
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphoester bond hydrolysis by a discrete zirconium-oxo cluster: mechanistic insights into the central role of the binuclear ZrIV–ZrIV active site†
Edinara
Luiz
 ab,
Francisco
de Azambuja
*a,
Albert
Solé-Daura
*c,
Jordi
Puiggalí-Jou
c,
Angelo
Mullaliu
a,
Jorge J.
Carbó
c,
Fernando R.
Xavier
d,
Rosely A.
Peralta
*b and
Tatjana N.
Parac-Vogt
*a
ab,
Francisco
de Azambuja
*a,
Albert
Solé-Daura
*c,
Jordi
Puiggalí-Jou
c,
Angelo
Mullaliu
a,
Jorge J.
Carbó
c,
Fernando R.
Xavier
d,
Rosely A.
Peralta
*b and
Tatjana N.
Parac-Vogt
*a
aDepartment of Chemistry, KU Leuven, Celestijnenlaan 200F, Leuven, 3001, Belgium. E-mail: francisco.deazambuja@kuleuven.be; tatjana.vogt@kuleuven.be
bDepartamento de Química, Universidade Federal de Santa Catarina, Florianópolis, Santa Catarina 88040-900, Brazil. E-mail: rosely.peralta@ufsc.br
cDepartament de Química Física i Inorgànica, Universitat Rovira i Virgili, Marcel·lí Domingo 1, Tarragona, 43007, Spain. E-mail: albert.soled@urv.cat
dDepartamento de Química, Universidade do Estado de Santa Catarina, Joinville, Santa Catarina 89219-710, Brazil
First published on 2nd October 2024
Abstract
Effective degradation of non-natural phosphate triesters (PTs) widely used in pesticides and warfare agents is of paramount relevance for human and environmental safety, particularly under acidic conditions where they are highly stable. Here, we present a detailed reactivity and mechanistic study pioneering discrete {Zr6O8} clusters, which are commonly employed as building blocks for Zr-MOFs and as non-classical soluble coordination compounds for the degradation of PTs using the pesticide ethyl paraoxon as a model. Combined computational studies, mechanistic experiments, and EXAFS analysis show that the reactivity of these clusters arises from their ZrIV–ZrIV bimetallic sites, which hydrolyze ethyl paraoxon under acidic conditions through an intramolecular pathway. Remarkably, the energetics of the reaction is dependent on the protonation state of the active sites, and a weakly acidic medium favors the reaction. Moreover, catalyst stability allowed for its recovery and reuse. Such a mechanism is in close analogy to enzymatic reactions and different from that previously reported for Zr-MOFs. These findings outline the potential of MIV–MIV active sites for PT degradation under challenging aqueous acidic conditions and contribute to the development of bioinspired catalysts and materials.
1. Introduction
Metallohydrolases are an essential component of nature's machinery to manipulate highly stable chemical bonds such as peptide and phosphate linkages that are ubiquitous in proteins and nucleic acids. However, they are inadequate to deal with unnatural man-made compounds such as the useful but hazardous phosphate triesters (PTs) widely present in pesticides and warfare agents.1 The high toxicity and wide use of PTs pose potential environmental and biological hazards, as the high chemical stability of P–O bonds renders them greatly persistent, particularly under neutral and acidic conditions.2 Thus, many synthetic analogs of active sites of metalloenzymes have been developed to degrade PT compounds in a safe and environmentally benign manner.3,4 These compounds have long relied on coordination complexes featuring a bimetallic active site similar to those found in naturally occurring phosphotriesterases (PTEs),1,5 one of the few enzymes capable of hydrolyzing PT compounds and purple acid phosphatases (PAPs).6–8 In general, these compounds perform better at pH > 5 and have greatly contributed to understanding enzymes' hydrolytic mechanisms2,9–12 but still exhibit poor reactivity towards phosphate triesters such as the broadly used pesticide ethyl paraoxon (diethyl-4-nitrophenylphosphate, 1).In this context, we envisioned that the stronger Lewis acid character of MIV–MIV active sites could offer distinctive reactivity for the degradation of phosphate triesters under mildly acidic conditions more relevant for environmental remediation and for potential applications in biotechnology and medicine.13–17 While many (nano)materials have been recently developed for the hydrolysis of phosphate esters,2,18,19 the hydrolysis of PTs in solutions at neutral pH or lower has been typically addressed using coordination compounds containing metal pair combinations such as MII–MII, MII–MIII, or MIII–MIII (MII = Zn, Mn, Co, Cu; MIII = Fe, Mn, Ga).9,10,20–25 However, compounds containing MIV–MIV active sites have rarely been explored.18 Critically, MIV–MIV active sites are present in Zr-based metal–organic frameworks (Zr-MOFs)26 like NU-1000 and UiO-66, and their ability to hydrolyze phosphate triesters has been broadly developed. Nevertheless, they have been always explored as PTE mimics,27–36 showing great efficiency in hydrolyzing the phosphate ester bond5,37 under strongly alkaline conditions (generally at pH ∼9).38–45 Meanwhile, the potential of these MIV–MIV active sites to mimic the reactivity of PAPs and hydrolyze PT compounds under non-alkaline conditions has been largely overlooked, leaving a critical knowledge gap for the development of technologies dependent on mildly acidic conditions, such as in environmental science and medicine.13–17 Thus, considering our interest in developing and understanding the molecular reactivity of metal oxo cluster based nanozymes towards biomolecules,19,46–48 we seized the opportunity to investigate the reactivity of discrete hexanuclear zirconium oxo clusters49 towards PTs as these rather underexplored species afford straightforward air and moisture stable MIV–MIV active sites and would allow us to study them under acidic conditions as potential acid phosphatase mimetic catalysts (Fig. 1).6,9,10,13,20,23,50–52
 | ||
| Fig. 1 (a) Metalloenzymes involved in P–O hydrolysis and their active sites; (b) examples of PAP and PTE biomimetic coordination compounds; (c) Zr-MOFs reported as catalysts for P–O bond hydrolysis (primarily under basic conditions); and (d) the unique design of discrete {Zr6O8} cluster catalysts studied in this work. | ||
In this work, we report a pioneering account of the catalytic activity of discrete Zr oxo clusters towards phosphate esters combining detailed kinetics and mechanistic experiments with density functional theory calculations (DFT). The substrate ethyl paraoxon (diethyl-4-nitrophenylphosphate, 1) was used as the model substrate, given its broad use as a pesticide. Remarkably, discrete {Zr6O8} clusters bear striking functional and mechanistic similarities to PAPs, which are effective catalysts even under rarely used acidic conditions. Under these conditions, they also surpassed the performance of zirconium salts and related Zr-MOFs without requiring additional co-catalysts or bases to control the acidity or boost the reactivity. Due to their soluble and discrete nature, such clusters also allowed for mechanistic insights which are difficult-to-obtain in heterogeneous reactions involving MOFs.53–55 Therefore, they provide key experimental and computational evidence complementing mechanisms previously addressed mostly with theoretical calculations31,38,56 and, in perspective, provide fertile ground for the molecular design of new types of bioinspired catalysts using cluster compounds.
2. Experimental section
More information about instrumentation, cluster characterization, EXAFS, DFT calculation and isotopic effects is provided in the ESI.†2.1 Synthesis of [Zr6O4(OH)4(C2H3O2)8(H2O)(OH)Cl3] (Zr6)
The [Zr6O4(OH)4(C2H3O2)8(H2O)(OH)Cl3] (Zr6) cluster was synthesized according to Dai et al. (2021).57 ZrCl4 (10 g) was added into a mixture of CH3COOH (15 mL) and isopropanol (25 mL); after homogenization, the reaction was stirred at 500 rpm under reflux in an oil bath at 120 °C for 60 min. After being cooled to room temperature, the cluster was collected through centrifugation at 5000 rpm for 30 min. The collected white solid was subsequently washed twice with acetone (15 mL for 2 hours each time) and allowed to air dry for 3 days before characterization. The product was characterized by PXRD, FTIR and 1H NMR of a 1 mol per L NaOD digested sample. The analysis results agreed with previously published characterization data.55,572.2 Hydrolysis reaction of paraoxon (diethyl-4-nitrophenylphosphate, 1) using clusters
In a 1.5 mL centrifuge tube, the Zr6 cluster (2.0 mg; 1.5 μmol) was dissolved in 1 mL of solvent (HEPES 0.1 mol L−1 pH = 7.0, or distilled water). The solution's pH was measured, and 6 μL of 1 (25 μmol) was added. The reaction was carried out in a thermos shaker at 60 °C. Samples (10 μL) were collected at specific time points after quickly centrifuging the reaction mixture (3 min, 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm), and immediately diluted with water (1 mL) and analyzed using a UV/vis spectrophotometer. The reaction was allowed to continue for 31 h, and the pH was measured again at this time point. The same procedure was followed for experiments carried out at 37 °C.
000 rpm), and immediately diluted with water (1 mL) and analyzed using a UV/vis spectrophotometer. The reaction was allowed to continue for 31 h, and the pH was measured again at this time point. The same procedure was followed for experiments carried out at 37 °C.
Experiments involving other Zr6 and Zr12 clusters and MOFs NU-1000 and UiO-66 were performed as described for Zr6 using 6 mol% catalyst in the solid state. Likewise, investigation of isotopic effects on the hydrolysis rate promoted by Zr6 was done under the same conditions described above but using deuterated water/buffer (HEPES 0.1 mol L−1 – pH 6.0) as the reaction solvent.
2.3 Hydrolysis reaction of 1 in the absence of clusters
Spontaneous hydrolysis was examined by carrying out reactions at 60 °C under identical conditions as described above for Zr6 but without the addition of any catalyst. 1 (6 μL, 25 μmol) was added into water (pH 3.0 corrected with HCl, 37%) or buffer (HEPES 0.1 mol L−1 pH = 7.0). As for Zr6, samples (10 μL) were collected at specific time points, diluted in water, and analyzed using a UV/vis spectrophotometer.3. Results and discussion
3.1 Paraoxon hydrolysis
To explore the reactivity of zirconium oxo clusters towards phosphate triester degradation, the [Zr6O4(OH)4(C2H3O2)8(H2O)(OH)Cl3] (Zr6) cluster (Fig. 1d) was selected as a model catalyst as it can be readily prepared from inexpensive ZrCl4 and acetic acid.57 This cluster has a {Zr6O4(OH)4} inorganic core equivalent to the nodes present in several Zr-MOFs previously shown to hydrolyze P–O bonds.58 It features an octahedral arrangement of six Zr centers bound to each other by μ3-O(H) groups situated in the faces of the octahedron and capped mostly by carboxylate ligands. The selected Zr6 cluster was synthesized as previously reported57 and characterized by infrared (IR) spectroscopy and powder X-ray diffraction (PXRD), and after basic digestion by 1H nuclear magnetic resonance (1H NMR) spectroscopy (see the ESI for details – Fig. S1–S3†). As a model reaction for this study, the hydrolysis of paraoxon (1), a phosphate triester largely used as a pesticide, was used, and the reaction progress was followed by UV/vis spectroscopy (Scheme 1a). To ensure the reliability of UV/vis measurements, we also confirmed by 1H and 31P NMR that hydrolysis of 1 using Zr6 only afforded products 2 and 3, and no side-products were detected (Fig. S6–S8†).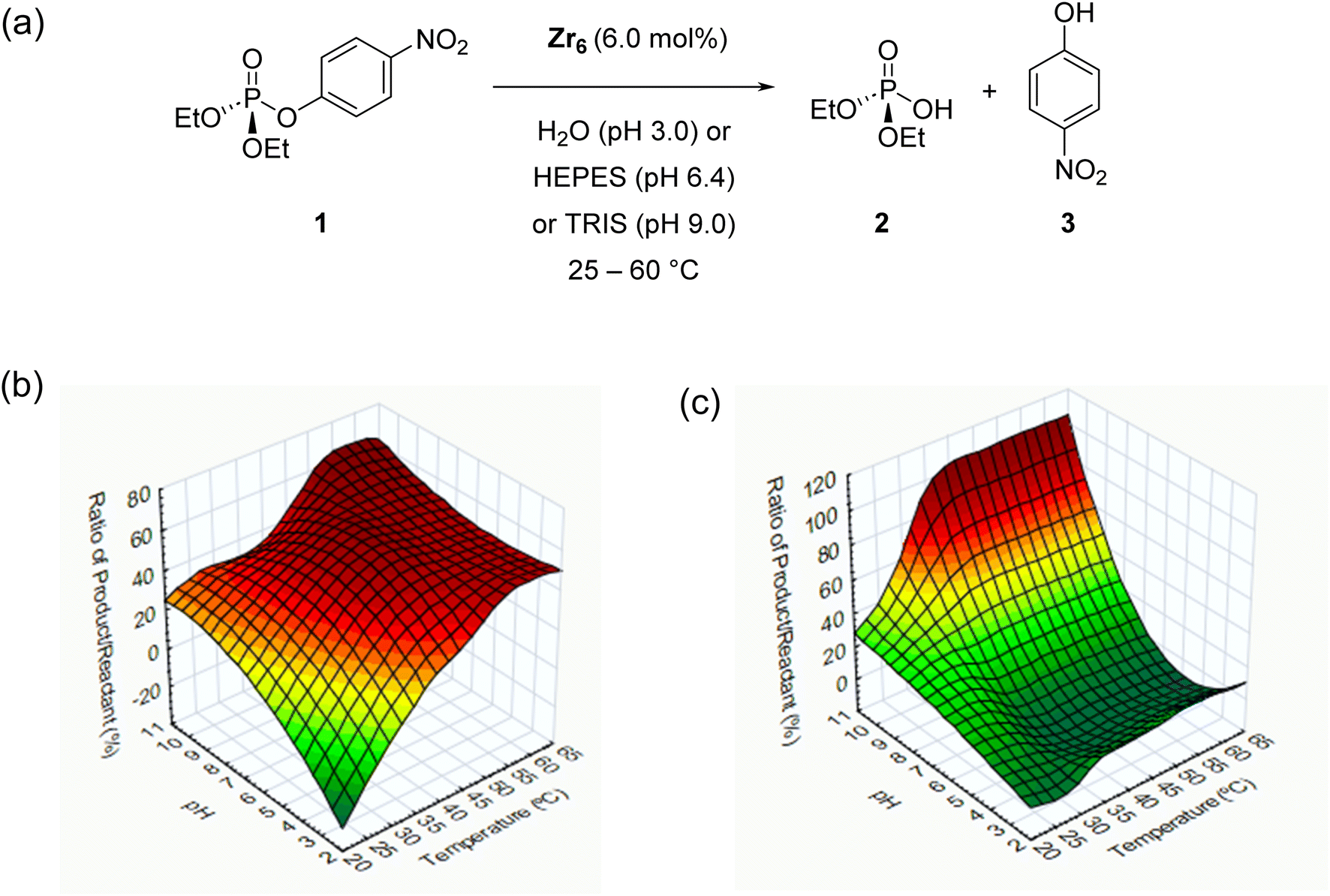 | ||
| Scheme 1 (a) Reaction conditions studied for the hydrolysis of paraoxon (1) with the Zr6 cluster. (b) Results of initial screening using Zr6 as the catalyst. (c) Results of spontaneous hydrolysis of (1) under the same screening conditions. Results are presented as the percentage ratio of product/reactant by integration of peaks in 1H NMR data. Green color represents lower yields, while red color represents higher yields. | ||
Considering that the hydrolytic activity of discrete zirconium oxo clusters (ZrOC) towards phosphoesters has not been studied previously, an initial screening of conditions using different temperatures (25, 37 or 60 °C) and pH (3.0, 6.4 or 9.0) was performed using a 6 mol% catalyst (see the ESI† for details). The reaction under acidic conditions was performed by simply adding the catalyst to water, as the deprotonation of water molecules coordinated to ZrIV centers promptly acidifies the solution to pH 3.0.59 For reactions at neutral and higher pH values, buffer solutions (HEPES 0.1 mol L−1, pH 7.0 and Tris 0.1 mol L−1, pH 9.8) were used. The presence of buffer was sufficient to minimize the influence of Zr6 induced acidification, resulting in pH 6.4 for the solution in HEPES buffer and pH 9.0 for Tris buffer.
Both temperature and acidity influence the reactivity of Zr6 towards 1, and in general, increasing both the temperature and pH afforded more product (Scheme 1b). Interestingly, hydrolysis was observed even under acidic conditions, which has rarely been reported for Zr-MOFs.31 More specifically, results have shown no hydrolytic activity at 25 °C and 37 °C at pH 3.0. However, the activity of the catalysts increased when the pH was increased to 6.4 or under basic conditions. The same trend was observed at 60 °C, but unlike for low temperatures, Zr6 showed hydrolytic activity even at pH 3.0. Control experiments in the absence of catalyst (Scheme 1c) under similar pH and temperature conditions revealed that uncatalyzed spontaneous hydrolysis of 1 is facile at high pH and temperature and does not need a catalyst to occur. Therefore, substantial influence of Zr6 on the hydrolysis of 1 is observed only in solutions incubated at 37 °C and pH 6.40, or at 60 °C and pH 3.0–6.4 (Scheme 1b).
Compared to alkaline conditions, acidic conditions have been rarely explored with Zr-MOFs, which is rather unusual, given that the spontaneous hydrolysis of 1 is significantly enhanced at high pH values.28,38,60,61 Moreover, reactions under alkaline conditions usually require basic buffers (typically ∼0.45 mol L−1N-ethylmorpholine) which also assist in the hydrolysis. Thus, such unique activity of Zr6 complements the reactivity developed with Zr-MOFs by providing the means to hydrolyze P–O bonds in a wide range of pH. Moreover, in Zr6, P–-O bonds do not require any additional basic additives or buffers (at pH 3.0) to enable or boost reactivity, significantly simplifying previously reported conditions. Therefore, based on these initial results, further reactions were carried out at pH 3.0 and 6.4 at 37–60 °C temperature (Scheme 1) to develop and understand the hydrolysis of phosphate triesters under non-alkaline conditions.
The temperature effect is more significant for reactions conducted at pH 3.0 than for those performed at pH 6.4, even though both reactions are enhanced at higher temperatures. To further investigate the influence of temperature on the hydrolysis of 1 by Zr6, the yields after 5 and 24 h of reaction were compared for reactions performed at 37 °C and 60 °C (Fig. 2). As expected, the reactions at 37 °C were less efficient than those at 60 °C under both pH conditions, and reactions at pH 6.4 were faster compared to those at pH 3.0, as evidenced by the yields observed after 5 hours. Nevertheless, at 60 °C, the reaction conducted at pH 3.0 significantly improved, and after 24 h it afforded a similar yield to the one carried out at pH 6.4. For the reaction at pH 3.0, the yield after 24 h is 12 times higher at 60 °C than at 37 °C. On the other hand, at pH 6.4, the yield of hydrolysis only increases by 3-fold upon changing the reaction temperature from 37 °C to 60 °C. Such a distinct influence of temperature on the reaction yield suggests that reactions performed at pH 3.0 and pH 6.4 follow slightly distinct reaction pathways. Interestingly, these temperature-induced enhancements were not observed in the absence of catalyst (white columns in Fig. 2), confirming that increasing temperature accelerates the catalytic reaction. These results ultimately suggest a transition state (TS) higher in energy at pH 3.0 than at pH 6.4. Although more experimental data are required to clarify this point, this observation aligns with both prior studies on phosphatase reaction mechanisms and with our computational analysis (vide infra).28,31
 | ||
| Fig. 2 Increasing the temperature enhances Zr6-catalyzed hydrolysis of 1 more significantly at 60 °C than at 37 °C. Yield of 1 hydrolysis after 5 (light colors) and 24 h (dark colors). Conditions: [Zr6] = 1.5 mmol L−1; [1] = 25 mmol L−1; solvent (1 mL – water (pH 3.0)) or HEPES 0.1 mol L−1 (pH 6.4). White columns correspond to the reaction performed without Zr6 (spontaneous hydrolysis). In the absence of catalysts, negligible amounts of product 3 were observed at 37 °C. | ||
Given the Zr6-induced acidification of aqueous solutions and the pH dependence of temperature effect, the pH of the reaction solution was re-measured after the reactions reached saturation. Unsurprisingly, a decrease in the pH of the solution was observed for all reactions, and in general, higher reaction temperatures caused bigger pH changes. For example, at 60 °C, the initial pH 3.0 for reactions carried out in water dropped to 2.0 after 31 h, while the pH for the hydrolysis conducted in HEPES buffer dropped from pH 6.4 at the beginning of the reaction to pH 4.2 at the end. However, at 37 °C, the changes were not as significant when buffer was used, with pH changing from 6.4 to 5.3. Under acidic conditions, the pH variation was even smaller, dropping from 3.0 to 2.7. These variations correlate well with the amount of product formed, which has a pKa of 1.42,62 and hint that the pH changes during reaction are related to the formation of diethyl phosphoric acid (2) (Scheme 1).
Interestingly, the influence of product formation on the pH reaction seems to affect the rate of product formation as well (Fig. 3a). At 60 °C, reactions performed at both pH 3.0 and pH 6.4 reached saturation at a similar concentration of the product (ca. 19% yield; 5.39 × 10−3 mol L−1; TON = 3.6). However, the initial rate of reaction at pH 6.4 was approximately 8 times higher, which reached saturation after only 6 h (TOF = 7.5 × 10−3 min−1), while the reaction at pH 3.0 needed 24 h to afford the same amount of product (TOF = 1.93 × 10−3 min−1). These results might be linked to the acidification of solution by product 2, as its deprotonation would afford a phosphate ligand which has greater affinity for ZrIV centers compared to the original carboxylate capping ligands present in Zr6.63,64 Notably, such limited reaction is not unusual in phosphotriester hydrolysis and has also been observed for Zr-MOFs (see discussion below).
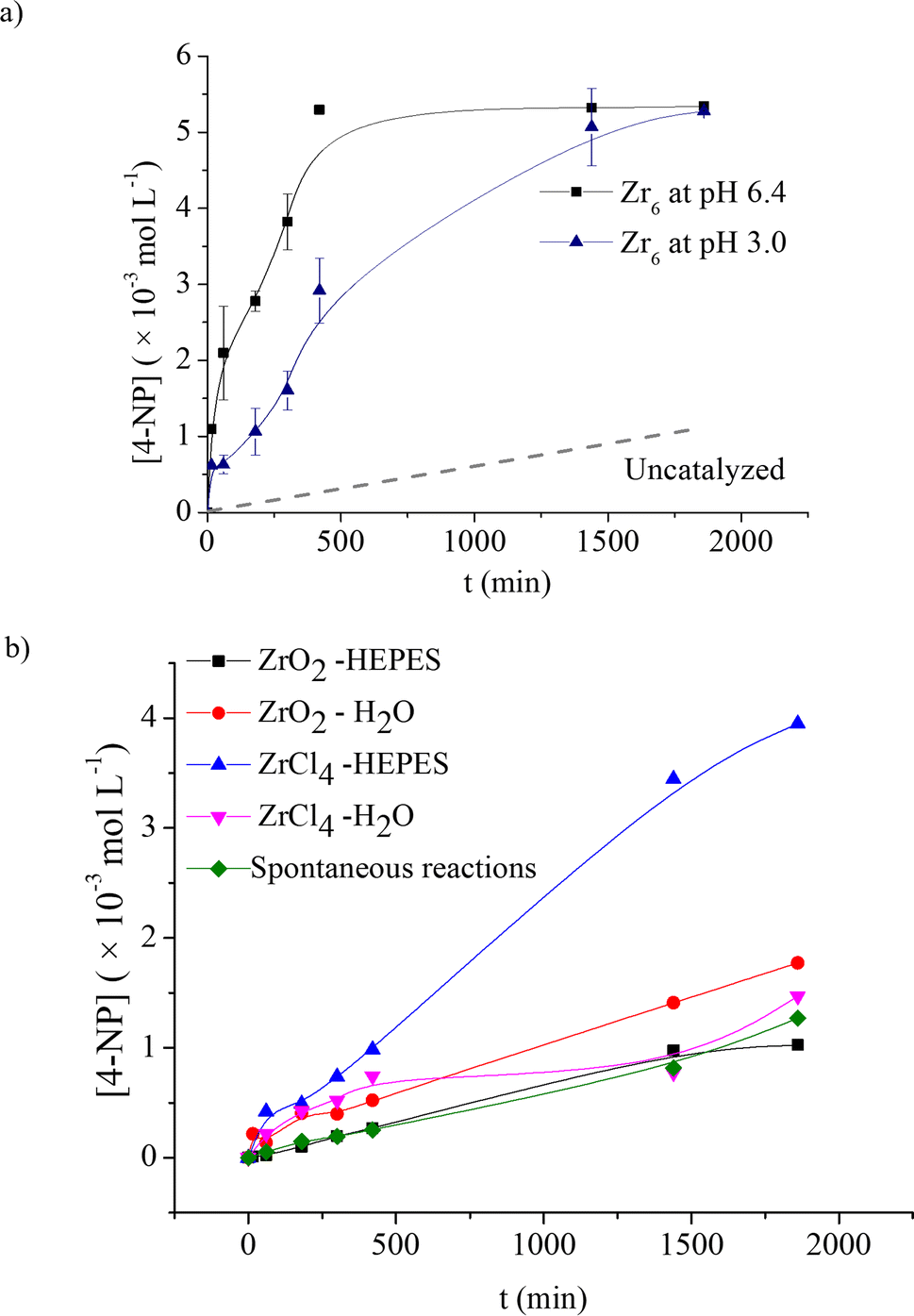 | ||
Fig. 3 (a) Zr6 catalyzed hydrolysis of 1 at 60 °C and pH 3.0 (blue  ) and 6.4 ( ) and 6.4 ( black). Conditions: [Zr6] = 1.5 mmol L−1; [1] = 25 mmol L−1 l; solvent (1 mL – water (pH 2.7–3.0) or HEPES 0.1 mol L−1 (pH 6.4–6.7)); 60 °C. (b) Control experiments featuring hydrolysis of 1 at 60 °C catalyzed by Zr salts (ZrO2 and ZrCl4) at pH 3.0 (ZrO2 – red circles black). Conditions: [Zr6] = 1.5 mmol L−1; [1] = 25 mmol L−1 l; solvent (1 mL – water (pH 2.7–3.0) or HEPES 0.1 mol L−1 (pH 6.4–6.7)); 60 °C. (b) Control experiments featuring hydrolysis of 1 at 60 °C catalyzed by Zr salts (ZrO2 and ZrCl4) at pH 3.0 (ZrO2 – red circles  ; ZrCl4 – pink triangles ; ZrCl4 – pink triangles  ) and pH 6.4 (ZrO2 – black squares ) and pH 6.4 (ZrO2 – black squares  ; ZrCl4 – blue triangles ; ZrCl4 – blue triangles  ). Spontaneous hydrolysis at both pH 7.0 (HEPES 0.1 mol L−1) and pH 3.0 is presented by green squares ( ). Spontaneous hydrolysis at both pH 7.0 (HEPES 0.1 mol L−1) and pH 3.0 is presented by green squares ( ). Conditions: [ZrCl4] = [ZrO2] = 1.5 mmol L−1; [1] = 25 mmol L−1 l; solvent (1 mL – water (pH 2.7–3.0) or HEPES 0.1 mol L−1 (pH 6.4–6.7)); 60 °C. ). Conditions: [ZrCl4] = [ZrO2] = 1.5 mmol L−1; [1] = 25 mmol L−1 l; solvent (1 mL – water (pH 2.7–3.0) or HEPES 0.1 mol L−1 (pH 6.4–6.7)); 60 °C. | ||
Analysis results of clusters recovered after the reaction are consistent with the affinity of product 2 for the catalyst, as strong P–O bands (1034 cm−1) were visible in the infrared spectrum (Fig. S12†), and a triplet at 1.28 ppm and an apparent quintet at 3.93 ppm corresponding to 2 were seen in the 1H NMR spectrum after basic digestion of the recovered solid (Fig. S13†). A change in buffer solution could be an option to overcome this catalyst inhibition, as suggested by the higher yield (56% after 24 h) we observed in bis-Tris pH 7.0, 0.1 mol L−1 solution when probing the effect of different buffer solutions on the reactivity (Fig. S9†). Even though this result strongly suggests that the low yield observed in our study is most likely a matter of reaction optimization to enhance catalyst turnover, for the sake of experimental consistency, we continued our study using a HEPES buffer solution for reactions at pH 6.4.
 | ||
| Fig. 4 Fourier transforms from the EXAFS extracted signals for recovered clusters after reaction at (a) pH 3.0 and (b) pH 6.4. The experimental signals are compared to the calculated ones, highlighting the good match between the experimental structure and adopted model. | ||
3.2 Catalyst structure vs. activity
Once the main aspects of Zr6 reactivity towards 1 were elucidated, a detailed investigation to probe the structure–activity relationship of Zr oxo clusters for the hydrolysis of phosphate triesters was carried out. To accomplish that, several other Zr oxo clusters featuring different nuclearities and carboxylate capping ligands were used to hydrolyze 1 at pH 3.0 and 60 °C, and the yields were measured after 5 and 24 h, as done previously (Fig. 5). Considering the unexpected saturation of reaction at mere 19% yield, we have also included Zr-based MOFs NU-1000 and UiO-66 in this stage of the study to probe the effect of the extended MOF network on the reactivity of Zr oxo clusters. | ||
| Fig. 5 Hydrolysis of 1 using different Zr oxo clusters. (a) Representative structure of Zr6 and Zr12 clusters tested (blue polyhedra - Zr6O8, gray circles - carbons); (b) capping ligands of Zr6 and Zr12 clusters tested; (c) yield (%) of 2 after 5 h (blue) and 24 h (black) for each cluster/MOF catalyst. Conditions: [cluster] = 1.5 mmol L−1; [MOF] = 1.5 mmol L−1; [1] = 25 mmol L−1; 60 °C; in water (1 mL, pH = 3.5–4.1). 19% conversion corresponds to 5.39 × 10−3 mol L−13. | ||
To investigate whether the cluster structure influences the reactivity, we focused on eight structurally related clusters (four Zr6 and four Zr12 clusters) to verify the effect of different nuclearities and capping ligands on the hydrolysis of 1 (Fig. 5b). Although several Zr oxo clusters featuring various carboxylate ligands have been reported, an exact comparison between hexanuclear clusters {Zr6O4(OH)4} and their dimeric derivatives {Zr6O4(OH)4}2 is difficult as the substitution pattern of capping ligands dictates their unique structural chemistry.65 In general, the hydrolysis of 1 is not influenced by cluster nuclearity. As for Zr6, a similar pH decrease was observed for all reactions, reinforcing the finding that this change arises from product 2 formation (Table S3†). More importantly, similar to Zr6, all clusters provided product 3 in the same 20% yield range after 24 h. Moreover, for clusters with similar capping ligands such as the Zr6/Zr12(OAc)24 and Zr6(OMc)12/Zr12(OAcr)24 pairs, a similar yield was observed. As all catalysts were used at the same concentration, meaning that the Zr12 cluster could afford twice as many Zr6 once present in the solution, similar yields suggest that Zr6 and Zr12 are not interconverting under the conditions used, which clearly indicates that the nuclearity has little effect on the catalytic activity. This is an interesting contrast to previous reports in organic medium where both Zr6–Zr12 undergo interconversion,65 and a slightly better performance of Zr12 has been reported.54 The similar behavior of Zr6 and Zr12 clusters is easily explained by evaluating their structure. Zr12 clusters are formed by dimerization of two Zr6 units connected by inter-cluster carboxylates,65 making the six zirconium atoms in the center unavailable to perform the hydrolytic reaction. This also suggests a similar general mechanism for all clusters, regardless of their organic ligand.
In contrast to the cluster nuclearity, the nature of capping ligands clearly affects the reactivity, and the difference in hydrolysis yields after 5 h suggests a capping ligand effect on reaction rates. For example, the acetate ligand affords the lowest 5 h yield among the series, regardless of the cluster nuclearity. On the other hand, aliphatic and acrylic acids afforded yields that were much closer to the 19% mark, though with small differences between α-substituted ligands (2-MeBuCOO vs. But; OMc vs. OAcr). This indicates that these reactions are faster compared to clusters with acetate ligands, which could arise from an easier ligand exchange during the reaction. Previously, enhanced ligand lability was attributed to acidification of the reaction medium.31 However, all reactions showed a similar pattern of pH variation (Table S3†), and the potential differences in ligand lability are more likely caused by the steric hindrance, as it is well established for ordinary coordination compounds.66 Curiously, Zr6(BzO)12 performed better than Zr6 (Zr6(OAc)8) after 5 h (Fig. 5c), in line with other clusters, but provided a lower yield after 24 h. This indicates that increasing the steric hindrance of capping ligands to speed up ligand exchange is beneficial only up to a certain size, after which it probably starts hampering access of the substrate to the cluster inorganic core.
More importantly, Zr-MOFs which feature Zr6 clusters as active sites also showed a similar behavior (Fig. 5c). Specifically, reactions under the same conditions performed with {Zr6O8}-based NU-1000 and UiO-66 catalysts, also showed saturation at ca. 20% yield, even at pH 6.40 (Fig. S16†). Although results showed that the nature of the MOF may affect the reaction, the similar yield limitation observed showcases how the intrinsic reactivity of Zr-MOFs and Zr oxo clusters follows related trends. Remarkably, even under alkaline conditions, the cluster and UiO-66 MOF showed similar tendency, that is, the reactions reached saturation after four hours, though yields increased to 29% and 43%, respectively (Fig. S17†). This hints that the outstanding reactivity of the MOF towards organophosphates reported elsewhere might be more related to the pH of the reaction medium or the presence of an additional base rather than to an intrinsic superior reactivity of the porous extended structure of MOFs.40,67 Notably, similar conversion limitation has also been observed for other types of well-established MOF catalysts featuring different nuclearities and substrates, despite their promising catalytic rates under similar pH conditions.68
3.3 Mechanistic considerations
Considering the reactivity observed under acidic conditions discussed above, we envision that the mechanism of hydrolysis of 1 by Zr6 could be similar to the one generally accepted for PAP enzymes (Fig. 1). In these enzymes, a phosphoester substrate coordinates to the MII center displacing a water ligand and positioning the substrate for an intramolecular attack. Even though there is some debate about the reaction nucleophile,11,13,69 a well-accepted mechanism relies on the established addition–elimination mechanism typical of phosphatases and suggests an intramolecular attack by the neighboring MIII–OH moiety.13 Likewise, 1 could coordinate to the Zr6 cluster displacing a water molecule in one ZrIV center and being attacked by a ZrIV–OH moiety next to it. Notably, this mechanism is conceptually distinct from the ones previously reported for MOF catalysts,27,60 where the substrate is also activated by coordination to a ZrIV center, but a general base mechanism operates due to the alkaline conditions used.31 However, since negligible amounts of hydroxide are present in the acidic conditions used in this work, we initially assumed that Zr6 probably follows an intramolecular path similar to PAP enzymes (Scheme 2). Different pieces of evidence comprising reactivity trends, structural features, titration experiments, and calculations presented below were consistent with this hypothesis.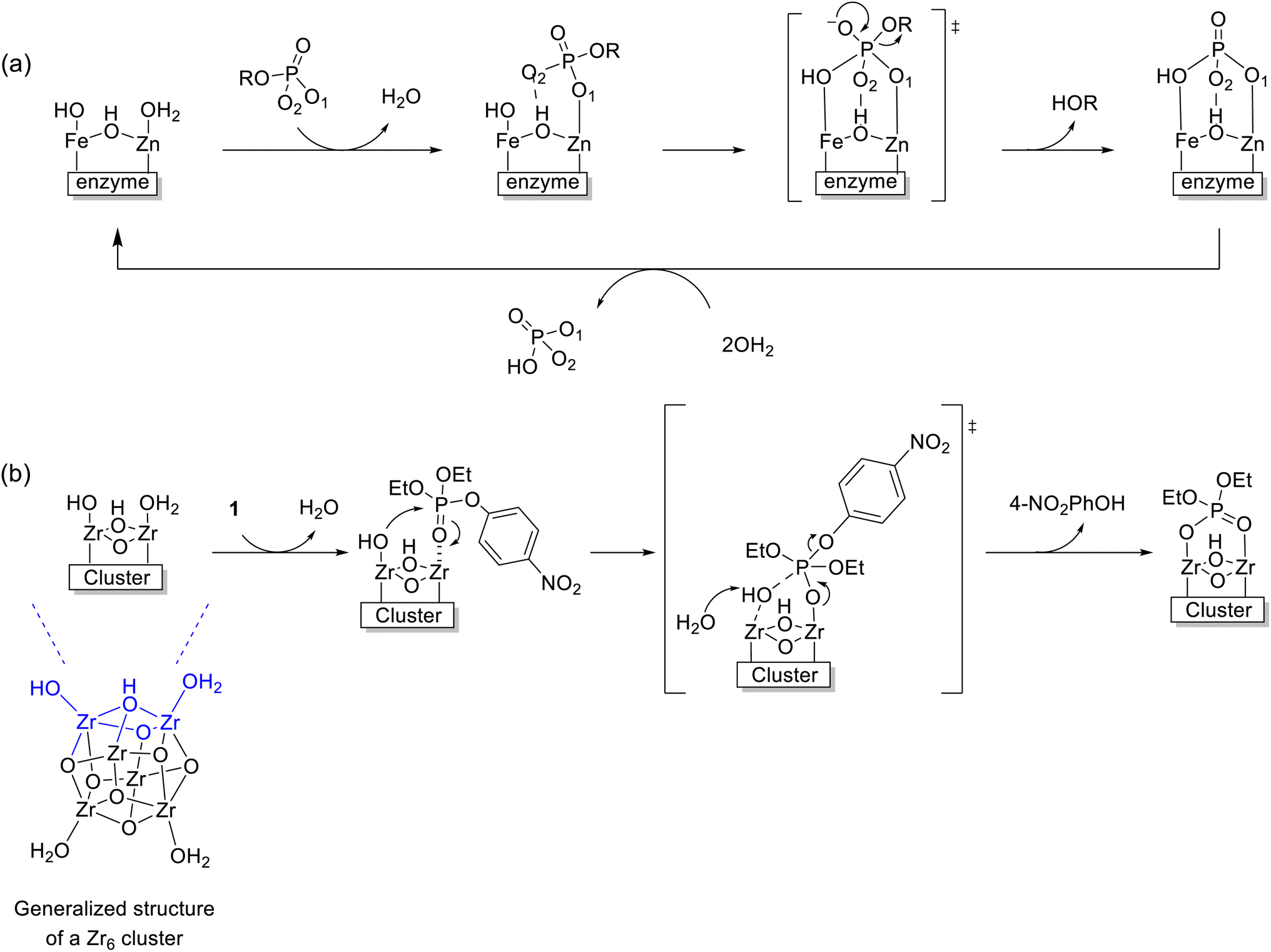 | ||
| Scheme 2 (a) Intramolecular hydrolysis mechanism of PAPs.13 (b) Proposed mechanism of hydrolysis of 1 catalyzed by the Zr6 cluster, for reaction at pH 3.0. | ||
Key structural similarities between Zr6 and the bimetallic cofactors of PAPs support the proposed mechanistic similarity. Specifically, both the cluster and PAPs exhibit comparable interatomic distance between the metallic centers.9,10,23,51,70 In the enzyme, the Fe–MII distance spans 3.1–3.5 Å,69 while for the cluster a Zr–Zr distance of 3.6 Å was measured using EXAFS. The same arrangement is also common in various biomimetic catalysts previously reported.13 Furthermore, the activity of PAPs peaks in mildly acidic solution (pH 5–7) due to the formation of active species essential to the mechanism, where the reaction nucleophile (FeIII–OH) is generated via a facile deprotonation of a water ligand coordinated to FeIII, next to a neighboring metal center also bound to water (Scheme 2a). Remarkably, a similar active site structure [(OH)Zr-μ(O(H))–Zr(OH2)] could be formed on the surface of Zr6 at both pH 3.0 and 6.4 (as detailed in Fig. 6), thus enabling the reaction to proceed through a mimicking mechanism. Additionally, the structural similarities between Zr6 and PAPs could, in principle, also explain the faster hydrolysis at pH 6.4 as more ZrIV–OH nucleophilic moieties would be available at this pH in comparison to the reaction at pH 3.0. This scenario was supported by a potentiometric titration of Zr6 aqueous solution (Fig. 6a), which has shown that the Zr6 cluster has three distinct pKa values at pKa-1 = 3.4, pKa-2 = 4.8 and pKa-3 = 7.4. However, a thorough computational study at the density functional theory (DFT) level suggests that other factors also play a role in this case (vide infra).
 | ||
| Fig. 6 (a) Zr6 cluster titration curve (red circles) and its first derivative (blue triangles). Conditions: [Zr6] = 1 × 10−3 mol L−1, [KCl] = 0.02 mol L−1, water (40 mL) pHi = 3.0, pHf = 11.5, titrant KOH – 0.1 mol L−1; (b) proposed acid–base equilibria for Zr6-(OH)4 species. DFT-derived assignment of the experimental pKa values are given in parentheses. Bridging acetate ligands are omitted for clarity. The stability of each species with regard to its structural isomers, which differ in the relative positions of protons, is evaluated in Scheme S3.† | ||
As shown in the free-energy profile of Fig. 7, the reaction at pH 3.0 starts with the replacement of an aqua ligand of Zr6-(OH)4 by substrate 1 forming species S. This ligand exchange is exergonic by 5.5 kcal mol−1 and activates the phosphorus atom of the substrate towards a nucleophilic attack. This is a consequence of ZrIV Lewis acidity,31,38,56,60,74 similarly to other transition metal-catalyzed hydrolysis reactions.75–80 However, the ensuing nucleophilic attack may take place through several mechanisms (depicted in different colors in Fig. 7), given the molecular complexity of the system. These mechanisms include two intramolecular mechanisms, whereby the attack is performed by a hydroxo ligand bound either on a neighboring (intra path, black lines) or the same Zr center (intra′ path, red lines) as the substrate. Alternatively, two intermolecular mechanisms are also possible, in which the nucleophilic attack is carried out by a solvent water molecule, assisted by distinct Brønsted basic Zr–OH moieties situated in a neighboring (inter path, blue lines) or the same position (inter′ path, green lines) where 1 binds.
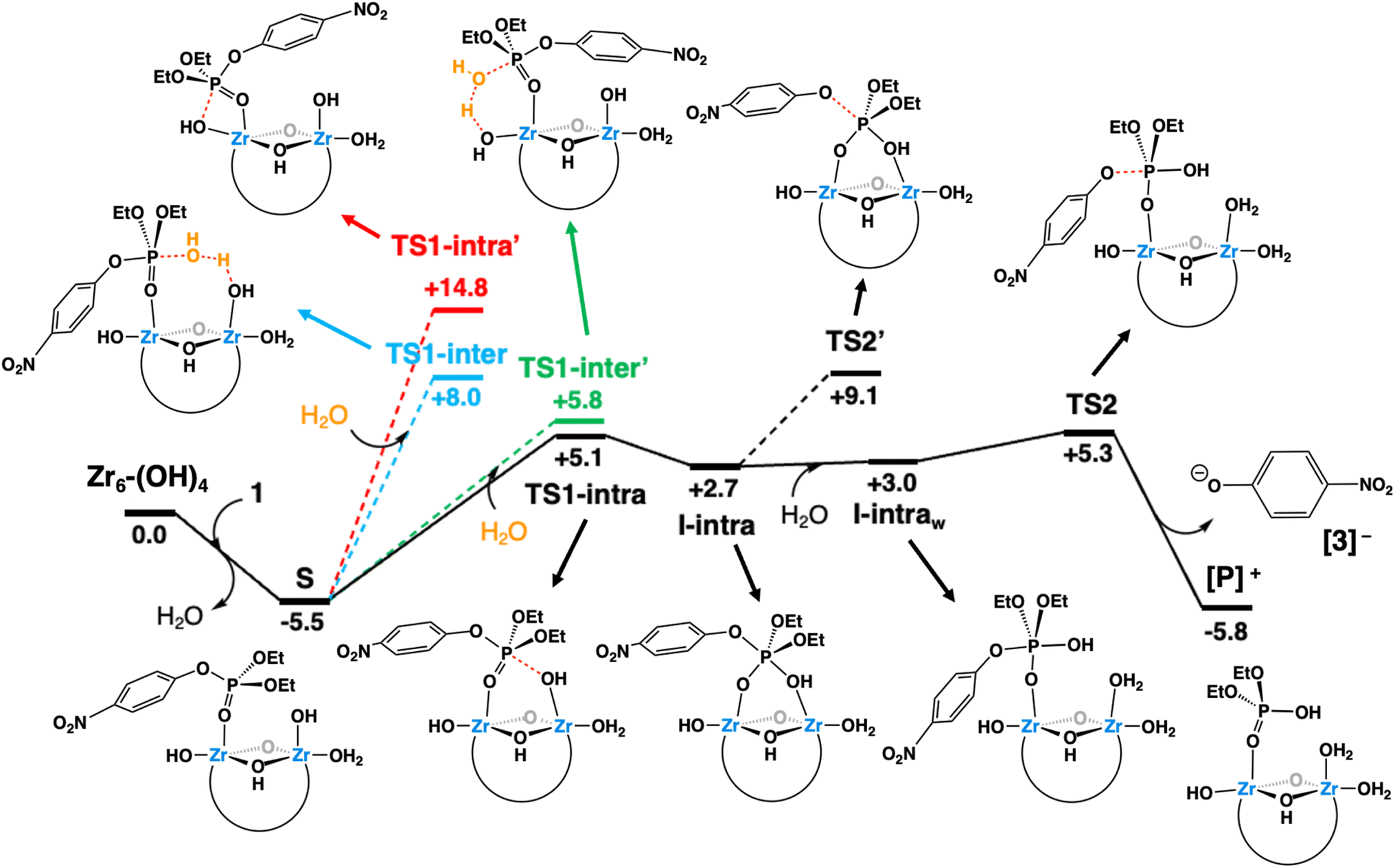 | ||
| Fig. 7 Gibbs free-energy profile (kcal mol−1) for the hydrolysis of 1 at 60 °C catalyzed by Zr6-(OH)4, the most abundant catalytic species at pH 3. Black solid lines represent the lowest free-energy pathway (intra path), while dashed lines denote alternative mechanisms that are higher in energy. | ||
Analysis of all possibilities for the nucleophilic attack revealed the intra path through TS1-intra (see Fig. 8a for a molecular representation) to be the most favorable, overcoming a free-energy barrier of 10.6 kcal mol−1 from S (vs. 11.3, 13.5 and 20.3 kcal mol−1 for inter′, inter, and intra paths, respectively). These differences can be rationalized as follows. On the one hand, TS-intra′ involves a highly strained 4-membered cycle, making it the least stable one.80,81 On the other hand, the slightly more stable nature of TS-intra compared to the TSs of intermolecular paths can be ascribed to: (i) a smaller entropic penalty due to its non-associative nature; (ii) the participation of Zr–OH as a nucleophile, which is stronger than the external water molecules participating in intermolecular paths, while releasing the strain existing in TS-intra′, thanks to the cooperation of two neighboring Zr centers.
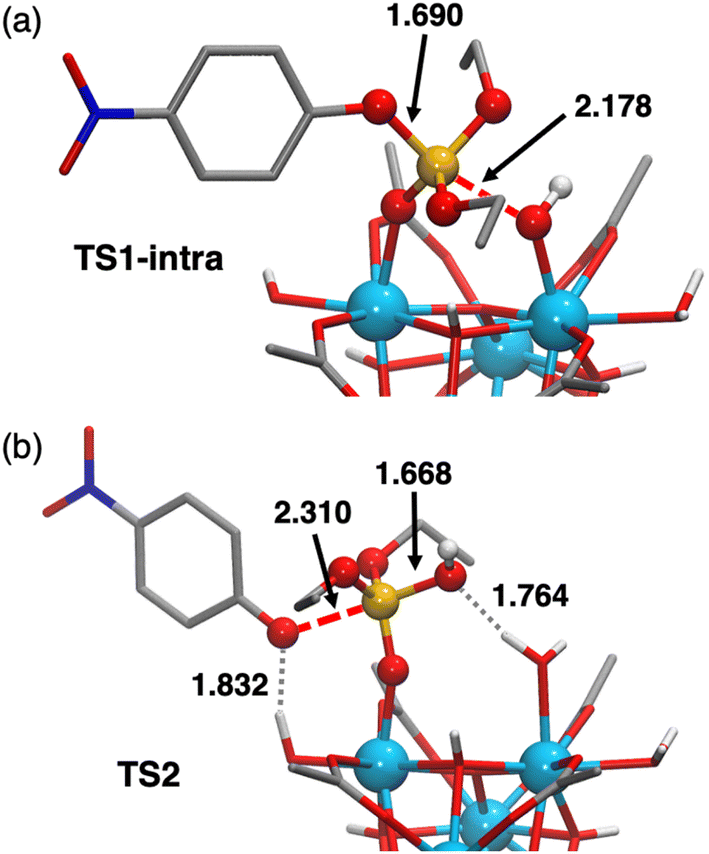 | ||
| Fig. 8 Ball-and-stick representation of the optimized geometries for TS1-intra (a) and TS2 (b). Red dashed lines represent bonds being formed or cleaved and gray dotted lines denote hydrogen bonds. Selected distances are given in Å. C-bound H atoms are omitted for clarity. Color code: Zr (light blue), C (gray), H (white) O (red), P (orange), and N (dark blue). | ||
Although the intra path was predicted to be slightly favored over the others, a contribution of an alternative mechanism involving TS1-inter′ (Fig. S19†) could not be discarded due to the small free-energy differences between both pathways. To distinguish between these two possibilities, reactions were conducted in deuterated solvents (see the ESI† for more details). The intermolecular general base mechanism, whereby a free hydroxyl performs an outer-sphere attack on the substrate, is generally slowed down under deuterated conditions as D2O is less labile than H2O.82 However, no significant difference was observed between the reactions conducted in deuterated or regular solvents. These findings are analogous to those reported for PAP enzymes, dismissing a general base mechanism,82 and indicate that the Zr6 catalyzed hydrolysis proceeds through an intramolecular ZrIV–OH addition.
The nucleophilic attack of a Zr–OH group at the phosphorus center of substrate 1 through TS1-intra leads to the cleavage of the P–O bond and subsequent formation of the products 2 and 3, with 2 still complexed to Zr6 (species [P]+, Fig. 7). At first, the intramolecular attack of a Zr–OH group on substrate 1 generates a trigonal bipyramidal pentavalent phosphorane intermediate I-intra, which lies 2.7 kcal mol−1 above the reactants (Zr6-(OH)4 + 1). Addition of water quickly converts I-intra into I-intraw (ΔG = +0.3 kcal mol−1) by dissociation of the PO(H)–Zr bond and triggers a facile P–OC6H4NO2 bond cleavage through TS2 (shown in Fig. 8b). This P–O bond cleavage requires overcoming a very low free-energy barrier of 2.3 kcal mol−1 from I-intraw and leads to the formation of [P]+ (ΔG = −5.8 kcal mol−1) upon dissociation of [3]−. The free-energy cost to reach TS2 from the most stable intermediate S in the energy profile is 10.8 kcal mol−1, which is very similar to the S → TS1-intra (10.6 kcal mol−1) conversion and shows that both P–O bond formation and cleavage steps require similar energy inputs to occur. It is worth noting that all our attempts to characterize transition states for a concerted process were unsuccessful. Furthermore, changes in the coordination of acetate ligands to the cluster during the reaction are thermodynamically accessible; however, these do not have a significant impact on the reaction mechanism (see the ESI† for details). An alternative pathway wherein μ3-O assists the product formation acting as a Brønsted base was found to be significantly higher in energy (Fig. S20†).
Species [P]+ spontaneously deprotonates (estimated pKa = −5.59) to afford highly stable neutral species P (Fig. 9a), from which the release of product [2]− requires a rather high free-energy cost of 24.7 kcal mol−1 (Fig. 9b, red dashed lines). However, the energy required to release phosphate product 2 from the catalysts largely decreases upon deprotonation of P. This species exists in equilibrium with its deprotonated anionic form [P]− (estimated pKa = 3.15, Fig. 9b), from which dissociation of [2]− and regeneration of initial species Zr6-(OH)4 are less energy demanding, requiring only 17.7 kcal mol−1. Importantly, an alternative bidentate coordination of species P to the cluster was also probed. However, it was found to be 1.9 kcal mol−1 less stable than the monodentate configuration shown in Fig. 9a. Hence, both configurations might exist in equilibrium, yet without affecting the kinetic picture inferred from Fig. 9b.
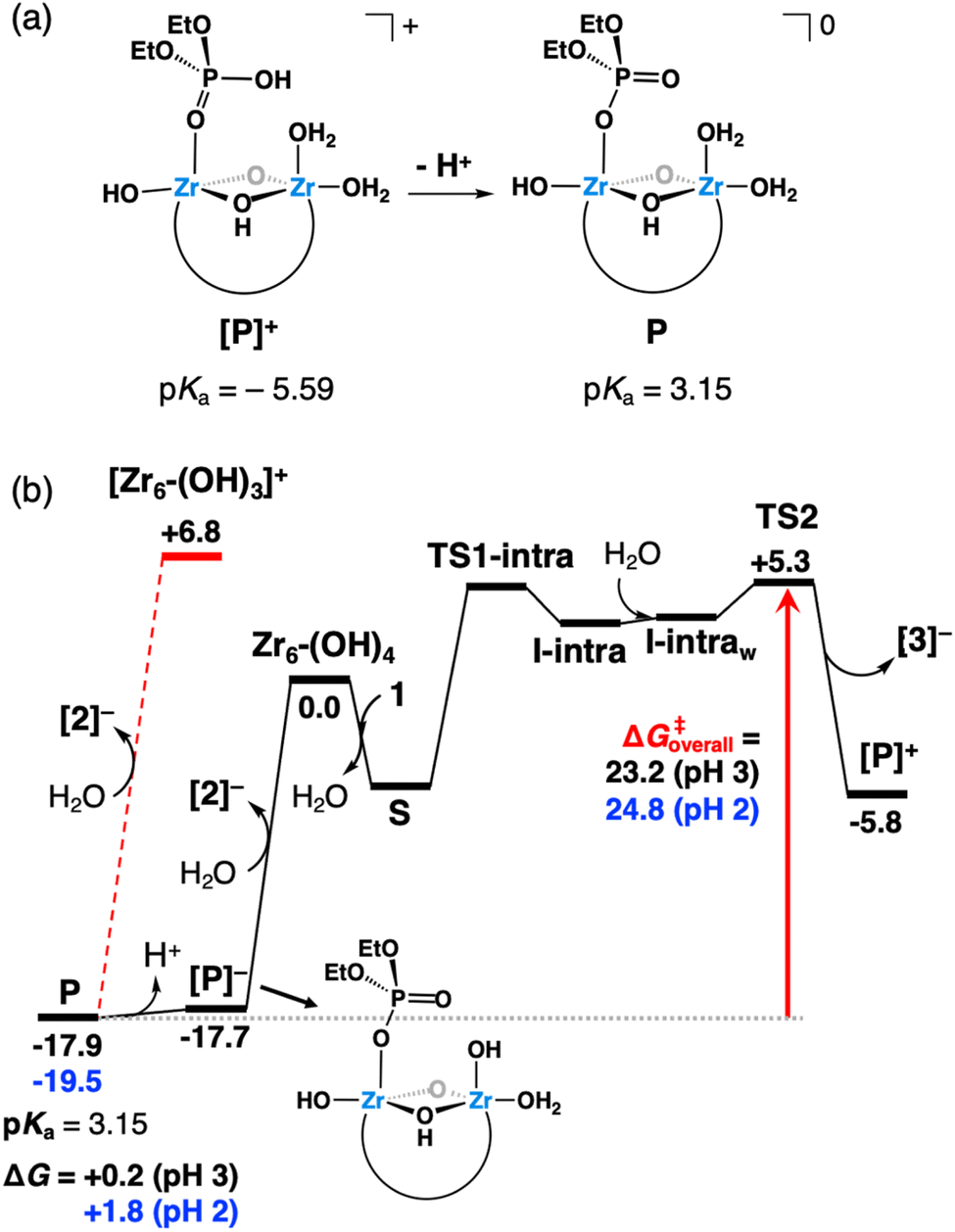 | ||
| Fig. 9 (a) Schematic representation of the spontaneous deprotonation of a Zr-coordinated 2 product molecule (species [P]+). (b) Gibbs free-energy profile (kcal mol−1), where a red arrow highlights the overall hydrolysis free-energy barrier from the resting state (P) to the highest transition state (TS2) as a function of the solution pH. Black values represent free energies at the initial pH of 3, whereas values in blue were estimated for a pH of 2, which is the experimental pH at the end of the reaction at 60 °C. Only the relative energies of key species are shown; the rest are given in Fig. 7. Red dashed lines represent an alternative, more energy-demanding process to regenerate the catalyst. | ||
Notably, the predicted stability of P and its energetically accessible deprotonated form ([P]−) agrees with the high affinity of the phosphate product to the ZrIV sites of the catalyst inferred from spectroscopic analyses (vide supra) and the general acidification observed after reaction (Table S3†). Moreover, it helps to understand the slow catalyst regeneration and the low reaction yield under the investigated conditions. Considering the catalyst regeneration to Zr6-(OH)4, our calculations indicate an overall energy barrier between 23.2 and 24.8 kcal mol−1 for the hydrolysis reaction depending on the solution acidity (Fig. 9). This is in line with the contrasting product yields observed at different temperatures under acidic conditions (Fig. 2), as the calculated barriers are more difficult to overcome at 37 °C than at 60 °C. Remarkably, the 12-fold higher yield observed experimentally (Fig. 2) is in good agreement with an estimated 13-fold increase in the reaction rate at 60 °C compared to 37 °C, if one assumes a pseudo-first order kinetics and downplay variations in the free-energy barrier due to temperature.
Finally, our mechanistic investigation also points out directions for further development of Zr oxo clusters as PAP analogues since DFT calculations also provided key insights that could overcome the limited yields observed and improve the turnover under acidic conditions. Critically, the acidity increase due to phosphate product 2 formation likely decreases the availability of active species (Zr6-(OH)4 or Zr6-(OH)6) in the reaction mixture (Fig. 6b) and favors less reactive protonated forms. Thus, the strong affinity of the phosphate product for Zr6, which was detected when characterizing the catalyst after reaction, may not be the only reason behind the low turnover observed. Overall, these results not only support the mechanistic analogy between PAP enzymes and zirconium oxo clusters proposed in this work but also suggests that identifying new ways to tune the proton topology of these clusters could also enhance their hydrolytic performance.
4. Conclusion
In summary, this work introduced discrete zirconium oxo clusters (ZrOC) as a new class of catalysts for the degradation of phosphate triesters under acidic conditions by demonstrating that ZrOC like the [Zr6O4(OH)4(CH3CO2)8(H2O)2Cl3] (Zr6) cluster are effective catalysts for the hydrolysis of the hazardous phosphate triester ethyl paraoxon. A combination of detailed DFT calculations with experimental approaches that combined kinetic measurements, structural characterization, and key mechanistic investigations revealed that the mechanism is different from the one generally accepted for Zr-MOFs. Specifically, zirconium oxo clusters hydrolyze phosphate triesters by an ‘intramolecular mechanism’ analogous to the one proposed for purple acid phosphatase (PAP) enzymes and can be seen as a new class of functional biomimetic catalysts for these enzymes featuring an unprecedented binuclear ZrIV–ZrIV active site, which provides a synergic Lewis acid center/nucleophilic Zr–OH paired moiety for the activation of phosphoester bonds. Additionally, these results showcase that a series of discrete homogeneous cluster catalysts are able to cleave toxic organophosphates under close to neutral and acidic conditions, which complement the well-established reactivity of Zr-MOFs in alkaline medium. Furthermore, by combining cluster speciation studies with theoretical calculations, we consolidate a unique option to gain further fundamental molecular insights into the hydrolytic reactivity of Zr-MOFs. Finally, this work presents an exciting opportunity to expand the research on bioinspired catalysts beyond classical coordination compounds towards discrete atomically precise inorganic clusters.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Edinara Luiz – investigation, methodology, formal analysis, validation, visualization, data curation, writing – original draft, writing – review & editing. Francisco de Azambuja – project administration, conceptualization, methodology, visualization, writing – original draft, writing – review & editing. Albert Solé-Daura – methodology, formal analysis, writing original draft, supervision, writing – review & editing. Jordi Puiggalí-Jou – methodology, formal analysis, data curation, writing – original draft. Angelo Mullaliu – formal analysis, data curation, writing – original draft. Jorge J. Carbó – methodology, formal analysis, data curation, writing – original draft. Fernando Roberto Xavier – supervision, resources, funding acquisition, writing – review & editing. Rosely A. Peralta – supervision, resources, funding acquisition, writing – review & editing. Tatjana N. Parac-Vogt – project administration, conceptualization, supervision, resources, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank KU Leuven (F. d. A., STG/23/022), Research Foundation Flanders (FWO, infrastructure grant I002720N and I001920N), and Conselho Nacional de Pesquisa (CNPq) (40269/2021-0) for generous funding. F. d. A. (1281921N) and A. M. (1228622N) thank the FWO for fellowships. E. L. thanks Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for the scholarship. R. A. P. thanks CNPq for the scholarship (305572/2020-2). R. A. P. thanks FAPESC and INCT-Catálise for funding. F. R. X. thanks CNPq for the scholarship (306851/2022-9). We also acknowledge Elettra Sincrotrone Trieste for providing access to its synchrotron radiation facilities (Project No. 20225383, A. M. as PI) and for financial support under the IUS internal project. In particular, we thank Dr Giuliana Aquilanti and Dr Giovanni Agostini for their help during the XAFS experiments. J. J. C., J. P.-J. and A. S.-D. are grateful for grants PID2021-128128NB-I00 and PID2020-112762GB-I00 funded by MINECO/AEI/10.13039/501100011033 and by “ERDFA way of making Europe” and the Generalitat de Catalunya (2021SGR00110). A. S.-D. also acknowledges the Spanish Ministry of Universities and the European Union – Next Generation EU for financial support through a Margarita Salas grant.References
- M. Thakur, I. L. Medintz and S. A. Walper, Front. Bioeng. Biotechnol., 2019, 7, 289 CrossRef PubMed.
- L. Jiang, Y. Sun, Y. Chen and P. Nan, ChemistrySelect, 2020, 5, 9492–9516 CrossRef.
- M. A. G. B. Gomes, C. Fernandes, L. R. Gahan, G. Schenk and A. Horn, Chem.–Eur. J., 2021, 27, 877–887 CrossRef CAS PubMed.
- S. Tang, P. T. Wong, J. Cannon, K. Yang, S. Bowden, S. Bhattacharjee, J. J. O'Konek and S. K. Choi, Chem. Biol. Interact., 2019, 297, 67–79 CrossRef CAS.
- Y.-H. Zhou, Z. Zhang, M. Patrick, F. Yang, R. Wei, Y. Cheng and J. Gu, Dalton Trans., 2019, 48, 8044–8048 RSC.
- T. Klabunde, H. Witzel and B. Krebs, J. Mol. Biol., 1996, 259, 737–748 CrossRef CAS PubMed.
- N. Sträter, T. Klabunde, P. Tucker, H. Witzel and B. Krebs, Science, 1995, 268, 1489–1492 CrossRef PubMed.
- D. Feder, R. P. McGeary, N. Mitić, T. Lonhienne, A. Furtado, B. L. Schulz, R. J. Henry, S. Schmidt, L. W. Guddat and G. Schenk, Plant Sci., 2020, 294, 110445 CrossRef CAS.
- C. Pathak, D. Kumar, M. K. Gangwar, D. Mhatre, T. Roisnel and P. Ghosh, J. Inorg. Biochem., 2018, 185, 30–42 CrossRef CAS.
- C. Pathak, M. K. Gangwar and P. Ghosh, Polyhedron, 2018, 145, 88–100 CrossRef CAS.
- G. Schenk, N. Mitić, G. R. Hanson and P. Comba, Coord. Chem. Rev., 2013, 257, 473–482 CrossRef CAS.
- G. Sharma, V. M. Jayasinghe-Arachchige, Q. Hu, G. Schenk and R. Prabhakar, ACS Catal., 2020, 10, 3684–3696 CrossRef CAS.
- L. A. Wilson, M. M. Pedroso, R. A. Peralta, L. R. Gahan and G. Schenk, J. Inorg. Biochem., 2023, 238, 112061 CrossRef CAS PubMed.
- V. M. Jayasinghe-Arachchige, L. F. Serafim, Q. Hu, C. Ozen, S. N. Moorkkannur, G. Schenk and R. Prabhakar, ACS Catal., 2023, 3131–3147 CrossRef CAS.
- L. F. Serafim, V. M. Jayasinghe-Arachchige, L. Wang, P. Rathee, J. Yang, S. M. N and R. Prabhakar, Chem. Commun., 2023, 59, 8911–8928 RSC.
- X.-R. Tian, Z.-Y. Jiang, S.-L. Hou, H.-S. Hu, J. Li and B. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202301764 CrossRef CAS PubMed.
- S.-L. Hou, J. Dong, X.-Y. Zhao, X.-S. Li, F.-Y. Ren, J. Zhao and B. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202305213 CrossRef CAS PubMed.
- M. Komiyama, Sci. Technol. Adv. Mater., 2023, 24, 2250705 CrossRef PubMed.
- C. Simms, A. Mullaliu, S. Swinnen, F. de Azambuja and T. N. Parac-Vogt, Mol. Syst. Des. Eng., 2023, 8, 270–288 RSC.
- F. R. Xavier, A. Neves, A. Casellato, R. A. Peralta, A. J. Bortoluzzi, B. Szpoganicz, P. C. Severino, H. Terenzi, Z. Tomkowicz, S. Ostrovsky, W. Haase, A. Ozarowski, J. Krzystek, J. Telser, G. Schenk and L. R. Gahan, Inorg. Chem., 2009, 48, 7905–7921 CrossRef.
- D. C. Durigon, L. Duarte, J. Fonseca, T. Tizziani, D. R. S. Candela, A. L. Braga, A. J. Bortoluzzi, A. Neves and R. A. Peralta, Polyhedron, 2022, 225, 116050 CrossRef.
- F. G. Maranha, G. A. dos Santos Silva, A. J. Bortoluzzi, E. Nordlander, R. A. Peralta and A. Neves, Inorg. Chim. Acta, 2020, 502, 119280 CrossRef.
- E. Luiz, G. Farias, A. J. Bortoluzzi, A. Neves, L. M. de Melo Mattos, M. D. Pereira, F. R. Xavier and R. A. Peralta, J. Inorg. Biochem., 2022, 236, 111965 CrossRef.
- N. A. Rey, A. Neves, A. J. Bortoluzzi, C. T. Pich and H. Terenzi, Inorg. Chem., 2007, 46, 348–350 CrossRef PubMed.
- C. Pereira, G. Farias, F. G. Maranha, N. Castilho, G. Schenk, B. de Souza, H. Terenzi, A. Neves and R. A. Peralta, JBIC J. Biol. Inorg. Chem., 2019, 24, 675–691 CrossRef PubMed.
- L. Feng, J. Pang, P. She, J. Li, J. Qin, D. Du and H. Zhou, Adv. Mater., 2020, 32, 2004414 CrossRef.
- A. M. Plonka, Q. Wang, W. O. Gordon, A. Balboa, D. Troya, W. Guo, C. H. Sharp, S. D. Senanayake, J. R. Morris, C. L. Hill and A. I. Frenkel, J. Am. Chem. Soc., 2017, 139, 599–602 CrossRef.
- M. J. Katz, R. C. Klet, S.-Y. Moon, J. E. Mondloch, J. T. Hupp and O. K. Farha, ACS Catal., 2015, 5, 4637–4642 CrossRef.
- J. A. Harvey, C. J. Pearce, M. G. Hall, E. J. Bruni, J. B. DeCoste and D. F. Sava Gallis, Dalton Trans., 2019, 48, 16153–16157 RSC.
- V. S. D. Devulapalli, M. Richard, T.-Y. Luo, M. L. De Souza, N. L. Rosi and E. Borguet, Dalton Trans., 2021, 50, 3116–3120 RSC.
- M. Xu, L. Feng, L.-N. Yan, S.-S. Meng, S. Yuan, M.-J. He, H. Liang, X.-Y. Chen, H.-Y. Wei, Z.-Y. Gu and H.-C. Zhou, Nanoscale, 2019, 11, 11270–11278 RSC.
- L. González, R. Gil-San-Millán, J. A. R. Navarro, C. R. Maldonado, E. Barea and F. J. Carmona, J. Mater. Chem. A, 2022, 10, 19606–19611 RSC.
- Y. Wen, M. Feng, P. Zhang, H.-C. Zhou, V. K. Sharma and X. Ma, ACS EST Eng., 2021, acsestengg.1c00051 Search PubMed.
- T. G. Grissom, A. M. Plonka, C. H. Sharp, A. M. Ebrahim, Y. Tian, D. L. Collins-Wildman, A. L. Kaledin, H. J. Siegal, D. Troya, C. L. Hill, A. I. Frenkel, D. G. Musaev, W. O. Gordon, C. J. Karwacki, M. B. Mitchell and J. R. Morris, ACS Appl. Mater. Interfaces, 2020, 12, 14641–14661 CrossRef.
- N. S. Bobbitt, M. L. Mendonca, A. J. Howarth, T. Islamoglu, J. T. Hupp, O. K. Farha and R. Q. Snurr, Chem. Soc. Rev., 2017, 46, 3357–3385 RSC.
- S. Rojas, A. Rodríguez-Diéguez and P. Horcajada, ACS Appl. Mater. Interfaces, 2022, 14, 16983–17007 CrossRef PubMed.
- C. Simms, A. Mullaliu, S. Swinnen, F. de Azambuja and T. N. Parac-Vogt, Mol. Syst. Des. Eng., 2023, 8, 270–288 RSC.
- J. E. Mondloch, M. J. Katz, W. C. Isley III, P. Ghosh, P. Liao, W. Bury, G. W. Wagner, M. G. Hall, J. B. DeCoste, G. W. Peterson, R. Q. Snurr, C. J. Cramer, J. T. Hupp and O. K. Farha, Nat. Mater., 2015, 14, 512–516 CrossRef.
- T. Islamoglu, A. Atilgan, S.-Y. Moon, G. W. Peterson, J. B. DeCoste, M. Hall, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 2672–2675 CrossRef.
- K. O. Kirlikovali, Z. Chen, T. Islamoglu, J. T. Hupp and O. K. Farha, ACS Appl. Mater. Interfaces, 2020, 12, 14702–14720 CrossRef.
- S. G. Ryu, M.-K. Kim, M. Park, S. O. Jang, S. H. Kim and H. Jung, Microporous Mesoporous Mater., 2019, 274, 9–16 CrossRef.
- M. R. Mian, X. Wang, X. Wang, K. O. Kirlikovali, H. Xie, K. Ma, K. M. Fahy, H. Chen, T. Islamoglu, R. Q. Snurr and O. K. Farha, J. Am. Chem. Soc., 2023, jacs.2c13887 Search PubMed.
- M. C. de Koning, G. W. Peterson, M. van Grol, I. Iordanov and M. McEntee, Chem. Mater., 2019, 31, 7417–7424 CrossRef.
- Z. Zhang, C.-A. Tao, J. Zhao, F. Wang, J. Huang and J. Wang, Catalysts, 2020, 10, 1086 CrossRef.
- B. Gibbons, E. C. Bartlett, M. Cai, X. Yang, E. M. Johnson and A. J. Morris, Inorg. Chem., 2021, 60, 16378–16387 CrossRef PubMed.
- F. de Azambuja, J. Moons and T. N. Parac-Vogt, Acc. Chem. Res., 2021, 54, 1673–1684 CrossRef.
- D. E. Salazar Marcano, N. D. Savić, S. A. M. Abdelhameed, F. De Azambuja and T. N. Parac-Vogt, JACS Au, 2023, 3, 978–990 CrossRef PubMed.
- D. E. S. Marcano, N. D. Savić, K. Declerck, S. A. M. Abdelhameed and T. N. Parac-Vogt, Chem. Soc. Rev., 2024, 53, 84–136 RSC.
- Y. Zhang, F. de Azambuja and T. N. Parac-Vogt, Coord. Chem. Rev., 2021, 438, 213886 CrossRef.
- L. Zhang, H. Wang, X. Liu, W. Zhou and Z. Rao, Biochem. Biophys. Res. Commun., 2019, 510, 224–229 CrossRef PubMed.
- A. Neves, M. Lanznaster, A. J. Bortoluzzi, R. A. Peralta, A. Casellato, E. E. Castellano, P. Herrald, M. J. Riley and G. Schenk, J. Am. Chem. Soc., 2007, 129, 7486–7487 CrossRef.
- C. Piovezan, R. Jovito, A. J. Bortoluzzi, H. Terenzi, F. L. Fischer, P. C. Severino, C. T. Pich, G. G. Azzolini, R. A. Peralta, L. M. Rossi and A. Neves, Inorg. Chem., 2010, 49, 2580–2582 CrossRef PubMed.
- Y. Zhang, F. de Azambuja and T. N. Parac-Vogt, Catal. Sci. Technol., 2022, 12, 3190–3201 RSC.
- Y. Zhang, I. Y. Kokculer, F. de Azambuja and T. N. Parac-Vogt, Catal. Sci. Technol., 2023, 13, 100–110 RSC.
- K. Declerck, N. D. Savić, M. A. Moussawi, C. Seno, R. Pokratath, J. De Roo and T. N. Parac-Vogt, J. Am. Chem. Soc., 2024, 146, 11400–11410 Search PubMed.
- H. Chen, P. Liao, M. L. Mendonca and R. Q. Snurr, J. Phys. Chem. C, 2018, 122, 12362–12368 CrossRef.
- S. Dai, C. Simms, I. Dovgaliuk, G. Patriarche, A. Tissot, T. N. Parac-Vogt and C. Serre, Chem. Mater., 2021, 33, 7057–7066 CrossRef.
- A. Loosen, F. de Azambuja and T. N. Parac-Vogt, Mater. Adv., 2022, 3, 2475–2487 RSC.
- J. Moons, F. de Azambuja, J. Mihailovic, K. Kozma, K. Smiljanic, M. Amiri, T. C. Velickovic, M. Nyman and T. N. Parac-Vogt, Angew. Chem., Int. Ed., 2020, 59, 9094–9101 CrossRef.
- Y. Liao, T. R. Sheridan, J. Liu, Z. Lu, K. Ma, H. Yang, O. K. Farha and J. T. Hupp, ACS Catal., 2024, 14, 437–448 CrossRef.
- M. C. de Koning, M. van Grol and T. Breijaert, Inorg. Chem., 2017, 56, 11804–11809 CrossRef.
- W. D. Kumler and J. J. Eiler, J. Am. Chem. Soc., 1943, 65, 2355–2361 CrossRef.
- L. Deblock, E. Goossens, R. Pokratath, K. De Buysser and J. De Roo, JACS Au, 2022, 2, 711–722 CrossRef CAS PubMed.
- E. Dhaene, S. Coppenolle, L. Deblock, K. De Buysser and J. De Roo, Chem. Mater., 2023, 35, 558–569 CrossRef.
- D. Van den Eynden, R. Pokratath, J. P. Mathew, E. Goossens, K. De Buysser and J. De Roo, Chem. Sci., 2023, 14, 573–585 RSC.
- D. T. Richens, Chem. Rev., 2005, 105, 1961–2002 CrossRef.
- S. J. Garibay, T. M. Tovar, I. O. Iordanov, G. W. Peterson and J. B. DeCoste, J. Mater. Chem. A, 2023, 11, 13300–13308 RSC.
- J. Dong, H.-D. An, Z.-K. Yue, S.-L. Hou, Y. Chen, Z.-J. Zhang, P. Cheng, Q. Peng and B. Zhao, ACS Cent. Sci., 2021, 7, 831–840 CrossRef PubMed.
- N. Mitić, S. J. Smith, A. Neves, L. W. Guddat, L. R. Gahan and G. Schenk, Chem. Rev., 2006, 106, 3338–3363 CrossRef.
- P. Karsten, A. Neves, A. J. Bortoluzzi, M. Lanznaster and V. Drago, Inorg. Chem., 2002, 41, 4624–4626 CrossRef CAS PubMed.
- N. Planas, J. E. Mondloch, S. Tussupbayev, J. Borycz, L. Gagliardi, J. T. Hupp, O. K. Farha and C. J. Cramer, J. Phys. Chem. Lett., 2014, 5, 3716–3723 CrossRef.
- R. C. Klet, Y. Liu, T. C. Wang, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 1479–1485 RSC.
- H. Chen, Chem. Phys. Lett., 2022, 800, 139658 CrossRef.
- T. K. N. Luong, P. Shestakova, T. T. Mihaylov, G. Absillis, K. Pierloot and T. N. Parac-Vogt, Chem. – Eur. J., 2015, 21, 4428–4439 CrossRef PubMed.
- A. Gil and J. J. Carbó, Front. Chem., 2022, 10, 876630 CrossRef PubMed.
- H. G. T. Ly, T. Mihaylov, G. Absillis, K. Pierloot and T. N. Parac-Vogt, Inorg. Chem., 2015, 54, 11477–11492 CrossRef PubMed.
- V. M. Jayasinghe-Arachchige, Q. Hu, G. Sharma, T. J. Paul, M. Lundberg, D. Quinonero, T. N. Parac-Vogt and R. Prabhakar, J. Comput. Chem., 2019, 40, 51–61 CrossRef CAS PubMed.
- H. G. T. Ly, T. T. Mihaylov, P. Proost, K. Pierloot, J. N. Harvey and T. N. Parac-Vogt, Chem. – Eur. J., 2019, 25, 14370–14381 CrossRef CAS.
- J. Lanuza, Á. Sánchez−González, N. A. G. Bandeira, X. Lopez and A. Gil, Inorg. Chem., 2021, 60, 11177–11191 CrossRef.
- A. Solé-Daura, A. Rodríguez-Fortea, J. M. Poblet, D. Robinson, J. D. Hirst and J. J. Carbó, ACS Catal., 2020, 10, 13455–13467 CrossRef.
- G. Norjmaa, A. Solé-Daura, M. Besora, J. M. Ricart and J. J. Carbó, Inorg. Chem., 2021, 60, 807–815 CrossRef PubMed.
- K. A. Deal, A. C. Hengge and J. N. Burstyn, J. Am. Chem. Soc., 1996, 118, 1713–1718 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03946g |
| This journal is © The Royal Society of Chemistry 2024 |