
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-cationic hyper-crosslinked ionic polymers with hierarchically ordered porous structures: facile synthesis and applications for highly efficient CO2 capture and conversion†
Bihua
Chen
 ,
Junfeng
Zeng
,
Shiguo
Zhang
* and
Yan
Zhang
*
,
Junfeng
Zeng
,
Shiguo
Zhang
* and
Yan
Zhang
*
College of Materials Science and Engineering, Hunan University, Changsha 410082, Hunan, China. E-mail: zhangsg@hnu.edu.cn; zyan1980@hnu.edu.cn
First published on 16th August 2024
Abstract
Hyper-crosslinked porous ionic polymers (HCPIPs) have garnered significant attention due to their unique ionic properties and high specific surface areas. However, the limited variety of monomers, low ionic density, and difficulty in functionalization restrict their development. Herein, a series of functionalized non-cationic HCPIPs with high ionic density are designed and directly synthesized via an innovative and straightforward approach – anion (and cation) hyper-crosslinking of tetraphenylborate-based ionic liquids (ILs). These HCPIPs offer controllable hydroxyl group content (0–2.40 mmol g−1), high IL content (1.20–1.78 mmol g−1), and large specific surface area (636–729 m2 g−1) with hierarchically ordered porous structures. These HCPIPs demonstrate exceptional CO2 adsorption capacities and CO2/N2 adsorption selectivities, reaching up to 2.68–3.01 mmol g−1 and 166–237, respectively, at 273 K and 1 bar. Furthermore, these ionic porous materials serve as highly efficient heterogeneous catalysts for CO2 cycloaddition to epoxides under mild conditions (1 bar CO2, 60–80 °C, 12–24 h). Notably, the CO2 adsorption performances and catalytic activities of these HCPIPs are regulated by the hydroxyl groups within their structures, with enhancements observed as the number of hydroxyl groups increases. This work presents a facile and widely applicable method for constructing high-performance and task-specific HCPIPs.
Introduction
Porous organic polymers (POPs) constitute an important class of functional porous materials, which are built by linking organic monomers using covalent bonds.1–4 Owing to their diverse structures, organic frameworks, and abundant porosity, POPs show great potential for use in various applications, such as adsorption, separation, catalysis, and energy conversion and storage.5–9 Precise control of pore chemistry and structure promotes the development of high-performance and task-specific POPs.4,10,11 Ionic liquids (ILs), which are molten salts composed of organic cations and organic or inorganic anions, with melting points below 100 °C, have been used for the preparation of ionic POPs.12,13 The activity and designability of ILs and the unique nanoporous ionic environment brought by ILs not only enhance the performance of POPs but also greatly expand their application scope, which has attracted widespread interest.12–16 The past decade has witnessed the development of various synthetic methods for ionic POPs. The post-modification method provides a facile way to introduce ionic sites but suffers from decreased porosity and pore accessibility.12,14 Templated synthetic strategies afford well-defined architecture, but inevitably sacrifice templates and rarely construct micropores.13,16 Overall, the current methodologies for fabricating ionic POPs still exhibit limitations.The Friedel–Crafts alkylation of aromatic building blocks is a straightforward and efficient method to prepare POPs.17 This method is characterized by low cost and mild operating conditions, and the resulting hyper-crosslinked polymers (HCPs) possess large specific surface areas, good chemical and thermal stability, and broad practical application foreground.17–19 Recently, Friedel–Crafts chemistry has been used to incorporate IL ionic sites into the porous architecture to construct hyper-crosslinked porous ionic polymers (HCPIPs) – a novel category of ionic POPs that combines the properties of HCPs and ILs.20 The specific synthetic strategies include post-grafting of HCPs,21,22 post-hyper-crosslinking of ionic polymers,23–25in situ generations of ionic sites during hyper-crosslinkage,26–29 and hyper-crosslinking of IL monomers.30–33 Among these strategies, in situ generations of ionic sites during hyper-crosslinkage and hyper-crosslinking of IL monomers facilitate one-step synthesis of HCPIPs while preserving pore structure integrity, offering significant advantages. Currently, the strategy of in situ generation of ionic sites during hyper-crosslinkage is restricted due to the limited availability of crosslinkers and monomers, which are benzyl halides and nitrogen- and phenyl-containing compounds (e.g., 2-phenylimidazoline, benzimidazole, and 4-phenylpyridine), respectively. The use of benzyl halides will result in the production of polluting halogenated acids during the preparation process. Additionally, due to the poor reactivity of these monomers33 and the high reactivity of benzyl halides, the obtained HCPIPs have ultra-low ionic density,26–29 which greatly limits their activities. The strategy of hyper-crosslinking of IL monomers provides an efficient method for synthesizing HCPIPs with high IL content. Nevertheless, this strategy currently faces the problem of selecting appropriate IL monomers. This is because the Friedel–Crafts alkylation reaction involves an electrophilic substitution step, making it very difficult for IL monomers with electron-withdrawing groups (e.g., N-heterocyclic cations of ILs) to hyper-crosslink.23,30 So far, only a few IL monomers containing a suitable number of phenyl groups in the cations have been successfully hyper-crosslinked.31–33 The phenyl groups on the cations provide hyper-crosslinking sites and weaken the influence of electron-withdrawing groups, thereby promoting the hyper-crosslinking. However, the phenyl groups also occupy the N sites of IL monomers, making it almost impossible to introduce other functional groups (e.g., hydroxyl, amino, and carboxyl) to further enhance the performance.31–33 To date, functionalized HCPIPs are rarely reported.
The structural diversity and properties of ILs mainly depend on their cations, particularly diverse functional groups in the cations.34–38 The currently reported synthetic strategies for HCPIPs are only suitable for hyper-crosslinking of a few monomers with specific structures, and the obtained HCPIPs are cationic type and lack ionic moieties and functional groups, which greatly limits the kinds of HCPIPs and the development of high-performance and task-specific HCPIPs. Consequently, there is a pressing need to explore a facile and widely applicable synthetic strategy to prepare novel HCPIPs.
In this work, we introduced a novel approach called anion (and cation) hyper-crosslinking to produce diverse HCPIPs. This method involved incorporating hyper-crosslinking sites into anions to facilitate hyper-crosslinking and introducing functional groups into cations to tailor properties. Specifically, we developed a distinctive class of IL monomers, denoted as 1-R1-3-R2-imidazolium tetraphenylborate ([R1R2Im]BPh4) [R1 = M (methyl) and Bn (benzyl); R2 = E (ethyl), HE (2-hydroxyethyl), and DHP (2,3-dihydroxypropyl)]. The BPh4− anion with a rigid tetrahedral structure not only effectively weakened the influence of the electron-withdrawing groups in the Friedel–Crafts alkylation reaction, but also provided abundant three-dimensional sites for the hyper-crosslinking. A series of non-cationic HCPIPs were directly synthesized by the simple anion (and cation) hyper-crosslinking of [R1R2Im]BPh4 (and [R1R2Im]Br). The prepared non-cationic HCPIPs possessed abundant, diverse, and controllable functional groups, high IL contents, hierarchically ordered porous structures, large specific surface areas, and excellent stabilities. Impressively, these HCPIPs exhibited superior CO2 adsorption capacities and CO2/N2 adsorption selectivities. Furthermore, they demonstrated remarkable catalytic activities in the CO2 cycloaddition to various epoxides under extremely mild conditions (1 bar CO2, 60–80 °C, 12–24 h). Notably, the CO2 adsorption performances and catalytic activities of these HCPIPs were regulated by the hydroxyl groups within their structures and gradually enhanced as the number of hydroxyl groups increased.
Experimental
Materials
CO2 (>99.999%) and N2 (>99.999%) were supplied by Changsha Saizhong Special Gas Co., Ltd. 1-Methylimidazole (99%), 1-benzylimidazole (98%), bromoethane (99%), 2-bromoethanol (98%), 1-allylimidazole (98%), benzyl bromide (99%), sodium tetraphenylborate (NaBPh4, 99%), anhydrous iron(III) chloride (FeCl3, 98%), 1,2-dichloroethane (water < 50 ppm, 99.5%), and tetrabutylammonium bromide (TBABr, 99%) were purchased from Adamas Reagent Co., Ltd. Potassium permanganate (99%) was obtained from Sinopharm Chemical Reagent Co., Ltd. Formaldehyde dimethyl acetal (FDA, 98%), styrene oxide (98%), epichlorohydrin (99%), epibromohydrin (97%), 1,2-epoxybutane (99%), 1,2-epoxyhexane (96%), allyl glycidyl ether (99%), n-butyl glycidyl ether (98%), and glycidyl phenyl ether (99%) were purchased from TCI (Shanghai) Development Co., Ltd. All other chemical reagents were of analytical grade and used without further purification.Synthesis of the non-cationic HCPIPs
IL monomers [R1R2Im]Br and [R1R2Im]BPh4 (R1 = M and Bn; R2 = E, HE, and DHP) were synthesized through quaternary ammonization reactions and BPh4− anion exchange reactions (Schemes S1–S9†).The non-cationic HCPIPs were synthesized from the Friedel–Crafts alkylation induced anion (and cation) hyper-crosslinking of IL monomers [R1R2Im]BPh4 (and [R1R2Im]Br) and crosslinker (FDA) catalyzed by a Lewis acid catalyst (anhydrous FeCl3). In a typical run, [BnDHPIm]BPh4 (2.5 mmol), [BnDHPIm]Br (2.5 mmol), FDA (20.0 mmol), and anhydrous FeCl3 (20.0 mmol) were added into a 100 mL two-necked round bottom flask. The flask was purged with argon (Ar). Then 20 mL 1,2-dichloroethane was injected into the flask. Under an Ar atmosphere, the reaction mixture was first heated to 45 °C and stirred for 3 h to form a network and then continued to increase to 80 °C for 21 h to complete the condensation reaction. The purpose of gradient heating is to create greater porosity.39,40 After cooling to room temperature, the resultant solid was isolated by filtration, washed with methanol (5 × 50 mL) and dichloromethane (5 × 50 mL), and purified by Soxhlet extraction with methanol (24 h) and dichloromethane (24 h) to remove FeCl3 and unreacted IL monomers and crosslinker. After drying under vacuum at 60 °C for 12 h, the brown solid was collected and denoted as H-[BnDHPIm]Br/BPh4-FDA.
Other non-cationic HCPIP samples featuring different IL monomers were synthesized similarly and the detailed synthesis conditions of each sample are listed in Table S1.†
Characterization
1H and 13C NMR spectra were recorded using an AVANCE III HD 400 spectrometer (Bruker Daltonics Inc., USA) at frequencies of 400 and 100 MHz, respectively. Solid-state CP/MAS 13C NMR spectra were collected with an AVANCE III HD 400 spectrometer equipped with a 4 mm ZrO2 rotor at a frequency of 100 MHz, a spinning rate of 5.0 kHz, and a recycling delay of 6.5 s. Fourier transform infrared (FT-IR) spectra were obtained on a Nicolet iS50 FT-IR spectrometer (Thermo Fisher Scientific, USA) using KBr pellets in the range of 4000–400 cm−1. For the in situ FT-IR measurement of CO2 adsorption, the H-[BnDHPIm]Br/BPh4-FDA sample was filled into an in situ IR cell and Ar and CO2 were introduced into the cell at room temperature. The sample was first purged with Ar for 30 min. Then, CO2 was purged for 15 min, followed by a slow purge of Ar for 15 min. For the in situ FT-IR measurement of the interaction between H-[BnDHPIm]Br/BPh4-FDA and styrene oxide, the H-[BnDHPIm]Br/BPh4-FDA sample with styrene oxide adsorbed was filled into a variable-temperature in situ IR cell, vacuumed, heated to 80 °C, and maintained at this temperature for 60 min. For the in situ FT-IR measurement of the cycloaddition reaction between CO2 and styrene oxide catalyzed by H-[BnDHPIm]Br/BPh4-FDA + TBABr, H-[BnDHPIm]Br/BPh4-FDA, TBABr, and styrene oxide were filled into a high-temperature reaction in situ IR cell, introduced with CO2, heated to 80 °C, and reacted for 15 min. X-ray photoelectron spectroscopy (XPS) measurements were performed on an ESCALAB Xi+ X-ray photoelectron spectrometer (Thermo Fisher Scientific, USA) with Al Kα irradiation at θ = 90° for X-ray sources, and the binding energies were calibrated using the C 1s peak at 284.8 eV. Elemental analyses for C, H, and N were performed on a UNICUBE organic elemental analyzer (Elementar, Germany). Powder X-ray diffraction (XRD) patterns were collected on a MiniFlex 600 X-ray diffractometer (Rigaku, Japan) with Cu Kα radiation. Scanning electron microscopy (SEM) measurements were carried out on an S-4800 field emission scanning electron microscope (Hitachi, Japan). Transmission electron microscopy (TEM) measurements were performed on an FEI Tecnai F20 transmission electron microscope (Thermo Fisher Scientific, USA). Thermogravimetric analysis (TGA) was performed by using a STA7200 thermogravimetric analyzer (Hitachi, Japan) at a heating rate of 10 °C min−1 in the range of 30–800 °C under a stream of Ar. N2 adsorption–desorption isotherms (at 77 K), N2 adsorption isotherms (at 273 K), and CO2 adsorption isotherms (at 273 and 298 K) were measured by using a JW-BK200C surface area and pore size analyzer (JWGB, China). Before the measurements, the samples were degassed at 100 °C for 6 h under vacuum. A temperature-programmed desorption of CO2 (CO2-TPD) experiment was conducted using an AutoChem II 2920 chemisorption apparatus (Micromeritics, USA) with a thermal conductivity detector (TCD). Before the measurement, the sample was degassed at 120 °C for 1 h under a He flow.CO2/N2 adsorption selectivity calculation
The CO2/N2 adsorption selectivity was determined using the Ideal Adsorbed Solution Theory (IAST) model and Henry's law.41–44The IAST model was utilized to predict the adsorption of gas mixtures from pure component isotherms presented by Myers and Prausnitz. IAST starts from the Raoult's law type of relationship between the fluid and adsorbed phase:
| Pi = Pyi = Pi0xi | (1) |
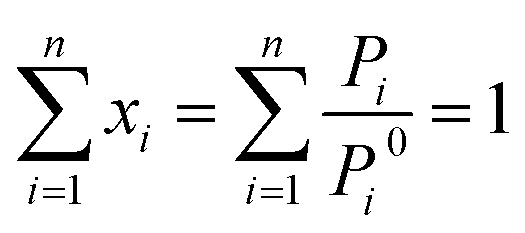 | (2) |
Based on IAST theory, it is assumed that the equality of spreading pressures for all components at equilibrium with the mixture spreading pressure, πi0 = πPi0, is defined using the Gibbs adsorption isotherm:
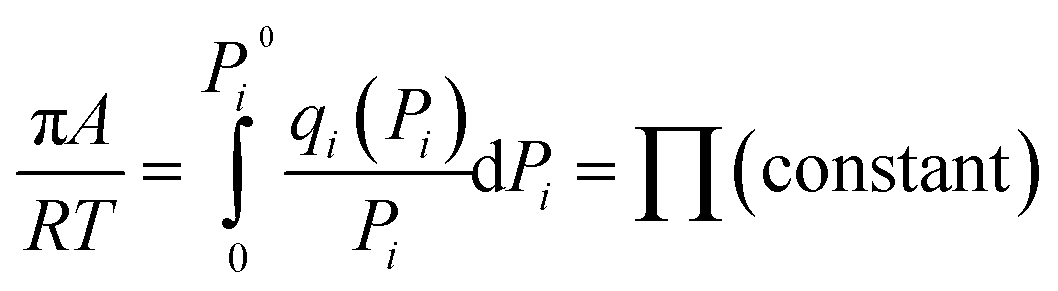 | (3) |
Since the dual-site Langmuir–Freundlich isotherm model is the most consistent with the experimental data, to achieve higher accuracy, the dual-site Langmuir–Freundlich model was used for IAST prediction. For this isotherm, the analytical expression for the integral can be described as:
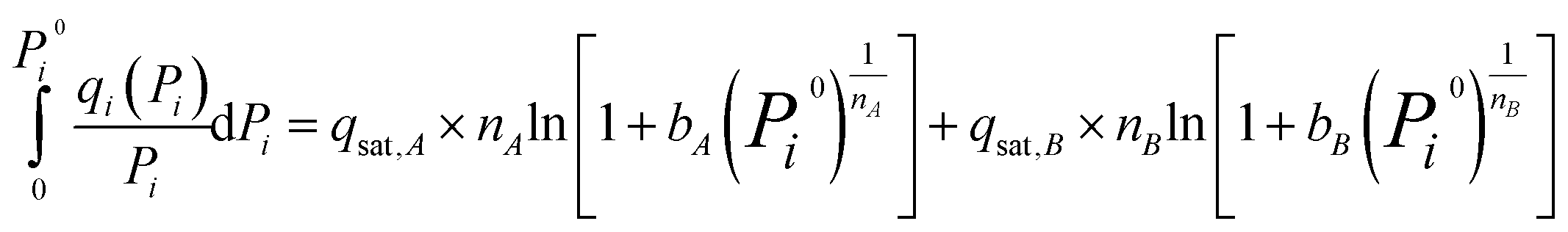 | (4) |
For a binary component system, Pi0 can be obtained by simultaneously solving eqn (1) and (3). The adsorbed amount for each component in a mixture is:
| qi = xiqT | (5) |
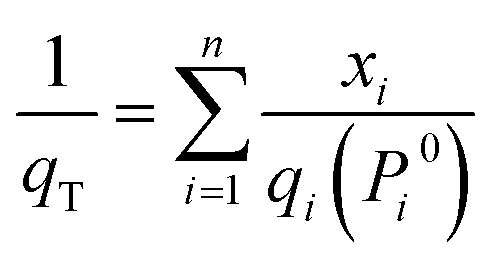 | (6) |
The trial-and-error procedure is continued to find all converged values of Π and Pi0. Then, the mole fractions at the adsorbed phase (xi) and the total amount adsorbed (qT) are obtained using eqn (1) and (6), respectively. Finally, the adsorption selectivity was calculated using the following equation:
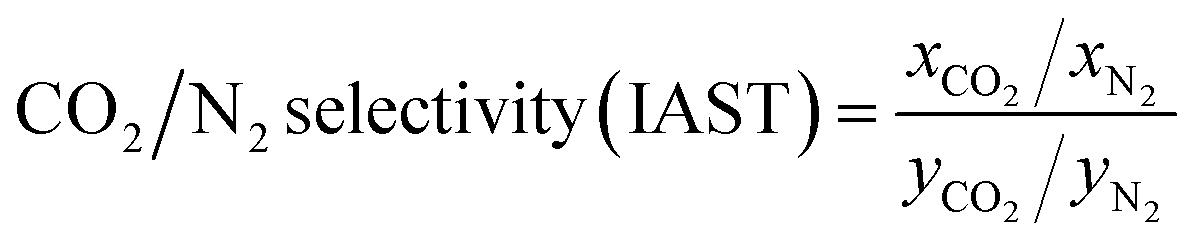 | (7) |
The Henry's law CO2/N2 adsorption selectivity was calculated according to the following eqn (8):
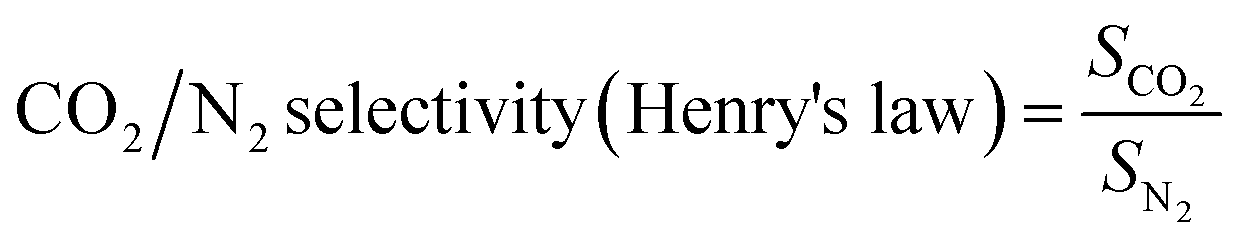 | (8) |
Isosteric heat of adsorption (Qst) calculation
The Qst for CO2 adsorption was calculated according to the Clausius–Clapeyron eqn (9),45 using the measured CO2 adsorption isotherms at 273 and 298 K.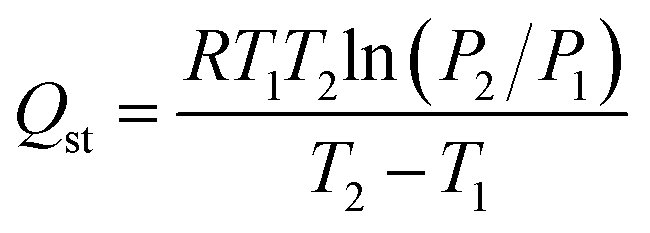 | (9) |
Catalytic cycloadditions of CO2 with epoxides at ambient pressure
The conversion of CO2 into cyclic carbonates at ambient pressure was carried out in a 10 mL Schlenk tube. In a typical run, styrene oxide (12.5 mmol), the non-cationic HCPIP catalyst (60.0 mg, 4 wt%), and TBABr (80.6 mg, 0.25 mmol, 2 mol%) were added into the tube connected with a CO2 balloon. To eliminate air while maintaining the epoxide, the assembled reactor was first frozen using liquid nitrogen, then subjected to vacuum pumping, and filled with CO2. This process was repeated three times to ensure thorough removal of air. Then, the reactor was placed in a preheated oil bath at a designated temperature while being allowed to stir for a designated time frame. After the reaction, the reactor was cooled to room temperature, and the remaining CO2 in the balloon was slowly vented. The reaction mixture was diluted with 20 mL acetone and analyzed by gas chromatography (GC) (GC-2014, Shimadzu, Japan) equipped with a capillary column and a flame ionization detector (FID), and 1,3,5-trimethylbenzene was used as the internal standard. For the recycling test, the non-cationic HCPIP catalyst was separated by centrifugation, washed with acetone, dried under vacuum at 60 °C for 12 h, and then reused for the next run.Computational details
DFT calculations were performed using the Gaussian 16 quantum chemistry package using B3LYP functionals and the 6-311++G(d,p) basis set.46,47 The minimum of the analyzed geometries on the potential energy surface was optimized. Grimme's DFT-D3(BJ) empirical dispersion correction was used to describe the long range van der Waals interactions.48Results and discussion
Design, synthesis, and characterization of the non-cationic HCPIPs
The Friedel–Crafts alkylation reaction is an electrophilic aromatic substitution reaction. Due to the electron-withdrawing effect of N-heterocyclic cations, at present, only a few IL monomers containing an appropriate number of phenyl groups in the cations can carry out the Friedel–Crafts alkylation reaction.31–33 A sufficient number of phenyl groups can weaken the influence of N-heterocyclic cations, thereby ensuring that the aromatic π–bonds have enough electron density to engage in a nucleophilic attack on the alkyl cation.The BPh4− anion is an ionic building block consisting of a central negatively charged boron atom with four phenyl groups. From a structural point of view, compared with the phenyl group in the cation, the BPh4− anion with a rigid tetrahedral structure can provide more hyper-crosslinking sites, can more effectively weaken the electron-withdrawing effect of the N-heterocyclic cation, can build a three-dimensional rigid polymer skeleton, and does not occupy the N sites of the cation. Therefore, BPh4− anion-based ILs would be promising monomers for the construction of various HCPIPs.
A series of BPh4− anion-based ILs [R1R2Im]BPh4 and corresponding Br− anion-based ILs [R1R2Im]Br (R1 = M and Bn; R2 = E, HE, and DHP) were meticulously designed and synthesized (Schemes S1–S9†). Subsequently, endeavors were made to fabricate HCPIPs utilizing these ILs as monomers and FDA as the crosslinker through FeCl3-catalyzed Friedel–Crafts alkylation reactions (Scheme 1 and Table S1†).
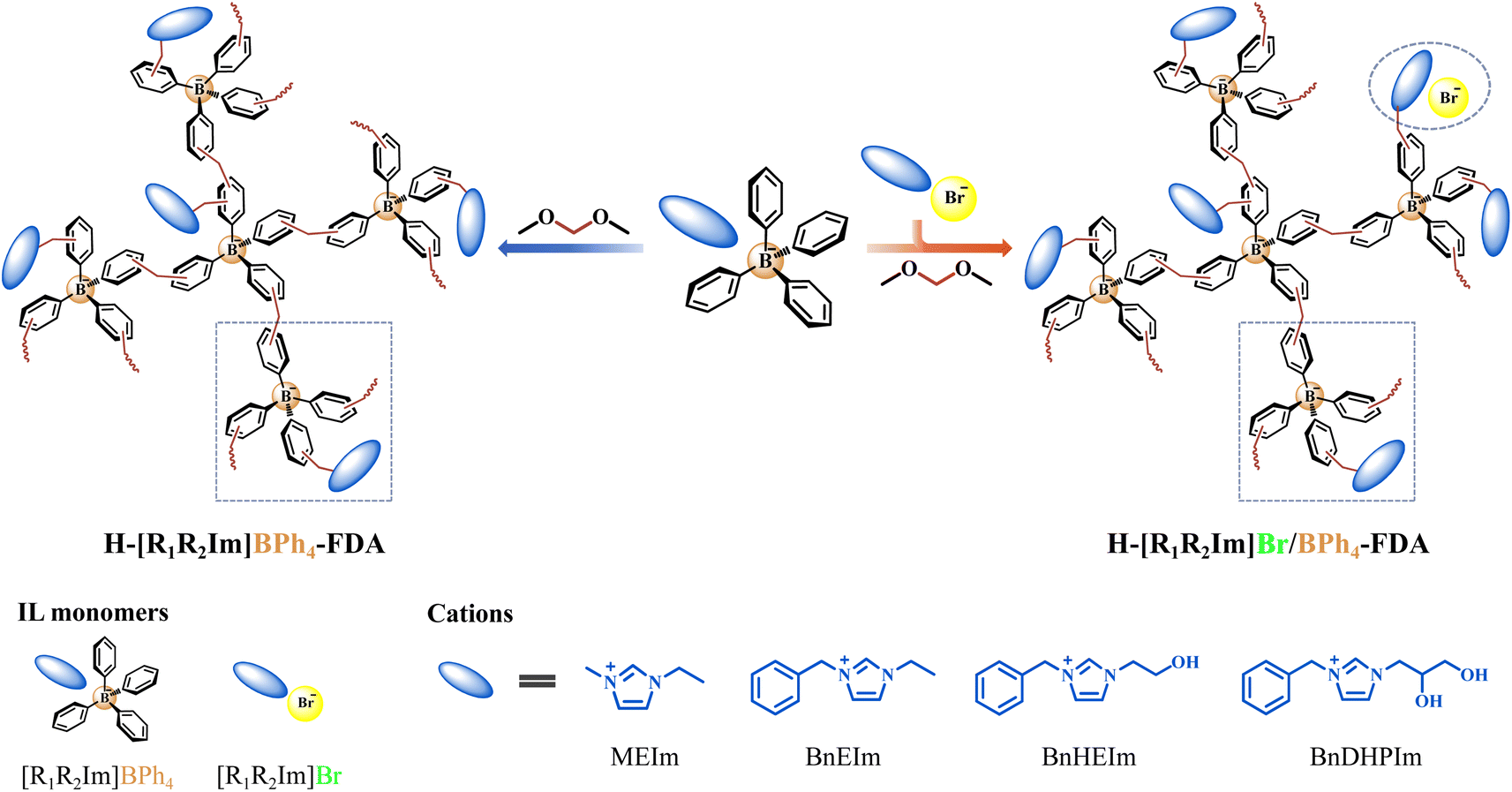 | ||
| Scheme 1 Synthesis of the non-cationic HCPIPs H-[R1R2Im]BPh4-FDA and H-[R1R2Im]Br/BPh4-FDA (R1 = M and Bn; R2 = E, HE, and DHP) via the anion (and cation) hyper-crosslinking strategy. | ||
Notably, HCPIP H-[MEIm]BPh4-FDA was successfully synthesized via the anion hyper-crosslinking of [MEIm]BPh4, exhibiting a specific surface area of 95 m2 g−1 (Table S2†). Conversely, [BnEIm]Br, containing a phenyl group in the cation, proved resistant to hyper-crosslinking (Table S1†). These results confirmed that the introduction of the BPh4− anion significantly promotes the Friedel–Crafts alkylation reaction of IL monomers compared to the introduction of a phenyl group in the cation. To enhance the porosity, a phenyl group was introduced into the cation of BPh4− anion-based IL monomers. The resulting H-[BnEIm]BPh4-FDA, formed through the anion and cation hyper-crosslinking of [BnEIm]BPh4, demonstrated a higher crosslinking density than H-[MEIm]BPh4-FDA, with a specific surface area of 137 m2 g−1. For the development of tailored functionalized HCPIPs, the anion and cation hyper-crosslinking of single and dual hydroxyl-functionalized ILs [BnHEIm]BPh4 and [BnDHPIm]BPh4 was performed. The corresponding HCPIPs H-[BnHEIm]BPh4-FDA and H-[BnDHPIm]BPh4-FDA were obtained with specific surface areas of 121 and 116 m2 g−1, respectively. As the volume of the IL monomer substituent group increased (E < HE < DHP), the specific surface area of the corresponding H-[BnR2Im]BPh4-FDA gradually decreased.
Individual [BnR2Im]Br cannot undergo hyper-crosslinking (Table S1†), and the H-[BnR2Im]BPh4-FDA obtained from the individual [BnR2Im]BPh4 anion and cation hyper-crosslinking had a low specific surface area. Surprisingly, the H-[BnR2Im]Br/BPh4-FDA obtained from the anion and cation hyper-crosslinking of [BnR2Im]Br and [BnR2Im]BPh4 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) exhibited a remarkably high specific surface area, measuring 729 (R2 = E), 708 (R2 = HE), and 636 m2 g−1 (R2 = DHP), respectively (Table S2†). This may occur because [BnR2Im]Br, which resists the Friedel–Crafts alkylation reaction, occupies specific spatial positions during the anion and cation hyper-crosslinking process, functioning similarly to a template agent.
:1 molar ratio) exhibited a remarkably high specific surface area, measuring 729 (R2 = E), 708 (R2 = HE), and 636 m2 g−1 (R2 = DHP), respectively (Table S2†). This may occur because [BnR2Im]Br, which resists the Friedel–Crafts alkylation reaction, occupies specific spatial positions during the anion and cation hyper-crosslinking process, functioning similarly to a template agent.
The pore structures of these non-cationic HCPIPs were further analyzed. Fig. 1a, b, and S1† depict their N2 adsorption–desorption isotherms. Following the IUPAC classification, these isotherms can be classified as a combination of type-I and type-IV isotherms.24,29,33 Specifically, the N2 adsorption sharply increased at relatively low pressure (P/P0 < 0.01), indicating that these non-cationic HCPIPs possess abundant microporous structures.29 Additionally, the observation of hysteresis loops at medium pressure suggested the existence of mesoporous structures.29 Furthermore, it was calculated that the specific surface areas of micropores and mesopores in these non-cationic HCPIPs are approximately equal (Table S2†). Fig. 1c, S2, and S3† display the pore size distributions of these materials. Compared with previously reported HCPIPs with disordered porous structures,20–33 these non-cationic HCPIPs demonstrated unique hierarchically ordered porous structures with narrow pore size distributions, predominantly centered around 0.55, 1.41, 2.76, and 3.77 nm. This might be attributed to the tendency of the BPh4− anion with a rigid tetrahedral structure to form stable three-dimensional regular porous frameworks.
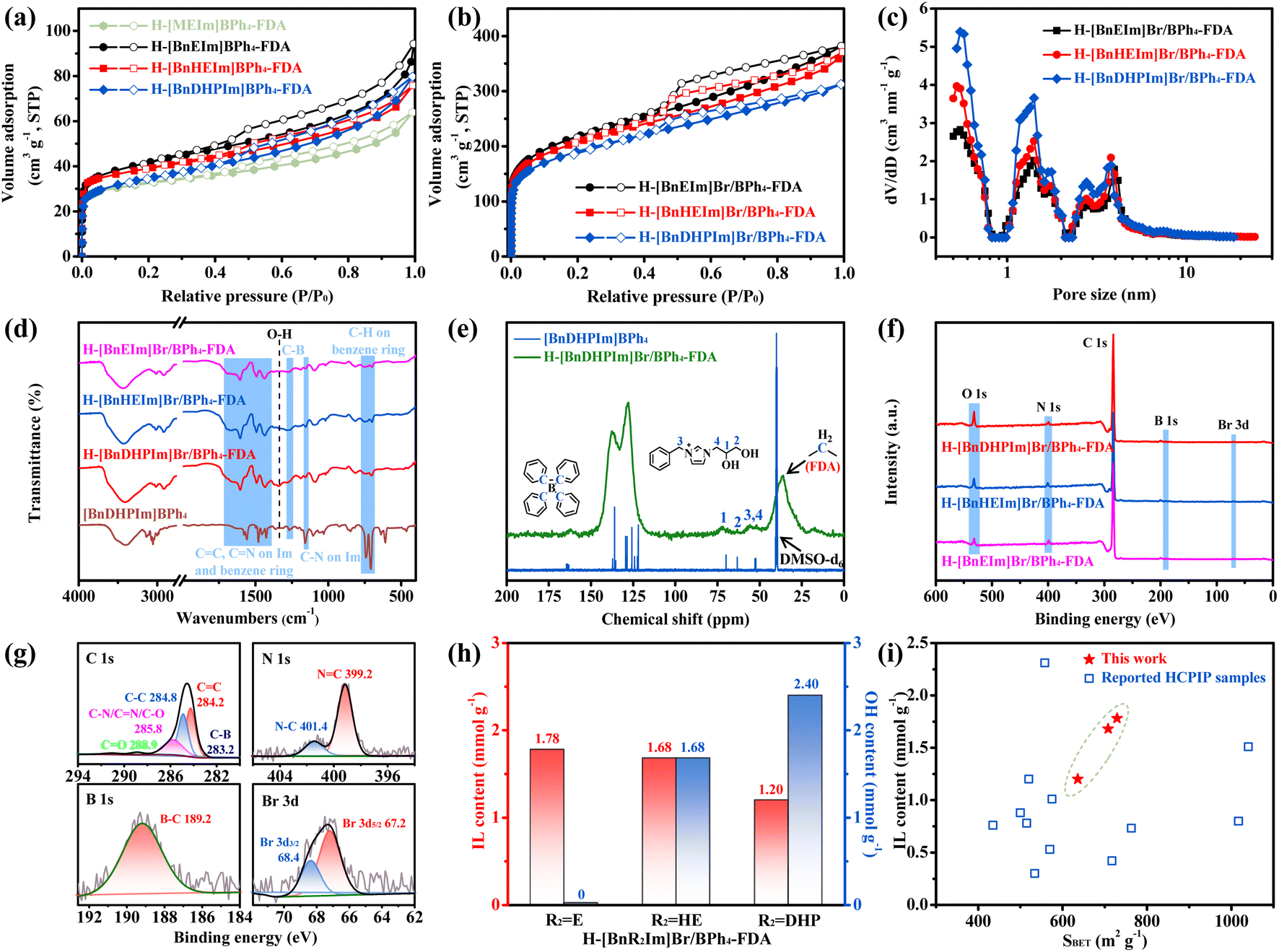 | ||
| Fig. 1 (a) N2 adsorption–desorption isotherms of H-[R1R2Im]BPh4-FDA (R1 = M and Bn; R2 = E, HE, and DHP). (b) N2 adsorption–desorption isotherms of H-[BnR2Im]Br/BPh4-FDA. (c) Pore size distributions of H-[BnR2Im]Br/BPh4-FDA. (d) FT-IR spectra of H-[BnR2Im]Br/BPh4-FDA and [BnDHPIm]BPh4. (e) Solid-state CP/MAS 13C NMR spectrum of H-[BnDHPIm]Br/BPh4-FDA and 13C NMR spectrum (in DMSO-d6) of [BnDHPIm]BPh4. (f) XPS spectra of H-[BnR2Im]Br/BPh4-FDA. (g) XPS spectra of C 1s, N 1s, B 1s, and Br 3d of H-[BnDHPIm]Br/BPh4-FDA. (h) IL content and OH content of H-[BnR2Im]Br/BPh4-FDA. (i) Comparison of H-[BnR2Im]Br/BPh4-FDA and reported HCPIP samples in terms of specific surface areas and IL contents (the original data are shown in Table S4†). | ||
Subsequently, these non-cationic HCPIPs were systematically characterized. The FT-IR spectra of the HCPIPs and IL monomers ([BnDHPIm]BPh4 and [BnDHPIm]Br) are depicted in Fig. 1d, S4, and S5†. The peaks at around 1660, 1567, and 1159 cm−1 were attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, CC, and C–N stretching vibrations of the imidazole (Im) rings (Fig. 1d and S4†).49–51 The peaks at around 1604, 1495, and 1436 cm−1 were associated with the benzene ring stretching vibrations.52–54 The peaks at around 749 and 700 cm−1 were assigned to the C–H out-of-plane bending vibrations of the benzene rings. The characteristic peak of C–B stretching vibration (at around 1268 cm−1) was observed (Fig. 1d, S4, and S5†). As expected, the spectra of H-[BnR2Im]Br/BPh4-FDA and H-[BnR2Im]BPh4-FDA (R2 = HE and DHP) showed the characteristic peak of O–H bending vibration (at around 1336 cm−1) of hydroxyl groups.55 Moreover, the intensity of the O–H bending vibration peaks in H-[BnDHPIm]Br/BPh4-FDA and H-[BnDHPIm]BPh4-FDA were stronger than those of H-[BnHEIm]Br/BPh4-FDA and H-[BnHEIm]BPh4-FDA, respectively, due to the higher number of hydroxyl groups in the corresponding IL monomer (DHP > HE). Given the same IL monomer [BnR2Im]BPh4, the spectra of H-[BnR2Im]Br/BPh4-FDA and H-[BnR2Im]BPh4-FDA (R2 = E, HE, and DHP) were similar (Fig. S6†). These results confirmed the successful synthesis of the non-cationic HCPIPs.
N, CC, and C–N stretching vibrations of the imidazole (Im) rings (Fig. 1d and S4†).49–51 The peaks at around 1604, 1495, and 1436 cm−1 were associated with the benzene ring stretching vibrations.52–54 The peaks at around 749 and 700 cm−1 were assigned to the C–H out-of-plane bending vibrations of the benzene rings. The characteristic peak of C–B stretching vibration (at around 1268 cm−1) was observed (Fig. 1d, S4, and S5†). As expected, the spectra of H-[BnR2Im]Br/BPh4-FDA and H-[BnR2Im]BPh4-FDA (R2 = HE and DHP) showed the characteristic peak of O–H bending vibration (at around 1336 cm−1) of hydroxyl groups.55 Moreover, the intensity of the O–H bending vibration peaks in H-[BnDHPIm]Br/BPh4-FDA and H-[BnDHPIm]BPh4-FDA were stronger than those of H-[BnHEIm]Br/BPh4-FDA and H-[BnHEIm]BPh4-FDA, respectively, due to the higher number of hydroxyl groups in the corresponding IL monomer (DHP > HE). Given the same IL monomer [BnR2Im]BPh4, the spectra of H-[BnR2Im]Br/BPh4-FDA and H-[BnR2Im]BPh4-FDA (R2 = E, HE, and DHP) were similar (Fig. S6†). These results confirmed the successful synthesis of the non-cationic HCPIPs.
The solid-state CP/MAS 13C NMR spectrum of H-[BnDHPIm]Br/BPh4-FDA and the 13C NMR spectrum of the corresponding IL monomer [BnDHPIm]BPh4 are presented in Fig. 1e. Within the spectrum of H-[BnDHPIm]Br/BPh4-FDA, the peak at around 36 ppm was attributed to the FDA methylene carbon (attached between two phenyl rings).56,57 Additionally, influenced by the inductive effect of the FDA methylene, the carbon of C–B in the BPh4− anion upfield shifted from 165 to 162 ppm.58 The peaks at around 128 and 137 ppm were assigned to the carbons of the benzene and Im rings, while other peaks at around 52, 56, 63, and 72 ppm were ascribed to the carbons of the DHP substituent group and methylene in the [BnDHPIm]+ cation. These results also suggested that the non-cationic HCPIPs were successfully synthesized.
The electronic structure and elemental composition of these non-cationic HCPIPs were confirmed by XPS (Fig. 1f, g, S7, and S8†). The survey scan spectra showed that H-[BnR2Im]Br/BPh4-FDA (R2 = E, HE, and DHP) contained C, N, O, B, and Br elements (Fig. 1f), while H-[BnR2Im]BPh4-FDA contained C, N, O, and B elements (Fig. S7†), consistent with their chemical compositions. The O content of H-[BnDHPIm]Br/BPh4-FDA and H-[BnHEIm]Br/BPh4-FDA was significantly higher than that of H-[BnEIm]Br/BPh4-FDA, which was due to the hydroxyl groups in their structures. Taking H-[BnDHPIm]Br/BPh4-FDA as an example (Fig. 1g), its C 1s spectrum could be deconvoluted into five peaks at around 283.2, 284.2, 284.8, 285.8, and 288.9 eV, which were assigned to the carbon signals for C–B, CC, C–C, C–N/CN/C–O, and CO bonds, respectively.59,60 The N 1s spectrum could be deconvoluted into two peaks at around 399.2 and 401.4 eV, which were derived from the Im rings.24,59,61 The peak at around 189.2 eV (B 1s) was attributed to the B–C bonds in BPh4− anions,62 and the peaks at around 67.2 eV (Br 3d5/2) and 68.4 eV (Br 3d3/2) belonged to the free Br− anions.49,61 Moreover, the peak at around 533.2 eV in the O 1s spectrum of H-[BnDHPIm]Br/BPh4-FDA indicated the existence of the OH–C bond (Fig. S8†).61 The above results demonstrated that the IL monomers were successfully incorporated into the non-cationic HCPIP frameworks, in good agreement with the results obtained from FT-IR and NMR spectra.
The N contents of these non-cationic HCPIPs were determined through elemental analysis, with results summarized in Table S3.† The N content of H-[BnR2Im]Br/BPh4-FDA was in the range of 3.35–4.99 wt%. Since N originated from the IL monomers, the IL content of the HCPIPs could be calculated, as high as 1.20 to 1.78 mmol g−1 (Fig. 1h). With the increase in the number of hydroxyl groups in the IL monomer (DHP > HE > E), the IL content of the corresponding H-[BnR2Im]Br/BPh4-FDA gradually decreased from 1.78 to 1.68 and 1.20 mmol g−1. Despite H-[BnDHPIm]Br/BPh4-FDA exhibiting the lowest IL content, it possessed the highest hydroxyl group content (2.40 mmol g−1) (Fig. 1h). Furthermore, the B/Br molar ratio in H-[BnR2Im]Br/BPh4-FDA ranged from 31.4 to 38.8, indicating that only a small amount of the IL monomer [BnR2Im]Br participated in the anion and cation hyper-crosslinking of [BnR2Im]BPh4 and [BnR2Im]Br, accounting for approximately 3% of the total IL content in H-[BnR2Im]Br/BPh4-FDA, which was due to its very poor reactivity in the Friedel–Crafts alkylation reaction.
Additionally, the IL contents of these non-cationic HCPIPs were compared with those of various previously reported HCPIPs. As illustrated in Fig. 1i and detailed in Table S4,† previously reported HCPIPs typically exhibited large specific surface areas but low IL contents.20 Notably, these non-cationic HCPIPs demonstrated both high IL contents and large specific surface areas, a distinctive feature likely attributable to the anion and cation hyper-crosslinking strategy.
The morphology and microstructure of the non-cationic HCPIPs were characterized by SEM and TEM. As shown in Fig. 2a, b, and S9†, H-[BnDHPIm]Br/BPh4-FDA was composed of irregular nanoparticles and layers and presented abundant pore channels. The TEM images of H-[BnDHPIm]Br/BPh4-FDA (Fig. 2c, d, S10a, and b†) were translucent and composed of interconnected and disordered wormhole structures, suggesting that it has abundant microporous and mesoporous structures.31,63 Moreover, elemental mapping images (Fig. 2e, S10c, and d†) demonstrated the uniform distribution of C, N, O, B, and Br throughout the polymer backbone of H-[BnDHPIm]Br/BPh4-FDA. The Br element content in H-[BnDHPIm]Br/BPh4-FDA was much lower than that of the B element, consistent with the XPS results (Table S3†). The powder XRD patterns of these non-cationic HCPIPs (Fig. S11†) exhibited broad and featureless peaks, indicating their amorphous and non-crystalline structures.
 | ||
| Fig. 2 (a and b) SEM images of H-[BnDHPIm]Br/BPh4-FDA. (c and d) TEM images of H-[BnDHPIm]Br/BPh4-FDA. (e) Elemental (O, B, and Br) mapping images of H-[BnDHPIm]Br/BPh4-FDA. (f) TGA curves of [BnDHPIm]Br, [BnDHPIm]BPh4, and H-[BnDHPIm]Br/BPh4-FDA. | ||
The thermal stability of polymer materials is one of the key factors that affects their practical applications. The thermal stabilities of these non-cationic HCPIPs were determined by TGA. As shown in Fig. 2f, H-[BnDHPIm]Br/BPh4-FDA exhibited only 5% weight loss at 400 °C, while the weight losses of the corresponding IL monomers [BnDHPIm]Br and [BnDHPIm]BPh4 were as high as 90% and 82%, respectively. This result indicated that H-[BnDHPIm]Br/BPh4-FDA had excellent thermal stability, which might be attributed to its highly crosslinked structures generated by the anion and cation hyper-crosslinking strategy. The maximum decomposition of H-[BnDHPIm]Br/BPh4-FDA occurred at around 550 °C, attributed to the degradation of the imidazolium skeletons and the destruction of the hyper-crosslinked polymer backbones.64 Similar TGA curves were observed for other non-cationic HCPIPs (Fig. S12†), indicating the same outstanding thermal stabilities.
CO2 capture
The CO2 adsorption performances of the non-cationic HCPIPs, specifically H-[BnR2Im]Br/BPh4-FDA (R2 = E, HE, and DHP), were examined at 273 and 298 K, with the corresponding adsorption isotherms depicted in Fig. 3a. Relatively high CO2 uptakes were observed in all the samples, with the order following H-[BnEIm]Br/BPh4-FDA < H-[BnHEIm]Br/BPh4-FDA < H-[BnDHPIm]Br/BPh4-FDA. In particular, dual hydroxyl-functionalized H-[BnDHPIm]Br/BPh4-FDA, possessing the highest hydroxyl group content (2.40 mmol g−1), exhibited the most significant CO2 uptakes, measuring 3.01 mmol g−1 at 273 K and 1.82 mmol g−1 at 298 K (both at 1 bar).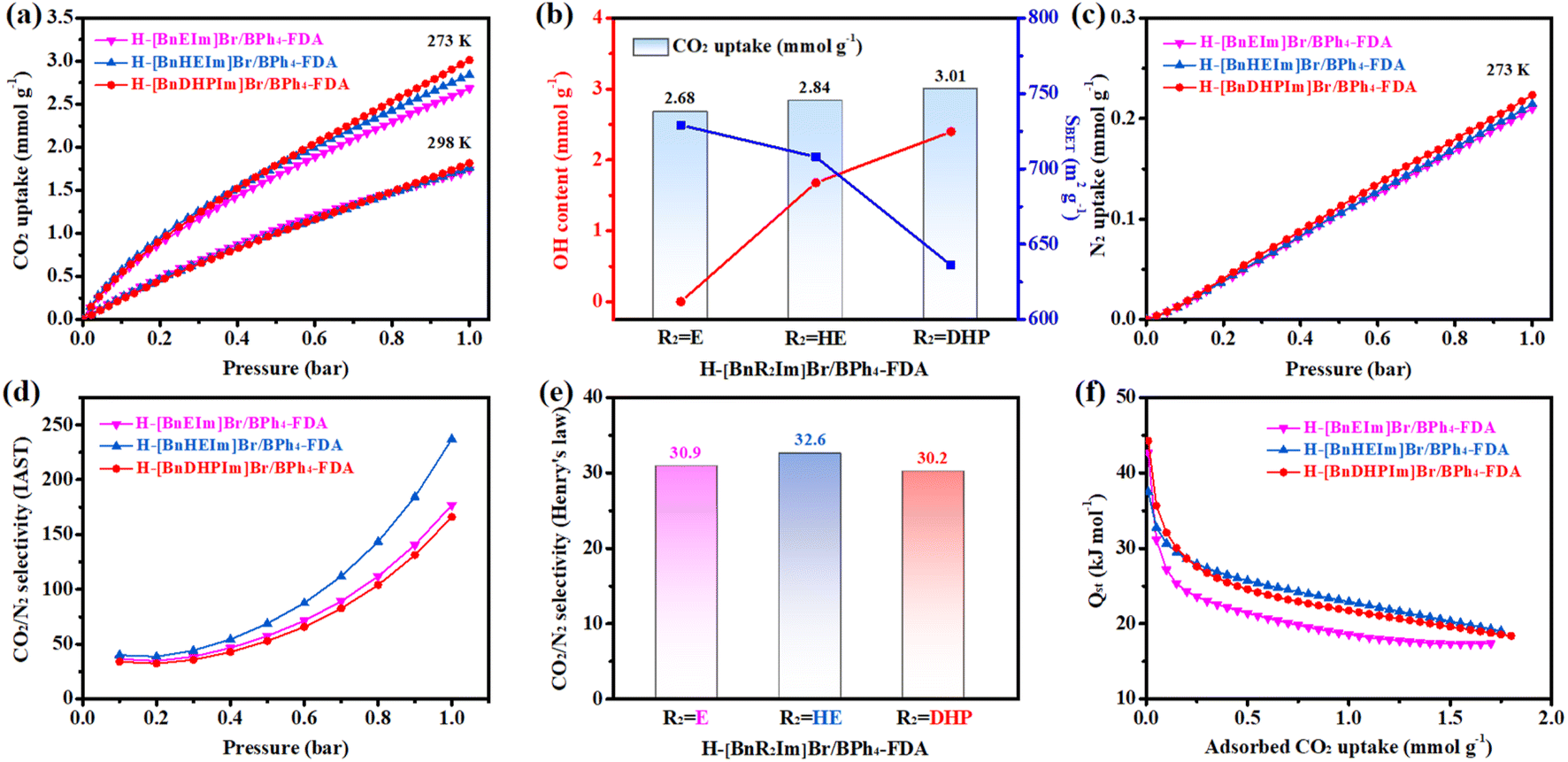 | ||
| Fig. 3 (a) CO2 adsorption isotherms of H-[BnR2Im]Br/BPh4-FDA (R2 = E, HE, and DHP) at 273 and 298 K. (b) The relationship between the CO2 uptake (at 273 K and 1 bar) of H-[BnR2Im]Br/BPh4-FDA and its OH content and specific surface area. (c) N2 adsorption isotherms of H-[BnR2Im]Br/BPh4-FDA at 273 K. (d) IAST CO2/N2 selectivity of H-[BnR2Im]Br/BPh4-FDA at 273 K. [Calculated by using the IAST model for the CO2/N2 (15% CO2 + 85% N2) mixture] (e) Henry's law CO2/N2 selectivity of H-[BnR2Im]Br/BPh4-FDA at 273 K. [Calculated by using the Henry's law constants in the linear low-pressure range (<0.11 bar)] (f) Qst of H-[BnR2Im]Br/BPh4-FDA for CO2 adsorption. | ||
Interestingly, while the CO2 adsorption capacity of previously reported HCPIPs typically correlates positively with specific surface area or IL content, the behavior of H-[BnR2Im]Br/BPh4-FDA diverges. Here, the CO2 adsorption capacity was positively correlated with hydroxyl group content but inversely correlated with specific surface area/IL content (Fig. 3b). This phenomenon suggested that the presence of hydroxyl groups significantly enhances the CO2 adsorption capacity of HCPIPs due to the dipole–dipole and hydrogen-bonding interactions with CO2 molecules.65 This point was also supported by the CO2 adsorption performance of H-[BnDHPIm]BPh4-FDA. Despite having a small specific surface area of 116 m2 g−1, H-[BnDHPIm]BPh4-FDA, which had a high hydroxyl group content (1.98 mmol g−1), exhibited good CO2 adsorption capacity, with values of 2.04 and 1.28 mmol g−1 at 273 and 298 K, respectively (Fig. S13†).
Using CO2-TPD, the interaction of CO2 with the non-cationic HCPIPs was investigated. Based on the TG analysis of H-[BnDHPIm]Br/BPh4-FDA, CO2-TPD was tested in the temperature range of 40–300 °C. As shown in Fig. S14,† multiple CO2 desorption peaks were observed within the range of 40–300 °C, mainly concentrated between 50–125 °C and 225–300 °C. This suggested that H-[BnDHPIm]Br/BPh4-FDA possesses two types of adsorption sites. One type of site might be associated with the hydroxyl groups and ionic moieties, exhibiting a high-affinity coefficient, while the other type corresponded to the neutral backbone, displaying a low-affinity coefficient.30,66
Noteworthy is the fact that the CO2 uptakes of H-[BnR2Im]Br/BPh4-FDA surpass those of most previously reported HCPIPs, as indicated in Table S5.† The outstanding CO2 adsorption capacity of the non-cationic HCPIPs was attributed to the cooperative effects of their hydroxyl groups (facilitating the dipole–dipole and hydrogen-bonding interactions with CO2), ionic moieties (enabling the electrostatic and dipole–quadruple interactions with CO2), and large specific surface areas along with hierarchically ordered porous structures.65,66
To gain deeper insights into the CO2 adsorption dynamics of H-[BnR2Im]Br/BPh4-FDA, various adsorption isotherm models were employed for fitting. The CO2 adsorption isotherms of H-[BnR2Im]Br/BPh4-FDA were excellently captured using the dual-site Langmuir–Freundlich isotherm model, showcasing remarkable fitting with R2 values exceeding 0.9999 (Fig. S15–S17†). This phenomenon also implied that there exist two types of CO2 adsorption sites.30,66 Furthermore, this result indicated that H-[BnR2Im]Br/BPh4-FDA exhibits physical adsorption and multilayer adsorption of CO2.24,67
The N2 adsorption capacities of H-[BnR2Im]Br/BPh4-FDA were also examined to estimate their CO2/N2 adsorption selectivities. The N2 uptakes were found to be quite low, measuring 0.210 (R2 = E), 0.214 (R2 = HE), and 0.224 mmol g−1 (R2 = DHP) at 273 K and 1 bar (Fig. 3c). Subsequently, the CO2/N2 selectivities of H-[BnR2Im]Br/BPh4-FDA at 273 K were determined utilizing the IAST model for the CO2/N2 (15% CO2 + 85% N2) mixture and the Henry's law constants in the linear low-pressure range (<0.11 bar) (Fig. 3d, e, S15–S20†). H-[BnR2Im]Br/BPh4-FDA exhibited high CO2/N2 adsorption selectivities, with IAST CO2/N2 selectivities of 36.4 (R2 = E), 40.1 (R2 = HE), and 34.0 (R2 = DHP) at 0.1 bar. Furthermore, as pressure increased, the IAST CO2/N2 selectivities increased, reaching 177, 237, and 166 at 1 bar, respectively. The Henry's law CO2/N2 selectivities of H-[BnR2Im]Br/BPh4-FDA were 30.9, 32.9, and 30.2 in the linear low-pressure range (<0.11 bar), respectively, which were consistent with their IAST CO2/N2 selectivities. Compared to other non-cationic HCPIPs, H-[BnHEIm]Br/BPh4-FDA showed higher CO2/N2 selectivity, possibly due to the more significant role of the microporous structures in CO2/N2 selective adsorption rather than other factors such as hydrogen-bonding, electrostatic, dipole–dipole, and dipole–quadrupole interactions.68 Importantly, the CO2/N2 selectivities of H-[BnR2Im]Br/BPh4-FDA were highly competitive, surpassing those of most HCPs and rivaling those of previously reported efficient HCPIPs (Table S5†), highlighting the significant potential of non-cationic HCPIPs in selective CO2 capture applications.
The Qst of CO2 adsorption for H-[BnR2Im]Br/BPh4-FDA was determined using the Clausius–Clapeyron equation. As shown in Fig. 3f, the initial Qst values of H-[BnR2Im]Br/BPh4-FDA were 42.66 (R2 = E), 37.46 (R2 = HE), and 44.25 kJ mol−1 (R2 = DHP), suggesting strong affinities toward CO2. Notably, the Qst values of H-[BnR2Im]Br/BPh4-FDA gradually decreased with increasing CO2 coverage, indicating non-uniform adsorption sites of the porous materials.24,25,67 Furthermore, overall, the Qst value of H-[BnEIm]Br/BPh4-FDA was significantly lower than that of H-[BnHEIm]Br/BPh4-FDA and H-[BnDHPIm]Br/BPh4-FDA, indicating a weaker interaction between H-[BnEIm]Br/BPh4-FDA and CO2.24 This might be due to the absence of hydroxyl groups in H-[BnEIm]Br/BPh4-FDA that could interact with CO2 molecules.
CO2 cycloaddition at ambient pressure
The cycloaddition of CO2 to epoxides is a 100% atomic utilization reaction, utilizing CO2 and yielding cyclic carbonates – a group of chemicals widely used as aprotic polar solvents, polycarbonate synthesis monomers, pharmaceutical intermediates, battery electrolytes, etc.69–72 Moreover, cyclic carbonates are highly stable liquid compounds, offering the potential for long-term CO2 sequestration. Among various cyclic carbonates, styrene carbonate (SC), produced from the cycloaddition of CO2 with styrene oxide (SO), has attracted extensive attention. The commercial value of SC is about 120 times that of SO.73 However, the current industrial manufacture of SC from CO2 typically requires harsh and energy-intensive conditions such as high temperature (10–150 °C) and pressure (10–30 bar).74 Here, we applied the non-cationic HCPIPs with notable CO2 adsorption capacities (2.68–3.01 mmol g−1), abundant hydroxyl groups (0–2.40 mmol g−1), high IL contents (1.20–1.78 mmol g−1), hierarchically ordered porous structures, large specific surface areas (636–729 m2 g−1), and excellent stabilities to catalyze the production of SC through CO2 cycloaddition under mild reaction conditions (60–80 °C and 1 bar CO2).The catalytic performances of H-[BnR2Im]Br/BPh4-FDA (R2 = E, HE, and DHP) in the cycloaddition reaction of CO2 and SO at ambient pressure were investigated. The TBABr co-catalyst was employed as a nucleophile to facilitate the ring opening of epoxides as done in previous studies.73 H-[BnDHPIm]Br/BPh4-FDA yielded 94.2% SC after a 15 h reaction at 80 °C under 1 bar CO2 (optimized reaction conditions, Fig. S21–S23†). The yield remained constant with further extension of the reaction time (Fig. S23†). The incomplete conversion observed was attributed to the slight condensation of SO on the inner wall of the reaction Schlenk tube during the reaction (Fig. S24†).
Importantly, the catalytic activity of H-[BnDHPIm]Br/BPh4-FDA + TBABr was significantly better than that of homogeneous [BnDHPIm]BPh4 + TBABr or TBABr alone (Fig. 4a). Under the optimized reaction conditions, [BnDHPIm]BPh4 + TBABr and TBABr alone exhibited considerably lower yields of 57.0% and 44.5%, respectively. The remarkable catalytic activity of H-[BnDHPIm]Br/BPh4-FDA primarily arose from its heterogeneous nature, large specific surface area, hierarchically ordered porous structures, and high CO2 adsorption capacity. Compared to the homogeneous [BnDHPIm]BPh4, the heterogeneous H-[BnDHPIm]Br/BPh4-FDA possesses a higher density of active centers.75,76 Furthermore, its large specific surface area, hierarchically ordered porous structures, and high CO2 adsorption capacity facilitate the enrichment of CO2 and SO near the active centers located in its hyper-crosslinked polymer backbones.76
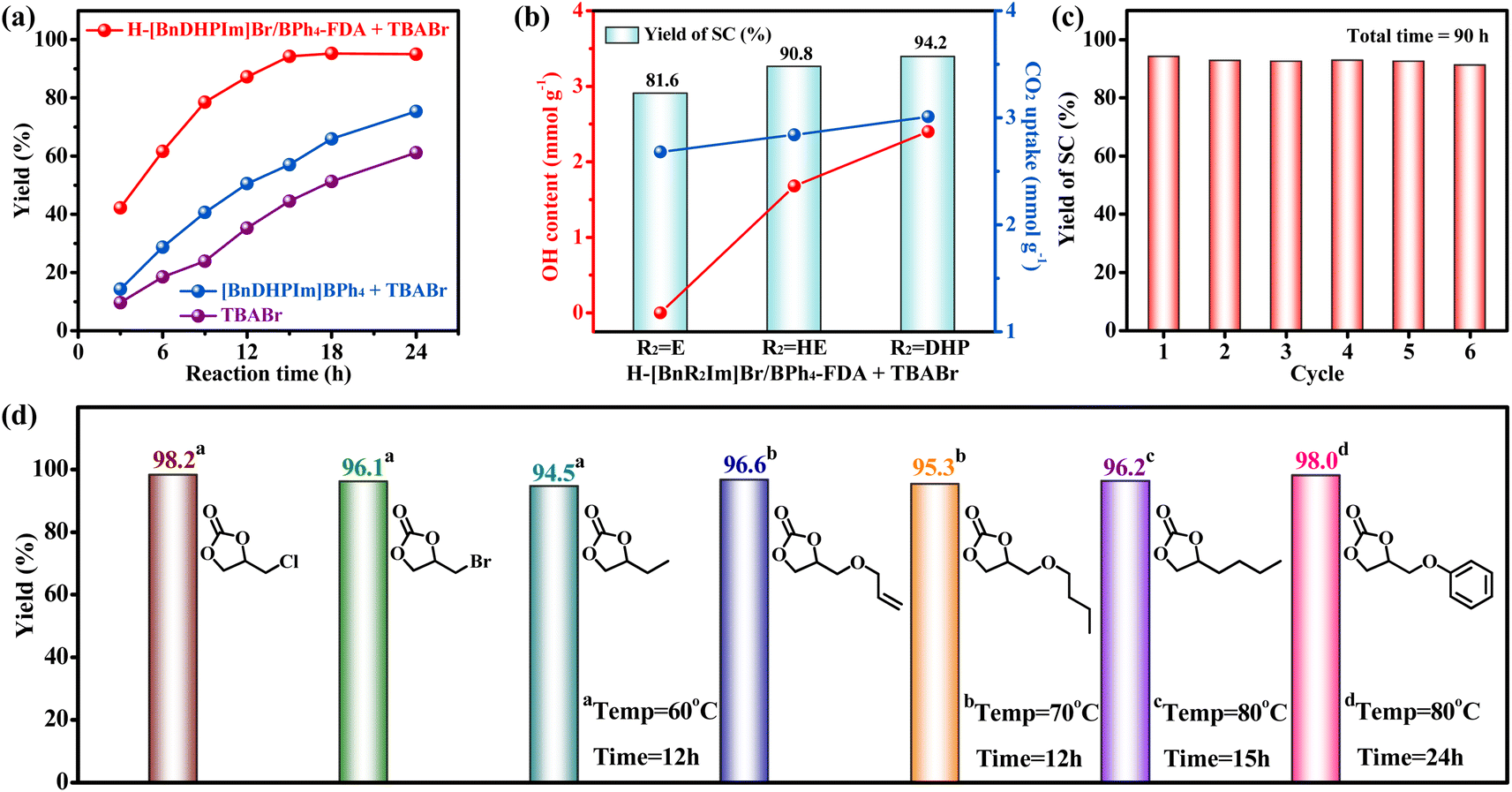 | ||
| Fig. 4 (a) Catalytic activities of H-[BnDHPIm]Br/BPh4-FDA + TBABr, [BnDHPIm]BPh4 + TBABr, and TBABr alone in the cycloaddition reaction of CO2 and SO at different reaction times. (b) The relationship between the catalytic activity of H-[BnR2Im]Br/BPh4-FDA (R2 = E, HE, and DHP) in the cycloaddition reaction of CO2 and SO and its OH content and CO2 uptake. (c) Cycling performance of H-[BnDHPIm]Br/BPh4-FDA. (d) The cycloaddition reactions of CO2 with various epoxides catalyzed by H-[BnDHPIm]Br/BPh4-FDA + TBABr under ambient conditions. Reaction conditions: 12.5 mmol epoxide, 1 bar (balloon) CO2, (a) H-[BnDHPIm]Br/BPh4-FDA (4 wt%) + TBABr (2 mol%), [BnDHPIm]BPh4 (4 wt%) + TBABr (2 mol%), TBABr (2 mol%), 80 °C; (b) H-[BnR2Im]Br/BPh4-FDA (4 wt%) + TBABr (2 mol%), 80 °C, 15 h; (c) H-[BnDHPIm]Br/BPh4-FDA (4 wt%) + TBABr (2 mol%), 80 °C, 15 h; (d) H-[BnDHPIm]Br/BPh4-FDA (4 wt%) + TBABr (2 mol%). | ||
H-[BnEIm]Br/BPh4-FDA and H-[BnHEIm]Br/BPh4-FDA afforded 81.6% and 90.8% yields of SC, respectively, under the optimized reaction conditions. As shown in Fig. 4b, the catalytic activity of H-[BnR2Im]Br/BPh4-FDA was positively correlated with its hydroxyl group content (0 < 1.68 < 2.40 mmol g−1) and CO2 uptake (2.68 < 2.84 < 3.01 mmol g−1). These results highlighted that the hydroxyl group can greatly enhance the catalytic activity of H-[BnR2Im]Br/BPh4-FDA, because the hydroxyl group can not only promote the adsorption and activation of CO2 but also activate SO through hydrogen-bonding interactions.23,65
Cycling stability remains a stringent challenge for many heterogeneous CO2 cycloaddition catalysts. The stability of H-[BnDHPIm]Br/BPh4-FDA was evaluated under the optimized reaction conditions. After reacting at 80 °C for 15 h as a cycle, the catalyst H-[BnDHPIm]Br/BPh4-FDA was retrieved, washed, dried, and then mixed with fresh SO and TBABr for subsequent reaction cycles. As shown in Fig. 4c, H-[BnDHPIm]Br/BPh4-FDA could be reused without noticeable loss of catalytic activity for six consecutive runs (equivalent to a 90 h test). FT-IR, N2 adsorption–desorption isotherm, and CO2 adsorption isotherm characterization studies of H-[BnDHPIm]Br/BPh4-FDA after the cycling stability test confirmed that it retains the same structure and the high specific surface area (585 m2 g−1) and CO2 adsorption capacity (2.80 mmol g−1 at 273 K and 1 bar) (Fig. S25–S27†). The outstanding reusability and stability of H-[BnDHPIm]Br/BPh4-FDA stemmed from its highly stable polymer skeleton structures formed by the anion and cation hyper-crosslinking of the rigid tetrahedral BPh4− anion-based IL monomers.
The universality of H-[BnDHPIm]Br/BPh4-FDA in the CO2 cycloaddition reaction under ambient conditions (1 bar CO2) was investigated. As shown in Fig. 4d, small-sized epoxides, including epichlorohydrin, epibromohydrin, and 1,2-epoxybutane, were efficiently converted into the corresponding cyclic carbonate products at 60 °C and 12 h with yields of 94.5–98.2%. As the size of the substituent increases, steric hindrance also increases, which decreases the reaction rate. Therefore, higher reaction temperatures or longer reaction times are required to promote the reaction. By increasing the reaction temperature to 70 °C, allyl glycidyl ether and n-butyl glycidyl ether achieved yields of 96.6 and 95.3%, respectively. On further increasing the temperature to 80 °C and prolonging the reaction time to 15 h, 1,2-epoxyhexane achieved a yield of 96.2%. Glycidyl phenyl ether, which contains a benzene ring, has the largest steric hindrance, requiring 80 °C and 24 h to achieve a yield of 98%. Overall, H-[BnDHPIm]Br/BPh4-FDA demonstrated good catalysis universality, achieving excellent conversion efficiencies for all epoxide substrates under mild conditions (1 bar CO2, 60–80 °C, 12–24 h). Moreover, in all cases, the selectivity towards the formation of cyclic carbonates was > 99%.
Numerous heterogeneous catalytic systems have been developed for the cycloaddition reactions of CO2 and epoxides. Table S6† summarizes the reported HCPIP catalysts and the highly efficient metal-based heterogeneous catalysts for the cycloaddition reaction of CO2 and SO. The reported HCPIPs typically require high CO2 pressure (10–30 bar) and reaction temperature (100–135 °C) to achieve efficient CO2 cycloaddition of SO (entries 2 and 5–10). To achieve efficient CO2 cycloaddition at ambient pressure, reported HCPIPs usually need a long reaction time (>36 h) or large amounts of co-catalyst (>17 mol% TBABr) (entries 3, 4, and 11). Similarly, highly active metal-based heterogeneous catalysts also require 2–31 mol% TBABr to achieve efficient cycloaddition of SO and CO2 at ambient pressure (entries 12–18). In this study, H-[BnDHPIm]Br/BPh4-FDA effectively catalyzed the cycloaddition reaction of CO2 (1 bar) and SO at 80 °C and 15 h with only 2 mol% TBABr. Its TOF and productivity were 10.9 h−1 and 2.15 g g−1 h−1, respectively, both outperforming many efficient heterogeneous catalytic systems. In particular, compared with most metal-based heterogeneous catalysts, H-[BnDHPIm]Br/BPh4-FDA exhibited superior catalytic performance without metal consumption.
Overall, the combination of ease of preparation and functionalization, high catalytic activity, excellent stability, and good catalysis universality makes H-[BnDHPIm]Br/BPh4-FDA a promising candidate for industrial CO2 fixation to produce high-value-added cyclic carbonates.
Effect of hydroxyl on CO2 capture and conversion
To gain deeper insights into the hydroxyl effect on CO2 capture and conversion, DFT calculations, 1H NMR titration, FT-IR, and in situ FT-IR characterization were performed. The interactions between H-[BnDHPIm]Br/BPh4-FDA and CO2 molecules were investigated through DFT calculations. The optimized geometrical structures of [BnDHPIm]+ and the complex of [BnDHPIm]+ with CO2 were simulated at the B3LYP/6-311++G(d,p) level of theory (Fig. 5a). The bond lengths of O1–H1, O2–H2, and C–H3 in [BnDHPIm]+ were elongated after complexation with CO2 (Table S7†), and hydrogen-bonds O1–H1⋯O, O2–H2⋯O, and C–H3⋯O were formed with lengths of 1.888, 1.915, and 2.220 Å, respectively. These results implied that the dual hydroxyl groups and ionic moieties (C–H3 on the Im ring) of H-[BnDHPIm]Br/BPh4-FDA facilitated CO2 adsorption through hydrogen-bonding interactions. Furthermore, when CO2 interacted with O1–H1, O2–H2, and C–H3 through hydrogen-bonds, the bond angle of CO2 bent from 180° to 177.9°, 179.6°, and 179.9°, respectively. This suggested that CO2 was successfully activated by H-[BnDHPIm]Br/BPh4-FDA.49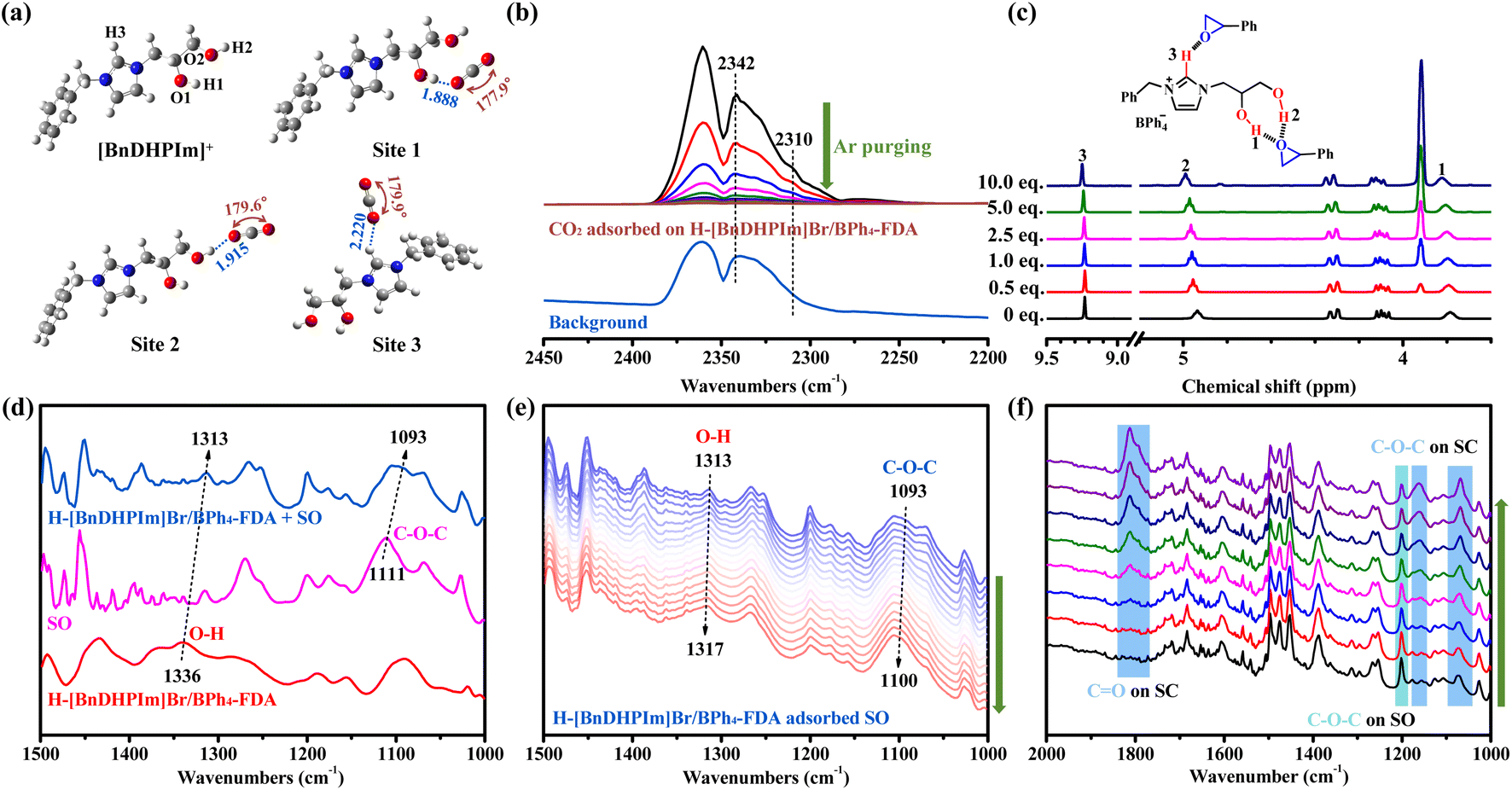 | ||
| Fig. 5 (a) DFT-optimized geometries of [BnDHPIm]+ and the complex of [BnDHPIm]+ and CO2. Bond lengths are reported in Å. (b) In situ FT-IR spectra of CO2 adsorbed on H-[BnDHPIm]Br/BPh4-FDA with Ar purging for 15 min at room temperature, and the FT-IR spectrum of the background. (c) Partial 1H NMR titration spectra of [BnDHPIm]BPh4 in DMSO-d6 with different amounts of SO (the complete spectra are shown in Fig. S28†). The values (0–10.0 eq.) in the spectra represent the molar ratios of SO/[BnDHPIm]BPh4. (d) FT-IR spectra of H-[BnDHPIm]Br/BPh4-FDA, SO, and H-[BnDHPIm]Br/BPh4-FDA with SO adsorbed. (e) In situ FT-IR spectra of H-[BnDHPIm]Br/BPh4-FDA with SO adsorbed at 80 °C for 60 min. (f) In situ FT-IR spectra of H-[BnDHPIm]Br/BPh4-FDA + TBABr catalyzing the cycloaddition reaction of SO and CO2 at ambient pressure and 80 °C for 15 min. | ||
Furthermore, the active ability of H-[BnDHPIm]Br/BPh4-FDA for CO2 was visually explored through in situ FT-IR spectroscopy of CO2 adsorption. In the in situ FT-IR spectra of CO2 adsorbed on H-[BnDHPIm]Br/BPh4-FDA (Fig. 5b), except for the CO characteristic peaks of free CO2 at around 2360, 2339, and 2331 cm−1, two new peaks assigned to the CO bending vibrations of CO2 interacting with dual hydroxyl groups and ionic skeletons at around 2342 and 2310 cm−1 were observed.24,49 With the purging of Ar for 15 min at room temperature, these two peaks were still visible. This phenomenon also suggested that CO2 could be adsorbed and activated by H-[BnDHPIm]Br/BPh4-FDA.
Subsequently, the interaction between H-[BnDHPIm]Br/BPh4-FDA and SO was investigated. A 1H NMR titration experiment was conducted by gradually adding SO to the IL monomer [BnDHPIm]BPh4. Fig. 5c shows the resonance shifts of the dual hydroxyl and C2-position (on the Im ring) H protons upon interaction with SO. As SO was progressively added, the chemical shifts of the dual hydroxyl and C2-position H protons gradually shifted downfield from the initial values of 3.786, 4.936, and 9.230 ppm to 3.823, 4.990, and 9.249 ppm, respectively. This shift is likely due to changes in the hydrogen-bond donor–acceptor interactions.77 In [BnDHPIm]BPh4, the dual hydroxyl and C2-position H act as hydrogen-bond donors interacting with the oxygen atom of SO, which acts as the hydrogen-bond acceptor. These findings suggested that SO could be activated by H-[BnDHPIm]Br/BPh4-FDA.
Fig. 5d shows the FT-IR spectra of H-[BnDHPIm]Br/BPh4-FDA, SO, and H-[BnDHPIm]Br/BPh4-FDA with SO adsorbed. Upon adsorption of SO by H-[BnDHPIm]Br/BPh4-FDA, the characteristic peaks of O–H bending vibration in H-[BnDHPIm]Br/BPh4-FDA and C–O–C asymmetric stretching vibration of SO shifted from 1336 to 1313 cm−1 and 1111 to 1093 cm−1, respectively, indicating the strong hydrogen-bonding interactions between H-[BnDHPIm]Br/BPh4-FDA and SO. The in situ FT-IR spectra of H-[BnDHPIm]Br/BPh4-FDA with SO adsorbed at 80 °C for 60 min showed that the peak intensity decreased overall (Fig. S29†), and the O–H bending vibration and C–O–C asymmetric stretching vibration characteristic peaks gradually shifted to higher wavenumbers (Fig. 5e), which was due to the gradual decrease of SO at 80 °C. Notably, the characteristic peaks of SO remained distinctly visible after 60 min at 80 °C, also implying the strong hydrogen-bonding interactions between H-[BnDHPIm]Br/BPh4-FDA and SO.
To investigate the catalytic reaction process, in situ FT-IR spectroscopy of H-[BnDHPIm]Br/BPh4-FDA + TBABr for the cycloaddition reaction of CO2 and SO at 80 °C and ambient pressure was performed. As shown in Fig. 5f, with the increase in reaction time (from 0 to 15 min), the characteristic peaks of CO stretching vibration (at around 1812 cm−1) and C–O–C symmetric and asymmetric stretching vibrations (at around 1161 and 1069 cm−1, respectively) belonging to the product SC appeared and gradually enhanced,49 and the characteristic peaks of reactant SO (e.g., C–O–C symmetric stretching vibration at around 1201 cm−1) gradually weakened, indicating the successful conversion of SO and CO2 into SC.
Based on the above characterization and experimental results and the previous literature,30,55 a plausible catalytic reaction mechanism for the cycloaddition reaction of CO2 and SO catalyzed by H-[BnDHPIm]Br/BPh4-FDA + TBABr under ambient conditions was proposed (Fig. 6). In this reaction, the dual hydroxyl groups and C2-position H on H-[BnDHPIm]Br/BPh4-FDA acted as hydrogen-bond donors to activate SO through hydrogen-bonding interactions, leading to the polarization of C–O bonds. Meanwhile, the Br− anions served as nucleophiles to attack the less sterically hindered carbon atom of the activated SO (I), resulting in the ring opening of SO and generating an oxyanion intermediate (II). Subsequently, the O− in the intermediate (II) attacked the carbon atom of the CO2 that has been activated by H-[BnDHPIm]Br/BPh4-FDA, forming a carbonate intermediate (III). Finally, the product SC was generated via the intramolecular O− nucleophilic attack and removal of Br− anions from the intermediate (III), and the catalyst H-[BnDHPIm]Br/BPh4-FDA was recovered.
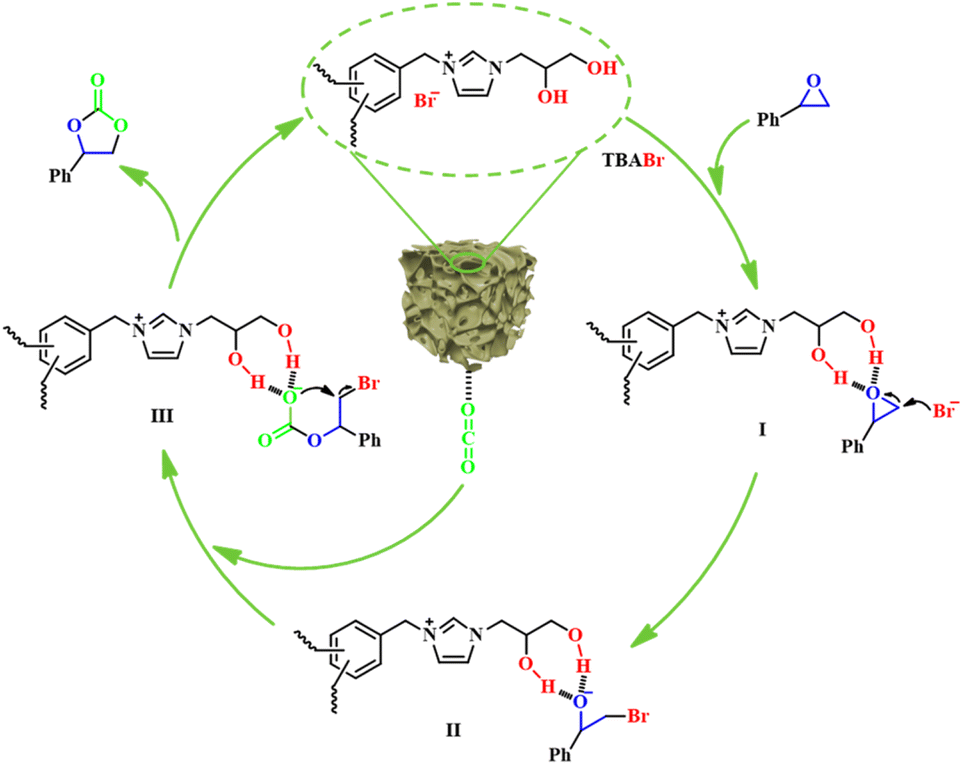 | ||
| Fig. 6 Proposed reaction mechanism for the cycloaddition reaction of CO2 and SO catalyzed by the dual hydroxyl-functionalized H-[BnDHPIm]Br/BPh4-FDA + TBABr. | ||
Conclusions
We developed and synthesized novel non-cationic HCPIPs through direct anion (and cation) hyper-crosslinking of the unique BPh4− anion-based IL monomers [R1R2Im]BPh4 (and [R1R2Im]Br) (R1 = M and Bn; R2 = E, HE, and DHP) using FDA as the crosslinker. The introduction of the rigid tetrahedral BPh4− anion not only effectively weakens the influence of strong electron-withdrawing groups in the Friedel–Crafts alkylation reaction but also provides abundant three-dimensional sites for the anion (and cation) hyper-crosslinking, ensuring the seamless synthesis of the non-cationic HCPIPs. The resulting non-cationic HCPIPs H-[BnR2Im]Br/BPh4-FDA exhibit controllable hydroxyl group content (0–2.40 mmol g−1), high IL content (1.20–1.78 mmol g−1), large specific surface area (636–729 m2 g−1) with hierarchically ordered porous structures, and excellent stability. Consequently, H-[BnR2Im]Br/BPh4-FDA demonstrates a good CO2 adsorption capacity of 2.68–3.01 mmol g−1 and high IAST CO2/N2 adsorption selectivity of 166–237 at 273 K and 1 bar. Serving as a heterogeneous catalyst, H-[BnR2Im]Br/BPh4-FDA is highly active in the cycloaddition of CO2 with epoxides under mild conditions (1 bar CO2, 60–80 °C, 12–24 h), showing remarkable catalytic activity with high yield, stable cycling stability, and broad substrate universality. Notably, the CO2 adsorption performance and catalytic activity of H-[BnR2Im]Br/BPh4-FDA are regulated by the hydroxyl groups within their structures and gradually enhanced as the number of hydroxyl groups increased. DFT calculations, 1H NMR titration, FT-IR, and in situ FT-IR characterization elucidate the role of hydroxyl groups in promoting the adsorption, enrichment, and activation of CO2, as well as in activating epoxides through hydrogen-bonding interactions. This work provides a novel and simple approach to the construction of high-performance and task-specific HCPIPs.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Bihua Chen: investigation, methodology, visualization, writing – original draft. Junfeng Zeng: formal analysis. Shiguo Zhang: supervision, writing – review & editing, funding acquisition. Yan Zhang: supervision, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (Grant No. 21872046, 52373206, and 52072118) and the Hunan Provincial Natural Science Foundation of China (Grant No. 2020JJ4174 and 2023JJ40114).References
- Y. Shi, Y. Wang, N. Meng and Y. Liao, Small Methods, 2024, 2301554 CrossRef PubMed.
- W. Wang, M. Zhou and D. Yuan, J. Mater. Chem. A, 2017, 5, 1334–1347 RSC.
- D. Luo, T. Shi, Q.-H. Li, Q. Xu, M. Strømme, Q.-F. Zhang and C. Xu, Angew. Chem., Int. Ed., 2023, 62, e202305225 CrossRef CAS PubMed.
- X.-H. Xu, Y.-X. Li, L. Zhou, N. Liu and Z.-Q. Wu, Chem. Sci., 2022, 13, 1111–1118 RSC.
- Y. Wang, Y. Yang, Q. Deng, W. Chen, Y. Zhang, Y. Zhou and Z. Zou, Adv. Funct. Mater., 2023, 33, 2307179 CrossRef CAS.
- D. Luo, M. Li, Q. Ma, G. Wen, H. Dou, B. Ren, Y. Liu, X. Wang, L. Shui and Z. Chen, Chem. Soc. Rev., 2022, 51, 2917–2938 RSC.
- Z. Cheng, H. Pan, H. Zhong, Z. Xiao, X. Li and R. Wang, Adv. Funct. Mater., 2018, 28, 1707597 CrossRef.
- L. Zou, Y. Sun, S. Che, X. Yang, X. Wang, M. Bosch, Q. Wang, H. Li, M. Smith, S. Yuan, Z. Perry and H.-C. Zhou, Adv. Mater., 2017, 29, 1700229 CrossRef PubMed.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- X. Suo, Y. Huang, Z. Li, H. Pan, X. Cui and H. Xing, Sci. China Mater., 2022, 65, 1068–1075 CrossRef CAS.
- X. Cui, K. Chen, H. Xing, Q. Yang, R. Krishna, Z. Bao, H. Wu, W. Zhou, X. Dong, Y. Han, B. Li, Q. Ren, M. J. Zaworotko and B. Chen, Science, 2016, 353, 141–144 CrossRef CAS PubMed.
- D. Xu, J. Guo and F. Yan, Prog. Polym. Sci., 2018, 79, 121–143 CrossRef CAS.
- S. Zhang, K. Dokko and M. Watanabe, Chem. Sci., 2015, 6, 3684–3691 RSC.
- W. Li, W. Xiao, Q. Luo, J. Yan, G. Zhang, L. Chen and J. Sun, Coord. Chem. Rev., 2023, 493, 215304 CrossRef CAS.
- S. Zhang, J. Zhang, Y. Zhang and Y. Deng, Chem. Rev., 2017, 117, 6755–6833 CrossRef CAS PubMed.
- J.-K. Sun, M. Antonietti and J. Yuan, Chem. Soc. Rev., 2016, 45, 6627–6656 RSC.
- L. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- Y. Gu, S. U. Son, T. Li and B. Tan, Adv. Funct. Mater., 2021, 31, 2008265 CrossRef CAS.
- W. Song, Y. Tang, B. Y. Moon, Q. Liao, H. Xu, Q. Hou, H. Zhang, D.-G. Yu, Y. Liao and I. Kim, Green Chem., 2024, 26, 2476–2504 RSC.
- R. Zhang, G. Cui, X. Wang, Y. Chen, X. Qiu, Q. Ke, D. Deng, C. Ge, H. Lu and S. Dai, Chem. Eng. J., 2024, 489, 151102 CrossRef CAS.
- J. Wang, W. Sng, G. Yi and Y. Zhang, Chem. Commun., 2015, 51, 12076–12079 RSC.
- Y. Sang, Z. Shu, Y. Wang, L. Wang, D. Zhang, Q. Xiao, F. Zhou and J. Huang, Appl. Surf. Sci., 2022, 585, 152663 CrossRef CAS.
- D. Jia, L. Ma, Y. Wang, W. Zhang, J. Li, Y. Zhou and J. Wang, Chem. Eng. J., 2020, 390, 124652 CrossRef CAS.
- K. Cai, P. Liu, Z. Chen, P. Chen, F. Liu, T. Zhao and D.-J. Tao, Chem. Eng. J., 2023, 451, 138946 CrossRef CAS.
- H. Lyu, X. Wang, W. Sun, E. Xu, Y. She, A. Liu, D. Gao, M. Hu, J. Guo, K. Hu, J. Cheng, Z. Long, Y. Liu and P. Zhang, Green Chem., 2023, 25, 3592–3605 RSC.
- J. Li, D. Jia, Z. Guo, Y. Liu, Y. Lyu, Y. Zhou and J. Wang, Green Chem., 2017, 19, 2675–2686 RSC.
- Y. Sang and J. Huang, Chem. Eng. J., 2020, 385, 123973 CrossRef CAS.
- C. Liu, L. Shi, J. Zhang and J. Sun, Chem. Eng. J., 2022, 427, 131633 CrossRef CAS.
- C. Li, H. Cai, X. Yang, F. Liu, C. Yang, P. Chen, Z. Chen and T. Zhao, J. CO2 Util., 2022, 64, 102203 CrossRef CAS.
- W. Zhang, F. Ma, L. Ma, Y. Zhou and J. Wang, ChemSusChem, 2020, 13, 341–350 CrossRef CAS PubMed.
- H. Song, Y. Wang, Y. Liu, L. Chen, B. Feng, X. Jin, Y. Zhou, T. Huang, M. Xiao, F. Huang and H. Gai, ACS Sustainable Chem. Eng., 2021, 9, 2115–2128 CrossRef CAS.
- Z. Zhang, D. Yang, J. Wang, C. Deng, H. Gai, M. Xiao, T. Huang, Q. Zhu and H. Song, Mol. Catal., 2023, 535, 112809 CrossRef CAS.
- X. Liao, Z. Wang, Z. Li, L. Kong, W. Tang, Z. Qin and J. Lin, Chem. Eng. J., 2023, 471, 144455 CrossRef CAS.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- P. A. Hunt, C. R. Ashworth and R. P. Matthews, Chem. Soc. Rev., 2015, 44, 1257–1288 RSC.
- M. Petkovic, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Chem. Soc. Rev., 2011, 40, 1383–1403 RSC.
- S. Koutsoukos, F. Philippi, F. Malaret and T. Welton, Chem. Sci., 2021, 12, 6820–6843 RSC.
- G. Kaur, H. Kumar and M. Singla, J. Mol. Liq., 2022, 351, 118556 CrossRef CAS.
- S. Yang, Z. Zhong, J. Hu, X. Wang and B. Tan, Adv. Mater., 2024, 36, 2307579 CrossRef CAS PubMed.
- B. Li, R. Gong, W. Wang, X. Huang, W. Zhang, H. Li, C. Hu and B. Tan, Macromolecules, 2011, 44, 2410–2414 CrossRef CAS.
- A. M. Najafi, S. Soltanali and H. Ghassabzadeh, Chem. Eng. J., 2023, 468, 143719 CrossRef CAS.
- M. Vorokhta, M. I. M. Kusdhany, D. Vöröš, M. Nishihara, K. Sasaki and S. M. Lyth, Chem. Eng. J., 2023, 471, 144524 CrossRef CAS.
- Y. Li, H. Chen, C. Wang, Y. Ye, L. Li, X. Song and J. Yu, Chem. Sci., 2022, 13, 5687–5692 RSC.
- E. Dautzenberg, S. van Hurne, M. M. J. Smulders and L. C. P. M. de Smet, Comput. Phys. Commun., 2022, 280, 108494 CrossRef CAS.
- S. D. Bao, W. Jian, J. H. Li, Y. S. Bao and A. Bao, Chem. Eng. J., 2023, 475, 146149 CrossRef CAS.
- M. Li, Y. Gao, D. Yu, Z. Hu, Z. Liu, X. Wang, Q. Weng, Y. Chen, Y. Zhang and S. Zhang, Energy Storage Mater., 2023, 59, 102753 CrossRef.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- L. Goerigk and S. Grimme, J. Chem. Theory Comput., 2011, 7, 291–309 CrossRef CAS PubMed.
- M. Qiu, J. Li, H. Wu, Y. Huang, H. Guo, D. Gao, L. Shi and Q. Yi, Appl. Catal., B, 2023, 322, 122125 CrossRef CAS.
- B. Chen, S. Zhang and Y. Zhang, Green Chem., 2023, 25, 7743–7755 RSC.
- A. Pourjavadi, S. H. Hosseini, M. Doulabi, S. M. Fakoorpoor and F. Seidi, ACS Catal., 2012, 2, 1259–1266 CrossRef CAS.
- B. Chen, T. Ding, X. Deng, X. Wang, D. Zhang, S. Ma, Y. Zhang, B. Ni and G. Gao, Chin. J. Catal., 2021, 42, 297–309 CrossRef CAS.
- B. Chen, M. Wang, X. Wang, Q. Zhao, Y. Wang and G. Gao, Polym. Chem., 2021, 12, 2731–2742 RSC.
- R. Shen, X. Yan, Y.-J. Guan, W. Zhu, T. Li, X.-G. Liu, Y. Li and Z.-G. Gu, Polym. Chem., 2018, 9, 4724–4732 RSC.
- Z. Guo, Q. Jiang, Y. Shi, J. Li, X. Yang, W. Hou, Y. Zhou and J. Wang, ACS Catal., 2017, 7, 6770–6780 CrossRef CAS.
- P. Puthiaraj, S. Ravi, K. Yu and W.-S. Ahn, Appl. Catal., B, 2019, 251, 195–205 CrossRef CAS.
- L. Jiang, P. Wang, Y. Wang, Y. Wang, X. Li, Q. Xia and H. Ren, J. Colloid Interface Sci., 2021, 582, 631–637 CrossRef CAS PubMed.
- X. Jiang, S. Li, S. He, Y. Bai and L. Shao, J. Mater. Chem. A, 2018, 6, 15064–15073 RSC.
- S. Aldroubi, B. Larhrib, L. Larbi, I. B. Malham, C. M. Ghimbeu, L. Monconduit, A. Mehdi and N. Brun, J. Mater. Chem. A, 2023, 11, 16755–16766 RSC.
- Y. Zhao, H. Huang, H. Zhu and C. Zhong, Microporous Mesoporous Mater., 2022, 329, 111526 CrossRef CAS.
- G. Chen, Y. Zhang, J. Xu, X. Liu, K. Liu, M. Tong and Z. Long, Chem. Eng. J., 2020, 381, 122765 CrossRef CAS.
- R. L. Tapping, Inorg. Chem., 1982, 21, 1691–1692 CrossRef CAS.
- S. Zhang, T. Mandai, K. Ueno, K. Dokko and M. Watanabe, Nano Energy, 2015, 13, 376–386 CrossRef CAS.
- K. Cai, P. Liu, T. Zhao, K. Su, Y. Yang and D.-J. Tao, Microporous Mesoporous Mater., 2022, 343, 112135 CrossRef CAS.
- M. Yin, L. Wang and S. Tang, ACS Catal., 2023, 13, 13021–13033 CrossRef CAS.
- J. Cao, W. Shan, Q. Wang, X. Ling, G. Li, Y. Lyu, Y. Zhou and J. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 6031–6041 CrossRef CAS PubMed.
- J. Gu, Y. Yuan, T. Zhao, F. Liu, Y. Xu and D.-J. Tao, Sep. Purif. Technol., 2022, 301, 121971 CrossRef CAS.
- J. Zhang, Z.-A. Qiao, S. M. Mahurin, X. Jiang, S.-H. Chai, H. Lu, K. Nelson and S. Dai, Angew. Chem., Int. Ed., 2015, 54, 4582–4586 CrossRef CAS PubMed.
- G. Li, S. Dong, P. Fu, Q. Yue, Y. Zhou and J. Wang, Green Chem., 2022, 24, 3433–3460 RSC.
- G. A. Bhat and D. J. Darensbourg, Green Chem., 2022, 24, 5007–5034 RSC.
- L. Guo, K. J. Lamb and M. North, Green Chem., 2021, 23, 77–118 RSC.
- W. Liu, L. Li, W. Shao, H. Wang, Y. Dong, M. Zuo, J. Liu, H. Zhang, B. Ye, X. Zhang and Y. Xie, Chem. Sci., 2023, 14, 1397–1402 RSC.
- J. Xu, H. Xu, A. Dong, H. Zhang, Y. Zhou, H. Dong, B. Tang, Y. Liu, L. Zhang, X. Liu, J. Luo, L. Bie, S. Dai, Y. Wang, X. Sun and Y. Li, Adv. Mater., 2022, 34, 2206991 CrossRef CAS PubMed.
- B.-H. Xu, J.-Q. Wang, J. Sun, Y. Huang, J.-P. Zhang, X.-P. Zhang and S.-J. Zhang, Green Chem., 2015, 17, 108–122 RSC.
- D.-J. Tao, F. Liu, L. Wang and L. Jiang, Appl. Catal., A, 2018, 564, 56–63 CrossRef CAS.
- Y. Zhao, Y. Peng, C. Shan, Z. Lu, L. Wojtas, Z. Zhang, B. Zhang, Y. Feng and S. Ma, Nano Res., 2022, 15, 1145–1152 CrossRef CAS.
- T. Zhu, Y. Xu, Z. Li, J. He, X. Yuan, D. Qian, T. Chang, L. Lu, B. Chi and K. Guo, J. Org. Chem., 2024, 89, 7408–7416 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03708a |
| This journal is © The Royal Society of Chemistry 2024 |