
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon–metal versus metal–metal synergistic mechanism of ethylene electro-oxidation via electrolysis of water on TM2N6 sites in graphene†
Yun-Jie
Chu
a,
Chang-Yan
Zhu
a,
Chun-Guang
Liu
 *b,
Yun
Geng
a,
Zhong-Min
Su
c and
Min
Zhang
*a
*b,
Yun
Geng
a,
Zhong-Min
Su
c and
Min
Zhang
*a
aInstitute of Functional Material Chemistry, Faculty of Chemistry, National & Local United Engineering Laboratory for Power Batteries, Northeast Normal University, Changchun 130024, China. E-mail: mzhang@nenu.edu.cn
bDepartment of Chemistry, Faculty of Science, Beihua University, Jilin City 132013, P. R. China. E-mail: liucg407@163.com
cState Key Laboratory of Supramolecular Structure and Materials, Institute of Theoretical Chemistry, College of Chemistry, Jilin University, Changchun, 130021, P. R. China
First published on 2nd August 2024
Abstract
Acetaldehyde (AA) and ethylene oxide (EO) are important fine chemicals, and are also substrates with wide applications for high-value chemical products. Direct electrocatalytic oxidation of ethylene to AA and EO can avoid the untoward effects from harmful byproducts and high energy emissions. The most central intermediate state is the co-adsorption and coupling of ethylene and active oxygen intermediates (*O) at the active site(s), which is restricted by two factors: the stability of the *O intermediate generated during the electrolysis of water on the active site at a certain applied potential and pH range; and the lower kinetic energy barriers of the oxidation process based on the thermo-migration barrier from the *O intermediate to produce AA/EO. The benefit of two adjacent active atoms is more promising, since diverse adsorption and flexible catalytic sites may be provided for elementary reaction steps. Motivated by this strategy, we explored the feasibility of various homonuclear TM2N6@graphenes with dual-atomic-site catalysts (DASCs) for ethylene electro-oxidation through first-principles calculations via thermodynamic evaluation, analysis of the surface Pourbaix diagram, and kinetic evaluation. Two reaction mechanisms through C–TM versus TM–TM synergism were determined. Between them, a TM–TM mechanism on 4 TM2N6@graphenes and a C–TM mechanism on 5 TM2N6@graphenes are built. All 5 TM2N6@graphenes through the C–TM mechanism exhibit lower kinetic energy barriers for AA and EO generation than the 4 TM2N6@graphenes through the TM–TM mechanism. In particular, Pd2N6@graphene exhibits the most excellent catalytic activity, with energy barriers for generating AA and EO of only 0.02 and 0.65 eV at an applied potential of 1.77 V vs. RHE for the generation of an active oxygen intermediate. Electronic structure analysis indicates that the intrinsic C–TM mechanism is more advantageous than the TM–TM mechanism for ethylene electro-oxidation, and this study also provides valuable clues for further experimental exploration.
1 Introduction
Acetaldehyde (AA) and ethylene oxide (EO) are important fine chemicals, which can also be further converted into other successive high-value chemical products.1,2 Acetaldehyde, as one of the most important aldehydes, is produced in excess of 1 × 106 tons per year worldwide and is widely used as a starting material for the synthesis of acetic acid, 2-ethylhexanol, acetate esters, pentaerythritol and other industrial chemicals.2,3 Ethylene oxide produced at about 2 × 106 tons per year is widely used in the automotive industry, medicine, and other fields, and in major consumer products for antifreeze, pharmaceuticals, detergents, and plastics.1,4 The industrial production of AA and EO is dependent mainly on the oxidation reaction of ethylene. The main industrial process for AA production is the Wacker process, in which CuCl2/PdCl2 is utilized as a catalyst for the oxidation of ethylene in aqueous solution. Ethylene is oxidized to AA, accompanied by the reduction of Pd(II) to Pd (0), and then Pd (0) is further re-oxidized to Pd(II) by CuCl2 in a simultaneous co-catalytic cycle.5 Toxic and corrosive liquid byproducts are produced in this technique. Industrial production of EO from ethylene utilizes an Ag-based catalyst at 230–270 °C and 1–3 MPa to activate O2 and accompanied by some thermodynamic side reactions, such as the excessive oxidation of ethylene.1,6,7 Hence, alternative economic and environmental oxidation strategies for ethylene are desirable.The green oxidation of alkenes through renewable electricity as a driving force is an attractive approach to achieve its chemical conversion, which has received widespread attention as it can effectively reduce carbon emissions.8 In the past few decades, electrochemical oxidation methods for alkenes have been reported and it has proven to be a substantial strategy.8–13 In 1975, Holbrook et al. reported that ethylene and propylene could be partially oxidized on an Ag anode, and the corresponding products were ethylene glycol and propylene oxide.9 In 2010, Šebera et al. investigating the electrochemical oxidation of ethylene on an Au electrode indicated that polycrystalline Au shows selectivity for the formation of AA.10 In 2020, Sargent et al. reported a one-step route for the electro-oxidation of ethylene to ethylene glycol under mild conditions on a gold-doped palladium catalyst as the anode with a Faraday efficiency of approximately 80% for ethylene glycol, in which the tuning of the OH binding energy is crucial for product formation.11 Moreover, propylene electro-oxidation has also made significant progress in recent years. In 2022, Geng et al. developed an efficient Ag3PO4 catalyst for the oxidation of propylene to propylene oxide starting from water electrolysis under ambient conditions, and the Ag3PO4 (100) facets achieved a product yield rate of 5.3 gPO m−2 h−1 at 2.4 V vs. RHE.12 Nørskov et al. proved that electro-epoxidation of propylene is facile through water electrolysis to provide an oxygen source at the catalyst surface when atomic oxygen pre-exists on a catalyst surface with several weakly bound oxygens.13 These studies indicate great potential for the selective anodic oxidation of alkenes to AA/EO in aqueous solution under mild conditions. However, an essential understanding of the electro-oxidation process of alkenes is currently lacking, in particular a typical research case study on the direct oxidation reaction using an active oxygen intermediate (*O) generated by starting from water splitting as an oxygen source.
For the direct oxidation of ethylene, the crucial step is to introduce an oxygen atom to ethylene, which is the vital intermediate for the selective oxidation of ethylene to form AA or EO.14–16 It is well known that the water electrolysis process can produce an *O intermediate on the anode surface under mild conditions.17,18 It can be speculated that there will be a potential–pH (U–pH) range for *O intermediates to exist stably under electrolysis of water if the applied potential is high enough to generate *O intermediates but lower than the necessary potential value to carry out the subsequent elementary reactions or to generate other intermediates.13,19 The restrictive *O intermediate provides the foreground to directly oxidize ethylene at appropriate sites. More importantly, this approach avoids the use of molecular oxygen, thereby avoiding the high energy barrier of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O cleavage and greatly reducing the hazardous complete oxidation of ethylene.
O cleavage and greatly reducing the hazardous complete oxidation of ethylene.
Dual-atomic-site catalysts (DASCs) by introducing an alternative active site, as an extension of single-atom catalysts (SACs), have recently drawn a surge in attention, which possessing flexible active sites when acting in isolation, offer greater probability of creating a synergistic effect, thereby producing a higher yield and achieving a higher Faraday efficiency.20–25 Meanwhile, the top region over carbon sites in the dipyridyl subunit also exhibits a preference for binding to oxygen-containing species besides that over two metals for several homonuclear TM2N6@graphenes.26,27 Hence, the TM2N6 moiety, which has been prepared experimentally28 with the advantage of greater possibilities for the co-adsorption of ethylene and reactive oxygen species from the two homonuclear metals and the bridge-carbon atoms adjacent to two metals, was explored in this study (as shown in Fig. 1). Furthermore, 19 homonuclear TM2N6@graphenes (Fig. 1 and S1†), with thermodynamic and electrochemical stability reported in the previous studies,26,27,29 were investigated here for their application to ethylene electro-oxidation. Notably, our calculated data indicates that 9 TM2N6@graphenes among them can exist in the U–pH range of *O intermediates, and two diverse subsequent reaction mechanisms through C–TM versus TM–TM synergism were determined, including 4 TM2N6@graphenes through the TM–TM mechanism and 5 TM2N6@graphenes through the C–TM mechanism. In addition, all 5 TM2N6@graphenes through the C–TM mode exhibit lower kinetic energy barriers for AA and EO generation than the 4 other TM2N6@graphenes. In particular, Pt2N6@graphene possesses the best reaction activity for the generation of AA and EO due to the lowest energy barriers for the ethylene oxidation reaction.
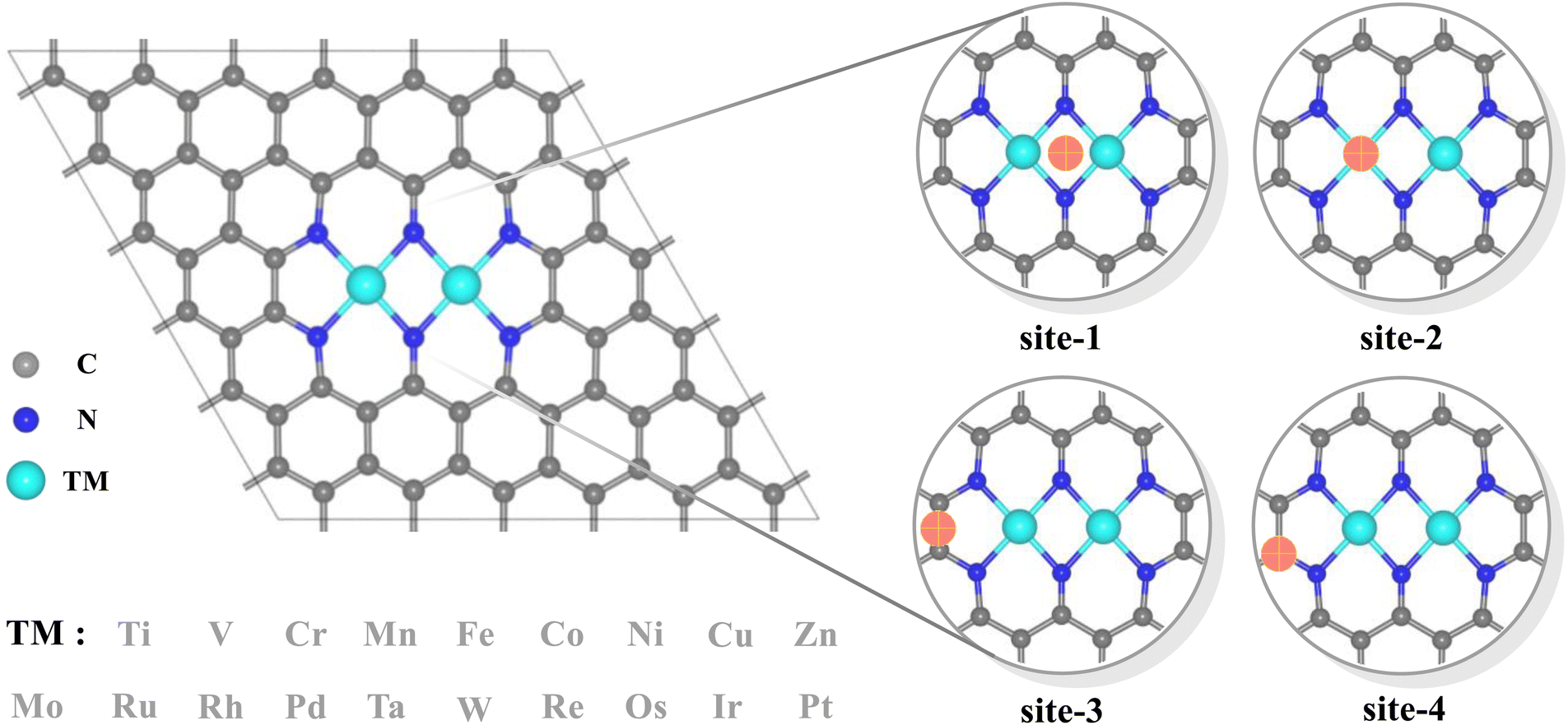 | ||
| Fig. 1 Structural model of 19 N-doped graphene-supported homonuclear transition metals, and possible adsorption sites of oxygen containing species. The metal, carbon, and nitrogen atoms, and potential adsorption sites are shown in cyan, gray, blue, and orange, respectively. | ||
2 Computational methods
All spin-polarized DFT calculations were carried out with the Vienna ab initio simulation package (VASP).30 The Perdew–Burke–Ernzerhof (PBE) exchange correlation function of the generalized gradient approximation (GGA) was used to describe the electron interactions, and the projection augmented wave (PAW) pseudopotential was employed to treat the core electrons.31,32 The DFT-D3 method was adopted for the van der Waals interaction between reaction intermediates and potential catalysts.33 A 5 × 5 graphene supercell was employed to investigate the catalytic process and a 20 Å vacuum region was created to avoid the interaction between mirror structures. During structural optimization, the energy cutoff, and convergence criteria for the energy and force were set to 500 eV, 1 × 10−5 eV and 0.02 eV Å−1, respectively. The convergence criterion of energy was improved to 1 × 10−7 eV for frequency calculation. Electron spin polarization was considered in all calculations. A Monkhorst–Pack k-mesh of 3 × 3 × 1 was set for structural optimization and frequency calculation, and an improved 5 × 5 × 1 grid was set for electronic structure calculation. The implicit Poisson–Boltzmann solvation model34,35 was used to simulate the solvation effect for all reaction paths to evaluate the accuracy of the calculations, in which the dielectric constant of water was taken as 78.4, except for ab initio molecular dynamics (AIMD) simulations. AIMD simulations at 300 K and 500 K for 10 ps with a time step of 1 fs under NVT ensemble were made to demonstrate the stability of potential catalysts.36The Gibbs free energy change (ΔG) of each elementary step in the OER process was obtained from the calculated hydrogen electrode (CHE) model proposed by Nørskov et al.37 The adsorption free energy of a reaction intermediate was calculated as follows:
ΔG = ΔEDFT + ΔEZPE − TΔS − neU − nkBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(10) × pH ln(10) × pH | (1) |
| ΔG = ΔEDFT + ΔEZPE − TΔS − neU | (2) |
The Pourbaix diagram reveals the thermodynamically stable structures in an electrochemical system as a function of pH and applied electrode potential (U)38–40 by calculating the ΔG for possible intermediates in OER. Moreover, the transition states of the ethylene oxidation reactions underwent a preliminary search by the climbing-image nudged elastic band (CI-NEB) method and then by the DIMER method for further identification.41,42 The DIMER calculations of energy and force convergence were set to 10−7 eV and 0.02 eV Å−1. Post processing analysis of VASP was conducted with the help of the VASPKIT package.43
3 Results and discussions
3.1 Active oxygen intermediates (*O) and Pourbaix diagrams
Various 2D layered materials, such as graphene, graphene nitride carbon (g-C3N4) and other nitrogen-doped carbons, have emerged in recent years as supporting substrates for capturing metal dimers in experiments.20–23,44–46 Among these materials, nitrogen-doped graphene is regarded as one of the most promising substrates for stabilizing DASC due to its compatible ability to anchor metal, excellent catalytic performance and relatively easy preparation. In the present work, 19 homonuclear TM2N6@graphenes were chosen to explore the feasibility of ethylene electro-oxidation (Fig. 1 and S1†) and to study their intrinsic mechanism, for the following reasons: (1) this pattern of TM2N6@graphene with the five member-ring adjacent to pyridine nitrogen units has been prepared experimentally.28 (2) The corresponding carbon sites in the five member-ring possess the ability to adsorb oxygen containing species,26,27 providing more flexible adsorption sites for active oxygen and ethylene in some cases. (3) These 19 homonuclear TM2N6@graphenes possess thermodynamic and electrochemical stability, as proven in previous studies.26,27,29 The distance between two metal atoms is in the range of 2.23 Å (Fe2N6@graphene) to 2.67 Å (Pt2N6@graphene) in our simulated level, and most of these structures maintain planar characteristics except for a few metals with a large atomic radius that protrude upward.According to the scheme proposed in this work, the ethylene oxidation reaction starts from ethylene and *O intermediates (generated by electrolysis of water) to produce EO or AA. The total reaction equation for ethylene oxidation is as follows:
| *O + C2H4 → * + EO/AA | (3) |
Notably, the carbons of the five-member ring connected with pyridine and the metals in homonuclear TM2N6@graphene can all be utilized as potential adsorption sites for oxygen-containing species. In fact, this phenomenon has also been observed in a couple of TMN4 moieties in graphene, possibly due to doping-induced charge redistribution.47 Hence, four kinds of possible active sites were selected during electrolysis of water for oxygen-containing species, as shown in the right-hand part of Fig. 1, namely a metal–metal bridge site (site-1), a metal top site (site-2), a carbon–carbon bridge site (site-3), and a carbon top site (site-4). Firstly, for the *O intermediates of the 19 TM2N6@graphenes, the structures of the four potential sites are shown in Fig. S2–S5† and the relative energies are listed in Table S1.† The total thermodynamic Gibbs free energy changes (ΔG) of ethylene oxidation initiated by stable *O intermediates to AA and EO is shown in Fig. 2, and the relevant data are also listed in Table S2.† The ethylene oxidation reaction requires effective adsorption of an ethylene molecule on the *O intermediate, which is an exothermic or slightly endothermic process, followed by the formation of AA or EO on the catalyst surface after undergoing one corresponding transition state, and subsequent desorption. The transition-state energy barriers must in general be higher than the Gibbs free energies of the initial to final states. If the Gibbs free energies of the final states are higher, the barrier energies of their transition states must be even higher. As shown in Fig. 2 and Table S2,† there are 12 TM2N6@graphenes (TM = Mn, Fe, Co, Ni, Cu, Zn, Ru, Rh, Pd, Os, Ir, and Pt) which almost possess negative ΔG of the final states for generating AA and EO simultaneously, except for the slightly endothermic generation of EO on Mn2@graphenes (0.23 eV) and Os2N6@graphenes (0.13 eV). Hence, these 12 TM2N6@graphenes were further investigated in detail for ethylene electro-oxidation.
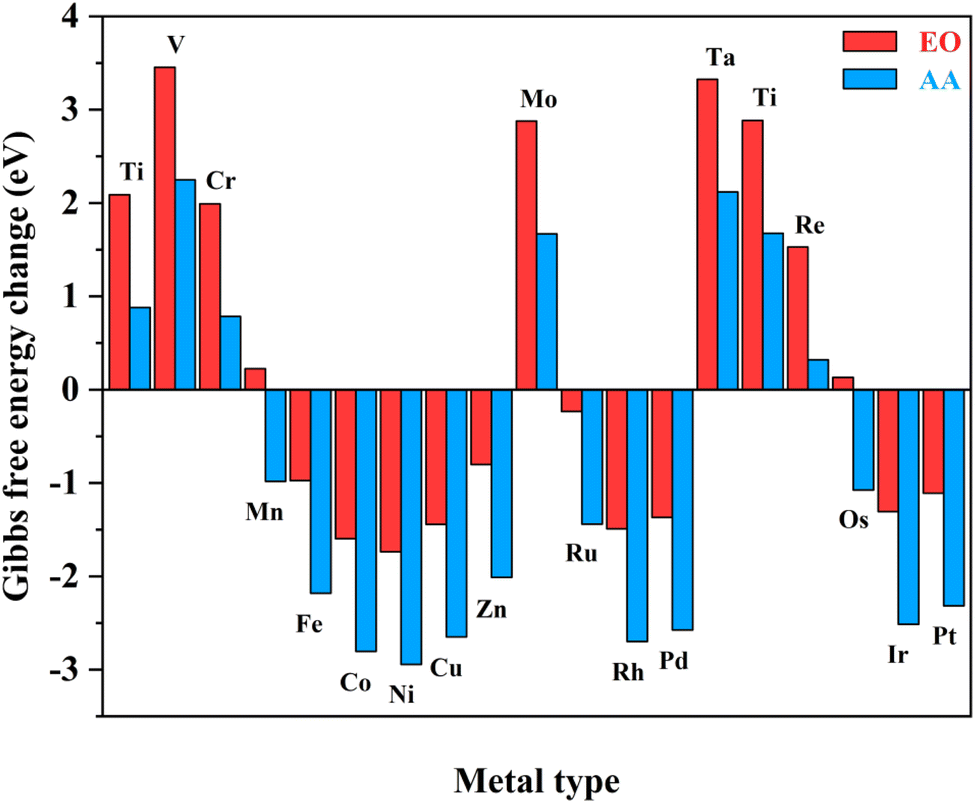 | ||
| Fig. 2 Total Gibbs free energy changes of the ethylene oxidation reaction to AA and EO initiated by *O intermediates for various TM2N6@graphenes. | ||
As mentioned above, the source of active oxygen species, namely the *O intermediates, is assumed to be generated by the electrolysis of water, thus avoiding the traditional drawbacks caused by OO bond breakage. Therefore, a wide potential–pH (U–pH) range is optimal and desirable to stabilize the *O intermediate in the electrochemical process. Specifically, only *O intermediates can exist, i.e. ΔG*O < 0, and ΔG*O < ΔG*OOH, which means that the generation of *O intermediates cannot be overwhelmed by the generation of *OOH during OER under the certain applied potential and pH conditions. Here, possible adsorption intermediates were considered and the optimized structures are shown in Fig. S2–S10,† and the Pourbaix diagrams of these 12 TM2N6@graphenes were constructed (Fig. 3) with the most thermodynamically stable intermediates during OER in our calculations. As shown in Fig. 3, the thermodynamically stable range for the 12 TM2N6@graphene surfaces during OER was explored under the given applied potential values of 0–3 V against the SHE and a pH of 0–14. Firstly, for Mn2-, Ru2-, and Os2N6@graphenes (Fig. 3a, g, and j), there exists a very wide range to stabilize the *O intermediate, and the lowest applied potential for generating the *O intermediates are only 0.72, 0.97, and 0.76 V, respectively, at pH = 0. All of Fe2-, Cu2-, Rh2-, Ir2-, and Pt2N6@graphenes (Fig. 3b, e, h, and k), have a moderate U–pH range to stabilize the *O intermediate, and the applied potentials for generating *O intermediates of these surfaces are below 2.0 V, lower than the reaction of propylene electro-epoxidation at 2.4 V vs. RHE on the Ag3PO4 (100) facets.12 Secondly, for Co2N6@graphene (Fig. 3c), there is only a very narrow U–pH range with *O intermediate, and its area ratio is only 0.67% for the given range. However, for completeness of the data, we also conducted the following studies on Co2N6@graphene. Thirdly, for Ni2- and Zn2N6@graphenes (Fig. 3d and f), there is no U–pH range that can stabilize the *O intermediate. The applied potential necessary to generate the *OH intermediate (from * → *OH, pH = 0, U = 1.94 V, or pH = 14, U = 1.11 V) can directly complete the OER process on Ni2N6@graphene. The applied potential for generating the *O intermediate (from *O → *OH, pH = 0, U = 1.92 V, or pH = 14, U = 1.09 V) are higher than the applied potential for generating the *OOH intermediate from *O (from *O → *OOH, pH = 0, U = 1.65 V, or pH = 14, U = 0.82 V) on Zn2N6@graphene. Therefore, although both Ni2-, and Zn2N6@graphenes exhibit good OER catalytic performance with applied overpotentials of 0.71 and 0.69 V at pH = 0, they are not suitable for the ethylene electro-oxidation reaction. Meanwhile, it should be noted that surface Pourbaix diagrams have been constructed for all 12 TM2N6@graphene surfaces, on which the *OH intermediates can be adsorbed on three potential sites except for site-3, and the relevant structures and the relative energies are shown in Fig. S6–S8† and listed in Table S3.† The *OOH and *OO intermediates cannot be adsorbed around the C sites, and the most stable structures are shown in Fig. S9 and S10.† (It should be noted that it is a desorption thermochemical process from *OO to *+O2, and the ΔG of O2 desorption from TM2N6@graphenes are 0.71 eV from Mn2-, 0.05 eV from Fe2-, −0.35 eV from Co2-, −0.42 eV from Ni2-, −0.36 eV from Cu2-, −0.02 eV from Zn2-, 0.24 eV Ru2-, −0.50 eV from Rh2-, −0.18 eV from Pd2-, 0.21 eV from Os2-, −0.46 eV from Ir2-, and −0.11 eV from Pt2N6@graphenes, respectively), and there are no U–pH ranges of *OOH intermediates for the 12 TM2N6@graphenes. In summary, 10 potential TM2N6@graphenes are eligible for ethylene electro-oxidation based on their Pourbaix diagrams.
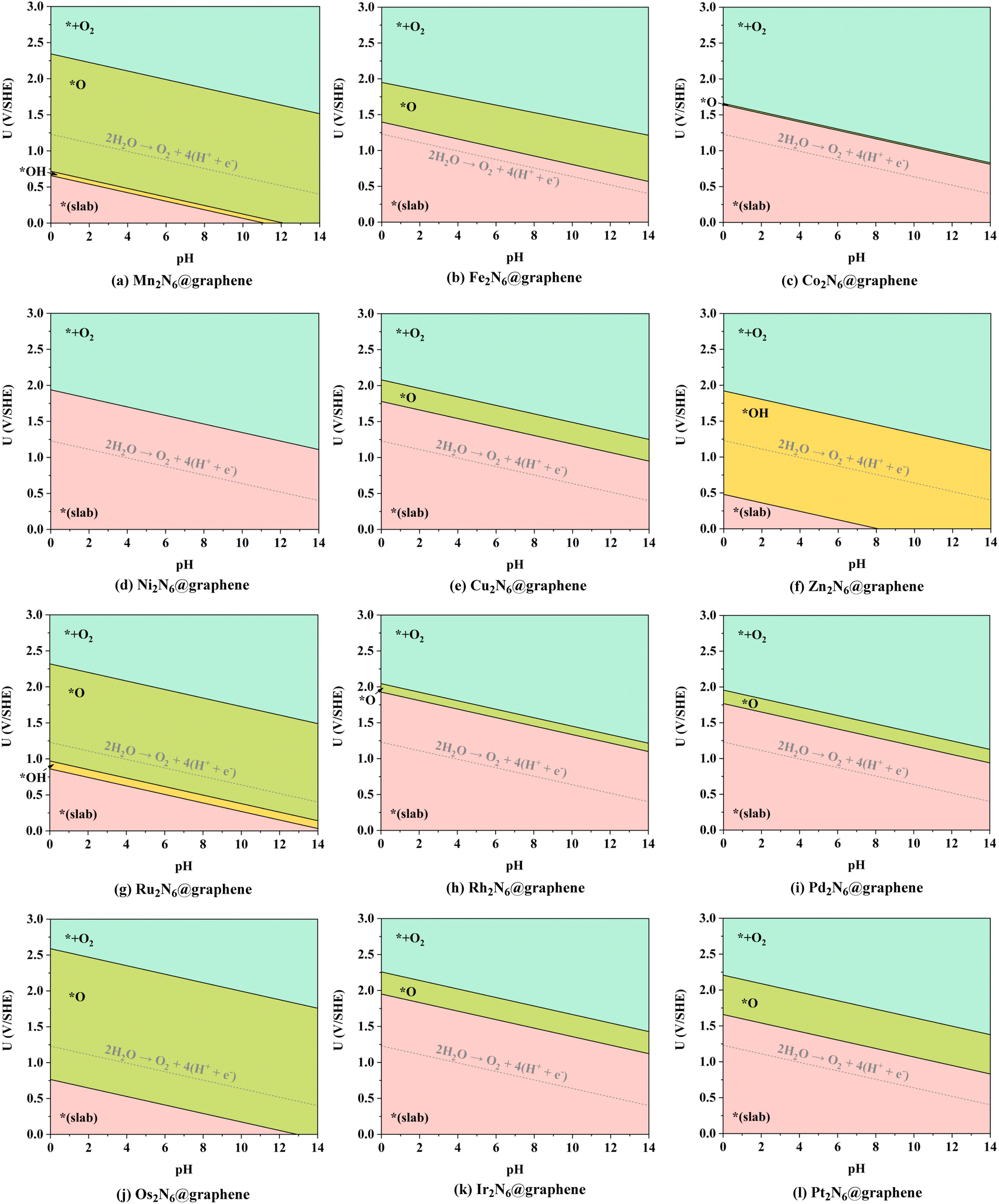 | ||
| Fig. 3 Pourbaix diagrams on the surfaces of 12 TM2N6@graphenes under the given applied potential value of 0–3 V against the SHE and pH scale of 0–14 at their most stable adsorption sites. Pink, yellow, green, and cyan represent the U–pH ranges in which the intermediates of *(slab), *OH, *O, and *+O2 can be stabilized, respectively. | ||
3.2 Ethylene oxidation during OER
For the 10 TM2N6@graphenes to be further studied in detail, the most stable *O intermediates on 4 surfaces are the O adsorption on the metal sites (site-1 for Fe2N6@graphene, and site-2 on Mn2-, Ru2-, and Os2N6@graphenes), and those on 6 surfaces are the C adsorption of O on C sites (site-4 on Co2-, Rh2-, Ir2-, Pd2-, Pt2- and Cu2N6@graphenes). Therefore, the difference in their most stable O adsorption sites on various TM2N6@graphene surfaces leads to the decoupling difference of C–O bonding and TM–O bonding and the subsequent coupling oxidation progress between O and ethylene.The ethylene oxidation reaction also requires the effective adsorption of an ethylene molecule on the adjacent sites with the *O intermediate as mentioned above, so the stable configurations of ethylene adsorption on the *O intermediates of the 10 TM2N6@graphenes are approached and shown in Fig. 4. Therefore, the ethylene oxidation was divided into two mechanisms for the formation of AA or EO in this study through different O adsorption sites, namely the metal–metal (TM–TM) synergistic mechanism and the carbon–metal (C–TM) synergistic mechanism, which are very similar to the oxametallacycle intermediate (OMME).1,48 Notably, the calculations show that the O moves from site 1 to site 2 on Fe2N6@graphene while stabilizing the adsorption structure of ethylene. The calculated adsorption energies (ΔGad) of ethylene on *O intermediates through the TM–TM mode are −0.62, −1.10, −0.65, and −0.17 eV for Mn2-, Fe2-, Ru2-, and Os2N6@graphenes, respectively. The corresponding ΔGad through the C–TM mode are −0.42. −0.28, −0.21, −0.13, and −0.17 eV for Co2-, Rh2-, Ir2-, Pd2-, Pt2N6@graphenes, respectively. Only the corresponding ΔGad is an endothermic process of 0.42 eV through the C–TM mode for Cu2N6@graphene, which may be a very weak physical adsorption mode and may not possess a comparable potential for ethylene electro-oxidation. Hence, the focus will be on the two kinds of ethylene electro-oxidation on the other 9 TM2N6@graphenes through C–TM and TM–TM modes in the following part.
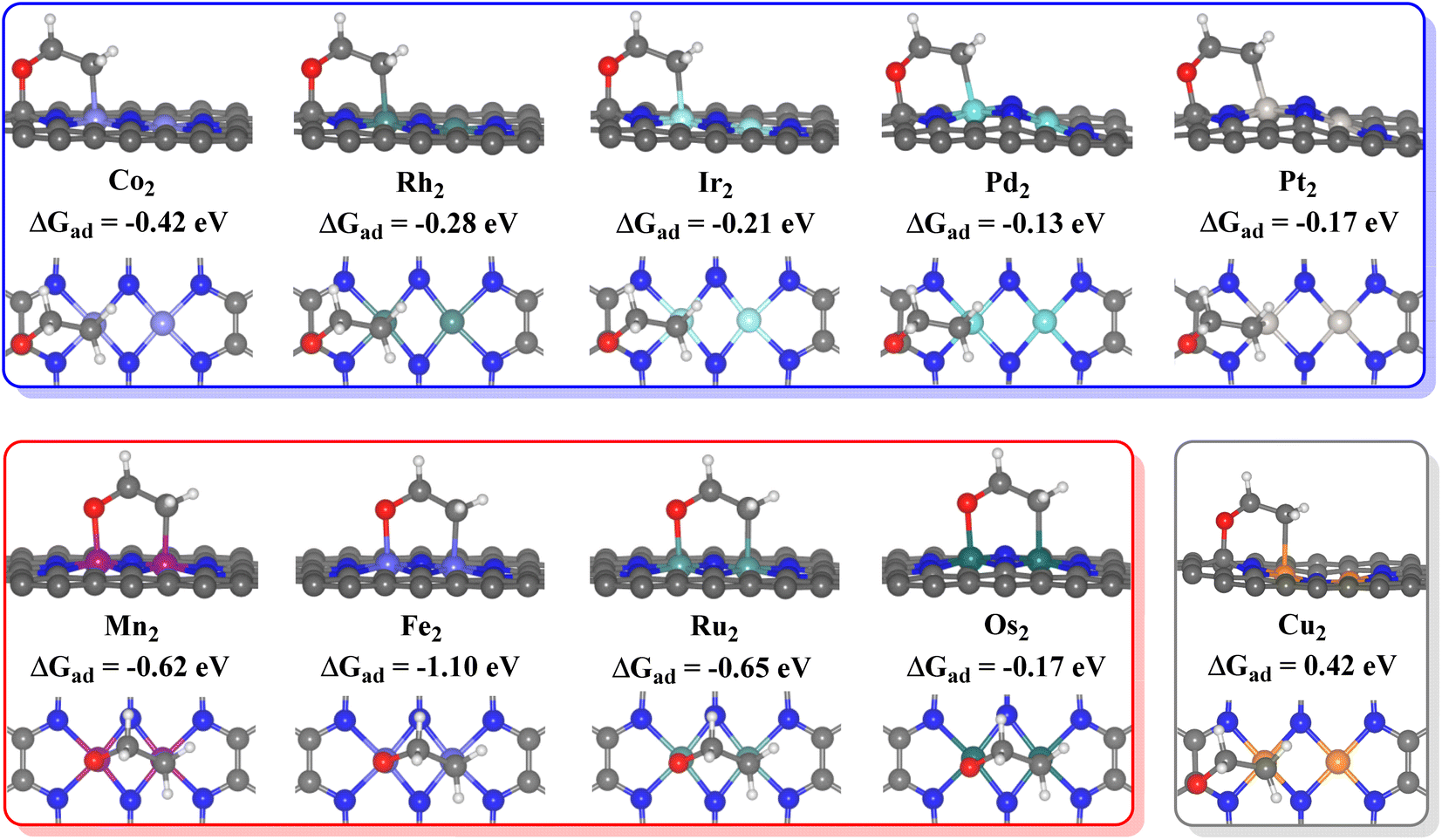 | ||
| Fig. 4 Side and top views of adsorption structures of ethylene on the *O surface of 10 TM2N6@graphenes, and their adsorption energies (ΔGad) in eV. | ||
The ethylene electro-oxidation reaction can be divided into two continuous parts: a water electrolysis process and a thermochemical ethylene oxidation process. The first half is the electrochemical process of water electrolysis at the lowest applied potential to generate the *O intermediates, while the second half is the thermochemical process for ethylene oxidation to AA and EO, including ethylene adsorption adjacent to the *O intermediates on TM2N6@graphenes, the production of *AA and *EO, and then their final desorption. The thermodynamic steps and the free energy changes (ΔG) of electrochemistry and thermochemistry on 9 selected TM2N6@graphenes are shown in Fig. 5, 6 and S11.† The adsorption states of ethylene on the *O intermediate towards AA (*AA as shown in Fig. S12†) are exothermic processes through the TM–TM mode for Fe2-, Ru2-, and Os2N6@graphenes, except for a slightly endothermic process for Mn2N6@graphenes. The following desorptions of AA from the catalyst surface are all exothermic. The adsorption states of EO (*EO as shown in Fig. S13†) are all endothermic processes for the 4 TM2N6@graphenes, and then the corresponding desorptions of EO are exothermic. In contrast, the adsorption states of ethylene towards *AA or *EO are all obviously exothermic processes through the C–TM mode of Rh2-, Ir2, Pd2-, Pt2-, and Co2N6@graphenes. Subsequently, the desorption of AA from Pd2-, Pt2- and Co2N6@graphenes and the desorption of EO from Pt2-, and Co2N6@graphene are slightly endothermic, but the desorption of other products from TM2N6@graphenes is exothermic.
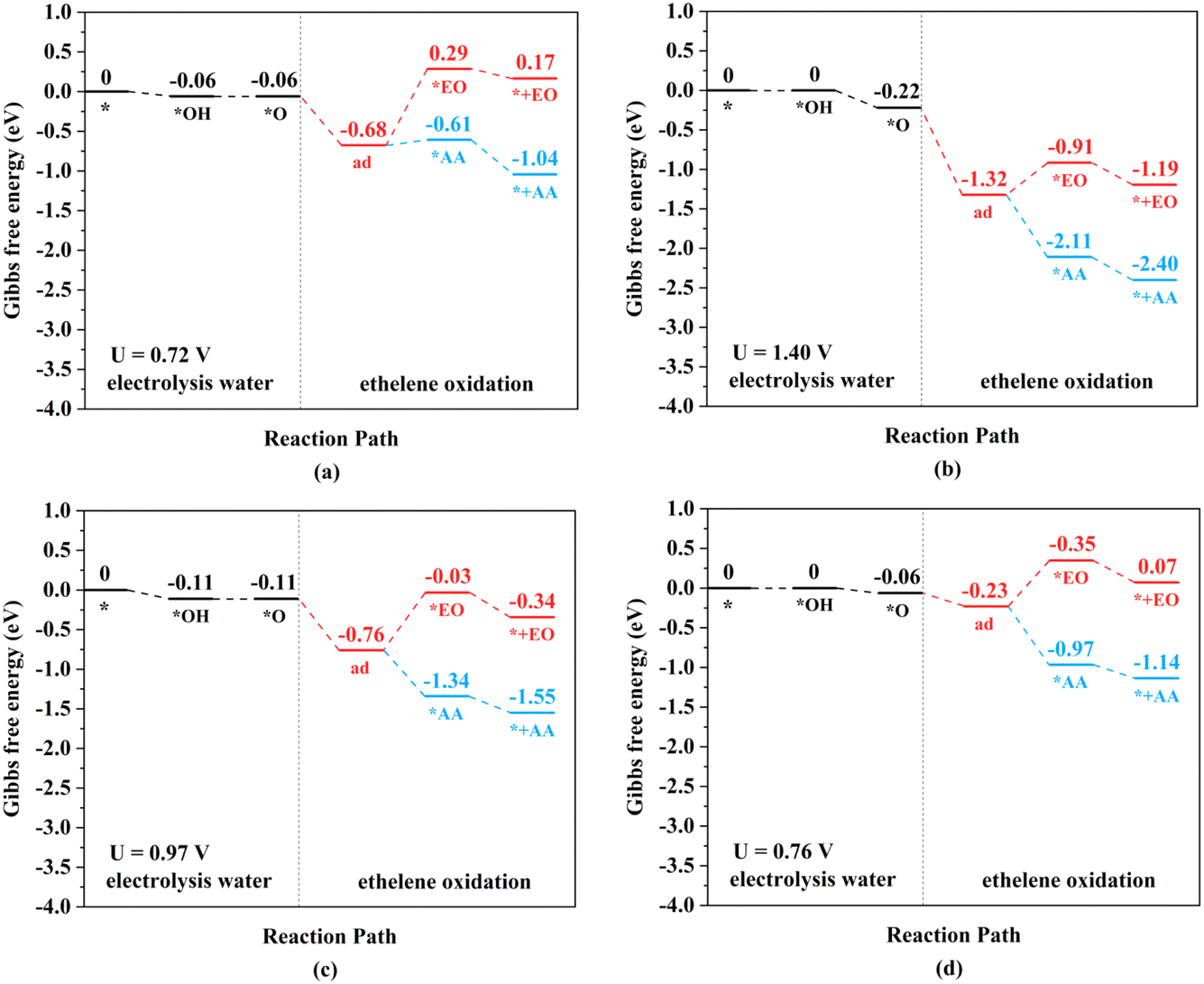 | ||
| Fig. 5 Gibbs free energy change diagrams for ethylene electro-oxidation on (a) Mn2-, (b) Fe2-, (c) Ru2-, and (d) Os2N6@graphenes at the applied potential at which the *O intermediates can generated at pH = 0. | ||
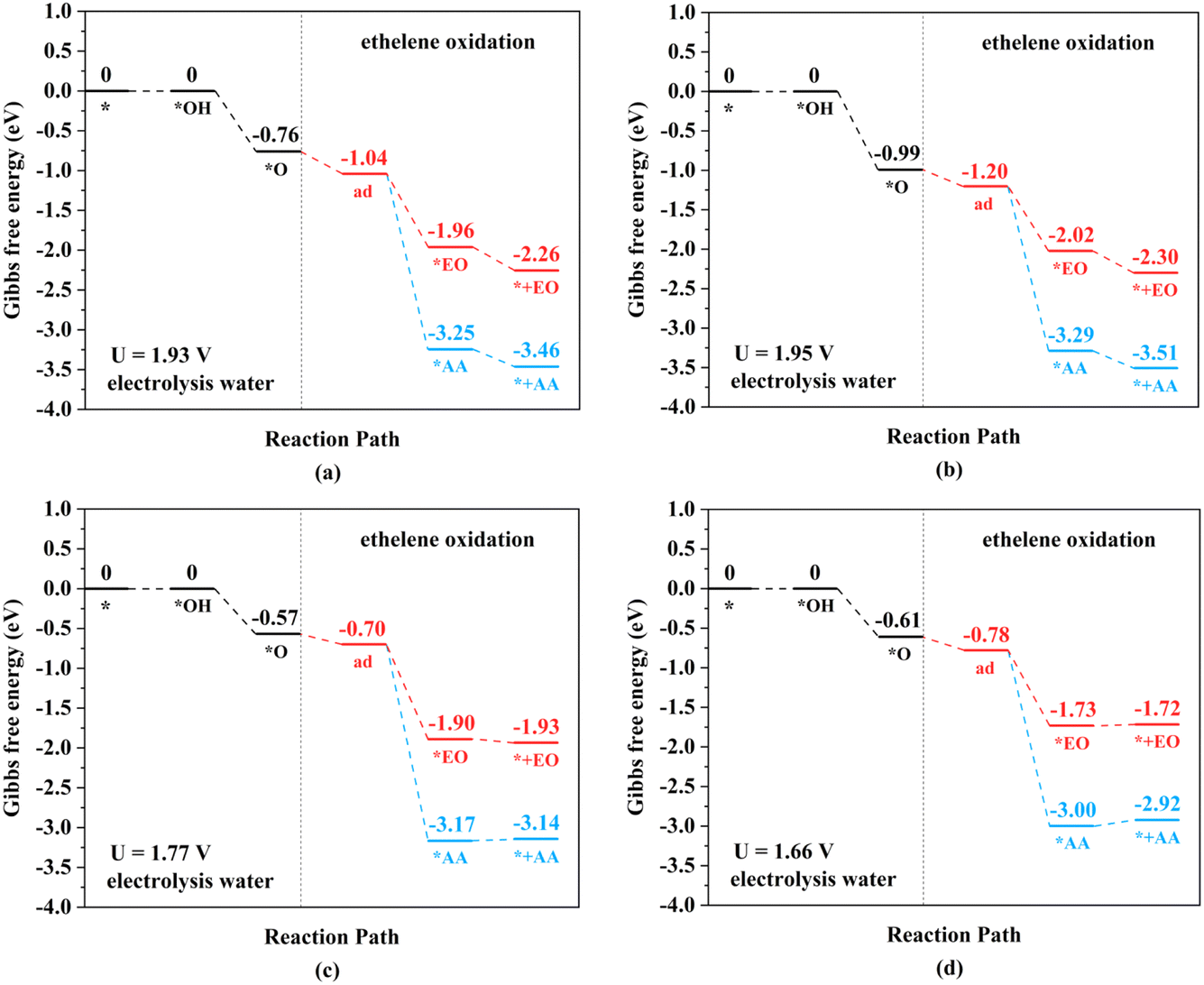 | ||
| Fig. 6 Gibbs free energy change diagrams for ethylene electro-oxidation on (a) Rh2-, (b) Ir2-, (c) Pd2-, and (d) Pt2N6@graphenes at the lowest applied potential to generate *O intermediates at pH = 0. | ||
The TM–TM mechanism exhibits a lower applied potential for the generation of *O intermediates during their electrochemical process than in the previous discussion, but the C–TM mechanism is thermodynamically more favorable for a thermochemical process. More importantly, their kinetic energy barriers of ethylene oxidation are decisive for catalyst activity and final product selectivity, since they cannot be controlled by the applied potential. The kinetic energy barriers to AA or EO were further investigated to evaluate the catalytic activity and selectivity, which are given in Fig. 7, 8 and S14.†
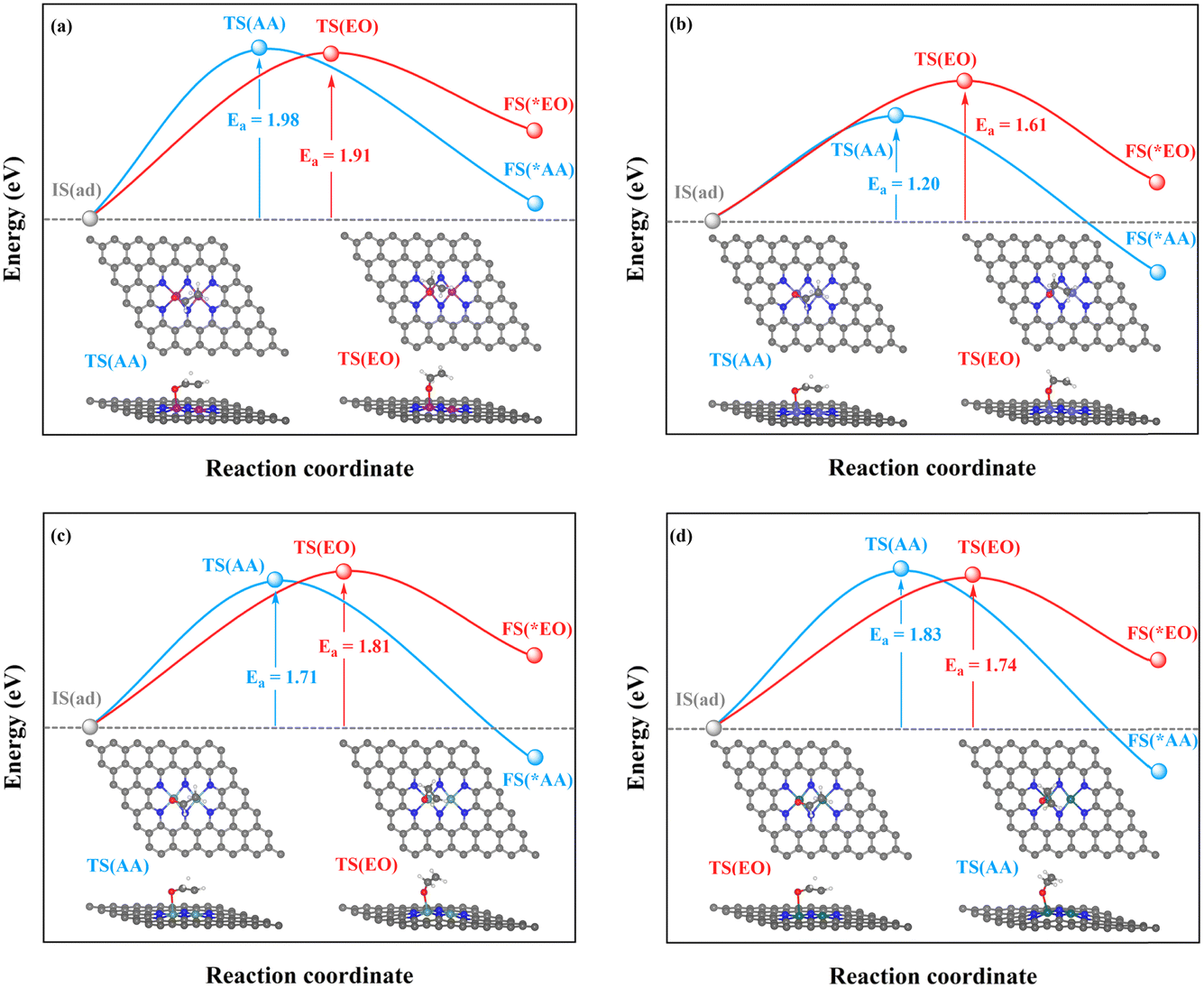 | ||
| Fig. 7 Kinetic energy barriers and transition-state structures of AA and EO generation through the TM–TM mechanism on (a) Mn2-, (b) Fe2-, (c) Ru2-, and (d) Os2N6@graphenes. | ||
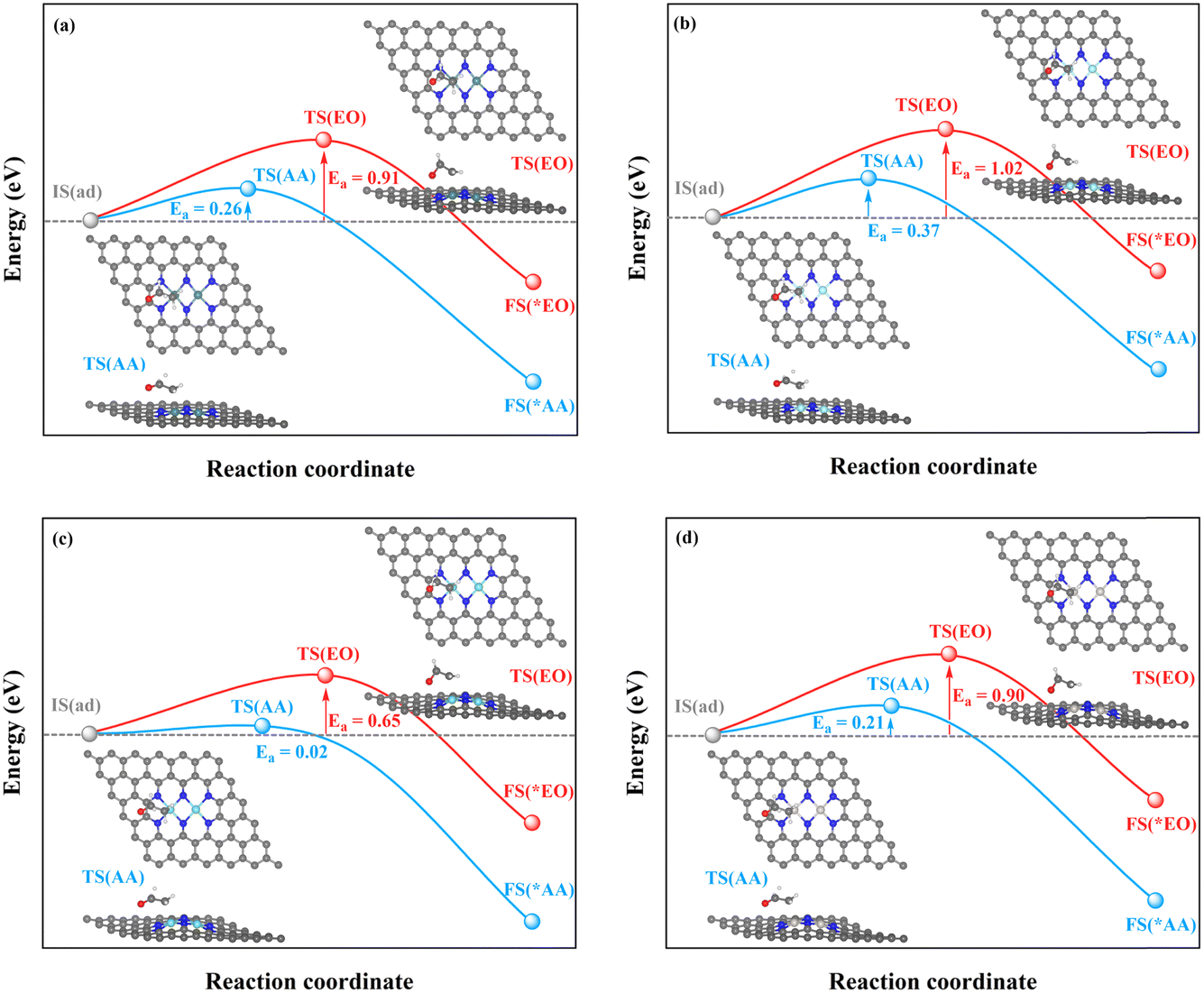 | ||
| Fig. 8 Kinetic energy barriers and transition-state structures of AA and EO generation through the C–TM synergistic mechanism on (a) Rh2-, (b) Ir2-, (c) Pd2-, and (d) Pt2N6@graphenes. | ||
Through the TM–TM mode the barriers for the formation of EO (1.91 and 1.74 eV) are lower than those of AA (1.98 and 1.83 eV) on Mn2- and Os2N6@graphenes, and the barriers for the formation of AA (1.20 and 1.71 eV) are lower than those of EO (1.61 and 1.81 eV) on Fe2- and Ru2N6@graphenes. Through the C–TM mode the barriers of AA (0.26 eV on Rh2-, 0.37 eV on Ir2-, 0.02 eV on Pd2-, and 0.21 eV on Pt2-, and 0.36 eV on Co2N6@graphenes, respectively) are lower than those of EO (0.91 eV on Rh2-, 1.02 eV on Ir2-, 0.65 eV on Pd2-, and 0.90 eV on Pt2-, and 1.06 eV on Co2N6@graphenes, respectively). Hence, these data indicate that the thermochemical ethylene oxidation process through the C–TM synergistic mechanism is superior to that through the TM–TM synergistic mechanism. In particular, both AA and EO generation on Pd2N6@graphene present very low kinetic energy barriers.
3.3 The origin of TM–TM and C–TM synergistic mechanisms
The intrinsic nature of ethylene electro-oxidation in our scheme is the stable existence of *O intermediates on active sites of TM2N6@graphene surfaces in a certain U–pH range during the electrolysis water (Fig. 3), the formation of the *OC2H4 intermediate through the TM–TM and C–TM synergistic mechanisms after the introduction of ethylene, and the execution of ethylene oxidation reactions towards *AA or *EO. During the whole process, it is crucial to understand *O stability on TM sites and C sites, which determines the following TM–TM and C–TM synergistic mechanisms on TM2N6@graphene surfaces.On the other hand, it is well known that the d-band center of a transition metal is an effective descriptor related to the adsorption behavior and the corresponding energy between the adsorbate and the substrate.49,50 The projected density of states (PDOS) of the d orbitals of the two metals in the 9 promising TM2N6@graphenes involved in the two reaction mechanisms are shown in Fig. S15.† In general, compared to the d orbital distributions of the 4 TM2N6@graphenes through the TM–TM mode (Fig. S15a–d†), those of the 5 TM2N6@graphenes (Fig. S15e–i†) through the C–TM mode are further away from the Fermi level. Therefore, the distinctive *O stability on different TM2N6@graphene surfaces can be attributed to their diversity of d orbital distributions in the metals, which is consistent with previous reports.26,27
To further explore the intrinsic correlation between the distribution of d orbitals and the adsorption energy of O at different sites, the relationship between the d-band center of metals in various TM2N6@graphenes and the energy difference of the most stable adsorption structure of O on the metal and C sites is shown in Fig. 9. There exists an approximately linear correlation between these two values on various TM2N6@graphenes except on Zn2N6@graphene, where f(x) = 0.86x + 2.88, R2 = 0.83. Moreover, OH is also adsorbed on these sites, and the relationship between the d-band center of the metals in the 9 promising TM2N6@graphenes involved in the two mechanisms in this study and the energy difference of the most stable adsorption structure of OH on these sites is another approximately linear correlation, as shown in Fig. S16,† where f(x) = 0.83x + 2.35, R2 = 0.76. This is close to the trend shown in Fig. 9. In fact, the narrow difference in energy between the occupied higher d-band center (closer to the Fermi level) of the metal and the unoccupied higher anti-bonding orbitals of the adsorbed molecule (closer to the Fermi level) will lead to stronger interaction and higher energy release after molecule adsorption.51 In addition, some of the electrons transfer from the metal to the O atom when the O atom is adsorbed on the metal, so the d-band center being much closer to the Fermi level results in many more electrons transferring to the O atom, which further stabilizes the adsorption of the O atom to the metal. These two reasons can explain why the metal sites of TM2N6@graphenes studied in this work exhibit lower competitive adsorption ability than C sites for active oxygen species as the d-band center of the metals decreases. As a result, a higher the d-band center results in stronger ability to bind an O atom. Moreover, the O atom must be separated from the binding site to generate AA or EO for the ethylene oxidation process. Therefore, the C–TM synergistic mechanism exhibits lower kinetic energy barriers than the TM–TM mechanism. These are the trends which correspond to the overall catalytic activity in Fig. 9 and S16,† where TM2N6@graphenes through the C–TM mode locate at the lower-left part around the lines, and TM2N6@graphenes through the TM–TM mode locate at the middle and upper-right part around the lines. That is, these data strongly support the proposition that ethylene electro-oxidation on TM2N6@graphenes through the C–TM synergistic mechanism possess higher catalytic activity and lower kinetic energy barrier than the process through the TM–TM synergistic mechanism. It is hoped that our findings on the C–TM versus the TM–TM synergistic mechanism can provide potential help to explore and screen other promising electro-catalysts.
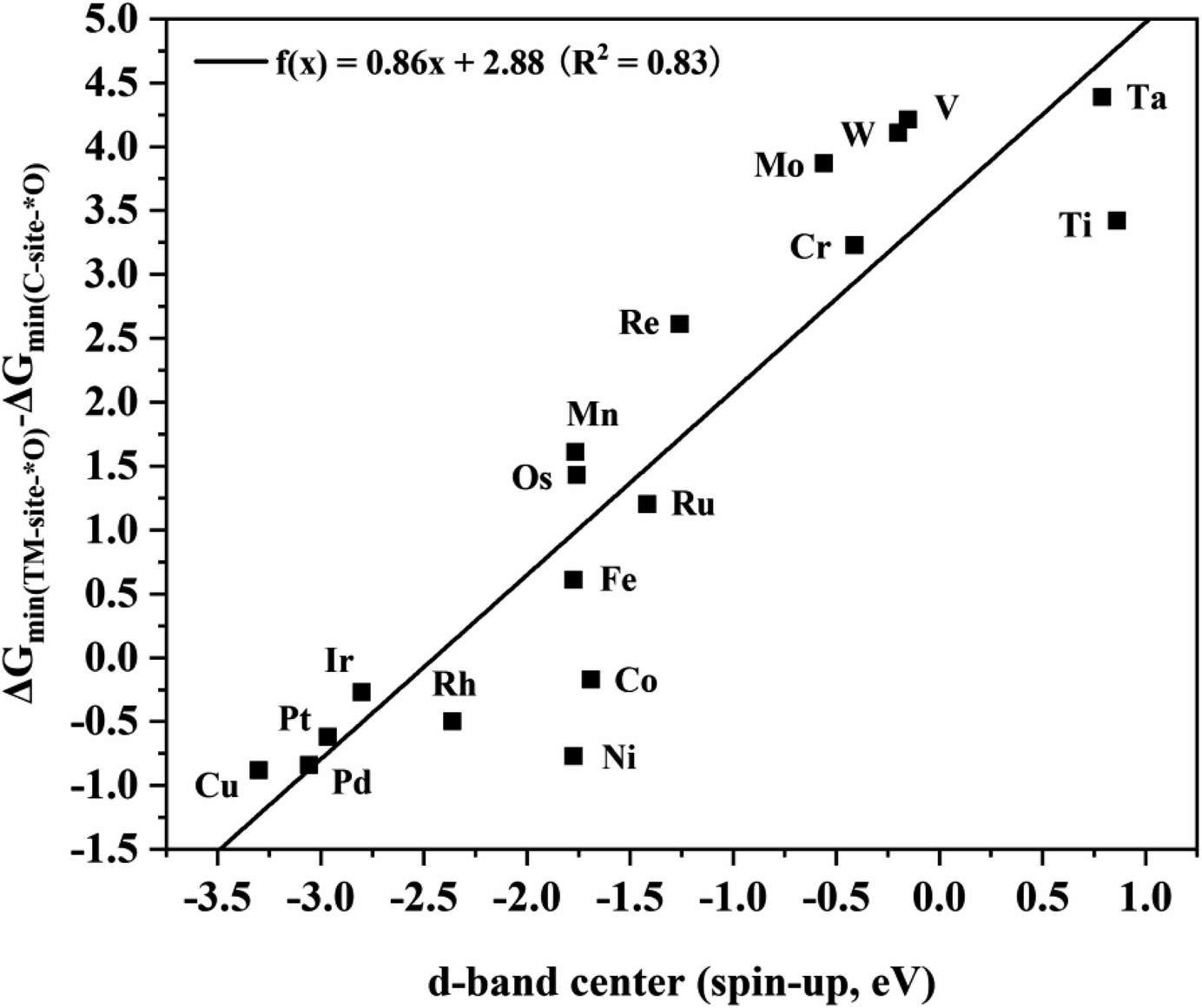 | ||
| Fig. 9 Linear relationship between the d-band center of metals in TM2N6@graphenes and the energy difference of the most stable O adsorption sites between the metal and C sites. | ||
Finally, the 9 promising TM2N6@graphene catalysts were studied by AIMD simulations at 300 K and 500 K for 10 ps, as shown in Fig. S17 and S18.† The frameworks of these catalysts were well maintained in their original configurations with the TMN6 moiety in the final snapshots of the AIMD simulations. These results further demonstrate their outstanding thermal stability at high temperature.
4 Conclusions
In this work, the feasibility of 19 homonuclear TM2N6@graphenes with dual-atomic-site catalysts for ethylene electro-oxidation to AA and EO through active oxygen intermediates generated by electrolysis of water was investigated in detail. Most of all, a reasonable strategy for generating an active oxygen intermediate is proposed that can exist stably during the electrolysis of water, and further oxidation of ethylene, thereby avoiding the potential drawbacks of traditional industrial processes. Fine promising TM2N6@graphenes including four through the metal–metal synergistic mechanism and five through the carbon–metal synergistic mechanism were selected according to the adsorption sites of oxygen-containing species, Pourbaix diagram analysis on their surfaces, the lowest applied potential for *O intermediate generation, and their thermodynamic and kinetic evaluation. For the TM–TM mode, the kinetic energy barriers are superior for EO formation on Fe2-, and Os2N6@graphenes, while they are more favorable for AA formation on Mn2-, and Ru2N6@graphenes. For the C–TM mode, all Rh2-, Ir2-, Pd2-, Pt2-, and Co2N6@graphenes possess lower formation barriers to both AA and EO. In particular, Pd2N6@graphenes have very low kinetic energy barriers of 0.02 and 0.65 eV for both AA and EO formation at an applied potential of 1.77 V vs. RHE for the generation of an *O intermediate. Electronic structure analysis indicates that there exists an approximately linear correlation between the d-band center of metals in TM2N6@graphenes and the energy difference of the most stable adsorption structure of O on the metal and C sites, where f(x) = 0.86x + 2.88, R2 = 0.83. The data reflect the intrinsic variance caused by the ability of the metal to adsorb the O atom, quantified by the d-band center of the metals decreasing, where TM2N6@graphenes through the C–TM mode locate at the lower-left part around the lines. Therefore, we believe that this work provides valuable clues for further experimental exploration and will stimulate more research to further explore the electro-oxidation of hydrocarbons.Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
Y. J. C. and M. Z. designed the research. Y. J. C. and C. Y. Z. demonstrated the initial idea and collected all the data. Y. J. C. wrote the paper. C. G. L., Y. G., Z. M. S. and M. Z. revised the paper and all authors commented on it.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (No. 21673036, 32130073 and 21373043), the Youth Development Foundation of Jilin Prov. (No. 20230508183RC), the China Postdoctoral Science Foundation (No. 2023M730539), the Natural Science Foundation of Jilin Province (YDZJ202201ZYTS540), the Fundamental Research Funds for the Central Universities (No. 2412022ZD050, No. 2412023QD012). Some computations were carried out on TianHe-2 at LvLiang Cloud Computing Center of China.Notes and references
- T. Pu, H. Tian, M. E. Ford, S. Rangarajan and I. E. Wachs, ACS Catal., 2019, 9, 10727–10750 CrossRef CAS.
- J. Shan, N. Janvelyan, H. Li, J. Liu, T. M. Egle, J. Ye, M. M. Biener, J. Biener, C. M. Friend and M. Flytzani Stephanopoulos, Appl. Catal., B, 2017, 205, 541–550 CrossRef CAS.
- W. Jeevapong, J. Sittiwong, M. Probst, B. Boekfa, C. Wattanakit, T. Maihom and J. Limtrakul, J. Phys. Chem. C, 2023, 127, 8473–8481 CrossRef CAS.
- K. Weissermel and H. Arpe, Industrial Organic Chemistry, John Wiley & Sons, 4th edn, 2003 Search PubMed.
- F. L. Quéméner, S. Barman, N. Merle, M. A. Aljuhani, M. K. Samantaray, Y. Saih, K. C. Szeto, A. D. Mallmann, Y. Minenkov, K. W. Huang, L. Cavallo, M. Taoufik and J. M. Basset, ACS Catal., 2018, 8, 7549–7555 CrossRef.
- S. Linic and M. A. Barteau, J. Catal., 2003, 214, 200–212 CrossRef CAS.
- M. Huš and A. Hellman, J. Catal., 2018, 363, 18–25 CrossRef.
- M. Chung, K. Jin, J. S. Zeng, T. N. Ton and K. Manthiram, J. Am. Chem. Soc., 2022, 144(38), 17416–17422 CrossRef CAS PubMed.
- L. L. Holbrook and H. wise, J. Catal., 1975, 38, 294–298 CrossRef CAS.
- J. Šebera, H. Hoffmannová, P. Krtil, Z. Samec and S. Záliš, Catal. Today, 2010, 158, 29–34 CrossRef.
- W. R. Leow, Y. Lum, A. Ozden, Y. Wang, D. H. Nam, B. Chen, J. Wicks, T. T. Zhuang, F. Li, D. Sinton and E. H. Sargent, Science, 2020, 368, 1228–1233 CrossRef CAS PubMed.
- J. Ke, J. Zhao, M. Chi, M. Wang, X. Kong, Q. Chang, W. Zhou, C. Long, J. Zeng and Z. Geng, Nat. Commun., 2022, 13, 932–940 CrossRef CAS PubMed.
- H. Li, C. S. Abraham, M. Anand, A. Cao and J. K. Nørskov, J. Phys. Chem. Lett., 2022, 13, 2057–2063 CrossRef CAS PubMed.
- H. Li, A. Cao and J. K. Nørskov, ACS Catal., 2021, 11, 12052–12057 CrossRef CAS.
- D. Chen, P. L. Kang and Z. P. Liu, ACS Catal., 2021, 11, 8317–8326 CrossRef CAS.
- E. A. Carbonio, T. C. R. Rocha, A. Y. Klyushin, I. Píš, E. Magnano, S. Nappini, S. Piccinin, A. Knop-Gericke, R. Schlögl and T. E. Jones, Chem. Sci., 2018, 9, 990–998 RSC.
- X. Fan, S. Tan, J. Yang, Y. Liu, W. Bian, F. Liao, H. Lin and Y. Li, ACS Energy Lett., 2022, 7, 343–348 CrossRef CAS.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- Y. J. Chu, C. Y. Zhu, X. Y. Zuo, C. G Liu, Y. Geng, Z. M. Su and M. Zhang, Appl. Surf. Sci., 2024, 652, 159362–159370 CrossRef CAS.
- W. Zhang, Y. Chao, W. Zhang, J. Zhou, F. Lv, K. Wang, F. Lin, H. Luo, J. Li, M. Tong, E. Wang and S. Guo, Adv. Mater., 2021, 33, 2102576–2102592 CrossRef CAS PubMed.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- Q. P. Zhao, W. X. Shi, J. Zhang, Z. Y. Tian, Z. M. Zhang, P. Zhang, Y. Wang, S. Z. Qiao and T. B. Lu, Nat. Synth., 2024, 3, 497–506 CrossRef.
- X. Zhao, F. Wang, X. P. Kong, R. Fang and Y. Li, J. Am. Chem. Soc., 2021, 143, 16068–16077 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2023, 123, 4855–4933 CrossRef CAS PubMed.
- J. Ren, B. Wang, H. Q. Yin, P. Zhang, X. H. Wang, Y. Quan, S. Yao, T. B. Lu and Z. M. Zhang, Adv. Mater., 2024, 2403101–2403110 CrossRef CAS PubMed.
- Y. Meng, C. Yin, K. Li, H. Tang, Y. Wang and Z. Wu, ACS Sustainable Chem. Eng., 2019, 7, 17273–17281 CrossRef CAS.
- Y. Meng, Y. Gao, K. Li, H. Tang, Y. Wang and Z. Wu, J. Phys. Chem. C, 2020, 124, 9142–9150 CrossRef CAS.
- Y. Zhou, W. Yang, W. Utetiwabo, Y. M. Lian, X. Yin, L. Zhou, P. Yu, R. Chen and S. Sun, J. Phys. Chem. Lett., 2020, 11, 1404–1410 CrossRef CAS PubMed.
- F. Li and Q. Tang, J. Mater. Chem. A, 2021, 9, 8761–8771 RSC.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- B. Hammer, L. B. Hansen and J. K. Norskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Chem. Phys., 2005, 319, 178–184 CrossRef CAS.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Henning, J. Chem. Phys., 2014, 140, 084106–084114 CrossRef PubMed.
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- H. A. Hansen, J. Rossmeisl and J. K. Nørskov, Phys. Chem. Chem. Phys., 2008, 10, 3722–3730 RSC.
- H. A. Hansen, I. C. Man, F. Studt, F. Abild-Pedersen, T. Bligaard and J. Rossmeisl, Phys. Chem. Chem. Phys., 2010, 12, 283–290 RSC.
- T. Lim, G. Y. Jung, J. H. Kim, S. O. Park, J. Park, Y. T. Kim, S. J. Kang, H. Y. Jeong, S. K. Kwak and S. H. Joo, Nat. Commun., 2020, 11, 412–422 CrossRef PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS.
- V. Wang, N. Xu, J. C. Liu, G. Tang and W. T. Geng, Comput. Phys. Commun., 2021, 267, 108033–108052 CrossRef CAS.
- M. He, W. An, Y. Wang, Y. Men and S. Liu, Small, 2021, 20, 2104445–2104452 CrossRef PubMed.
- H. Liu, Q. Huang, W. An, Y. Wang, Y. Men and S. Liu, J. Energy Chem., 2021, 61, 507–516 CrossRef CAS.
- P. Zhou, X. Hou, Y. Chao, W. Yang, W. Zhang, Z. Mu, J. Lai, F. Lv, K. Yang, Y. Liu, J. Li, J. Ma, J. Luo and S. Guo, Chem. Sci., 2019, 10, 5898–5905 RSC.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Zi. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 CrossRef CAS.
- S. Linic and M. A. Barteau, J. Am. Chem. Soc., 2002, 124, 310–317 CrossRef CAS PubMed.
- B. Hammer and J. K. Nørskov, Adv. Catal., 2000, 45, 71–129 CAS.
- H. Xin, A. Vojvodic, J. Voss, J. K. Nørskov and F. Abild-Pedersen, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 11514 CrossRef.
- Z. Ma, X. Liu, X. Wang, Z. Luo, W. Li, Y. Nie, L. Pei, Q. Mao, X. Wen and J. Zhong, Chem. Eng. J., 2023, 468, 143569–143577 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03944k |
| This journal is © The Royal Society of Chemistry 2024 |