectricity of (3-F-BTAB)PbBr3. When the frequency is 50 Hz, the result shows that the Ec and Ps of (3-F-BTAB)PbBr3 are 1.78 kV cm−1 and 7.18 μC cm−2 at room temperature, respectively, which indicates that the ferroelectric can undergo polarization reversal even with a rather low Ec. To our knowledge, the Ps is currently the highest among those reported for 1D lead-based HOIP ferroelectrics (Fig. 4f). Following the experimental validation of ferroelectricity, we carried out ferroelectric polarization calculations utilizing the Berry phase method to understand its ferroelectricity further. Based on the crystal structures of (3-F-BTAB)PbBr3 in LTP and HTP, we constructed a series of transition states (Fig. 4d). The dynamic evaluation path from the ferroelectric phase to the paraelectric phase is essential for estimating the theoretical polarization value. Through rotation and displacement of (3-F-BTAB)PbBr3 to estimate reasonably, the results show that there is not much difference between the theoretical macroscopic polarization value (Ps = 10.66 μC cm−2) and the measurement value (Fig. 4e).
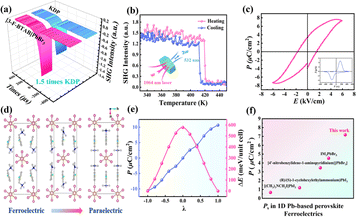 |
| | Fig. 4 (a) SHG intensity of (3-F-BTAB)PbBr3 and KDP. (b) Temperature-dependent SHG response. (c) Polarization–electric field (P–E) hysteresis loop and I–V curve at 298 K by the Sawyer–Tower method. (d) Berry phase calculation of polarization from ferroelectric phase structures to constructed paraelectric phase structures of (3-F-BTAB)PbBr3. (e) Calculated macroscopic polarization and attributions and changes of energy (ΔE) from diverse cations. (f) Comparison of Ps with those reported for 1D organic–inorganic hybrid lead-based ferroelectrics. | |
Photoluminescence properties
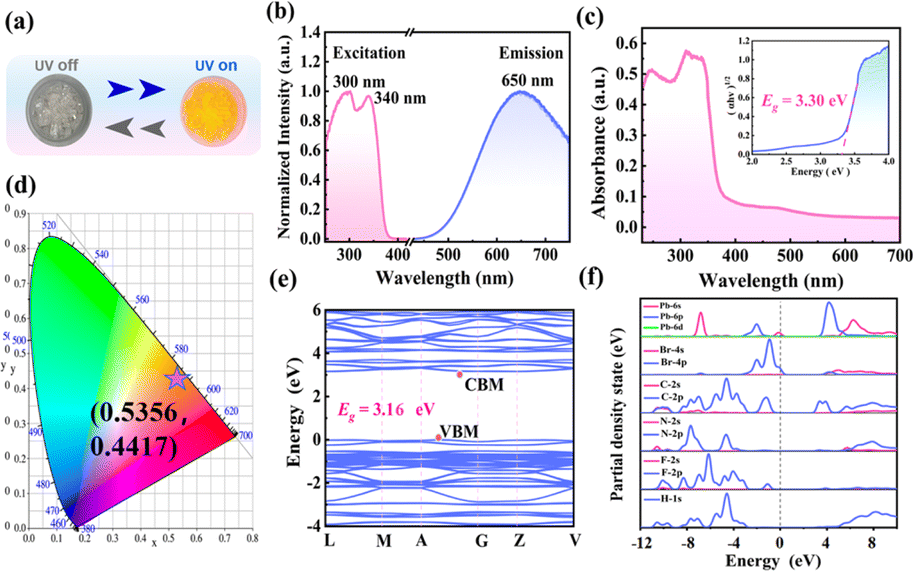
The 1D lead-based HOIPs generally exhibit optoelectronic properties. In this work, the photoluminescence (PL) characteristics of (3-F-BTAB)PbBr3 are also studied. The compound shows strong orange luminescence under ultraviolet (UV) lamps of 254 and 365 nm (Fig. 5a), which is in accordance with the orange chromaticity coordinates as determined by the Commission Internationale de l′Eclairage (CIE) coordination (Fig. 5d). Under the excitation of 300 or 340 nm, obvious broadband emission peaks with the same intensity at 650 nm were observed (Fig. 5b). (3-F-BTAB)PbBr3 has broadband emission and a large Stokes shift, which may be due to self-trapping excitons (STE) formed by the strong electron–phonon coupling of 1D octahedral chains.52 The PL decay curve at 650 nm was measured using an excitation wavelength of 340 nm and the PL decay lifetime was 61.91 ns (Fig. S6†). According to the measured results, the photoluminescence quantum yield of (3-F-BTAB)PbBr3 is 3.12% (Fig. S7†).
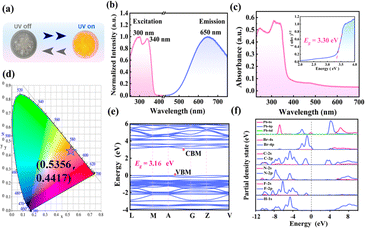 |
| | Fig. 5 (a) Crystal under ambient light and UV light irradiation at room temperature. (b) Normalized excitation and emission spectra of (3-F-BTAB)PbBr3 at room temperature. (c) UV-visible absorption of (3-F-BTAB)PbBr3 and optical band gaps were calculated using the Tauc equation. (d) CIE chromaticity coordinates (e) Band structure of (3-F-BTAB)PbBr3. (f) Partial density of states (PDOS) of (3-F-BTAB)PbBr3. | |
Semiconductor properties
Hybrid lead halide materials are potential semiconductor materials. To further understand the semiconductor properties of (3-F-BTAB)PbBr3, UV-visible absorption spectroscopy and theoretical calculations were conducted to study the band structure. From the UV-absorption spectrum, it can be observed that there is strong absorption at the band of 320 nm, which is consistent with the characteristics of indirect semiconductors. Meanwhile, according to the Tauc equation (αhν)1/n = A(hν − Eg), the optical bandgap of (3-F-BTAB)PbBr3 is calculated to be 3.30 eV (Fig. 5c). The band structure and partial density of states (PDOS) of (3-F-BTAB)PbBr3 were computed utilizing density functional theory (DFT) and the theoretical bandgap of (3-F-BTAB)PbBr3 is 3.16 eV, close to the experimental bandgap (Fig. 5e). The conduction band minimum (CBM) and valence band maximum (VBM) values are situated at different positions in the Brillouin zone, so (3-F-BTAB) PbBr3 belongs to an indirect bandgap semiconductor. According to the data of PDOS, the VBM is occupied by the electronic states of Br 4p, while the CBM is contributed by the Pb 6s state (Fig. 5f), so the bandgap of (3-F-BTAB)PbBr3 is mainly contributed by the inorganic frameworks.
Conclusions
In summary, we successfully synthesized a novel 1D HOIP ferroelectric (3-F-BTAB)PbBr3, with high performance through the implementation of the H/F substitution strategy. Compared with other harsh conditions requiring high temperature and pressure, (3-F-BTAB)PbBr3 demonstrates the capability to reverse polarization at a low Ec (1.78 kV cm−1) at ambient temperature, while also displaying a notably large Ps of 7.18 μC cm−2. To our knowledge, this is the highest Ps reported for 1D HOIP lead-based ferroelectrics. Moreover, (3-F-BTAB)PbBr3 exhibits remarkable nonlinear optical properties (signal intensity is up to 1.5 times KDP), photoluminescence, semiconductor properties and high Curie temperature (420 K), making it a high-performance ferroelectric. As a result, the precise molecular design the through H/F substitution strategy provides a novel perspective for future research on the synthesis of high-performance HOIP ferroelectrics.
Experimental
Synthesis and growth of crystals
All chemical reagents used in this work were obtained from commercial sources without further purification. The synthesis of the ligands BTAB and 3-F-BTAB can be referred to previous reports. 2 mmol of BTAB was weighed in a beaker and dissolved with hydrobromic acid, and then 2 mmol of PbBr2 was added. After complete dissolution, the solution was slowly evaporated at room temperature, and after a few weeks, a colorless block crystal of (BTAB)PbBr3 was obtained. The crystal of (3-F-BTAB)PbBr3 was obtained in the same way.
Characterization
Measurement methods for Single-crystal X-ray Diffraction (SXRD), dielectric measurement, Differential Scanning Calorimetry (DSC), Powder XRD (PXRD), ferroelectric measurement and Second Harmonic Generation (SHG) are described in detail in the ESI†. Density functional theory (DFT) calculation methods and data are also shown in the ESI†.
Data availability
CCDC 2350277, 2350294–2350295 contain the supplementary crystallographic data for this paper.
Author contributions
J.-Y. L. conceived the experiments and wrote the paper. M.-M. L. and J.-Y. L. performed theoretical calculations. Z.-J. W. and K. D. assisted in the auxiliary analysis of data. D.-W. F., H.-F. L. and Y. Z. guided and supervised this work.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant 21991141, 22371258 and 22375182) and the research fund of Southeast University. We thank the Big Data Computing Center of Southeast University for providing the facility support on the numerical calculations in this paper.
Notes and references
- H. Y. Zhang, Y. Y. Tang, Z. X. Gu, P. Wang, X. G. Chen, H. P. Lv, P. F. Li, Q. Jiang, N. Gu, S. Ren and R. G. Xiong, Science, 2024, 383, 1492–1498 Search PubMed.
- Q. Q. Jia, H. F. Lu, J. Q. Luo, Y. Y. Zhang, H. F. Ni, F. W. Zhang, J. Wang, D. W. Fu, C. F. Wang and Y. Zhang, Small, 2024, 20, e2306989 Search PubMed.
- Z. X. Zhang, H. Wang, H. F. Ni, N. Wang, C. F. Wang, P. Z. Huang, Q. Q. Jia, G. Teri, D. W. Fu, Y. Zhang, Z. An and Y. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202319650 Search PubMed.
- K. Ding, H. Ye, C. Su, Y.-A. Xiong, G. Du, Y.-M. You, Z.-X. Zhang, S. Dong, Y. Zhang and D.-W. Fu, Nat. Commun., 2023, 14, 2863 Search PubMed.
- J.-Y. Li, T. Zhang, M.-M. Lun, Y. Zhang, L.-Z. Chen and D.-W. Fu, Small, 2023, 19, 2301364 Search PubMed.
- T. Zhang, K. Xu, J. Li, L. He, D. W. Fu, Q. Ye and R. G. Xiong, Natl. Sci. Rev., 2023, 10, nwac240 Search PubMed.
- H.-F. Ni, L.-K. Ye, P.-C. Zhuge, B.-L. Hu, J.-R. Lou, C.-Y. Su, Z.-X. Zhang, L.-Y. Xie, D.-W. Fu and Y. Zhang, Chem. Sci., 2023, 14, 1781–1786 Search PubMed.
- H. Peng, J.-C. Qi, X.-J. Song, R.-G. Xiong and W.-Q. Liao, Chem. Sci., 2022, 13, 4936–4943 Search PubMed.
- Q.-Q. Jia, G. Teri, J.-Q. Luo, H.-F. Ni, P.-Z. Huang, M.-M. Lun, Z.-X. Zhang, Y. Zhang and D.-W. Fu, J. Am. Chem. Soc., 2024, 146, 21120–21128 Search PubMed.
- R. Clarke and A. M. Glazer, Ferroelectrics, 1976, 14, 695–697 Search PubMed.
- J. Valasek, Phys. Rev., 1921, 17, 475–481 Search PubMed.
- Z. Zhao, V. Buscaglia, M. Viviani, M. T. Buscaglia, L. Mitoseriu, A. Testino, M. Nygren, M. Johnsson and P. Nanni, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 024107 Search PubMed.
- Y. Ai, W. Q. Liao, Y. R. Weng, H. P. Lv, X. G. Chen, X. J. Song, P. F. Li and R. G. Xiong, J. Am. Chem. Soc., 2023, 145, 23292–23299 Search PubMed.
- Z. J. Feng, Y. A. Xiong, W. C. Sun, T. T. Sha, J. Yao, Q. Pan, H. Hu, S. Dong, R. G. Xiong and Y. M. You, Adv. Mater., 2024, 36, e2307518 Search PubMed.
- H. H. Jiang, X. J. Song, H. P. Lv, X. G. Chen, R. G. Xiong and H. Y. Zhang, Adv. Mater., 2024, 36, e2307936 Search PubMed.
- Y. Du, W. Q. Liao, Y. Li, C. R. Huang, T. Gan, X. G. Chen, H. P. Lv, X. J. Song, R. G. Xiong and Z. X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202315189 Search PubMed.
- N. Zhang, W. Sun, Y. Zhang, H. H. Jiang, R. G. Xiong, S. Dong and H. Y. Zhang, Nat. Commun., 2023, 14, 5854 Search PubMed.
- D. W. Fu, H. L. Cai, Y. Liu, Q. Ye, W. Zhang, Y. Zhang, X. Y. Chen, G. Giovannetti, M. Capone, J. Li and R. G. Xiong, Science, 2013, 339, 425–428 Search PubMed.
- M. M. Lun, C. Y. Su, Q. Q. Jia, Z. X. Zhang, J. Li, H. F. Lu, Y. Zhang and D. W. Fu, Inorg. Chem. Front., 2023, 10, 5026–5034 Search PubMed.
- J. Q. Luo, Q. Q. Jia, G. Teri, M. M. Lun, Z. J. Wang, Z. X. Zhang, Y. Zhang and D. W. Fu, ACS Mater. Lett., 2023, 6, 452–460 Search PubMed.
- Q. F. Luo, H. F. Ni, P. Z. Huang, M. Zhu, C. F. Wang, Q. H. Zhuo, D. W. Fu, Y. Zhang and Z. X. Zhang, Mater. Chem. Front., 2023, 7, 6247–6253 Search PubMed.
- J. Q. Luo, M. M. Lun, Q. Q. Jia, Z. J. Wang, H. F. Lu, Y. Zhang and D. W. Fu, Chin. J. Chem., 2024, 42, 1706–1712 Search PubMed.
- X.-T. Sun, H.-F. Ni, Y. Zhang and D.-W. Fu, Chin. J. Struct. Chem., 2024, 43, 100212 Search PubMed.
- B.-W. Deng, Z.-P. Rao, M.-J. Shen, K.-W. Liang, Y. Zhu, Z.-J. Wang, K. Ding, C.-Y. Su, M.-M. Lun, Z.-X. Zhang, Y. Zhang and D.-W. Fu, J. Mater. Chem. C, 2024, 12, 6098–6105 Search PubMed.
- Y. Yu Zhang, J.-Q. Luo, Y. Han, W.-Y. Zhang, Y. Zhang, H.-F. Lu and D.-W. Fu, Chin. Chem. Lett., 2024 DOI:10.1016/j.cclet.2024.109530.
- Z.-J. Wang, M.-J. Shen, Z.-P. Rao, P.-Z. Huang, M.-M. Lun, B.-W. Deng, J.-Y. Li, C.-F. Wang, H.-F. Lu, D.-W. Fu and Y. Zhang, Inorg. Chem. Front., 2024, 11, 2290–2299 Search PubMed.
- M.-M. Lun, H.-F. Ni, Z.-X. Zhang, J.-Y. Li, Q.-Q. Jia, Y. Zhang, Y. Zhang and D.-W. Fu, Angew. Chem., Int. Ed., 2024, 136, e202313590 Search PubMed.
- M.-M. Lun, J.-Q. Luo, Z.-X. Zhang, J. Li, L.-Y. Xie, H.-F. Lu, Y. Zhang and D.-W. Fu, Chem. Eng. J., 2023, 475, 145969 Search PubMed.
- M.-M. Lun, C.-Y. Su, J. Li, Q.-Q. Jia, H.-F. Lu, D.-W. Fu, Y. Zhang and Z.-X. Zhang, Small, 2023, 19, 2303127 Search PubMed.
- Z.-J. Wang, H.-F. Ni, T. Zhang, J. Li, M.-M. Lun, D.-W. Fu, Z.-X. Zhang and Y. Zhang, Chem. Sci., 2023, 14, 9041–9047 Search PubMed.
- H.-F. Ni, J.-H. Lin, G. Teri, Q.-Q. Jia, P.-Z. Huang, H.-F. Lu, C.-F. Wang, Z.-X. Zhang, D.-W. Fu and Y. Zhang, Chin. Chem. Lett., 2024 DOI:10.1016/j.cclet.2024.109690.
- D. W. Fu, J. X. Gao, W. H. He, X. Q. Huang, Y. H. Liu and Y. Ai, Angew. Chem., Int. Ed., 2020, 59, 17477–17481 Search PubMed.
- Y. Zhang, X. J. Song, Z. X. Zhang, D. W. Fu and R. G. Xiong, Matter, 2020, 2, 697–710 Search PubMed.
- Y. Y. Tang, P. F. Li, W. Y. Zhang, H. Y. Ye, Y. M. You and R. G. Xiong, J. Am. Chem. Soc., 2017, 139, 13903–13908 Search PubMed.
- P. P. Shi, Y. Y. Tang, P. F. Li, H. Y. Ye and R. G. Xiong, J. Am. Chem. Soc., 2017, 139, 1319–1324 Search PubMed.
- Y. Y. Tang, W. Y. Zhang, P. F. Li, H. Y. Ye, Y. M. You and R. G. Xiong, J. Am. Chem. Soc., 2016, 138, 15784–15789 Search PubMed.
- X. J. Song, Z. X. Zhang, X. G. Chen, H. Y. Zhang, Q. Pan, J. Yao, Y. M. You and R. G. Xiong, J. Am. Chem. Soc., 2020, 142, 9000–9006 Search PubMed.
- J. Ahn, E. Lee, J. Tan, W. Yang, B. Kim and J. Moon, Mater. Horiz., 2017, 4, 851–856 Search PubMed.
- Y. Ai, H. P. Lv, Z. X. Wang, W. Q. Liao and R. G. Xiong, Trends Chem., 2021, 3, 1088–1099 Search PubMed.
- X. N. Hua, W. Q. Liao, Y. Y. Tang, P. F. Li, P. P. Shi, D. W. Zhao and R. G. Xiong, J. Am. Chem. Soc., 2018, 140, 12296–12302 Search PubMed.
- W. Y. Zhang, Y. Y. Tang, P. F. Li, P. P. Shi, W. Q. Liao, D. W. Fu, H. Y. Ye, Y. Zhang and R. G. Xiong, J. Am. Chem. Soc., 2017, 139, 10897–10902 Search PubMed.
- C.-Y. Su, Y.-F. Yao, Z.-X. Zhang, Y. Wang, M. Chen, P.-Z. Huang, Y. Zhang, W.-C. Qiao and D.-W. Fu, Chem. Sci., 2022, 13, 4794–4800 Search PubMed.
- H. Ye, X. X. Chen, D. X. Liu, B. Q. Zhao, Y. B. Li, Y. Zeng, W. X. Zhang and X. M. Chen, Chem. Sci., 2022, 13, 14124–14131 Search PubMed.
- T. T. Sha, Y. A. Xiong, Q. Pan, X. G. Chen, X. J. Song, J. Yao, S. R. Miao, Z. Y. Jing, Z. J. Feng, Y. M. You and R. G. Xiong, Adv. Mater., 2019, 31, 1901843 Search PubMed.
- P. P. Shi, S. Q. Lu, X. J. Song, X. G. Chen, W. Q. Liao, P. F. Li, Y. Y. Tang and R. G. Xiong, J. Am. Chem. Soc., 2019, 141, 18334–18340 Search PubMed.
- K. Aizu, J. Phys. Soc. Jpn., 1969, 27, 387–396 Search PubMed.
- H. F. Ni, J. H. Lin, C. F. Wang, Q. F. Luo, P. Z. Huang, Z. X. Zhang, D. W. Fu and Y. Zhang, Inorg. Chem. Front., 2023, 10, 7231–7237 Search PubMed.
- S. P. Zhao, Y. Guo, J. Wang, H. Xu, Q. Qiao and R. Y. Huang, Polyhedron, 2020, 178 Search PubMed.
- Y. Hu, F. Florio, Z. Chen, W. A. Phelan, M. A. Siegler, Z. Zhou, Y. Guo, R. Hawks, J. Jiang, J. Feng, L. Zhang, B. Wang, Y. Wang, D. Gall, E. F. Palermo, Z. Lu, X. Sun, T. M. Lu, H. Zhou, Y. Ren, E. Wertz, R. Sundararaman and J. Shi, Sci. Adv., 2020, 6, eaay4213 Search PubMed.
- W. Zhang and R. G. Xiong, Chem. Rev., 2012, 112, 1163–1195 Search PubMed.
- L. Jin, F. Li and S. Zhang, J. Am. Ceram. Soc., 2014, 97, 1–27 Search PubMed.
- M. M. Lun, F. L. Zhou, D. W. Fu and Q. Ye, J. Mater. Chem. C, 2022, 10, 11371–11378 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, Fig. S1–S9 and Tables S1–S3. CCDC 2350277, 2350294–2350295. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03571b |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Hai-Feng
Lu
*a,
Hai-Feng
Lu