
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStabilizing ultra-close Pt clusters on all-in-one CeO2/Al2O3 fibril-in-tubes against sintering†
Wanlin
Fu
 a,
Kuibo
Yin
b,
Zhihui
Li
a,
Jun
Wang
a,
Mingyu
Tang
a,
Jilan
Tian
a,
Litao
Sun
b,
Yueming
Sun
b and
Yunqian
Dai
*a
a,
Kuibo
Yin
b,
Zhihui
Li
a,
Jun
Wang
a,
Mingyu
Tang
a,
Jilan
Tian
a,
Litao
Sun
b,
Yueming
Sun
b and
Yunqian
Dai
*a
aSchool of Chemistry and Chemical Engineering, Southeast University, Nanjing, Jiangsu 211189, China. E-mail: daiy@seu.edu.cn
bSEU-FEI Nano-Pico Center, Key Laboratory of MEMS of Ministry of Education, School of Electronic Science and Engineering, Southeast University, Nanjing, Jiangsu 211189, China
First published on 10th September 2024
Abstract
Metal sintering poses significant challenges for developing reliable catalytic systems toward high-temperature reactions, particularly those based on metal clusters with sizes below 3 nm. In this work, electrospun dual-oxide fibril-in-tubes consisting of CeO2 and Al2O3 are rationally designed in an all-in-one manner, to stabilize 2.3 nm Pt clusters with a Tammann temperature (sintering onset temperature) lower than 250 °C. The abundant pores and channels effectively stabilize the Pt clusters physically, while the strong support, CeO2, with high adhesion, pins Pt clusters firmly, and the adjacent weak support, Al2O3, with low adhesion, provides energy barriers to prevent the clusters and emitted Pt atom(s) from moving across the support. Therefore, the ultra-close 2.3 nm Pt clusters, featuring an average nearest neighboring distance of only 2.1 nm, were carefully stabilized against sintering at temperatures exceeding 750 °C, even in oxidative and steam-containing environments. In addition, this catalytic system can efficiently and durably serve in diesel combustion, a high-temperature exothermic reaction, showing no activity decline after 5 cycles. This work provides a comprehensive understanding of sinter-resistant catalytic systems, and presents new insights for the development of advanced nanocatalysts.
1 Introduction
In the past several decades, the development of nanoscience has enabled the precise manipulation of surfaces and interfaces of catalysts in the nanometer size regime and gives rise to a superior cornerstone for energy conversion and environmental protection.1 To maximize the mass-specific activity, bulky metals are generally dispersed into nanoparticles (typically >3 nm) and even isolated as clusters (usually <3 nm) or single atoms on specific supports.2 However, as the size of the metal decreases, the increasing surface energy presents a significant tendency for metal sintering (growing into larger clusters and/or particles).3 Taking 3 nm Pt nanoparticles as an example, the Tammann temperature, at which sintering initiates, could decrease to below 250 °C, which is extremely lower than that of the bulky Pt (749 °C).4 In this case, highly dispersed metal species suffered substantial deactivation during high-temperature reactions, such as the diesel combustion (600–800 °C), propane dehydrogenation (590–630 °C), and the dry reformation of methane (800–1000 °C).5 Besides, reaction exotherm can cause the local temperature of active sites far beyond that of the surroundings, leading to preferential damage of these most active atoms. Therefore, mitigating catalyst sintering is of significant importance for the successful translation of well-defined nanocatalysts from laboratory to industrial application, yet it remains highly challenging.The sintering pathways of metal nanoparticles are commonly dominated by both particle migration and coalescence (PMC) and Ostwald ripening (OR), either simultaneously or alternatively.3 Through the PMC pathway, metal nanoparticles would migrate across the support via Brownian-like motion, until they meet and merge into a larger particle.6 As for the OR mechanism, metal atoms leave the surface of one nanoparticle with a higher chemical potential to join another nanoparticle with a lower chemical potential.3 As such, the sinter-resistance relies deeply on the metal loading density on supports, because of the fact that mass transport through surface diffusion and/or migration of the metal species could be facilitated if the distance between adjacent species is shortened.7 Admittedly, employing porous supports with a high surface area could slow down the metal sintering by enlarging the neighboring distance. However, these porous supports would suffer from densification due to the excessive surface free energy, and then dramatically shorten the neighboring distance for supported metal species. Therefore, it is still an imperative challenge to stabilize metal clusters on reliable supports, especially for those in ultra-close proximity.
To mitigate sintering, great efforts have been devoted to stabilizing metals physically and/or chemically. So far, embedding metals in a hollow and/or porous matrix, such as silica,8–10 zeolites,11 or CeO2 (ref. 12) has been explored as an efficient approach toward sinter-resistant nanocatalysts. Alternatively, enhancing the metal–support interaction through tailoring the interface structure, composition, lattice match, charge rearrangement, etc, contributes to fascinating progress for stabilizing metals without any protective shell.13 However, recent theoretical work revealed that too-strong metal–support interaction triggered rapid OR, whereas too-weak metal–support interaction stimulated facile PMC, both of which severely worsened the thermal stability.6 Dual-oxide supports hold great promise to mitigate the metal sintering, but are still overwhelmed by the challenge in facile fabrication with controlled composition and dispersity at the nanoscale.
In this work, 2.3 nm Pt clusters in ultra-close proximity were well stabilized on CeO2/Al2O3 fibril-in-tubes, achieving boosted sintering resistance over 750 °C in an oxidative and steam-containing atmosphere. As shown in Fig. 1, the abundant inter-grain pores and elegant fibril-in-tube structure effectively stabilize the Pt clusters physically. Chemically, the strong support, CeO2, with higher adhesion pins Pt clusters firmly (i.e., mitigating cluster migration and coalescence), while the integration of adjacent weak support, Al2O3, with lower adhesion to Pt, provides energy barriers at the dual-oxide interfaces, and thus energetically prevent the clusters and emitted Pt atom(s) from moving across the support. The sintering behavior of Pt clusters on CeO2/Al2O3 fibril-in-tubes was dynamically monitored by in situ HAADF-STEM investigation. Moreover, we also demonstrated the superior performance of the Pt@CeO2/Al2O3 catalyst for diesel combustion, in terms of both activity and durability.
 | ||
| Fig. 1 Schematic illustration of the physical and chemical stabilization mechanisms for Pt clusters on the CeO2/Al2O3 fibril-in-tubes in close proximity. | ||
2 Experimental section
2.1 Materials
Polyvinylpyrrolidone (PVP, Mw ≈ 1.3 × 106 or 5.5 × 104), chloroplatinic acid hydrate (H2PtCl6·xH2O, 99.995%), cerium acetylacetone (Ce(acac)3), and aluminum acetylacetonate (Al(acac)3) were obtained from Alfa Aesar. The model soot used is Printex-U (Degussa Corporation). All chemicals were used as received. The water used in all the experiments was filtered through a Millipore filtration system with a resistivity of 18 MΩ cm.2.2 Fabrication of fibril-in-tube CeO2/Al2O3 nanofibers by single-spinneret electrospinning
The composite nanofibers were prepared by electrospinning a precursor containing 0.187 g of Al(acac)3, 0.3 g of Ce(acac)3, 0.4 g of PVP (Mw ≈ 1.3 × 106), and 6 mL of ethanol with a flow rate of 0.5 mL h−1, at 15 kV. Before electrospinning, the precursor was sonicated for 20 min, to achieve better homogeneity. The as-spun Ce(acac)3/Al(acac)3/PVP nanofibers were converted to CeO2/Al2O3 fibril-in-tubes after calcination at 500 °C for 2 h in air with a ramping rate of 4.2 °C min−1.2.3 Fabrication of CeO2 and Al2O3 nanofibers
The CeO2 nanofibers were prepared by electrospinning a precursor containing 0.3 g of Ce(acac)3, 0.6 g of PVP (Mw ≈ 1.3 × 106), 3 mL of acetone, and 3 mL of ethanol with a flow rate of 0.5 mL h−1, at 15 kV. The as-spun Ce(acac)3/PVP nanofibers were then calcined at 500 °C for 2 h in air with a ramping rate of 4.2 °C min−1. The Al2O3 nanofibers were prepared by electrospinning a precursor containing 0.3 g of Al(acac)3, 0.3 g of PVP (Mw ≈ 1.3 × 106), 3 mL of acetone, and 2 mL of ethanol with a flow rate of 0.3 mL h−1, at 15 kV. The as-spun Al(acac)3/PVP nanofibers were kept in air overnight followed by calcination at 500 °C for 2 h in air with a ramping rate of 4.2 °C min−1.2.4 Synthesis of Pt clusters
Pt clusters were prepared by a polyol method. Typically, 4 mL of ethylene glycol was added to a glass vial and heated in an oil bathtub at 110 °C for 30 min. Then, 22.5 mg of PVP (Mw ≈ 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and 16.5 mg of H2PtCl6 were dissolved separately in 2 mL of ethylene glycol at room temperature. 0.5 mL of each solution was then added simultaneously into the system at a rate of 0.67 mL min−1. The reaction continued with heating at 110 °C and was stopped when the color changed from yellow to light brown. The as-obtained Pt clusters were then cooled down to room temperature.
000) and 16.5 mg of H2PtCl6 were dissolved separately in 2 mL of ethylene glycol at room temperature. 0.5 mL of each solution was then added simultaneously into the system at a rate of 0.67 mL min−1. The reaction continued with heating at 110 °C and was stopped when the color changed from yellow to light brown. The as-obtained Pt clusters were then cooled down to room temperature.
2.5 Loading of Pt clusters
The as-prepared Pt clusters were loaded on the surface of different nanofibers by a simple impregnation method. First, 0.2 mL of Pt suspension was diluted into 2 mL of ethanol. Then, 5 mg of nanofibers were immersed in the 0.2 mL diluted Pt suspension, followed by a gentle stirring at room temperature for 2 h. The as-prepared sample was washed with ethanol six times. The centrifugation speed for washing was generally 6000–10000 rpm and each centrifugation lasted for 3 min. After centrifugation, the samples were dried in an oven at 40 °C.2.6 Dynamic observation
Dynamic observation of thermal stability of Pt@CeO2/Al2O3 was conducted by an in situ heating experiment under an aberration-corrected transmission electron microscope (TEM, FEI Titan operating at 300 kV). In a typical procedure, a drop (3 μL) of catalyst-in-ethanol suspension was dropped on an electrical chip (E-chip). After the sample was dried, the E-chip was transferred to a heating holder (Aduro, Protochips Company) and inserted into the microscope chamber. As the nominal ramp rate of the heating holder can reach as high as 1000 °C ms−1, the temperatures of the sample are assumed to reach the set value immediately after switching on the current. The images were acquired in scanning-TEM (STEM) mode and a high-angle annular dark-field (HAADF) detector at low beam densities (typically, 60 A cm−2) was used to reduce the irradiation-induced sputtering effects.2.7 Catalytic elimination of soot particulates
A simulated model diesel soot sample was prepared by mixing Printex-U (the soot) with catalysts using a spatula for 5 min. The weight percent of soot was controlled to be around 10 wt%. To evaluate the soot combustion performance, a thermogravimetric analysis (TGA) of the soot combustion was performed using a thermogravimetric analyzer (Netzsch, STA449 F3) at a temperature ranging from room temperature to 900 °C with a flow rate of 100 mL min−1. The heating rate was 10 °C min−1 in air (21% O2 and 79% N2). For the cycling reaction, a certain amount of Printex-U was added into the collected catalyst after each cycle of reaction, and the weight percent of soot was also controlled to be around 10 wt%.2.8 Characterization
Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images were collected using transmission electron microscopes (Tecnai G2 T20, Talos, and Titan, FEI). Energy dispersive X-ray (EDX) mapping and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) analysis were performed using a Talos. Aberration-corrected HAADF-STEM (AC-HAADF-STEM) images were obtained on a FEI Titan G2 60-300 STEM. SEM images were obtained using a field emission scanning electron microscope (FEI, Inspect F50) after coating the samples with Au. The ultra-thin slices were prepared by ultramicrotomy (LEICA EM UC7). Before characterization, non-woven CeO2/Al2O3 fibril-in-tubes were cut into ultra-thin slices and dropped onto a Cu-grid coated with a carbon membrane. The crystal structure information was obtained with X-ray diffraction (XRD) (Bruker, D8 advance using Cu-Kα radiation, λ = 1.5406 Å). The Pt mass was determined using an Inductively Coupled Plasma Optical Emission Spectrometer (ICP-OES) (Optima 7300DV, PerkinElmer Corporation). The Brunauer–Emmett–Teller (BET) surface area was obtained using a Nova 1200e (Quantachrome, U.S.A.). The binding energies were characterized using an X-ray photoelectron spectrometer (XPS, Escalab 250Xi, Thermo Fisher Scientific). The XPS spectra were analyzed by using Thermo Avantage software, based on the conventional Gaussian-based protocols. The defects were detected by Electron Paramagnetic Resonance (EPR) spectroscopy (A300, Bruker).3 Results and discussion
3.1 Synthesis and structural characterization of CeO2/Al2O3 fibril-in-tubes
The unusual CeO2/Al2O3 fibril-in-tubes, with a Ce/Al dosage atomic ratio of 1.2, were conveniently fabricated by a cost-effective and scalable single-spinneret electrospinning method, starting from a carefully designed precursor solution containing Ce(acac)3, Al(acac)3, PVP, and ethanol, followed by calcination at 500 °C in air (Fig. 2). According to the typical scanning electron microscope (SEM) and transmission electron microscopy (TEM) images (Fig. 2a, b and S1†), the external tube has an average diameter of ca. 275.9 ± 54.0 nm, whereas the size of the fibril in the center is ca. 109.6 ± 23.5 nm. The open channels between the fibril and tube, as well as the naturally formed inter-fiber pores (highlighted by the dashed lines in Fig. 2a), can greatly improve the mass transfer and enhance the trapping for reactants, thus being capable of accelerating the reaction kinetics. When serving as a substrate for metal species, the migration, and diffusion of metal species across different nanofibers, various surfaces within an individual fibril-in-tube, as well as adjacent accessible pores on the surface of the fibril-in-tube, can be hindered, thereby effectively enhancing the sinter-resistance of metal species. | ||
| Fig. 2 (a) SEM image of CeO2/Al2O3 fibril-in-tubes. The dashed lines indicate the inter-fiber pores. (b) Typical TEM image of one individual CeO2/Al2O3 fibril-in-tube. (c) TEM image collected on the cross-section of a single fibril-in-tube prepared with ultramicrotomy. (d) Inverse-FFT image of the HRTEM collected at the dashed squared area in (c). CeO2 and Al2O3 were colored in red and blue respectively, for a better demonstration. (e) HAADF-STEM and the elemental mapping results investigated on the cross-section. | ||
In sharp contrast, the single-component CeO2 and Al2O3 nanofibers exhibited no fibril-in-tube morphology (Fig. S2†). When introducing a minute quantity of Al(acac)3, an observable transformation in the morphology of the obtained nanofibers (with a Ce/Al dosage molar ratio of 2.5) can be witnessed, transitioning from conventional columnar structures to desired fibril-in-tube configurations. However, when the Ce/Al molar ratio decreased to 0.8, the surface of dual-oxide nanofibers became extremely porous and rough, being more like the porous Al2O3 nanofibers. Based on the thermogravimetry analysis and the temperature-dependent morphological evolution (Fig. S3†), it could be hypothesized that the formation of the fibril-in-tube structure might be contributed by the rapid and vigorous decomposition of Ce(acac)3 and Al(acac)3 together with the PVP matrix within the incredibly narrow temperature range. The rapid release of a large amount of gas could cause a huge pressure inside the columnar nanofibers, therefore, leaving a special channel within the fibril-in-tube structure.
The powder X-ray diffraction (XRD) results verified that the CeO2/Al2O3 fibril-in-tubes are composed of fluorite CeO2 and amorphous Al2O3 (Fig. S4†).14 Moreover, the Al3+ cations may somehow dope into the CeO2 crystal lattice, indicated by the peak shift of (111) toward a lower angle.15 The calculated CeO2 crystal size within CeO2/Al2O3 fibril-in-tubes is as small as 5.75 nm. Moreover, the CeO2/Al2O3 fibril-in-tubes were endowed with enriched mesopores with diameters concentrated at 2.5 nm, 8.0 nm, and 20.5 nm (Fig. S5†). Specifically, the 2.5 nm mesopores, if accessible, could allow Pt clusters with a size below 3 nm to sit in steadily and therefore be isolated from each other. Meanwhile, the restricted small size of CeO2 could contribute to abundant grain boundaries, which could favor the robust anchoring of metal species.16
A careful investigation of the cross-section of CeO2/Al2O3 fibril-in-tubes prepared by ultramicrotomy gave more detailed information about the spatial distribution of the two oxides (Fig. 2c–e). As shown, the cross-section of fibril-in-tubes whose axial direction is almost completely perpendicular to the ultra-thin slice exhibited a concentric circular morphology (Fig. 2c). The high-resolution TEM (HRTEM) image (Fig. S6†) and the corresponding inverse Fast Fourier Transform (FFT) image (Fig. 2d) demonstrated that the CeO2 exhibits a highly crystalline structure with lattice fringes of 0.312 and 0.274 nm, which can be indexed to the {111} and {200} planes of fluorite CeO2, respectively.17 However, the interstitial spaces between neighboring CeO2 nanograins are filled with amorphous Al2O3. The HAADF-STEM image and the elemental mappings suggested homogeneous distributions of Ce, Al, and O elements (Fig. 2e). This can be understood by considering the inter-doping of Al and Ce atoms into the lattices of their respective oxide nanocrystals, which has been observed in our previous study on TiO2/Al2O3 nanofibers.18 Therefore, the highly and uniformly distributed two oxides provide abundant and evenly dispersed energy barriers at the dual-oxide interfaces throughout the entire fibril-in-tubes, representing a promising candidate to stabilize metal species.
3.2 Sintering resistance of Pt@CeO2/Al2O3 fibril-in-tubes
As shown in Fig. 3a and S7,† sub-3 nm Pt clusters can be uniformly loaded onto the surfaces of the dual-oxide fibril-in-tubes via the widely used wet-impregnation approaches for industrial catalysts. The Pt loading density was controlled to be 1 wt%, as confirmed by inductively coupled plasma (ICP) analysis and energy-dispersive X-ray spectroscopy (EDX) spectra presented in Table S1.† To carefully evaluate the thermal stability, the Pt@CeO2/Al2O3 fibril-in-tubes were aged at a series of harsh temperatures in dry and humid air. Generally, if exposed to steam, metal sintering could be accelerated due to the formation of volatile compounds coordinated with water.19 Based on the ex situ TEM analysis (Fig. 3 and S8†), including the elemental mappings, no observable agglomeration of Pt clusters was visualized after the aging upon 500, 600, and 700 °C in dry air for 2 h, and even at 700 °C in humid air (10 vol% of steam) for 2 h or 750 °C in dry air for 10 h. The XRD patterns of Pt@CeO2/Al2O3 fibril-in-tubes before and after ex situ aging at a series of high temperatures show exclusively reflections of CeO2 (Fig. S9†). The absence of diffractions indexed to Pt long-range periodicity further confirmed the small size and high dispersity of Pt clusters on these dual-oxide fibril-in-tubes.20 More specifically, the AC-HAADF-STEM images of Pt@CeO2/Al2O3 fibril-in-tubes after aging at 750 °C in dry air for 10 h clearly demonstrated that there is no aggregated Pt cluster or nanoparticle that could be visualized across the substrate (Fig. S10†). | ||
| Fig. 3 (a–c) HAADF-STEM images of Pt@CeO2/Al2O3 (a) before and after aging in dry air at (b) 500 and (c) 600 °C for 2 h. (d–f) HAADF-STEM images and elemental mappings of Pt@CeO2/Al2O3 after aging (d) in dry air at 700 °C for 2 h, (e) in humid air (10 vol% of water) at 700 °C for 2 h, and (f) in dry air at 750 °C for 10 h. | ||
The sintering resistance of Pt clusters on CeO2/Al2O3 nanofibers with different Ce/Al molar ratios was also investigated. Due to the inherently stronger binding energy between Pt and CeO2 compared to that with Al2O3,21 Pt clusters exhibited significantly enhanced sinter-resistance on CeO2 nanofibers in comparison to those on Al2O3 nanofibers. As shown in Fig. S11,† Pt clusters maintained their small size on columnar CeO2 nanofibers after being aged at 700 °C for 2 h, while they underwent further aggregation and increased in diameter to 9.3 nm after aging at 750 °C for 10 h. However, on Al2O3 nanofibers, Pt clusters were observed to grow twice their initial size at a relatively low temperature of only 600 °C (Fig. S12†). Dual-oxide supports were endowed with efficiently promoted capability for stabilizing Pt clusters, in comparison with the single-component nanofibers. As shown, Pt clusters slightly grew from 2.3 nm to 4.2 nm on CeO2/Al2O3-2.5 after being aged at 750 °C in air for 10 h (Fig. S13a and b†). However, when the Ce/Al atomic ratio decreased to 0.8, the loaded 2.3 nm-Pt clusters sintered to reach a size of 4.5 nm after being aged at 750 °C in air for 10 h (Fig. S13c and d†).
In addition to sintering, leaching, which refers to the diffusion of metal into the surrounding environment, poses another threat to the lifespan of the catalyst. Exposure to an oxidative atmosphere can facilitate the oxidation of metallic Pt into more volatile PtOx, leading to irreversible metal leaching.22 Taking advantage of the naturally formed network structure in electrospun nanofibers, the emitted PtOx species from Pt clusters located on the outer surface of tubes could be trapped by neighboring fibril-in-tubes. Moreover, in comparison with the case of Pt clusters supported on conventional CeO2 nanofibers (Fig. S11†), the retention rate of Pt (determined by ICP) after aging at 700 °C in air for 2 h was increased by the CeO2/Al2O3 fibril-in-tubes from 61.4% to 71.4%. The promoted leaching resistance can be ascribed to the synergistic stabilization by the dual-oxide composition and unique fibril-in-tube nanostructure. In particular, the diffused Pt species can be effectively trapped by the multilayered surfaces as shown in Fig. S14,† therefore contributing to the high dispersion of Pt clusters after harsh aging processes, as demonstrated by the elemental mapping in Fig. 3d–f. These results sufficiently illustrated the elegant all-in-one design of CeO2/Al2O3 fibril-in-tubes, in terms of both the structure and component, for boosting the thermal stability of the whole catalytic system.
As an emerging modern technology to unravel the underlying sintering resistance mechanism, in situ microscopic observations can provide in-depth information on the dynamic changes of both the metal and support upon aging in different atmospheres.23 To exclude the complex impacts of the atmosphere, we recorded the time-series HAADF-STEM images of Pt@CeO2/Al2O3 at temperatures ranging from room temperature to 900 °C under vacuum, with regard to the thermal stress only (Fig. 4a, S15 and S16†). For quantitatively evaluating the thermal stability, we tracked the size change of Pt clusters (Fig. S17†) and summarized the results in Fig. 4b. During the harsh aging process, no observable sintering was visualized. The Pt clusters with an initial average diameter of merely 2.3 nm kept a size smaller than twice their initial size and sat on the support individually in ultra-close proximity, until being exposed at 800 °C for 2 min, strongly suggesting the superb sinter-resistance. After considering the desirable diameters in the range of industrial catalysts (that is 1.5–4.0 nm),7 the limit of sinter-resistance of Pt@CeO2/Al2O3 can be roughly regarded as 800 °C (denoted as Ts as highlighted in Fig. 4b).
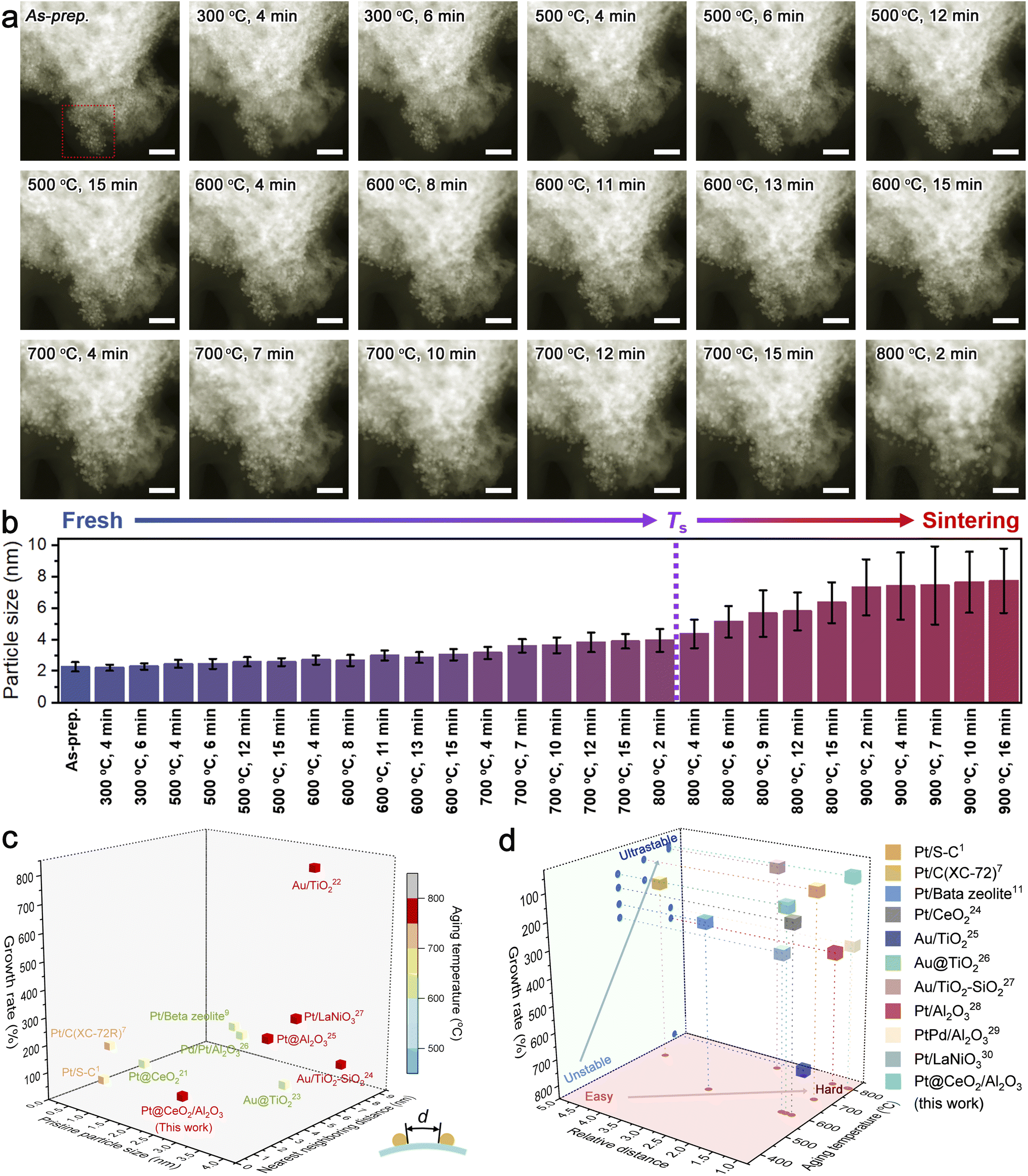 | ||
| Fig. 4 (a and b) In situ HAADF-STEM observation of Pt@CeO2/Al2O3 at elevated temperatures (a) and the corresponding size evolution of Pt clusters (b). The scale bars in (a) indicate 20 nm. The HAADF-STEM images were colored for a better demonstration. (c and d) Ashby chart plotting (c) pristine particle size, nearest neighboring distance versus the growth rate at a certain aging temperature, and the relative distance, (d) the growth rate versus the aging temperature for Pt@CeO2/Al2O3 and other reported sinter-resistant catalyst systems. The inset in (c) illustrates the neighboring distance. | ||
To date, there is rarely a reported method for impartially quantifying the anti-sintering capability in diverse systems. By taking the minimum value of the distance between one cluster and any neighboring cluster (illustrated in the inset of Fig. 4c and S18†), the intrinsic dispersity of metal species on their supports can be carefully studied. On CeO2/Al2O3 fibril-in-tubes, the average nearest neighboring distance of Pt clusters could be measured to be only around 2.1 nm, which is shorter than the pristine diameter of one Pt cluster. Such an ultra-close distribution causes a great challenge for stabilizing Pt clusters, as discussed above.
For a synthetic comparison, we summarized the pristine size, the average nearest neighboring distance, and the growth rate at a certain aging temperature of several recently reported sinter-resistant catalyst systems with different well-designed compositions and structures in Fig. 4c.1,7,11,24–30 The growth rate of the metal species is defined as the difference between the diameter after growth and the initial diameter as a percentage of the initial diameter. All the involved systems were based on metal catalysts with pristine sizes similar to the Pt clusters used in this work. Moreover, we correlated the average nearest neighboring distance with the pristine particle size (defined as relative distance) and plotted it versus the aging temperature and growth rate (Fig. 4d). As shown, although the Pt clusters on the CeO2/Al2O3 fibril-in-tubes were in ultra-close proximity with the average nearest neighboring distance of merely 0.93 of the pristine particle diameter, the catalytic system can maintain a small growth rate below 100% at 800 °C for a while. The boosted sintering resistance of Pt clusters can be largely attributed to the all-in-one stabilizing effects of the CeO2/Al2O3 fibril-in-tubes as illustrated in Fig. 1: (i) the built-in energy barriers energetically stabilized these Pt clusters; (ii) the pores and channels physically impeded the migration of Pt; and (iii) the promoted adhesion between the metal and support chemically anchored Pt.
To shed light on how the whole system failed eventually, the in situ heating temperature was further elevated (Fig. S16†). It is worth mentioning that the sintered Pt nanoparticles maintained a well-crystallized structure at temperatures up to 900 °C, which is somewhat higher than its melting point (727 °C, for 3 nm-Pt).3 However, the nanofibrous support was subjected to reconstruction during heating over 800 °C, in terms of the densification and the emergence of voids and cracks. Once the support suffered from reconstruction, the metal sintering would be severely accelerated, mainly because of the significantly shortened neighboring distance. Therefore, the in situ HAADF-STEM observation revealed that promoting the thermal stability of support has a profound impact on boosting the sinter-resistance of the catalytic systems, and should be highly addressed for the future design of durable catalysts with high metal loading.
According to the X-ray photoelectron spectroscopy (XPS) results (Fig. S19a†), the binding energies of Ce3+ and Ce4+ shifted to lower positions by 0.2–0.5 eV after being aged at high temperatures in air. The atomic ratio of Ce3+ to Ce4+ declined accordingly. No obvious chemical shift of Al 2s spectra was visualized (Fig. S19b†). The Pt 4d spectra exhibited an increased proportion of positively charged platinum species (Ptδ+) after aging at high temperatures, along with corresponding shifts towards higher binding energy (Fig. S19c†). This observation suggests a more pronounced interfacial charge transfer from Pt to the support at the interface.31 Moreover, the peak assigned to the lattice oxygen in Ce2O3 and CeO2 shifted to higher binding energies (Fig. S19d†), which could be ascribed to the generation of oxygen vacancies (Ov). The gradually increased indicator value of g peaks at 2.003 in the electron paramagnetic resonance (EPR) spectra (Fig. S19e†) further confirmed the enriched oxygen vacancies during heat treatment in an oxidative atmosphere.
In principle, the number of Ov is proportional to the concentration of Ce3+, where one oxygen defect can be generated accompanied by the formation of two Ce3+ ions.32 The exceptional increase of Ov in Pt@CeO2/Al2O3 should be largely ascribed to the incorporation of metal species into CeO2, including both Ptδ+ and Al3+.33 Consequently, the abundant oxygen vacancies provide ample anchoring sites for stabilizing Pt species, namely Pt–Ov–Ce sites, by improving the adhesion between Pt and CeO2.31 Moreover, the abundant Pt–Ov–Ce sites are supposed to be present not only within CeO2 nanodomains but also at dual-oxide interfaces and even in Al2O3 nanodomains as well, by considering the doping of Ce atoms into Al2O3 as we discussed above. Therefore, ultra-close Pt clusters with a size below 3 nm could be carefully stabilized against sintering on CeO2/Al2O3 fibril-in-tubes, becoming a promising catalyst for high-temperature reactions.
3.3 Catalytic soot combustion performance
Diesel particulate, consisting mainly of carbon nanoparticles with 20 nm in size, has seriously threatened public health in recent decades.33 To address this issue, employing efficient catalysts to remove the diesel particles from the source and further reduce the combustion temperature is extremely urgent. As shown in Fig. 5a, the naturally overlapped fibril-in-tubes with open channels can effectively trap the diesel particulates and thereby significantly promote the catalyst–diesel contact efficiency. On a more specific scale (Fig. 5b), the abundant oxygen vacancies at the metal/support interfaces can favor the activation of gaseous oxygen and the migration of active oxygen species to the surface, and thus promote the reaction thermodynamics significantly.34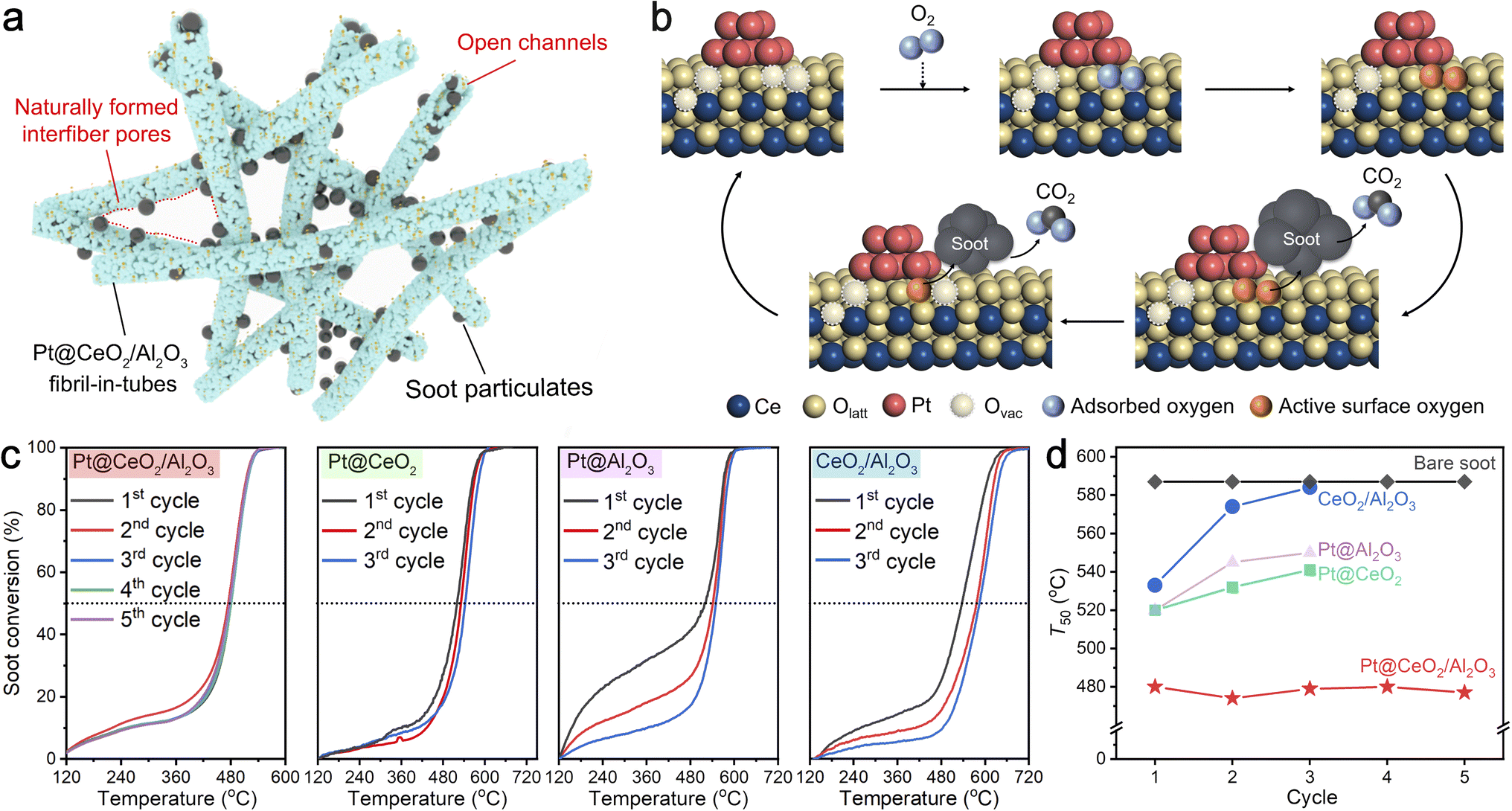 | ||
| Fig. 5 (a) Schematic illustration of Pt@CeO2/Al2O3 fibril-in-tubes physically trapping diesel soot. (b) Activation pathways for O2 at the Pt/CeO2 interface. Note that these structures are provided for illustration but are not calculated structures. (c) Soot conversion over Pt@CeO2/Al2O3 fibril-in-tubes, Pt@CeO2 nanofibers, Pt@Al2O3 nanofibers, and bare CeO2/Al2O3 fibril-in-tubes without Pt. (d) T50 of Pt@CeO2/Al2O3 within 5 cycles and Pt@CeO2 nanofibers, Pt@Al2O3 nanofibers, and CeO2/Al2O3 fibril-in-tubes within 3 cycles. | ||
To evaluate the catalytic performance, diesel combustion with a temperature-programmed oxidation method was conducted in a loose contact mode, which is the main fashion under practical conditions.35 The soot conversion curves over the Pt@CeO2/Al2O3 fibril-in-tubes, Pt@CeO2 nanofibers, Pt@Al2O3 nanofibers, and the bare CeO2/Al2O3 fibril-in-tubes without Pt for several cycles are shown in Fig. 5c. The catalytic activity was quantitatively evaluated from the values of T10, T50, and T90, which were defined as the temperatures at 10%, 50%, and 90% of soot conversion, respectively. Compared with the soot particles without any catalyst (Fig. S20,†T10, T50, and T90 of 501, 587, and 672 °C, respectively), the presence of CeO2/Al2O3 fibril-in-tubes alone can decrease the T10, T50, and T90 to 258, 533, and 607 °C at its first reaction cycle. After being loaded with only 0.8 wt% of Pt clusters, the Pt@CeO2/Al2O3 fibril-in-tubes effectively lowered the T10, T50, and T90 to 247, 480, and 518 °C further, being superb compared to Pt@CeO2 nanofibers and Pt@Al2O3 nanofibers. In particular, the ignition temperature (i.e., T10) of Pt@CeO2/Al2O3 was 254 °C lower than that of the reaction without catalysts. Such a low soot conversion temperature at a relatively high ramping rate of 10 °C min−1 is competitive with many recently reported catalyst systems (Table S2†).36–40
More specifically, the catalytic performance of Pt@CeO2/Al2O3 fibril-in-tubes, especially the T50, did not decline during five consecutive rounds of high-temperature and exothermic reactions but instead showed a stable and slightly fluctuating trend (summarized in Fig. 5d). The HAADF-STEM images of Pt@CeO2/Al2O3 fibril-in-tubes after each cycle of soot combustion are summarized in Fig. S21.† Despite the exothermic nature of the reaction (ΔH = −393.5 kJ mol−1), which leads to a localized increase in temperature around the active sites, specifically at the Pt/CeO2 interface, both the support and Pt clusters demonstrate exceptional thermal stability. However, the catalytic performances of bare CeO2/Al2O3 fibril-in-tubes, Pt@CeO2 nanofibers, and Pt@Al2O3 nanofibers were reduced somehow. As shown, the loaded Pt clusters sintered significantly to 12.1 nm on the CeO2 nanofibers and 17.6 nm on the Al2O3 nanofibers after 3 cycles of diesel combustion (Fig. S22†). Therefore, these observations unequivocally demonstrate the exceptional sinter-resistance of Pt@CeO2/Al2O3 fibril-in-tubes under reaction conditions, which presents a significantly greater challenge than merely stabilizing metal species against thermal stress.
4 Conclusions
In summary, we have demonstrated all-in-one designed CeO2/Al2O3 fibril-in-tubes to boost the sinter-resistance of supported 2.3 nm Pt clusters in ultra-close proximity. The strong support, CeO2, effectively immobilizes Pt clusters with strong adhesion, and the adjacent weak support, Al2O3, with low adhesion, constructs energy barriers to impede the migration of Pt species across the entire substrate. Moreover, the hierarchical pores and channels physically confine the Pt clusters. Therefore, the sinter-resistance of these 2.3 nm Pt clusters can be boosted to over 750 °C, under an oxidative and even steam-containing atmosphere. The in situ HAADF-STEM observation revealed that significant sintering of Pt clusters started at 800 °C, coinciding with the reconstruction of the supports. In diesel combustion, the Pt@CeO2/Al2O3 fibril-in-tubes decreased the ignition temperature by about 254 °C and showed reliable stability within 5 cycles. This work represents a rational design of sinter-resistant catalytic systems with high activity and durability by integrating multiple advances into one.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Wanlin Fu: investigation, writing, data analysis; Kuibo Yin: methodology, data analysis, supervision; Zhihui Li: investigation, writing; Jun Wang: investigation; Mingyu Tang: investigation; Jilan Tian: investigation; Litao Sun: supervision, project administration; Yueming Sun: supervision, project administration; Yunqian Dai: supervision, project administration, conceptualization, funding acquisition.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2022YFA1505700), the National Natural Science Foundation of China (22475044, 21975042 and 12174050), Pre-Research Fund of Ministry of Education of China (No. 8091B022212), the Project of Qinglan Talent of Jiangsu, and the Priority Academic Program Development of Jiangsu Higher Education Institutions.References
- C. L. Yang, L. N. Wang, P. Yin, J. Liu, M. X. Chen, Q. Q. Yan, Z. S. Wang, S. L. Xu, S. Q. Chu, C. Cui, H. Ju, J. Zhu, Y. Lin, J. Shui and H. W. Liang, Science, 2021, 374, 459–464 CrossRef PubMed.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121, 567–648 CrossRef PubMed.
- Y. Dai, P. Lu, Z. Cao, C. Campbell and Y. Xia, Chem. Soc. Rev., 2018, 47, 4314–4331 RSC.
- E. Toulkeridou, J. Kioseoglou and P. Grammatikopoulos, Nanoscale Adv., 2022, 4, 4819–4828 RSC.
- J. A. Rodriguez, D. C. Grinter, Z. Liu, R. M. Palomino and S. D. Senanayake, Chem. Soc. Rev., 2017, 46, 1824–1841 RSC.
- S. Hu and W. X. Li, Science, 2021, 374, 1360–1365 CrossRef CAS PubMed.
- P. Yin, S. Hu, K. Qian, Z. Wei, L. Zhang, Y. Lin, W. Huang, H. Xiong and W. Li, Nat. Commun., 2021, 12, 4865 CrossRef CAS PubMed.
- Y. Zhang, J. Zhang, B. Zhang, R. Si, B. Han, F. Hong, Y. Niu, L. Sun, L. Li, B. Qiao, K. Sun, J. Huang and M. Haruta, Nat. Commun., 2020, 11, 558 CrossRef CAS PubMed.
- L. Shang, T. Bian, B. Zhang, D. Zhang, L. Wu, C. Tung, Y. Yin and T. Zhang, Angew. Chem., Int. Ed., 2014, 53, 250–254 CrossRef CAS PubMed.
- Z. Yang, X. Xie, G. Peng, L. Shang and T. Zhang, Acc. Mater. Res., 2024, 5, 194–205 CrossRef CAS.
- J. Zhang, L. Wang, B. Zhang, H. Zhao, U. Kolb, Y. Zhu, L. Liu, Y. Han, G. Wang, C. Wang, D. S. Su, B. C. Gates and F. S. Xiao, Nat. Catal., 2018, 1, 540–546 CrossRef CAS.
- A. L. A. Marinho, R. C. Rabelo-Neto, F. Epron, N. Bion, F. S. Toniolo and F. B. Noronha, Appl. Catal., B, 2020, 268, 118387 CrossRef CAS.
- J. Zhou, G. Xiang, T. Zhai, Z. Liu, W. Zhao, X. Liang and L. Wang, Nat. Commun., 2022, 13, 327 CrossRef CAS PubMed.
- H. Chen, W. Lin, Z. Zhang, Z. Yang, K. Jie, J. Fu, S. Yang and S. Dai, Chem. Sci., 2020, 11, 5766–5771 RSC.
- A. M. Gänzler, M. Casapu, F. Maurer, H. Störmer, D. Gerthsen, G. Ferré, P. Vernoux, B. Bornmann, R. Frahm, V. Murzin, M. Nachtegaal, M. Votsmeier and J. D. Grunwaldt, ACS Catal., 2018, 8, 4800–4811 CrossRef.
- J. Lee, S. Kang, E. Lee, M. Kang, J. Sung, T. J. Kim, P. Christopher, J. Park and D. H. Kim, J. Mater. Chem. A, 2022, 10, 7029–7035 RSC.
- Y. Liu, A. J. McCue, P. Yang, Y. He, L. Zheng, X. Cao, Y. Man, J. Feng, J. A. Anderson and D. Li, Chem. Sci., 2019, 10, 3556–3566 RSC.
- W. Fu, W. Xu, K. Yin, X. Meng, Y. Wen, L. Peng, M. Tang, L. Sun, Y. Sun and Y. Dai, Mater. Horiz., 2023, 10, 65 RSC.
- H. Xiong, D. Kunwar, D. Jiang, C. E. G. Vargas, H. L. C. Du, G. Canning, X. I. P. Hernandes, Q. Wan, S. Lin, S. C. Purdy, J. T. Miller, K. Leung, S. S. Chou, H. H. Brongersma, R. Veen, J. Huang, H. Guo, Y. Wang and A. K. Datye, Nat. Catal., 2021, 4, 830–839 CrossRef CAS.
- C. Wang, S. Mao, Z. Wang, Y. Chen, W. Yuan, Y. Ou, H. Zhang, Y. Gong, Y. Wang, B. Mei, Z. Jiang and Y. Wang, Chem, 2020, 6, 752–765 CAS.
- N. J. O’ Connor, A. S. M. Jonayat, M. J. Janik and T. P. Senftle, Nat. Catal., 2018, 1, 531–539 CrossRef.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. P. Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS.
- Z. Chen, H. Cheng, Z. Cao, J. Zhu, T. Blum, Q. Zhang, M. Chi and Y. Xia, Nano Lett., 2024, 24, 1392–1398 CrossRef CAS.
- J. S. Du, T. Bian, J. Yu, Y. Jiang, X. Wang, Y. Yan, Y. Jiang, C. Jin, H. Zhang and D. Yang, Adv. Sci., 2017, 4, 1700056 CrossRef.
- H. Tang, F. Liu, J. Wei, B. Qiao, K. Zhao, Y. Su, C. Jin, L. Li, J. Liu, J. Wang and T. Zhang, Angew. Chem., Int. Ed., 2016, 5, 10606–10611 CrossRef PubMed.
- S. Liu, W. Xu, Y. Niu, B. Zhang, L. Zheng, W. Liu and J. Wang, Nat. Commun., 2019, 10, 5790 CrossRef PubMed.
- Y. Zhang, J. Zhang, B. Zhang, R. Si, B. Han, F. Hong, Y. Niu, L. Sun, L. Li, B. Qiao, K. Sun, J. Huang and M. Haruta, Nat. Commun., 2020, 11, 558 CrossRef PubMed.
- A. Aitbekova, C. Zhou, M. L. Stone, J. S. Lezama-Pacheco, A. C. Yang, A. S. Hoffman, E. D. Goodman, P. Huber, J. F. Stebbins, K. C. Bustillo, P. Ercius, J. Ciston, S. R. Bare, P. N. Plessow and M. Cargnello, Nat. Mater., 2022, 21, 1290–1297 CrossRef.
- J. Oh, A. Beck, E. D. Goodman, L. T. Roling, A. Boucly, L. Artiglia, F. Abild-Pedersen, J. A. Bokhoven and M. Cargnello, ACS Catal., 2023, 13, 1812–1822 CrossRef.
- S. Zhang, W. Zhou, J. Mao, K. An, N. Li, T. Qin, L. Chen, X. Liu, B. Mei, Z. Jiang, Z. Wang, Y. Yamauchi and Y. Liu, J. Mater. Chem. A, 2022, 10, 8227–8237 RSC.
- J. Yu, X. Qin, Y. Yang, M. Lv, P. Yin, L. Wang, Z. Ren, B. Song, Q. Li, L. Zheng, S. Hong, X. Xing, D. Ma, M. Wei and X. Duan, J. Am. Chem. Soc., 2024, 146, 1071–1080 CrossRef CAS PubMed.
- S. Pollitt, V. Truttmann, T. Haunold, C. Garcia, W. Olszewski, J. Llorca, N. Barrabés and G. Rupprechter, ACS Catal., 2020, 10, 6144–6148 CrossRef CAS PubMed.
- X. Mei, X. Zhu, Y. Zhang, Z. Zhang, Z. Zhong, Y. Xin and J. Zhang, Nat. Catal., 2021, 4, 1002–1011 CrossRef CAS.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2017, 358, 1419–1423 CrossRef CAS PubMed.
- Y. Li, T. Qin, Y. Wei, J. Xiong, P. Zhang, K. Lai, H. Chi, X. Liu, L. Chen, X. Yu, Z. Zhao, L. Li and J. Liu, Nat. Commun., 2023, 14, 7149 CrossRef PubMed.
- M. J. Kim, G. H. Han, S. H. Lee, H. W. Jung, J. W. Choung, C. H. Kim and K. Y. Lee, J. Hazard. Mater., 2020, 384, 121341 CrossRef PubMed.
- Y. Chen, G. Shen, Y. Lang, R. Chen, L. Jia, J. Yue, M. Shen, C. Du and B. Shan, J. Catal., 2020, 84, 96–105 CrossRef.
- J. H. Lee, S. H. Lee, J. W. Choung, C. H. Kim and K. Y. Lee, Appl. Catal., B, 2019, 246, 356–366 CrossRef.
- N. S. Portillo-Vélez and R. Zanella, Chem. Eng. J., 2020, 385, 123848 CrossRef.
- W. Ren, T. Ding, Y. Yang, L. Xing, Q. Cheng, D. Zhao, Z. Zhang, Q. Li, J. Zhang, L. Zheng, Z. Jiang and X. Li, ACS Catal., 2019, 9, 8772–8784 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04001e |
| This journal is © The Royal Society of Chemistry 2024 |