
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSlightly Li-enriched chemistry enabling super stable LiNi0.5Mn0.5O2 cathodes under extreme conditions†
Siqi
Chen
a,
Ping
Zhang
a,
Xin
Zhou
a,
Wenbin
Wu
a,
Xiaohong
Liu
*a,
Yifeng
Liu
b,
Guilin
Feng
 c,
Bin
Zhang
d,
Wangyan
Xing
d,
Meihua
Zuo
d,
Ping
Zhang
d,
Genpin
Lv
e,
Yao
Xiao
*b,
Shixue
Dou
f and
Wei
Xiang
*ag
c,
Bin
Zhang
d,
Wangyan
Xing
d,
Meihua
Zuo
d,
Ping
Zhang
d,
Genpin
Lv
e,
Yao
Xiao
*b,
Shixue
Dou
f and
Wei
Xiang
*ag
aCollege of Materials and Chemistry & Chemical Engineering, Chengdu University of Technology, Chengdu 610059, Sichuan, China. E-mail: xiangwei@cdut.edu.cn
bCollege of Chemistry and Materials Engineering, Wenzhou University, Wenzhou, 325035, PR China. E-mail: xiaoyao@wzu.edu.cn; xhl@cdut.edu.cn
cResearch Institute of Frontier Science, Southwest Jiaotong University, Chengdu 610031, PR China
dYibin Libode New Materials Co., Ltd, Yibin 644200, PR China
eRuyuan HEC New Energy Materials Co., Ltd, Ruyuan 512700, PR China
fInstitute of Energy Materials Science, University of Shanghai for Science and Technology, Shanghai 200093, PR China
gTianfu Yongxing Laboratory, Chengdu 610213, PR China
First published on 9th August 2024
Abstract
High voltage/high temperature operation aggravates the risk of capacity attenuation and thermal runaway of layered oxide cathodes due to crystal degradation and interfacial instability. A combined strategy of bulk regulation and surface chemistry design is crucial to handle these issues. Here, we present a simultaneous Li2WO4-coated and gradient W-doped 0.98LiNi0.5Mn0.5O2·0.02Li2WO4 cathode through modulating the content of the exotic dopant and stoichiometric lithium salt during lithiation calcination. Benefiting from the slightly Li-enriched chemistry induced by the hetero-epitaxially grown Li2WO4 surface, the 0.98LiNi0.5Mn0.5O2·0.02Li2WO4 cathode demonstrates superior electrochemical performance to W-doped LiNi0.49Mn0.49W0.02O2 and WO3 coated 0.98LiNi0.5Mn0.5O2·0.02WO3 cathodes without a Li-enriched phase. Specifically, when cycled in the potential range of 2.7–4.5 V at 30 °C, the 0.98LiNi0.5Mn0.5O2·0.02Li2WO4 cathode possesses a high discharge capacity of 199.2 and 156.5 mA h g−1 at 0.1 and 5C and a capacity retention of 92.88% after 300 cycles at 1C. Even at a high cut-off voltage of 4.6 V, it still retains a capacity retention of 91.15% after 200 cycles at 1C and 30 °C. Compared with LiNi0.5Mn0.5O2, the enhanced performance of 0.98LiNi0.5Mn0.5O2·0.02Li2WO4 can be attributed to its robust bulk and stable interface, inhibited lattice oxygen release, and improved Li+ transport kinetics. Our work emphasizes the significance of the slightly Li-enriched chemistry and bulk modulation strategy in stabilizing cathodes and hence unlocks vast possibilities for future cathode design.
1. Introduction
Owing to their high specific capacity, LiNiO2 and nickel-rich oxides such as LiNixM1−xO2 (where M = Mn, Co, Al, etc.) are highly promising cathodes for lithium-ion batteries with high energy density.1–5 However, high Ni content also means high cost (necessary but costly Co and Ni), high risk under extreme conditions (fast charging, high voltage or high temperature operation, etc.) and inferior thermal safety.6–8 Co-free alternatives such as LiNi1−xMnxO2, with a medium nickel content and appropriate manganese content, show enhanced structure stability due to the presence of Ni2+/Mn4+ charge separation.9 In the solid solution system, Ni2+ remains electrochemically active, and Mn4+ acts as a stabilizer for the structure, leading to improved cycling performance and thermal stability compared to LiNiO2.10,11 To manage the defect of the low discharge capacity brought by decreased Ni content, high voltage operation has been proven efficient in embedding and releasing more lithium ions, thereby enhancing discharge capacity and overall energy density.High voltage operation represents deep lithium-ion de-intercalation and sufficient redox reactions of the active transition metal, which cause chemical instability for the interface and bulk structure.12–14 The extraction of massive lithium ions would induce the generation of lithium vacancies followed by the transfer of transition metal ions to lithium sites, further aggravating cation disorder.15–18 In addition, the deepened delithiation reaction would intensify the volume expansion and contraction of the layered structure.19,20 More importantly, the deep redox reaction could generate more highly reactive and unstable Ni4+, which tends to obtain electrons to form Ni2+, especially at the surface of the cathode materials. This behavior would induce the release of lattice oxygen, resulting in a phase transition from layered to spinel and then to a rock salt phase.20–23 What's more, oxygen loss gradually penetrates the bulk of active particles, destroying the structural stability of the crystal. A similar phenomenon happened at high-temperature operation; faster lithiation/delithiation reactions and chaotic mass transfer at the electrode/electrolyte interface would also amplify the possibility of parasitic side reactions,24 gas release and local heating runaway.25,26 Hence, it is urgent to develop a rational and feasible strategy to enhance the bulk and interfacial stability, improving the cycling stability and rate capability of the cathode.
Since structure collapse always occurs at the crystal surface during high voltage operation, reported strategies, such as lattice doping and surface coating, are expected to retain the integrity of the cathode. In the former situation, exotic atoms, including Al, Ti, Nb, W, Mo, etc., are likely to be introduced to the crystal lattice near the particle's surface, promoting the structure robustness of the material during extended cycles.27–30 Lately, a family of W-doped Ni-rich cathodes has displayed improved cycle performance and thermal and structural stability.19,21,23,31–34 However, introducing electrochemically inert elements would cause capacity reduction. Although high-valent dopants could effectively trap labile oxygen in the layered structure, the introduction of some high-valent dopants also induces harmful cation mixing near the surface, which is detrimental for Li+ exchange.19,31,35 Thus, rational design should be considered to reduce capacity loss and alleviate harmful cation mixing caused by high-valent dopants. The latter surface coating with good mechanical strength could dramatically improve the interface properties of layered oxides by avoiding direct contact between the electrode and electrolyte. Except for mechanic strength, another critical factor is the limited Li+ transfer rate across the interface induced by coatings.36 Following this trend, a fast lithium-ion conductor tends to be selected owing to its high lithium-ion transfer coefficient.37–39 For the mechanism of lattice doping and surface coating in the enhancement of Li+ lithiation/delithiation, it's not difficult to see that most lattice doping works on the bulk, while surface coating places special emphasis on the surface/interface characters, whereas, the discrepancy between lattice doping and surface coating is not clear enough. Following this trend, the surface coating and doping integrated strategy is an ideal choice to save layered oxides from predicament. Considering the complex synthesis of layered cathodes and surface reactivity of layered oxides with moisture/air, easily scalable routes should be taken into consideration. In this context, all in one design including gradient doping and surface chemistry modulating should be adopted to enhance the electrochemical performance of Ni/Mn based layered oxides.
Therein, we designed simultaneous gradient W doped and Li2WO4-coated 0.98LiNi0.5Mn0.5O2·0.02Li2WO4 cathodes by modulating the content of the exotic dopant and stoichiometric lithium salt during calcination. Pristine LiNi0.5Mn0.5O2, W doped LiNi0.5Mn0.5O2 and WO3 coated LiNi0.5Mn0.5O2 cathodes were prepared as references to reveal the working mechanism of the 0.98LiNi0.5Mn0.5O2·0.02Li2WO4 cathode. Compared with W doped or WO3 coated cathodes without a Li-enriched phase, the 0.98LiNi0.5Mn0.5O2·0.02Li2WO4 cathode with a gradient W doped lattice and uniform Li2WO4 coated surface demonstrates superior electrochemical performance. This work resolves the two knotty issues of crystal stability and interfacial stability of high-voltage cathodes and emphasises the significance of a slightly Li-enriched surface/interface for high-energy cathodes.
2. Results and discussion
A series of (1 − x)LiNi0.5Mn0.5O2·xLi2WO4 (x = 0, 0.01, 0.02, 0.03, 0.05; denoted as NM50, NM50-LW1, NM50-LW2, NM50-LW3, and NM50-LW5) cathodes, along with LiNi0.49Mn0.49W0.02O2 (NM50-W2) and 0.98LiNi0.5Mn0.5O2·0.02WO3 (NM50-WO2) cathodes, were synthesized by a high-temperature solid-state method using a co-precipitated Ni0.5Mn0.5(OH)2 precursor. As shown in Fig. S1,† the Ni0.5Mn0.5(OH)2 precursor and subsequent calcined cathodes consist of spherical secondary particles composed of agglomerated lamellar primary particles. When x exceeds 0.03, the coating layer becomes visible on the surface of particles.X-ray diffraction (XRD) patterns of NM50, NM-W2, NM50-LW1, NM50-LW2, NM50-LW3, NM50-LW5 and NM-WO2 show that all peaks correspond to the layered α-NaFeO2 structure with a space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (Fig. 1a and S2†).40,41 Notably, the peak positions of (003) and (104) reflections gradually shift to lower angles and Li2WO4 peaks become stronger as the amount of Li2WO4 increases, indicating the bigger d spacing for (003)/(104) and the formation of Li2WO4 in the modified cathodes. Additionally, the shift of the (003) peak for NM50-W2, NM50-LW2 and NM50-WO2 to lower angles indicates that the (003) layer spacing of the modified cathodes is wider than that of NM50 (Fig. S3†). XRD Rietveld refinement and the corresponding lattice parameters are presented in Fig. 1b, c, S4 and Table S1.† Considering the relatively larger radius of W6+ (0.60 Å for W6+vs. 0.56 Å for Ni3+ and 0.53 Å for Mn4+), the increase of the c-axis for the W modified cathodes suggests the successful incorporation of W into the lattice of the crystal.34,42 Moreover, antisite defects where Ni atoms occupy Li sites were determined to be 8.6%, 8.1%, 7.3%, and 8.2% for NM50, NM50-W2, NM50-LW2, and NM50-WO2, respectively. And the cation mixing of NM50-LW1, NM50-LW2, NM50-LW3, and NM50-LW5 is lower than that of NM50, indicating the positive effect of Li2WO4 on the ordered layered structure.19,29
m (Fig. 1a and S2†).40,41 Notably, the peak positions of (003) and (104) reflections gradually shift to lower angles and Li2WO4 peaks become stronger as the amount of Li2WO4 increases, indicating the bigger d spacing for (003)/(104) and the formation of Li2WO4 in the modified cathodes. Additionally, the shift of the (003) peak for NM50-W2, NM50-LW2 and NM50-WO2 to lower angles indicates that the (003) layer spacing of the modified cathodes is wider than that of NM50 (Fig. S3†). XRD Rietveld refinement and the corresponding lattice parameters are presented in Fig. 1b, c, S4 and Table S1.† Considering the relatively larger radius of W6+ (0.60 Å for W6+vs. 0.56 Å for Ni3+ and 0.53 Å for Mn4+), the increase of the c-axis for the W modified cathodes suggests the successful incorporation of W into the lattice of the crystal.34,42 Moreover, antisite defects where Ni atoms occupy Li sites were determined to be 8.6%, 8.1%, 7.3%, and 8.2% for NM50, NM50-W2, NM50-LW2, and NM50-WO2, respectively. And the cation mixing of NM50-LW1, NM50-LW2, NM50-LW3, and NM50-LW5 is lower than that of NM50, indicating the positive effect of Li2WO4 on the ordered layered structure.19,29
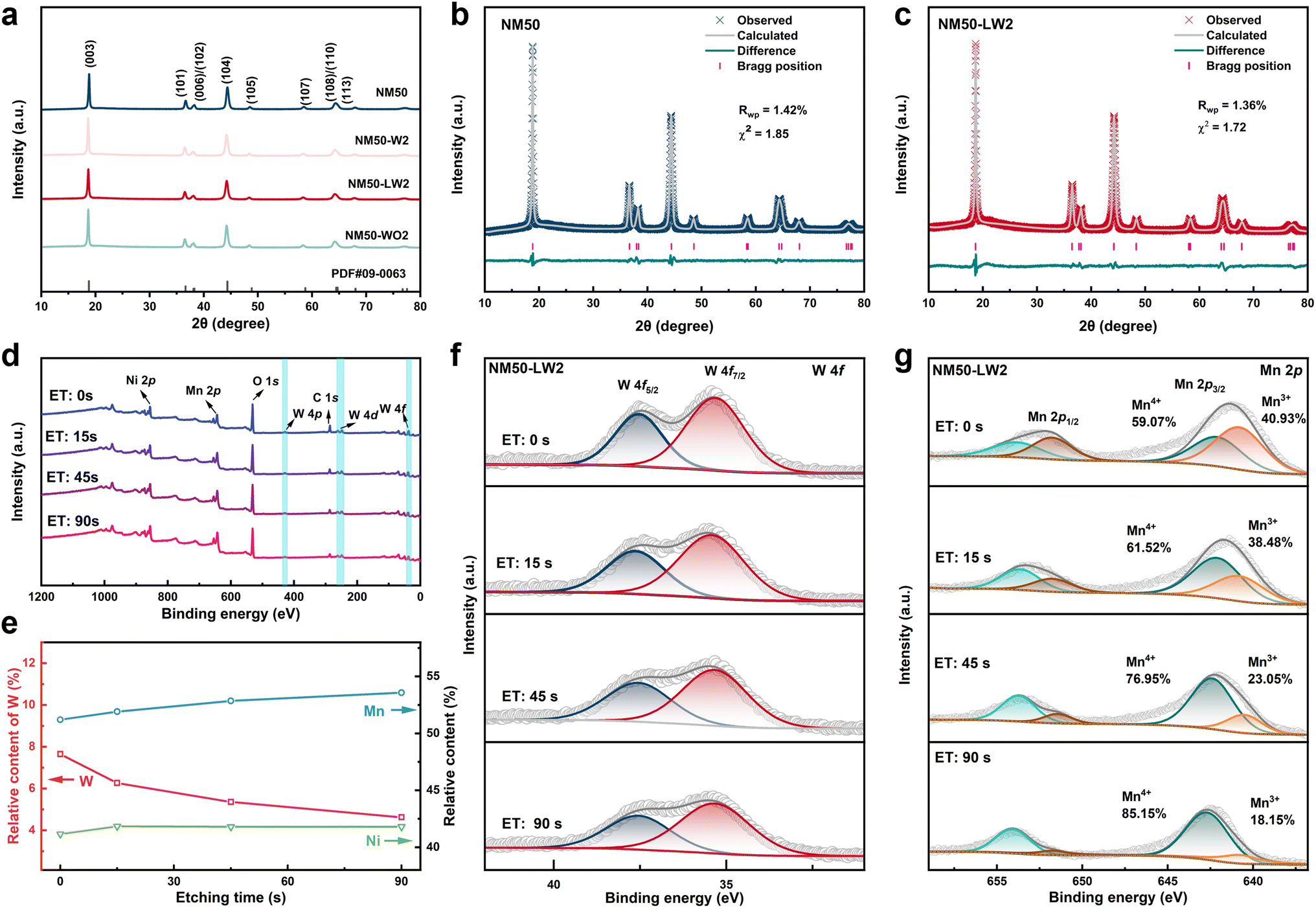 | ||
| Fig. 1 (a) XRD pattern of NM50, NM50-W2, NM50-LW2, and NM50-WO2; (b and c) Rietveld refinements of the XRD pattern of NM50 (b) and NM50-LW2 (c). (d) XPS full spectrum of NM50-LW2 obtained with different etching times; (e) relative content of Ni, Mn and W elements obtained by XPS with different etching times; (f and g) XPS depth profiles of W 4f (f) and Mn 2p (g) with different etching times. | ||
X-ray photoelectron spectroscopy (XPS) was utilized to investigate the valence states and relative content of the elements in NM50 and NM50-LW2 (Fig. 1d–g and S5†). Compared with pristine NM50, NM50-LW2 exhibits a reduced content of surface oxygen and a higher content of lattice oxygen (Fig. S5b and c†), which can be attributed to the stabilized active surface by the Li2WO4 layer. Additionally, the peak area of C–O–C and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O for NM50-LW2 is smaller than that for NM50, suggesting reduced residual Li compounds (such as Li2CO3) after Li2WO4 modification. Compared with the absence of the W 4f signal for NM50, an obvious W 4f signal can still be detected for NM50-LW2 after etching for 90 s (Fig. 1f and S5d†). The shift of Ni 2p and Mn 2p core levels to lower binding energy indicates that the content of Mn3+ and Ni2+ in NM50-LW2 is higher than that in NM50 (Fig. S5e and f†), which is due to the charge compensation induced by W6+ gradient doping.43 According to the relative content of Ni, Mn and W obtained after different etching times, the W content gradually decreases, while the Mn and Ni content increases from the surface to the inside, confirming the gradient distribution of W inside the crystal (Fig. 1e and f). Furthermore, the Mn 2p peak gradually shifts towards higher binding energy (Fig. 1g), suggesting a progressive increase in the valence state of Mn from the surface to the bulk, which further verifies the gradient distribution of W6+ from the surface to the bulk structure.
O for NM50-LW2 is smaller than that for NM50, suggesting reduced residual Li compounds (such as Li2CO3) after Li2WO4 modification. Compared with the absence of the W 4f signal for NM50, an obvious W 4f signal can still be detected for NM50-LW2 after etching for 90 s (Fig. 1f and S5d†). The shift of Ni 2p and Mn 2p core levels to lower binding energy indicates that the content of Mn3+ and Ni2+ in NM50-LW2 is higher than that in NM50 (Fig. S5e and f†), which is due to the charge compensation induced by W6+ gradient doping.43 According to the relative content of Ni, Mn and W obtained after different etching times, the W content gradually decreases, while the Mn and Ni content increases from the surface to the inside, confirming the gradient distribution of W inside the crystal (Fig. 1e and f). Furthermore, the Mn 2p peak gradually shifts towards higher binding energy (Fig. 1g), suggesting a progressive increase in the valence state of Mn from the surface to the bulk, which further verifies the gradient distribution of W6+ from the surface to the bulk structure.
High angle annular dark field scanning transmission electron microscopy (HAADF-STEM) was employed to characterize the microstructure of NM50 and NM50-LW2. As shown in Fig. 2a, a continuous phase transition from the ordered layered phase to the disordered rock salt phase can be observed from the inside to the surface region of NM50. The transition metal layers are distinctly discernible inside NM50, with a layer spacing of d = 4.73 Å, corresponding to the (003) crystal plane of the layered phase. Fast Fourier transform (FFT) analysis confirms that this region belongs to the Rm space group (Fig. 2a(I)). In the subsurface region, a defective spinel-like structure, with transition metals occupying lithium sites gradually emerges (Fig. 2a(II)). Ultimately, the phase in the surface region transforms into a disordered Fmm rock salt structure (Fig. 2a(III)). In contrast, NM50-LW2 exhibits a phase transition from the ordered layered phase to a slightly disordered layered structure, and a coating layer approximately 5 nm can be observed in the outer surface region (Fig. 2b). The bulk phase of NM50-LW2 demonstrates a clear layered structure, with the layer spacing d = 4.78 Å, which corresponds to the (003) crystal plane of the layered phase (Fig. 2b(I)). The larger layer spacing of NM50-LW2 than that of NM50 could be attributed to the incorporation of larger W6+ into the lattice, which generates wider channels for rapid Li+ diffusion and ensures structural stability during repeated Li+ insertion/extraction.44–50 Compared with NM50, a significant alleviation of Li+/Ni2+ cation mixing could be observed for the subsurface region of NM50-LW2 (Fig. 2b(II)). This improvement is possibly due to the Li-enriched conditions, which reduce the lithium vacancies near the surface region and inhibit migration of Ni2+ to the lithium layer in the corresponding region. Fig. 2b(III) provides a magnified view of the outer surface region of NM50-LW2. Obviously, there exists a hetero-epitaxially grown coating layer with a distinguishing layer spacing of d = 2.21 Å, which corresponds to the (152) crystal plane of Li2WO4, further confirming the successful construction and integration of Li2WO4. Thus, it can be concluded that the phase of NM50-LW2 transitions from a W doped layered phase to a spinel phase, then to a rock salt phase and finally to a Li2WO4 compound from the bulk to the surface (Fig. 2c).
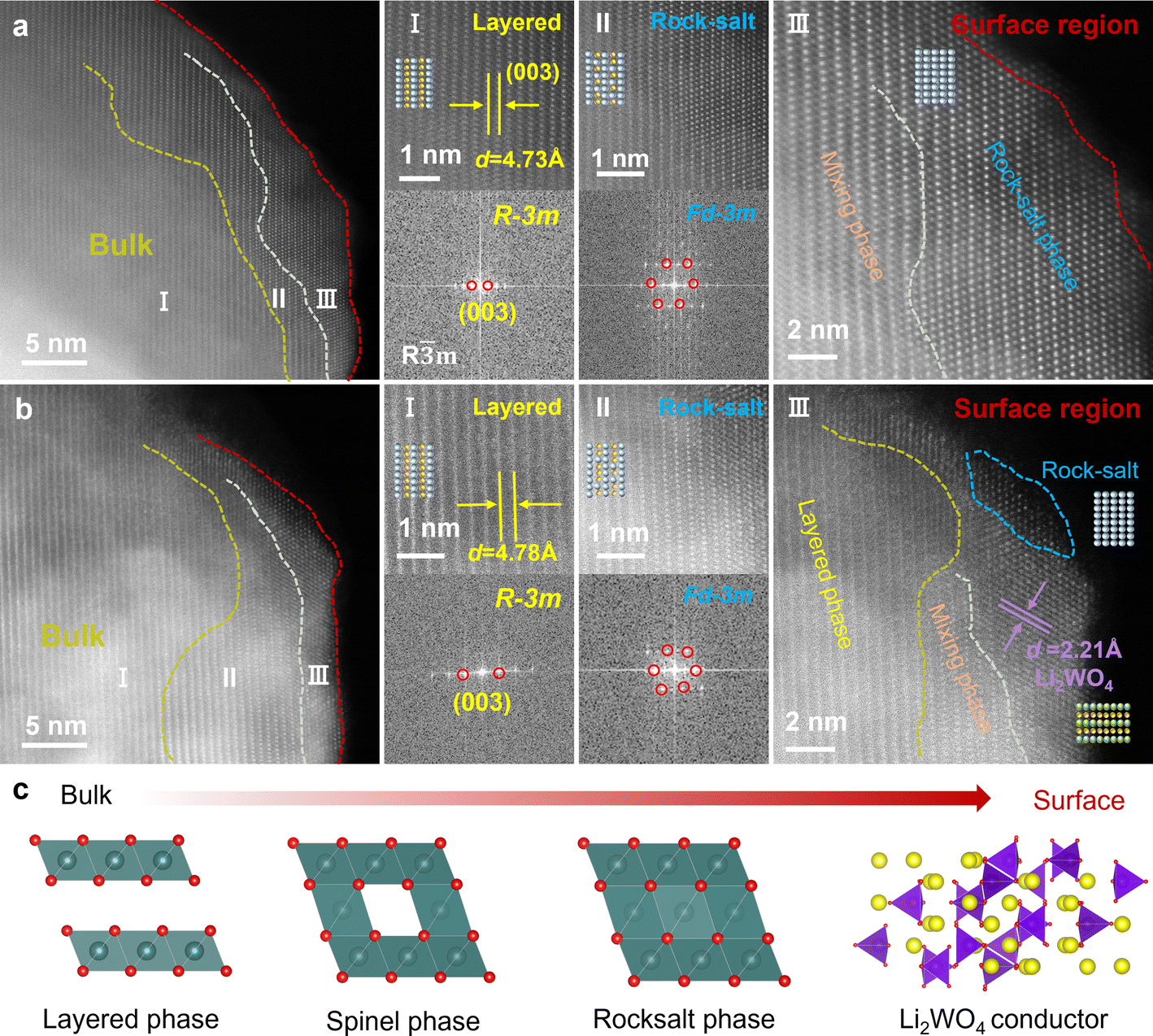 | ||
| Fig. 2 (a and b) High-angle annular dark-field (HAADF) and fast Fourier transform (FFT) images of NM50 (a) and NM50-LW2 (b). (c) Schematic diagram of the phase transitions for NM50-LW2 from the bulk to the surface. | ||
The electrochemical performance of the cathodes was evaluated using a half cell in the potential range of 2.7–4.5 V at 30 °C using galvanostatic charging/discharging (GCD) tests with 1C defined as 170 mA g−1. As shown in Fig. S6,† from the consideration of discharge capacity, rate capacity and cycle life, NM50-LW2 shows relatively superior electrochemical performance among the cathodes (1 − x)LiNi0.5Mn0.5O2·xLi2WO4 (x = 0, 0.01, 0.02, 0.03, 0.05) with various Li2WO4 contents. Obviously, excessive Li2WO4 would hinder the insertion/extraction of Li+ and goes against the electrochemical performance. In addition, as shown in Fig. 3a, NM50-LW2 also exhibits a much higher initial discharge capacity (199.17 mA h g−1) and initial coulombic efficiency (95.32%) than NM50 (196.97 mA h g−1 and 90.72%), NM50-W2 (191.27 mA h g−1 and 91.64%), and NM-WO2 (195.08 mA h g−1 and 93.54%). As shown in Fig. 3b, NM50-LW2 shows much improved rate performance than NM50, NM50-W2, and NM-WO2. At 5C, a high discharge capacity of 156.5 mA h g−1 can still be obtained for NM50-LW2, which outperformed 113.3 mA h g−1 for NM50, 137.6 mA h g−1 for NM50-W2, and 152.1 mA h g−1 for NM50-WO2. As shown in Fig. 3c, NM50-LW2 exhibits a superior capacity retention of 92.88% and a discharge capacity of 170.8 mA h g−1 after 300 cycles at 1C in the potential range of 2.7–4.5 V, which outperformed 87.01% and 152.3 mA h g−1 for NM50, 91.67% and 162.7 mA h g−1 for NM50-W2, and 91.84% and 165.7 mA h g−1 for NM50-WO2. When cycled at a high temperature of 55 °C, NM50-LW2 still possesses a capacity retention of 91.44% after 300 cycles (Fig. 3d), which is higher than 85.10% for NM50, 88.59% for NM50-W2 and 86.07% for NM50-WO2. Moreover, when cycled in the potential range of 2.7–4.6 V at 1C and 30 °C (Fig. 3e), NM50-LW2 can still exhibit a capacity retention of 91.15% after 200 cycles, which is higher than 88.39% for NM50, 90.81% for NM50-W2 and 90.05% for NM50-WO2. To further verify the excellent electrochemical performance induced by the slightly Li-enriched chemistry, the cycle performance of NM50-LW2 and NM50 was also measured using a full cell with graphite as the anode in the voltage range of 2.7–4.5 V at 1C and 30 °C. As shown in Fig. 3f, after 500 cycles, NM50-LW2 still exhibits a capacity of 169.4 mA h g−1, with a superior capacity retention of 97.47%, confirming the excellent cycle stability provided by the slightly Li-enriched chemistry. However, NM50 only exhibits a capacity of 118.9 mA h g−1, with a relatively lower capacity retention of 69.41%, which is significantly lower than that exhibited by NM50-LW2.
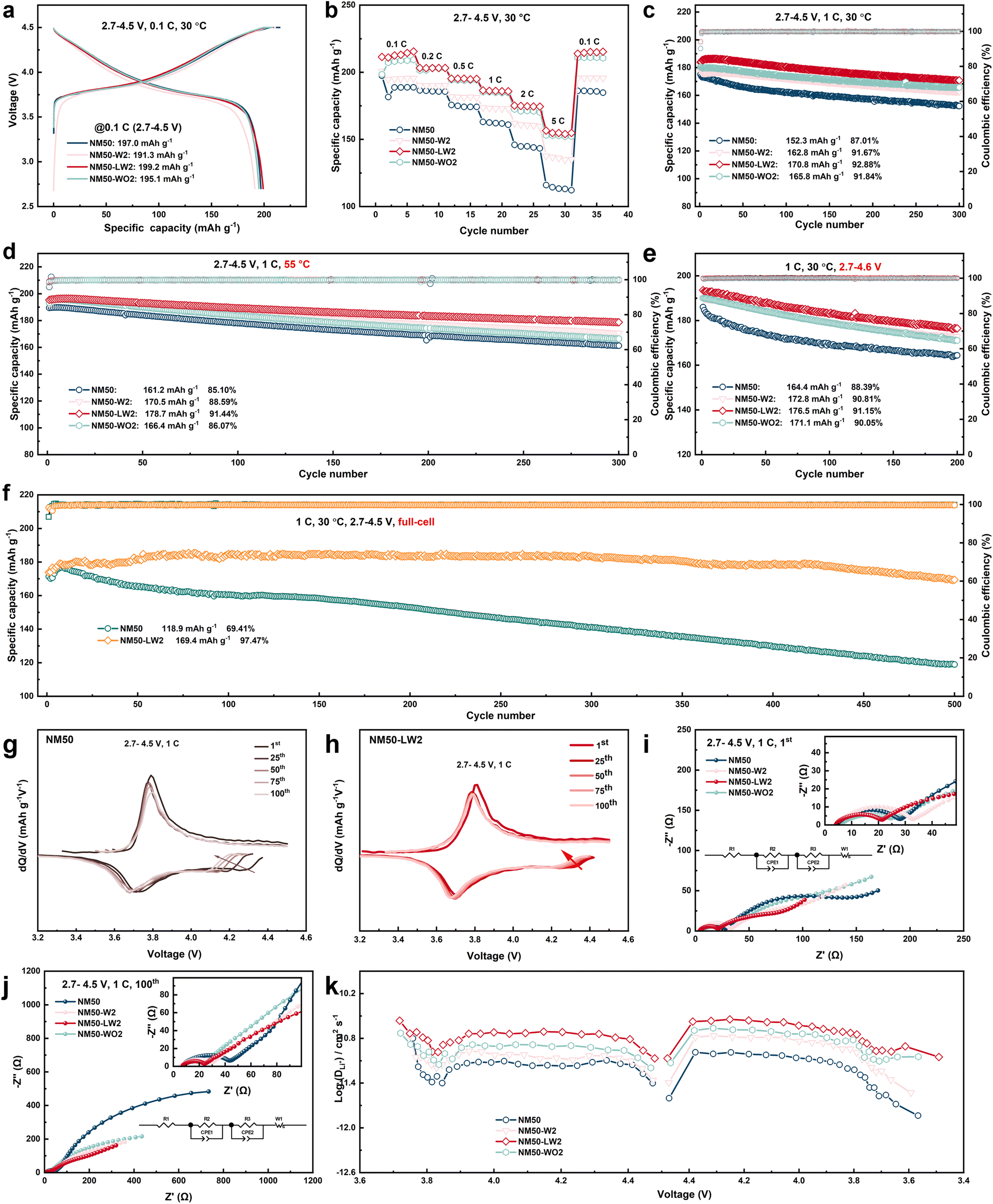 | ||
| Fig. 3 (a–e) The initial charging/discharging curve at 0.1C (a), rate capacity (b), and cycling performance of NM50, NM50-W2, NM50-LW2 and NM50-WO2 at 1C in the potential range of 2.7–4.5 V at 30 °C (c), at 1C in the potential range of 2.7–4.5 V at 55 °C (d), and at 1C in the potential range of 2.7–4.6 V at 30 °C (e). (f) Cycle performance of NM50-LW2 and NM50 in a full cell in the voltage range of 2.7–4.5 V at 1C and 30 °C. (g and h) The dQ/dV curves of NM50 (g) and NM50-LW2 (h). (i and j) Nyquist plots of NM50, NM-W2, NM50-LW2 and NM50-WO2 after one cycle (h) and 100 cycles (i); (k) Li+ diffusion coefficient of NM50, NM50-W2, NM50-LW2 and NM50-WO2 during the charge and discharge process. | ||
A comparison of the electrochemical performance of the cathodes reveals that both W doping and the slightly Li-enriched conditions are crucial for designing stable Ni/Mn layered oxides. Although 2 mol% WO3 coating and W doping both can enhance the cycling stability of the cathode, 2 mol% WO3 coating appears more effective for the enhancement of performance than W doping under mild conditions. However, under extreme conditions, the enhanced structural stability from lattice doping becomes more significant for long-term cycling. In our previous work, we also verified that surface stability is more critical than bulk stress-induced microcracks for long-life cycling in nickel-rich cathodes.51 Modification simply by doping can enhance the structure stability of LiNi0.5Mn0.5O2 but it doesn't create a stable surface to prevent electrolyte corrosion and metal dissolution. An inert WO3 coating layer can act as a robust barrier to isolate the cathode from the electrolyte but does not modify bulk structural properties. However, direct WO3 coating with a short calcination duration can induce lithium-deficient phases in the surface region due to the combination of W and Li+ to form LixWyOz compounds, leading to capacity attenuation.52 By modulating the content of the exotic dopant and stoichiometric lithium salt during lithiation calcination, the surface-enriched W element combines with Li+ to produce Li2WO4 compounds, forming a thin Li+ conductor layer and a slightly Li-enriched chemistry in the subsurface region, endowing NM50-LW2 with a robust bulk lattice and surface region as well as superior cycle stability.
The dQ/dV curves of the cathodes (Fig. 3g, h and S7†) across various cycles show a gradual shift of the oxidation peak to higher potential and a slight shift of the reduction peak to lower potential, indicating increased potential polarization due to structural degradation and surface-interface deterioration. Among these, NM50-LW2 demonstrates the smallest change, indicating the enhanced phase transition reversibility and stability introduced by Li2WO4. Moreover, the excellent structure stability of NM50-LW2 could be verified by the disparity in charge/discharge median voltage. As plotted in Fig. S8,† after 200 cycles NM50-LW2 only shows a small value of 0.189 V for the disparity in charge/discharge median voltage, which is far less than 0.241 V for NM50 and 0.201 V for NM50-W2. Nyquist plots of the cathodes after one cycle and 100 cycles were obtained and fitted to reveal the resistance evolution (Fig. 3i, j, S8b, S9 and Table S2†). Each curve consists of two semicircles and a slant line. The first semicircle in the high-frequency region, the second semicircle in the middle-frequency region, and the slant line in the low-frequency region represent the interface impedance (Rsf), charge transfer resistance (Rct), and Warburg impedance, respectively.53 Among these electrodes, NM50 shows the highest Rsf and Rct values (42.81 and 528.6 Ω) after 100 cycles, followed by NM50-W2 (28.69 and 207.8 Ω), NM50-WO2 (26.92 and 241.6 Ω), and NM50-LW2 (24.88 and 184.7 Ω). The lowest Rsf and Rct values for NM50-LW2 can be attributed to the surface Li2WO4 coating and the enlarged layer spacing in the layered bulk. The galvanostatic intermittent titration technique (GITT) was used to examine Li+ diffusion kinetics during lithiation/delithiation. Obviously, the average lithium diffusion rate of NM50-LW2 during the charging/discharging process is significantly higher than that of NM50, NM50-W2, and NM50-WO2, indicating that the modification facilitates quick Li+ migration (Fig. 3k).
To identify the structural evolution of NM50 and NM50-LW2 during the first cycle, in situ XRD was performed (Fig. 4). The (003) peak of NM50 and NM50-LW2 cathodes shifts to lower angles when charged from 2.7 to 4.3 V, labeling a gradual c-axis expansion. This can be attributed to a weakened shielding effect of the Li layer, which increases the repulsion between layers and subsequently enlarges the space for the Li–O layer.54 At around 4.3 V, as more lithium-ions are extracted from the lattice, the charge is equalized by oxidizing TM ions, leading to a sharp shrink in the TM–O layer spacing, that is the main reason for particle pulverization and microcrack generation.12,55 Additionally, NM50-LW2 almost completely returns to its original 2θ position after discharging, while NM50 shows a smaller offset. The maximum angular displacement of the (003) peak in NM50 (0.64°) is wider than that in NM50-LW2 (0.32°), suggesting that Li2WO4 surface modification and W-doping can effectively alleviate volume strain. The calculated lattice parameters from in situ XRD refinement, as a function of the charging state, show that the maximum c-axis shrinkage (Δc) of NM50 (2.61%) is higher than that of NM50-LW2 (1.79%). Additionally, the a-axis shrinkage degree (Δa) of NM50 and NM50-LW2 is 6.73% and 5.95%, respectively, further demonstrating that NM50-LW2 experiences less lattice distortion.56,57 The smaller pronounced lattice changes of NM50-LW2 demonstrate that the phase transition reversibility and stability are enhanced after introducing Li2WO4.
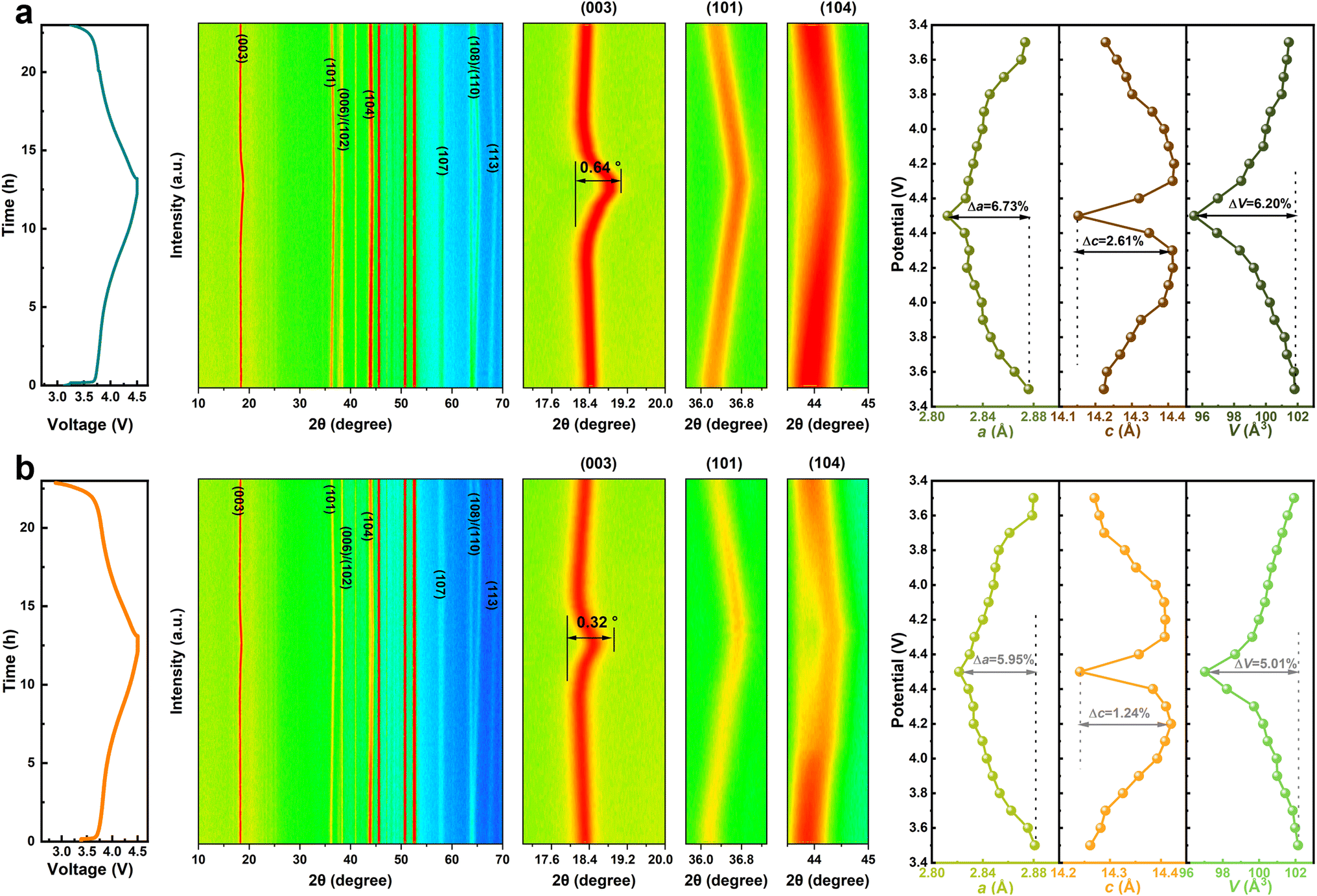 | ||
| Fig. 4 GCD curve (left), in situ XRD patterns, selected enlarged peak (medium) and corresponding variation of cell parameters (right) for NM50 (a) and NM50-LW2 (b). | ||
In situ differential electrochemical mass spectrometry (DEMS) was utilized to monitor the internal gas generation during the charging process (Fig. 5a and b). Without any modification, pristine NM50 releases O2 and CO2 gases after charging to 4.3 V. In contrast, the emission of O2 and CO2 gases was entirely restrained in NM50-LW2. The thermal stability of the fully charged NM50 and NM50-LW2 electrodes was also examined using differential scanning calorimetry (DSC) (Fig. 5c). The NM50-LW2 electrode exhibited a higher exothermic peak at 307.1 °C and a lower heat release of 71.38 J g−1, which are superior to 283.5 °C and 131.5 J g−1 exhibited by NM50. This indicates the reinforced stability provided by the surface lithium-rich Li2WO4 coating and gradient W-doped bulk and is consistent with the improved cycling performance at high temperature. Subsequently, XPS analysis of NM50 and NM50-LW2 after 300 cycles was conducted to gain further insights into the chemical composition of the cathode–electrolyte interface (CEI) layer near the cathode surface. In the O 1s spectrum (Fig. 5d), compared with NM50-LW2, the area of the ROCO2Li peak in NM50 is larger, suggesting that more severe electrolyte decomposition occurred in NM50. Notably, the TM–O peak is well-detected in NM50-LW2 but absent in NM50, indicating that a thinner CEI film forms on the surface of NM50-LW2. In addition, as shown in Fig. 5e, the content of LiF in NM50 is significantly higher than that in NM50-LW2. Moreover, the C–O and CO peaks in NM50-LW2 are smaller than those in NM50 (Fig. 5f), further demonstrating that the surface Li2WO4 coating and W-doped bulk inhibit side reactions.
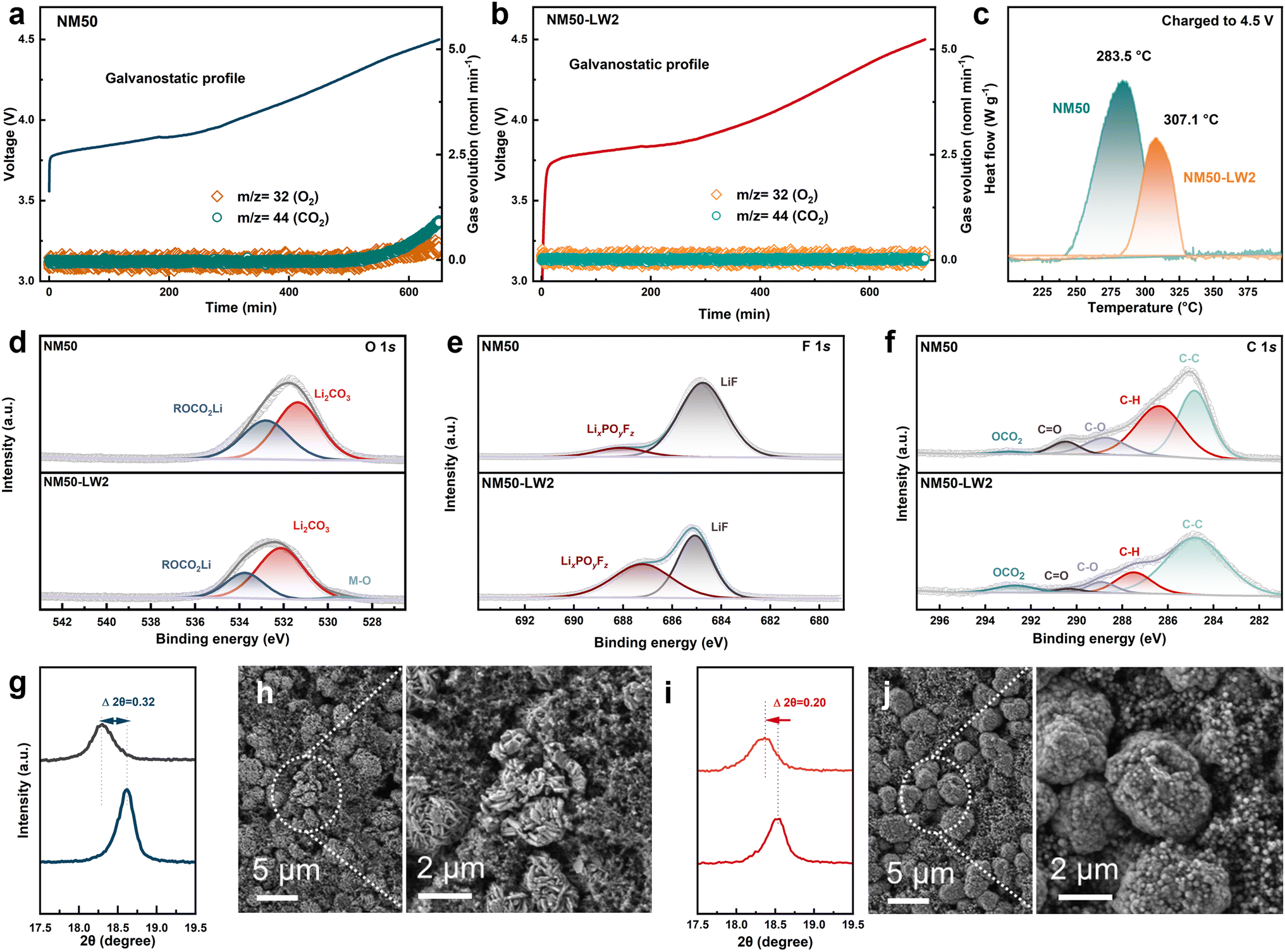 | ||
| Fig. 5 (a and b) In situ DEMS measurements of NM50 (a) and NM50-LW2 (b). (c) DSC measurements of NM50 and NM50-LW2 charged to 4.5 V. (d–f) XPS profiles of O 1s (d), F 1s (e), and C 1s (f) for NM50 and NM50-LW2 cathodes after 300 cycles. (g–j) Enlarged (003) reflections and SEM images of NM50 (g and h) and NM50-LW2 (i and j) after 300 cycles. | ||
The XRD patterns and SEM of the cycled electrodes were obtained to uncover the phase and morphology evolution of the cathodes during extended cycling. The XRD results (Fig. 5g and i) show that the (003) peak of NM50 exhibits a pronounced left shift with an offset of 0.32°, likely attributed to Li loss and corresponding phase degradation. In comparison, the shift magnitude of the (003) peak in NM50-LW2 (0.20°) is narrower, indicating a well-maintained layered structure. Within the repetitive charge–discharge process, the accumulation of anisotropic contraction/expansion in lattice parameters would induce breakage for secondary particles to buffer the volume strain. However, this breakage exposes more active surfaces to electrolyte, exacerbating side reactions and capacity fading.58 SEM images of NM50 and NM50-LW2 after 300 cycles demonstrate that NM50 has obvious microcracks, whereas NM50-LW2 basically maintains its sphere-like morphology without apparent microcracks (Fig. 5h and j). Therefore, the surface lithium-rich Li2WO4 coating and gradient doping integrated strategy strengthens the interface robustness and mitigates the strain aggregation. These results further support the surface Li-enriched Li2WO4 coating and gradient W-doping strategy could efficaciously alleviate structural degradation during long cycling.
In short, all W-modified materials demonstrate superior electrochemical performance compared to pristine LiNi0.5Mn0.5O2. Specifically, 0.98LiNi0.5Mn0.5O2·0.02Li2WO4 exhibits outstanding electrochemical performance under extreme conditions (Table S3†). The enhanced electrochemical performance of W-modified cathodes indicates that both lattice doping and surface coating are essential for improving the cycling stability and maintaining the bulk integrity of Ni/Mn based layered oxides. Although the direct introduction of WO3 coating or a W dopant during lithiation calcination can create W-rich surface regions on the surface of primary grains, the surface phase usually contains Li–W–O compounds and Li-poor regions due to the lack of stoichiometric Li sources. The surface lithium-deficient phase due to the lithium despoilment induced by W would inevitably intensify cation disordering and relatively inferior regions (Fig. 6a and b). In contrast, benefiting from the modulated content of the W dopant and stoichiometric lithium salt during lithiation calcination, a W-doped lattice and a Li-enriched surface region induced by hetero-epitaxially grown Li2WO4 would result in reduced cation disordering and enhanced structure stability during extended cycles (Fig. 6c). Kinetics measurements and structural evolution demonstrate that the Li-enriched surface and gradient doping strategy significantly facilitate lithium-ion conduction, alleviate strain accumulation, reduce parasitic side reactions from electrolyte decomposition, and improve the lattice O stability in high cut-off voltage and high temperature operation.
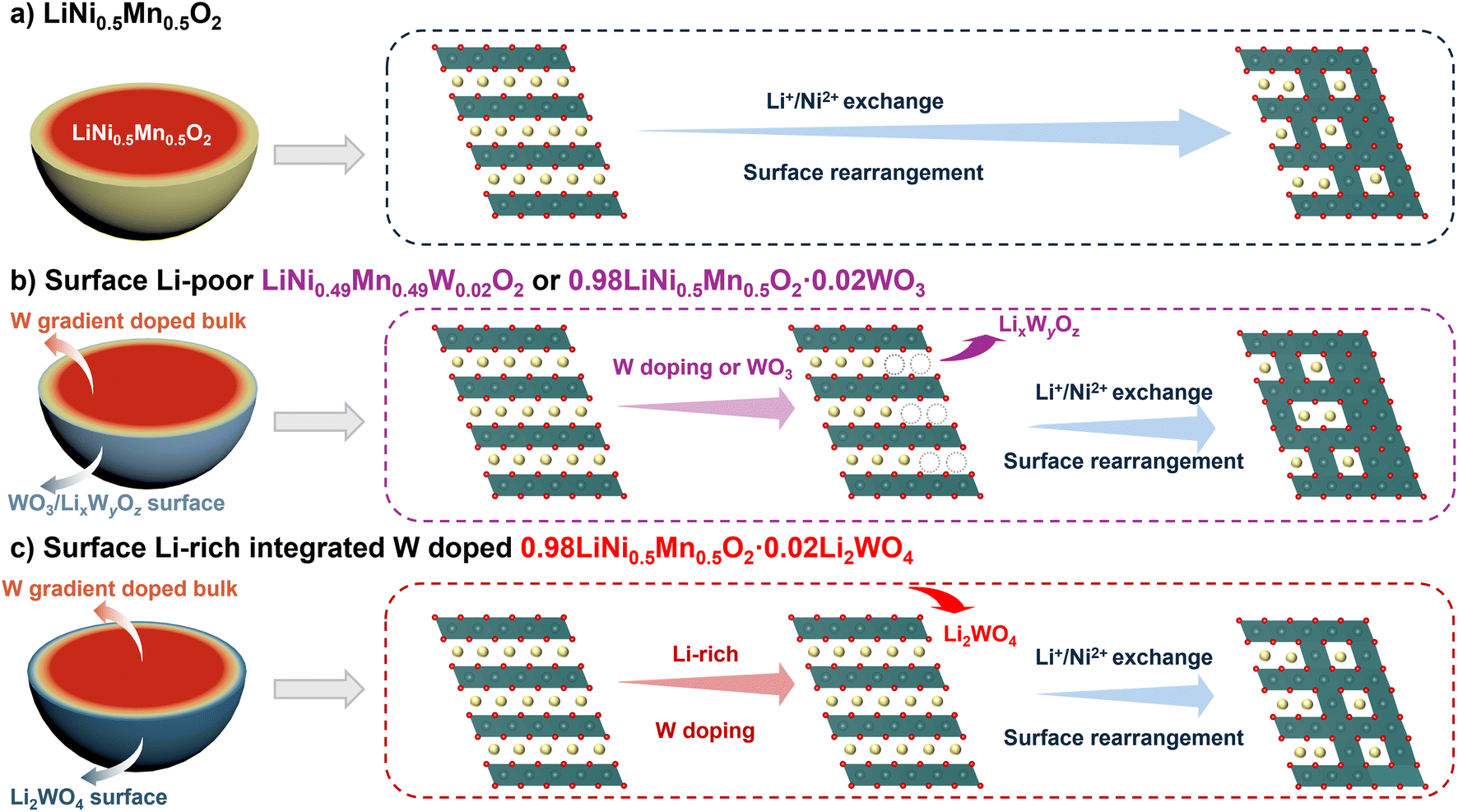 | ||
| Fig. 6 Schematic diagram for the mechanism of LiNi0.5Mn0.5O2 (a), surface Li-poor LiNi0.49Mn0.49W0.02O2 or 0.98LiNi0.5Mn0.5O2·0.02WO3 (b) and surface Li-enriched 0.98LiNi0.5Mn0.5O2·0.02Li2WO4 (c) during delithiation. | ||
3. Conclusions
In summary, a series of cathodes including LiNi0.5Mn0.5O2, (1 − x)LiNi0.5Mn0.5O2·xLi2WO4 (x = 0, 0.01, 0.02, 0.03, 0.05), LiNi0.49Mn0.49W0.02O2, and 0.98LiNi0.5Mn0.5O2·0.02WO3 were synthesized to investigate the role of Li-enriched conditions in the performance of Ni–Mn binary cathodes in high voltage/high temperature operation. Compared with lithium-deficient conditions induced by W doping and WO3 coating, the combination of a slightly Li-enriched Li2WO4 surface coating and gradient W-doping strategy can significantly enhance the performance of the 0.98LiNi0.5Mn0.5O2·0.02Li2WO4 cathode. Specifically, the 0.98LiNi0.5Mn0.5O2·0.02Li2WO4 cathode exhibits outstanding cycling performance even under harsh conditions, retaining 91.15% capacity after 200 cycles at 1C with a cut-off voltage of 4.6 V and maintaining 91.44% capacity after 300 cycles at elevated temperature. This study offers a scalable approach to design high-energy-density and high-safety cathodes for next generation lithium-ion batteries.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Siqi Chen: characterizations, investigation, formal analysis, writing – review & editing. Ping Zhang: characterizations. Xin Zhou: characterizations, investigation. Wenbin Wu: characterizations, investigation. Xiaohong Liu: editing, formal analysis. Yifeng Liu: editing. Guilin Feng: editing. Bin Zhang: supervision, resources. Wangyan Xing: supervision, resources. Meihua Zuo: supervision, resources. Ping Zhang: supervision, resources. Genpin Lv: resources. Yao Xiao: editing, formal analysis. Shixue Dou: resources. Wei Xiang: funding acquisition, supervision, investigation, resources, writing – review & editing, conceptualization, formal analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by a project from the National Natural Science Foundation of China (21805018), the Sichuan Science and Technology Program (2022ZHCG0018, 2023NSFSC0117, and 2023ZHCG0060), the Yibin Science and Technology Program (2022JB005), TianfuYongxing Laboratory Organized Research Project Funding (2023KJGG13) and the project funded by the China Postdoctoral Science Foundation (2022M722704). Specifically, we thank Ceshigo (https://www.ceshigo.com/) and Zhinanzhen (http://www.shiyanjia.com/) for characterization measurements.References
- J. T. Frith, M. J. Lacey and U. Ulissi, Nat. Commun., 2023, 14, 420 CrossRef CAS PubMed.
- J. Xiao, F. Shi, T. Glossmann, C. Burnett and Z. Liu, Nat. Energy, 2023, 8, 329–339 CrossRef.
- S.-T. Myung, F. Maglia, K.-J. Park, C. S. Yoon, P. Lamp, S.-J. Kim and Y.-K. Sun, ACS Energy Lett., 2017, 2, 196–223 CrossRef CAS.
- J. Xu, F. Lin, M. M. Doeff and W. Tong, J. Mater. Chem. A, 2017, 5, 874–901 RSC.
- E. M. Erickson, F. Schipper, T. R. Penki, J.-Y. Shin, C. Erk, F.-F. Chesneau, B. Markovsky and D. Aurbach, J. Electrochem. Soc., 2017, 164, A6341 CrossRef CAS.
- T. C. Liu, L. Yu, J. J. Liu, J. Lu, X. X. Bi, A. Dai, M. Li, M. F. Li, Z. X. Hu, L. Ma, D. Luo, J. X. Zheng, T. P. Wu, Y. Ren, J. G. Wen, F. Pan and K. Amine, Nat. Energy, 2021, 6, 277–286 CrossRef CAS.
- W. D. Li, S. Lee and A. Manthiram, Adv. Mater., 2020, 32, 2002718 CrossRef CAS PubMed.
- W. E. Gent, G. M. Busse and K. Z. House, Nat. Energy, 2022, 7, 1132–1143 CrossRef CAS.
- Y. Xia, L. Zhou, K. Wang, C. Lu, Z. Xiao, Q. Mao, X. Lu, J. Zhang, H. Huang, Y. Gan, X. He, W. Zhang and X. Xia, J. Solid State Electrochem., 2023, 27, 1363–1372 CrossRef CAS.
- S. Y. Tsai and K. Z. Fung, Molecules, 2023, 28, 794 CrossRef CAS PubMed.
- P. Periasamy and N. Kalaiselvi, J. Power Sources, 2006, 159, 1360–1364 CrossRef CAS.
- S. H. Song, M. Cho, I. Park, J. G. Yoo, K. T. Ko, J. Hong, J. Kim, S. K. Jung, M. Avdeev, S. Ji, S. Lee, J. Bang and H. Kim, Adv. Energy Mater., 2020, 10, 2000521 CrossRef CAS.
- Y. W. Mao, X. L. Wang, S. H. Xia, K. Zhang, C. X. Wei, S. Bak, Z. Shadike, X. J. Liu, Y. Yang, R. Xu, P. Pianetta, S. Ermon, E. Stavitski, K. J. Zhao, Z. R. Xu, F. Lin, X. Q. Yang, E. Y. Hu and Y. J. Liu, Adv. Funct. Mater., 2019, 29, 1900247 CrossRef.
- P. F. Yan, J. M. Zheng, M. Gu, J. Xiao, J. G. Zhang and C. M. Wang, Nat. Commun., 2017, 8, 14101 CrossRef CAS PubMed.
- D. Wang, R. Kou, Y. Ren, C.-J. Sun, H. Zhao, M.-J. Zhang, Y. Li, A. Huq, J. Y. P. Ko, F. Pan, Y.-K. Sun, Y. Yang, K. Amine, J. Bai, Z. Chen and F. Wang, Adv. Mater., 2017, 29, 1606715 CrossRef PubMed.
- C. S. Yoon, H.-H. Ryu, G.-T. Park, J.-H. Kim, K.-H. Kim and Y.-K. Sun, J. Mater. Chem. A, 2018, 6, 4126–4132 RSC.
- W. Li, B. Song and A. Manthiram, Chem. Soc. Rev., 2017, 46, 3006–3059 RSC.
- J. Zheng, Y. Ye, T. Liu, Y. Xiao, C. Wang, F. Wang and F. Pan, Acc. Chem. Res., 2019, 52, 2201–2209 CrossRef CAS PubMed.
- X. Wang, B. Zhang, Z. Xiao, L. Ming, M. Li, L. Cheng and X. Ou, Chin. Chem. Lett., 2023, 34, 10772 Search PubMed.
- D. Rathore, M. Garayt, Y. Liu, C. Geng, M. Johnson, J. R. Dahn and C. Yang, ACS Energy Lett., 2022, 7, 2189–2195 CrossRef CAS.
- L. Liu, Y. Zhao, G. Jiang, L. Shan, Z. Yang, Y. Ma, Y. Zhang, Q. Meng and P. Dong, Nano Res., 2023, 16, 9250–9258 CrossRef CAS.
- X. Liu, D. Ren, H. Hsu, X. Feng, G.-L. Xu, M. Zhuang, H. Gao, L. Lu, X. Han, Z. Chu, J. Li, X. He, K. Amine and M. Ouyang, Joule, 2018, 2, 2047–2064 CrossRef CAS.
- U. H. Kim, D. W. Jun, K. J. Park, Q. Zhang, P. Kaghazchi, D. Aurbach, D. T. Major, G. Goobes, M. Dixit, N. Leifer, C. M. Wang, P. Yan, D. Ahn, K. H. Kim, C. S. Yoon and Y. K. Sun, Energy Environ. Sci., 2018, 11, 1271–1279 RSC.
- O. Breuer, A. Chakraborty, J. Liu, T. Kravchuk, L. Burstein, J. Grinblat, Y. Kauffman, A. Gladkih, P. Nayak, M. Tsubery, A. I. Frenkel, M. Talianker, D. T. Major, B. Markovsky and D. Aurbach, ACS Appl. Mater. Interfaces, 2018, 10, 29608–29621 CrossRef CAS PubMed.
- K. Meng, Z. Wang, H. Guo and X. Li, Electrochim. Acta, 2017, 234, 99–107 CrossRef CAS.
- J. Wandt, A. T. S. Freiberg, A. Ogrodnik and H. A. Gasteiger, Mater. Today, 2018, 21, 825–833 CrossRef CAS.
- Y. Cheng, X. Zhang, Q. Leng, X. Yang, T. Jiao, Z. Gong, M.-S. Wang and Y. Yang, Chem. Eng. J., 2023, 474, 145869 CrossRef CAS.
- H. H. Sun, U.-H. Kim, J.-H. Park, S.-W. Park, D.-H. Seo, A. Heller, C. B. Mullins, C. S. Yoon and Y.-K. Sun, Nat. Commun., 2021, 12, 6552 CrossRef CAS PubMed.
- U.-H. Kim, G.-T. Park, B.-K. Son, G. W. Nam, J. Liu, L.-Y. Kuo, P. Kaghazchi, C. S. Yoon and Y.-K. Sun, Nat. Energy, 2020, 5, 860–869 CrossRef CAS.
- N. Liu, L. Chen, H. Wang, J. Zhao, F. Gao, J. Liu, J. Dong, Y. Lu, N. Li, Q. Shi, Y. Su and F. Wu, Chem. Eng. J., 2023, 472, 145113 CrossRef CAS.
- N. Zaker, C. Geng, D. Rathore, I. Hamam, N. Chen, P. Xiao, C. Yang, J. R. Dahn and G. A. Botton, Adv. Funct. Mater., 2023, 33, 2211178 CrossRef CAS.
- S. Yuan, H. Zhang, D. Song, Y. Ma, X. Shi, C. Li and L. Zhang, Chem. Eng. J., 2022, 439, 135677 CrossRef CAS.
- C. Geng, D. Rathore, D. Heino, N. Zhang, I. Hamam, N. Zaker, G. A. Botton, R. Omessi, N. Phattharasupakun, T. Bond, C. Yang and J. R. Dahn, Adv. Energy Mater., 2021, 12, 2103067 CrossRef.
- H.-H. Ryu, K.-J. Park, D. R. Yoon, A. Aishova, C. S. Yoon and Y.-K. Sun, Adv. Energy Mater., 2019, 9, 1902698 CrossRef CAS.
- Q. Zhang, Q. Deng, W. Zhong, J. Li, Z. Wang, P. Dong, K. Huang and C. Yang, Adv. Funct. Mater., 2023, 33, 2301336 CrossRef CAS.
- Q. Shi, F. Wu, H. Wang, Y. Lu, J. Dong, J. Zhao, Y. Guan, B. Zhang, R. Tang, Y. Liu, J. Liu, Y. Su and L. Chen, Energy Storage Mater., 2024, 67, 103264 CrossRef.
- S. Luo, F. Yang, Z. Xiong, Y. Wu, X. Ao, C. Li, Q. Chen and K. Wang, Chem. Eng. J., 2022, 448, 137663 CrossRef CAS.
- Z. Tan, Y. Li, X. Xi, S. Jiang, X. Li, X. Shen, P. Zhang and Z. He, Nano Res., 2022, 16, 4950–4960 CrossRef.
- Y. Liu, Q. Wang, L. Chen, Z. Xiao, X. Fan, S. Ma, L. Ming, A. Tayal, B. Zhang, F. Wu and X. Ou, Mater. Today, 2022, 61, 40–53 CrossRef CAS.
- C. X. Tian, F. Lin and M. M. Doeff, Acc. Chem. Res., 2018, 51, 89–96 CrossRef CAS PubMed.
- H. H. Ryu, K. J. Park, C. S. Yoon and Y. K. Sun, Chem. Mater., 2018, 30, 1155–1163 CrossRef CAS.
- J. Meng, L. Xu, Q. Ma, M. Yang, Y. Fang, G. Wan, R. Li, J. Yuan, X. Zhang, H. Yu, L. Liu and T. Liu, Adv. Funct. Mater., 2022, 32, 2113013 CrossRef CAS.
- G. T. Park, H. H. Ryu, N. Y. Park, C. S. Yoon and Y. K. Sun, J. Power Sources, 2019, 442, 227242 CrossRef CAS.
- D. Rathore, C. Liang, E. Zsoldos, M. Ball, S. Yu, C. Yang, Q. Wang and J. R. Dahn, J. Electrochem. Soc., 2023, 170, 020520 CrossRef CAS.
- F. Klein, C. Pfeifer, P. Scheitenberger, L. Pfeiffer, D. Zimmer, M. Wohlfahrt-Mehrens, M. Lindén and P. Axmann, J. Mater. Chem. A, 2023, 11, 5135–5147 RSC.
- Z. Jing, S. Wang, Q. Fu, V. Baran, A. Tayal, N. P. M. Casati, A. Missyul, L. Simonelli, M. Knapp, F. Li, H. Ehrenberg, S. Indris, C. Shan and W. Hua, Energy Storage Mater., 2023, 59, 102775 CrossRef.
- G. Ding, M. Yao, J. Li, T. Yang, Y. Zhang, K. Liu, X. Huang, Z. Wu, J. Chen, Z. Wu, J. Du, C. Rong, Q. Liu, W. Zhang and F. Cheng, Adv. Energy Mater., 2023, 13, 2300407 CrossRef CAS.
- T. Zhou, H. Wang, Y. Wang, P. Jiao, Z. Hao, K. Zhang, J. Xu, J.-B. Liu, Y.-S. He, Y.-X. Zhang, L. Chen, L. Li, W. Zhang, Z.-F. Ma and J. Chen, Chem, 2022, 8, 2817–2830 CAS.
- A. Liu, N. Phattharasupakun, R. Väli, D. Ouyang and J. R. Dahn, J. Electrochem. Soc., 2022, 169, 030538 CrossRef CAS.
- N. Phattharasupakun, M. M. E. Cormier, Y. Liu, C. Geng, E. Zsoldos, I. Hamam, A. Liu, M. B. Johnson, M. Sawangphruk and J. R. Dahn, J. Electrochem. Soc., 2021, 168, 110502 CrossRef CAS.
- X. Wang, X. Zhou, X. Liu, G. Feng, S. Wang, B. Zhang, P. Zhang, M. Zuo, W. Xing, W. Fan, H. Zhang, G. Lv and W. Xiang, Chem. Eng. J., 2024, 488, 150795 CrossRef CAS.
- F. Reissig, J. Ramirez-Rico, T. J. Placke, M. Winter, R. Schmuch and A. Gomez-Martin, Batteries, 2023, 9, 245 CrossRef CAS.
- W. Xiong, Y. Liu, G. Zhu, Y. Wang and H. Zheng, J. Energy Storage, 2023, 73, 109086 CrossRef.
- G.-T. Park, N.-Y. Park, T.-C. Noh, B. Namkoong, H.-H. Ryu, J.-Y. Shin, T. Beierling, C. S. Yoon and Y.-K. Sun, Energy Environ. Sci., 2021, 14, 5084–5095 RSC.
- X. Chao, J. R. Philip, J. Quentin and P. G. Clare, Adv. Energy Mater., 2020, 11, 2003404 Search PubMed.
- Z. Lu, J. Zhang, Q. Zhang, D. Wong, W. Yin, N. Zhang, Z. Chen, L. Gu, Z. Hu and X. Liu, Adv. Sci., 2023, 10, 2206442 CrossRef CAS.
- W. Li, H. Y. Asl, Q. Xie and A. Manthiram, J. Am. Chem. Soc., 2019, 141, 5097–5101 CrossRef CAS PubMed.
- H. Gao, X. Zeng, Y. Hu, V. Tileli, L. Li, Y. Ren, X. Meng, F. Maglia, P. Lamp, S.-J. Kim, K. Amine and Z. Chen, ACS Appl. Energy Mater., 2018, 1, 2254–2260 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03805c |
| This journal is © The Royal Society of Chemistry 2024 |