
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The missing pieces in the catalytic cycle of [FeFe] hydrogenases
Manon T.
Lachmann
a,
Zehui
Duan
b,
Patricia
Rodríguez-Maciá
 *a and
James A.
Birrell
*c
*a and
James A.
Birrell
*c
aSchool of Chemistry and Leicester Institute of Structural and Chemical Biology, University of Leicester, Leicester, LE1 7RH, UK. E-mail: prm28@leicester.ac.uk
bUniversity of Oxford, Department of Chemistry, Inorganic Chemistry Laboratory, South Parks Road, Oxford, OX1 3QR, UK
cSchool of Life Sciences, University of Essex, Colchester, CO4 3SQ, UK. E-mail: james.birrell@essex.ac.uk
First published on 7th August 2024
Abstract
Hydrogen could provide a suitable means for storing energy from intermittent renewable sources for later use on demand. However, many challenges remain regarding the activity, specificity, stability and sustainability of current hydrogen production and consumption methods. The lack of efficient catalysts based on abundant and sustainable elements lies at the heart of this problem. Nature's solution led to the evolution of hydrogenase enzymes capable of reversible hydrogen conversion at high rates using iron- and nickel-based active sites. Through a detailed understanding of these enzymes, we can learn how to mimic them to engineer a new generation of highly active synthetic catalysts. Incredible progress has been made in our understanding of biological hydrogen activation over the last few years. In particular, detailed studies of the [FeFe] hydrogenase class have provided substantial insight into a sophisticated, optimised, molecular catalyst, the active site H-cluster. In this short perspective, we will summarise recent findings and highlight the missing pieces needed to complete the puzzle.
 Manon T. Lachmann | Manon Lachmann studied chemistry at the University of Oxford where she graduated with an MChem in 2023. She is now undertaking a PhD at the University of Leicester under the supervision of Dr Rodríguez-Maciá. She works on semi-synthetic [FeFe] hydrogenases and carbon monoxide dehydrogenase enzymes. |
 Zehui Duan | Zehui Duan is a DPhil student at the University of Oxford, UK, co-supervised by Prof. Kylie Vincent and Dr Patricia Rodriguez Macia. She is a graduate of Imperial College London (M.Res. in Plastic Electronic Materials). Her studies have focussed on developing advanced spectroscopy techniques combined with electrochemistry, particularly applied to hydrogenases. |
 Patricia Rodríguez-Maciá | Patricia Rodriguez Macia studied Chemistry at the University of Alicante (Spain), before moving to Germany for her PhD at the Max Planck Institute for Chemical Energy Conversion (MPI-CEC) with Prof. Wolfgang Lubitz. She did postdocs with Prof. Serena DeBeer at MPI-CEC (2018–2020) and Prof. Kylie Vincent at University of Oxford (2020–2021). In 2021, she started her independent career as Glasstone Research Fellow in Oxford and, in 2023, she moved to the University of Leicester as a Lecturer (Assist Prof.) in the School of Chemistry. Her research focuses on metalloenzyme mechanistic investigations as inspiration for developing biohybrid catalysts for small-molecule activation. |
 James A. Birrell | James Birrell completed his PhD in 2012 at the MRC Mitochondrial Biology Unit and the University of Cambridge, UK, with Professor Judy Hirst FRS. He moved to Germany for postdoctoral work at the Max Planck Institute for Chemical Energy Conversion (MPI-CEC) with Professor Wolfgang Lubitz (2013–2017). He remained at MPI-CEC as a Group Leader in Professor Serena DeBeer's Bioinorganic Spectroscopy department (2017–2022). In 2022, James returned to the UK as a Lecturer at the University of Essex. James's present research interests are structural and functional aspects of anaerobic metabolism in bacteria and archaea. |
The big picture: renewable energy and the H2 economy
The Industrial Revolution marked an incredible time of human ingenuity and progress. Yet, it has also caused huge amounts of carbon dioxide, methane, and other potent greenhouse gases to accumulate in the atmosphere, leading to global warming and climate change. Now, efforts to curb greenhouse gas emissions require us to abandon fossil fuels and move to a completely circular energy economy. While electricity produced by renewable energy sources such as wind and solar power has the potential to supply all our current energy demands, the issue of energy storage remains a difficult problem to solve. Hydrogen could act as a suitable energy vector as it can be produced by water electrolysis and its combustion produces water as the sole byproduct (Fig. 1).1 Advantageously, hydrogen is incredibly energy-dense (120 MJ kg−1),2 however, its storage and transport prove problematic. Regardless, hydrogen is still a crucial reactant for several industrial processes, including oil refining and the Haber Bosch process for the reduction of nitrogen to ammonia, which each use around 30% of annual global hydrogen production.3 Presently, 98% of hydrogen is produced from fossil fuels, and of the remaining 2% produced by electrolysis, only a fraction is produced using renewable electricity.3 | ||
| Fig. 1 Schematic representation of a hydrogen economy. | ||
There are currently three major technologies for producing hydrogen via electrolysis: alkaline electrolysers, which use nickel-based electrodes and work at very large overpotentials, proton exchange membrane electrolysers, which use platinum, and solid oxide electrolysers, which use nickel-/yttria-stabilised zirconia.4 So, while water electrolysis is an established hydrogen production technology, there is still significant interest in developing more efficient, stable, and cheaper relevant materials. Since hydrogen has a relatively large bond dissociation energy (436 kJ mol−1),2 it is fairly inert and must be activated by a catalyst. As such, there has been incredible investment in understanding how nature uses hydrogen and catalyses this difficult reaction with high efficiency.
In nature, hydrogen is predominantly produced and consumed by hydrogenases, of which there are three phylogenetically distinct classes: the [NiFe] hydrogenases, the [FeFe] hydrogenases and the [Fe] hydrogenases. All three groups heterolytically split hydrogen, with [Fe] hydrogenases requiring a cosubstrate, methenyl-tetrahydromethanopterin, which they hydrogenate by direct hydride transfer. Meanwhile, [NiFe] and [FeFe] hydrogenases reversibly oxidise hydrogen to protons and electrons and transfer these electrons to electron carriers like ferredoxin. Nitrogenases and a plethora of other enzymes have been demonstrated to oxidise or produce hydrogen, albeit at substantially lower rates than the true hydrogenases.5–11
Studying how hydrogenases work to understand the crucial mechanistic principles of efficient hydrogen activation has been an active area of research for well over 50 years. But how far have we come and what is left to understand? In this perspective article, we will address these questions, with a particular focus on [FeFe] hydrogenases. We will also discuss where we see the field going over the next years and what the main challenges are for uncovering the ‘missing pieces’ in the catalytic cycle.
The unique structure of the [FeFe] hydrogenase active site
Since 1996 it has been known that [FeFe] hydrogenases contain an active site, referred to as the H-cluster, which is a unique type of iron–sulfur cluster coordinated by cyanide and carbon monoxide ligands.12 However, its intricacies were unknown13 until Peters and coworkers published the X-ray crystal structure of the [FeFe] hydrogenase from Clostridium pasteurianum (CpI) in 1998.14 For the first time, a unique [2Fe]H subcluster, coordinated by diatomic ligands and an additional unprecedented bridging ligand was observed covalently attached to a [4Fe–4S]H cubane cluster. This was immediately suggested as the site of hydrogen activation. Initially, the unknown bridging ligand was identified to contain two sulfurs, which directly coordinate each Fe. The electron density between them was modelled as water. A later structure from Nicolet and coworkers of the [FeFe] hydrogenase from Desulfovibrio desulfuricans (DdHydAB) modelled the bridging ligand as a propane-1,3-dithiolate ligand (PDT).15 A subsequent publication from the same group revised this to a 2-azapropane-1,3-ditholate ligand (ADT, also known as di(thiomethyl)amine or DTMA).16 This latter idea was supported by some groups through density functional calculations17 but others proposed a more likely bridging ligand to be 2-oxapropane-1,3-dithiolate (ODT, also known as di(thiomethyl)ether or DTME).18 After years of intense debate, the composition of the dithiolate as ADT is now well established, both through spectroscopic studies19–22 and reconstitution studies.23 Thus, today we have a fairly clear picture of the active site structure of [FeFe] hydrogenases (Fig. 2).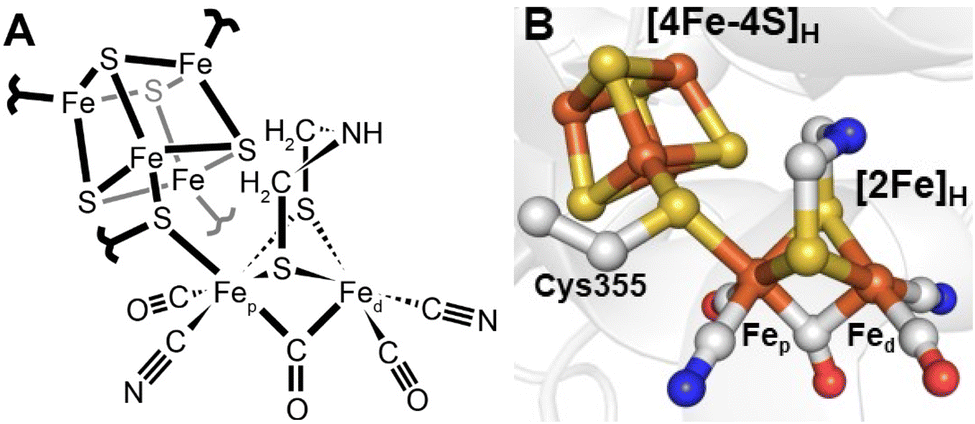 | ||
| Fig. 2 Structure of the active site H-cluster of [FeFe] hydrogenase in (A) a ChemDraw style representation and (B) a Pymol style representation. Residue numbering is from the C. pasteurianum enzyme. The image was prepared using PDB ID 4XDC.24 | ||
The [FeFe] hydrogenase H-cluster provides a rare and interesting example of two metal clusters electronically coupled by a simple covalent linkage (Fig. 3). The [4Fe–4S]H cluster in the absence of the [2Fe]H subcluster is stable and functions as a simple redox cofactor.23 As is the [2Fe]H subcluster which can also undergo protonation and deprotonation. However, the [2Fe]H subcluster alone is an extremely poor hydrogen conversion catalyst; it is only by inserting it into the [FeFe] hydrogenase protein scaffold that high activity is achieved. Substitution of the sulfur in the [4Fe–4S]H cluster with selenium alters the cluster redox potential but has very little effect on activity.25 Meanwhile, mutation of various amino acids surrounding the [2Fe]H subcluster produces highly inactive enzymes,26 suggesting this factor is most crucial for enzyme activity. Interestingly though, mutation of the ligands coordinated to [4Fe–4S]H (e.g., from cysteine to histidine) affects activity, shifting the catalytic bias and increasing the overpotential.27,28
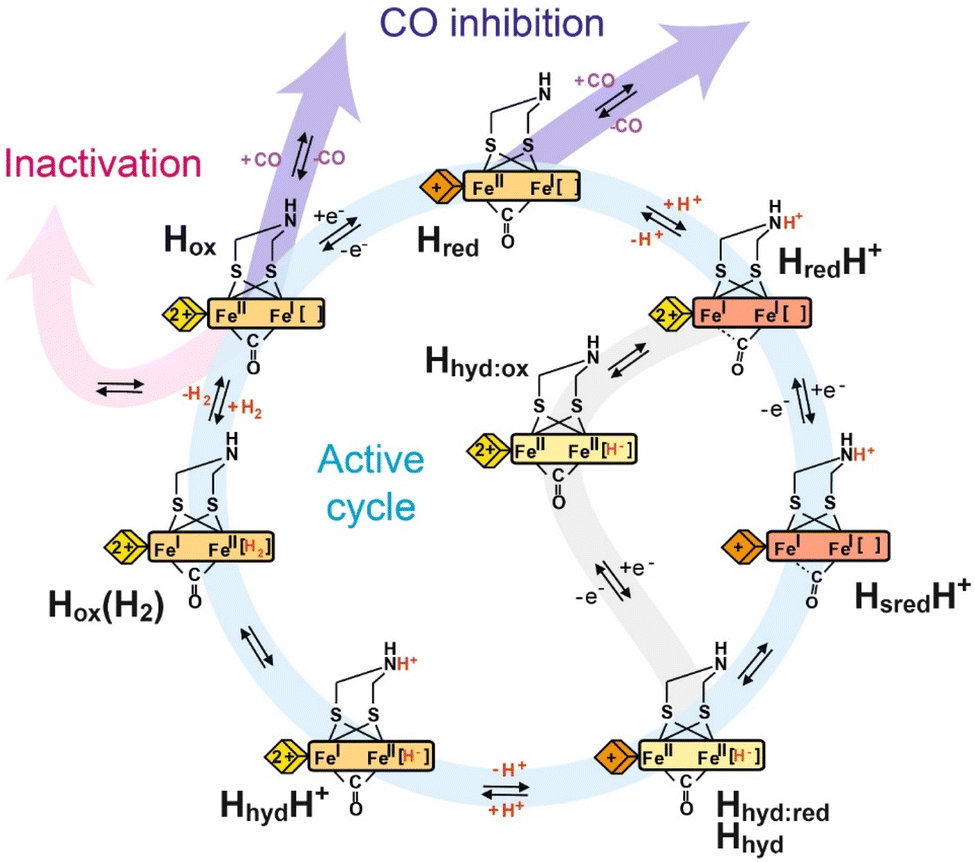 | ||
| Fig. 3 Proposed catalytic cycle of [FeFe] hydrogenase. The blue pathway indicates the active cycle, while the pink pathway indicates inactivation by binding to inhibitors like H2S and HCN, the purple pathway indicates reversible inhibition by CO, and the gray pathway indicates an alternative route from the HredH+ state to the Hhyd:red state under less reducing conditions. The yellow, orange and red rectangles represent the [2Fe]H cluster in the Fe(II)Fe(II), Fe(II)Fe(I) and Fe(I)Fe(I) oxidation states, respectively, with the terminal CO and CN- ligands omitted for clarity. The yellow and orange diamonds represent the [4Fe–4S]H cluster in the +2 and +1 oxidation states, respectively. | ||
General mechanistic principles: proton-coupled electron transfer and hydride formation
In 2006, Roseboom and coworkers investigated the [FeFe] hydrogenase from Desulfovbrio desulfuricans using infrared (IR) spectroscopy coupled with electrochemistry.29 This enzyme is unusual because it can be purified under air in an inactive over-oxidised state known as Hinact. In their titrations from high potential to low potential they observed conversion of Hinact to an intermediate state Htrans followed by formation of the active oxidised state, Hox. Further reduction formed the one-electron-reduced Hred state, followed by the formation of a two-electron-reduced, or super-reduced, Hsred state. Based on the positions of the IR bands as well as the known electron paramagnetic resonance (EPR) properties of each state, the Hinact state was described as a [4Fe–4S]2+-[Fe(II)Fe(II)] state. Reduction to Htrans involved reduction of the [4Fe–4S]H cluster and yielded a [4Fe–4S]+-[Fe(II)Fe(II)] state. Hox was thought to be isoelectronic with Htrans with a [4Fe–4S]2+-[Fe(II)Fe(I)] state, where the electron moved from the [4Fe–4S]H cluster to the [2Fe]H cluster. Reduction of Hox to Hred was thought to occur at the [2Fe]H cluster yielding a [4Fe–4S]2+-[Fe(I)Fe(I)] state, and formation of Hsred was thought to involve reduction of the [4Fe–4S]H cluster giving a [4Fe–4S]+-[Fe(I)Fe(I)] state. The last transition was observed to be not completely reversible. These transitions were all pH-dependent, indicating the important role of proton-coupled electron transfer for the enzymes' high catalytic activities.In 2009, Silakov and coworkers performed similar experiments with the smaller, simpler [FeFe] hydrogenase from Chlamydomonas reinhardtii (CrHydA1), made similar observations and came to similar conclusions.30 However, one interesting finding was a large difference in the IR bands of the one-electron-reduced Hred state between the two enzymes. This issue was later resolved when it was discovered that the one-electron reduced state in CrHydA1 consisted of two different states, whose population depended on the pH, having roughly equal populations of both at neutral pH.31 Thus, these two states were dubbed the Hred and HredH+ states, where the former was observed predominantly at high pH and the latter at low pH. The Hred state had small red-shifts in the IR bands relative to the Hox state whereas the HredH+ state had large red-shifts, indicating that the Hred state had a [4Fe–4S]+-[Fe(II)Fe(I)] electronic structure while the HredH+ state had a [4Fe–4S]2+-[Fe(I)Fe(I)] electronic structure. It was assumed that the ADT ligand bridging the two Fe atoms was protonated in the HredH+ state facilitating electron transfer by charge neutralisation effects. Using a non-protonatable propane-1,3-dithiolate (PDT) ligand prevented the formation of HredH+, supporting the assignment.32 Furthermore, photolysis experiments were able to enrich the Hred state.33 Resonance Raman (RR) spectroscopy of CrHydA1 revealed a species containing a reduced [4Fe–4S]+ cluster and a Hox-like [2Fe]H site, likely derived from HredH+. The RR experimental conditions, laser illumination at low temperature, could facilitate electron transfer from the [2Fe]H subcluster to [4Fe–4S]H, generating a [4Fe–4S]+-[Fe(II)Fe(I)] state, which was named Hred’ at the time. Katz et al. proposed a proton transfer between the H-cluster and a near base (Cys169)33 as the driving force for electron transfer between the [4Fe–4S]H and [2Fe]H sites. Therefore, the populations of the isoelectronic states Hred and HredH+ were controlled by the protonation states of the ADT ligand through charge compensation of the [FeFe] subcluster. This was later supported by IR spectroelectrochemical titrations.31
Interestingly, the [FeFe] hydrogenase from D. desulfuricans only shows the HredH+ state at neutral pH.29 More carefully analysed redox titrations revealed that initial reduction of the Hox state generates a mixture of both Hred states but that further reduction converts the Hred state into the HredH+ state. In this case, what appears to happen is the reduction of the accessory iron–sulfur clusters triggers the electron to move from the [4Fe–4S]H subcluster to the [2Fe]H subcluster by charge repulsion. This then increases the pKa on the H-cluster forcing it to be completely protonated at neutral pH.34
Despite having very little catalytic activity the [FeFe] hydrogenases in which the ADT cofactor is substituted with PDT or oxapropane-1,3-dithiolate (ODT) have proved incredibly useful for understanding the catalytic mechanism. The PDT variant can only exist in two oxidation states: an Hox-like state with [4Fe–4S]2+-[Fe(II)Fe(I)] and an Hred-like state with [4Fe–4S]+-[Fe(II)Fe(I)]. The missing nitrogen bridgehead seems to prevent the formation of states in which [2Fe]H is reduced to Fe(I)Fe(I) as its reduction potential is too negative without coupled protonation due to charge imbalance. If, as has been suggested by others,35 the HredH+ and HsredH+ states contain bridging hydrides rather than protonated amines, then it is not clear why these states would not form in the PDT variant; there may be slower kinetics involved but eventually, these states should form. It is also interesting to note that terminal hydrides have yet to be detected in the PDT variant, which hints that the differences between the ADT and PDT variants are more complicated than simply the loss of the bridging amine. It should also be noted that the redox potential for the Hox/Hred transition has been reported to be pH-dependent, indicating that reduction of [4Fe–4S]H involves PCET.36 However, this result has been disputed37 and argued to be an artefact due to the presence of sodium dithionite in the preparations.38
While the PDT variant was useful for understanding PCET at the H-cluster, the ODT variant has been useful for understanding the formation of terminal Fe-hydrides. The ODT variant forms a fairly stable state with the lowest CO-band at 1867 cm−1,23 which was thought to have a [4Fe–4S]+-[Fe(II)Fe(II)]H− structure. The reason for this is still not entirely clear. It has been argued that the ether group in ODT, being much less basic than the amine group of ADT, is a poor proton relay so allows kinetic trapping of the Fe-hydride intermediate.39 Another possibility is that, like the PDT variant, the ODT variant does not form the HredH+ state, but, unlike the PDT variant, the ODT variant undergoes PCET with protonation of Fed. Regardless, Reijerse et al. directly observed the terminal hydride on Fed by studying the 57Fe labeled CrHydA1 ODT variant with nuclear resonance vibrational spectroscopy (NRVS).40
A final point that required investigation was the observation that both the HredH+ and HsredH+ states appeared to be lacking a bridging CO band in the IR spectra, and instead appeared to have an additional terminal CO at approximately 1960 cm−1 or 1950 cm−1. This finding has been interpreted to mean that the bridging CO becomes terminal leading to the formation of bridging hydride states. Under some circumstances, however, the bridging CO appeared to be retained e.g. at low temperature41–43 and in sensory enzymes.44 Furthermore, the 1960 cm−1 and 1950 cm−1 bands have been suggested to be due to small amounts of terminal hydride-containing states, which can be populated at low temperatures by photoexcitation. Therefore, the structural assignments of the HredH+ and HsredH+ states remain contentious.
Overall, catalysis at the H-cluster appears to proceed as follows: in the proton reduction direction, the active oxidised Hox state can be reduced by one electron. This gives an H-cluster with a pKa of approximately 7.2, so, around pH 7, a protonated (HredH+) and deprotonated (Hred) form are observed. The HredH+ state has an oxidised [4Fe–4S]H cluster and accepts an electron to form the HsredH+ state. Meanwhile, in the reverse direction, the Hred state can be oxidised to the Hox state by electron transfer from the [4Fe–4S]H cluster. This ensures a high level of catalytic reversibility as both redox events happen at very similar potentials, which are pH-dependent and close to the 2H+/H2 couple. In the HsredH+ state, both electrons needed for hydrogen production are loaded at the H-cluster and a proton is bound to the [2Fe]H subcluster. Electron and proton rearrangement occurs and the [2Fe]H subcluster is transformed to an isomeric state containing a hydride – the Hhyd state. The intermediates of this process were studied by light-induced spectroscopy under cryogenic temperature43 (detailed discussion in Section 7). The Hhyd state then accepts a second proton forming the transient HhydH+ state before hydrogen is formed and released from the active site. After H2 is released from HoxH2, the enzyme returns to Hox and the next turnover begins. This cycle operates in the H2 oxidation direction by an exact reversal of each of these steps. In our interpretation of the data, proton-couple electron transfer (PCET) is essential for the catalytic cycle. The [4Fe–4S]H cluster serves as an electron input module and protonation of the amine in the [2Fe]H cluster triggers electron transfer from [4Fe–4S]H to [2Fe]H.
Thermodynamic and kinetic considerations of the catalytic mechanism: pre-steady state and steady-state conditions
So far, most data used to understand the mechanism of [FeFe] hydrogenases have been collected under equilibrium (often near-equilibrium) or steady-state turnover conditions. More operando experiments are needed to confirm that the catalytic states identified are indeed catalytic. It has been argued that all states observed under equilibrium/steady-state conditions are thermodynamic sinks and, therefore, off-pathway non-catalytic intermediates.35,45,46 This logic would seem to contend that an enzyme with only catalytic states and no off-pathways would be spectroscopically silent, which is extremely unlikely. The energy landscape during catalysis is unlikely to be entirely flat and homogenous, thus, the most abundantly observed intermediates will be those that fall into energy troughs. It should be noted that under equilibrium conditions, a relatively small difference in Gibbs free energy (≈2.7 kcal mol−1 – on the order of a hydrogen bond) is required for a state to be 100-fold more abundant than another (eqn (1)).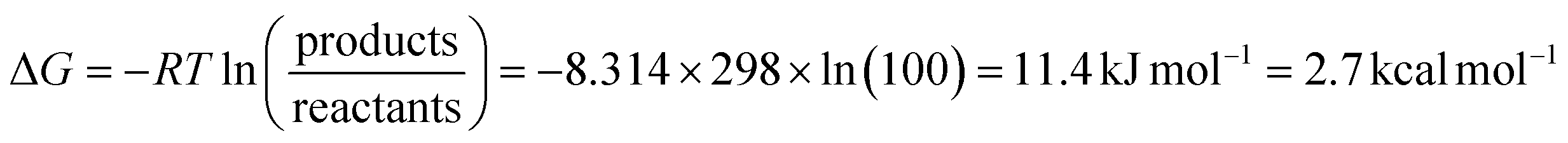 | (1) |
Small energy differences may be responsible for some catalytic intermediates remaining unobserved. However, that does not mean that every observed state is non-catalytic a priori. Steady-state turnover may reveal further catalytic states as rate-limiting steps start to populate higher energy intermediates, which accumulate faster than they decay. However, the ultimate proof of a state being catalytic will be its observation during pre-steady state kinetic analysis; the states that appear and disappear within a single turnover must, by definition, be involved in the catalytic cycle.
Despite the importance of such studies, relatively few publications have attempted to measure pre-steady state kinetics due to their experimental challenges. The [FeFe] hydrogenase from D. desulfuricans is suggested to turnover at least 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times per second.47 This indicates a catalytic cycle spanning 100 μs and requires a time resolution of at least 1 μs. This can be achieved by some spectroscopic techniques, including pump-probe IR spectroscopy to study the CO and CN band vibrations.48
000 times per second.47 This indicates a catalytic cycle spanning 100 μs and requires a time resolution of at least 1 μs. This can be achieved by some spectroscopic techniques, including pump-probe IR spectroscopy to study the CO and CN band vibrations.48
On top of this, experiments must be performed under conditions that allow turnover. This precludes (to a certain extent) the use of many spectroscopies (e.g. EPR spectroscopy) that require frozen samples. Turnover cannot be limited by exogenous factors such as diffusion of substrates to and from the active site. This adds complication as, during proton reduction, both protons and electrons are required by the hydrogenase. The latter requires an electron mediator, while the former can be mediated by water and buffer salts. Typical diffusion coefficients of small molecules in aqueous solution are on the order of 10−10 to 10−9 m2 s−1, while for protons they are estimated to be 105-fold higher based on the Grotthuss proton-hopping mechanism.49 Therefore, for efficient (sub-μs) electron transfer via an electron mediator, the average distance between the reduced mediator and the enzyme needs to be extremely short and the concentration of both the enzyme and electron mediator must be very high.
Several studies have attempted to measure pre-steady state kinetics in [FeFe] hydrogenases using photosensitisers and diffusible electron mediators for intermolecular electron transfer.48,50–55 However, diffusion limitations may still be an issue. To overcome these limitations, the hydrogenase needs to be covalently attached to the photosensitiser so that rapid intra-molecular electron transfer occurs. Several early studies indicated that this would be possible using thiol linkers56,57 but so far time-resolved studies using this approach have not been published. An alternative approach is to use the photosensitivity of the Hox–CO state. [FeFe] hydrogenases are inhibited by CO, forming the Hox–CO state, which is also reducible to the Hred–CO state. The Hox–CO state is known to be photosensitive (at least in the frozen state). An [FeFe] hydrogenase sample prepared under CO in the presence of H2 (to initiate H2 oxidation) or another reductant (to initiate H2 production) might be expected to become catalytically active upon photolysis of the Fe–CO bond. An open question here is whether CO rebinding is more rapid than catalysis.
Primary coordination sphere and contributions from the protein framework: from secondary coordination sphere to long-range interactions, proton transfer pathway and electron relay
Metallic Fe as well as various synthetic Fe-based materials do not come close to the efficiency or reversibility that is achieved by [FeFe] hydrogenases for hydrogen interconversion. The primary coordination sphere of the H-cluster finely tunes the electronic structure of the Fe ions in such a way that low oxidation states and spin states are stabilised in the enzyme. This is achieved through the combination of strong σ-donating (CN−) and π-accepting (CO) ligands, along with the covalent “soft” thiolate ligands. The π back-bonding interactions favor the lower valence Fe(I) and Fe(II) formal oxidation states as electron density is accepted from the metal 3dxy, yz, xz orbitals. Meanwhile the CN− ligands, which only weakly engage in π backbonding, but are much stronger σ donors, donate electron density into the metal 3dx2−y2 and 3dz2 orbitals, favoring higher valence formal oxidation states. It was recently observed that the binding of CN− to the H-cluster stabilised its overoxidised Fe(II)Fe(II) oxidation state,58,59 whereas CO binding maintains the Fe(II)Fe(I) oxidation state observed for Hox. It has not yet been possible to synthesise a [2Fe] analogue with extra CN− ligands, however, it has been possible to integrate a [2Fe] analogue where a CN− ligand was replaced by a CO ligand (i.e. MonoCN).60,61 In this case, spectroscopic data indicated stabilisation of the Fe(I)Fe(I) oxidation state. These results support the postulation that the balance of CN− and CO ligands in the H-cluster dramatically affects its electronic structure. There is a balance to be struck between the σ-donating and π-accepting character of the ligands to tune the electron density on the metals such that the redox transitions (II/I) fall into a potential range suited for hydrogen production and oxidation.Additional insight into this can be garnered from studies of synthetic complexes integrated into [FeFe] hydrogenases. In 2015, Siebel et al. attempted to reconstitute the H-cluster using 15 synthetic [2Fe] analogues, including the native precursor complex, as well as variations on the dithiolate bridging ligand and the number of CO/CN− ligands.61 Ten of these complexes were successfully inserted. Only two of the ten reconstituted enzymes showed significant catalytic activity, namely, the native ADT cofactor and a monocyanide (pentacarbonyl) form of the ADT complex, which presented with 47% H2 production activity and 41% H2 oxidation activity compared to the native enzyme (Table 1). Evidence provided by Lorenzi et al. suggests that the CN− ligand coordinates to Fep in the monocyanide variant of CrHydA1.60 So, although changing the relative σ-donating/π-accepting abilities of the ligands has a large effect on the electronic structure of the H-cluster, it seems to have no significant effect on the activity.60
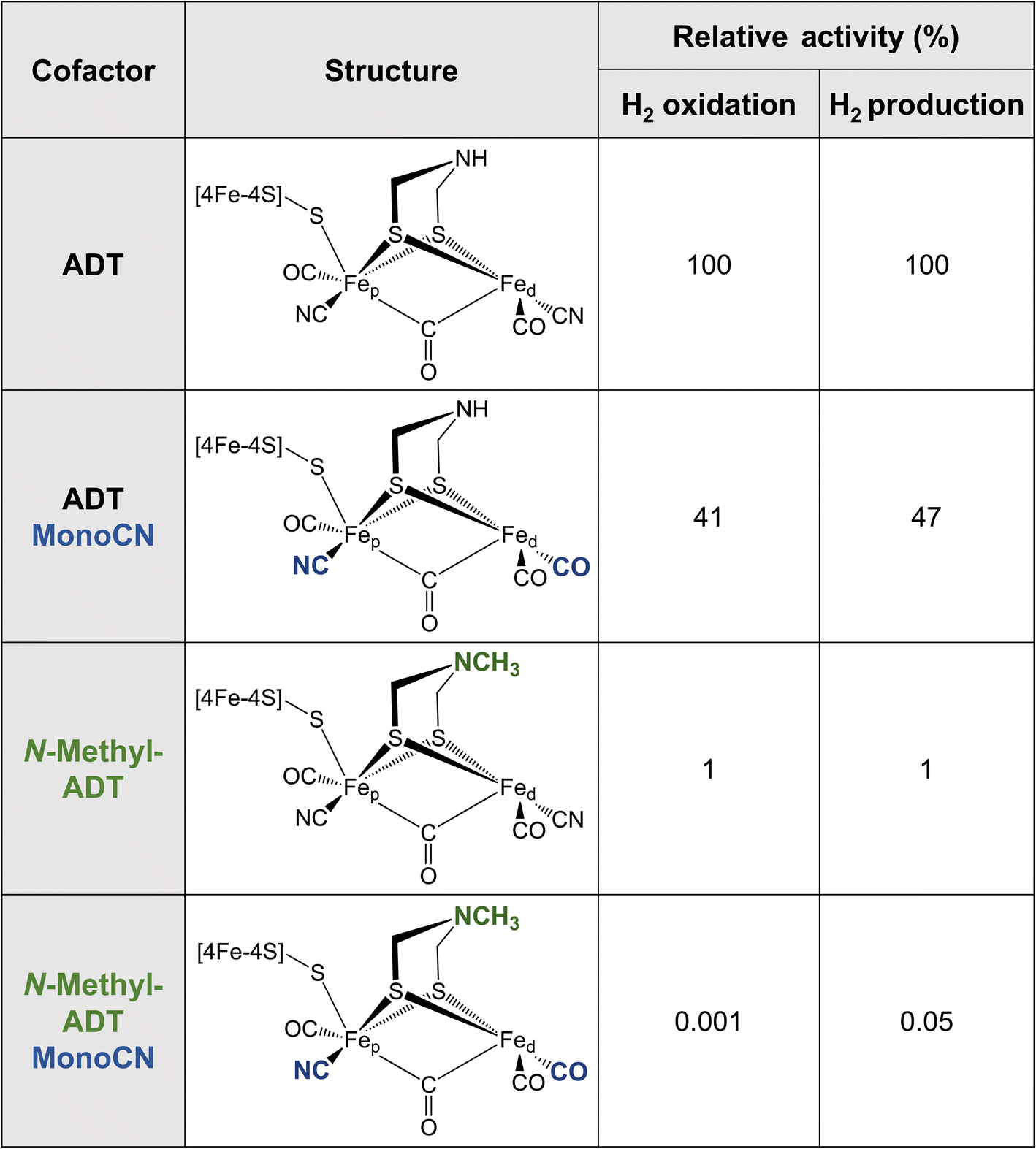
|
The enzyme containing an N-methylazadithiolate (N-methyl-ADT) bridge possessed 1% activity for H2 production and oxidation. This may indicate that the ability to form an ammonium ion, with a doubly protonated nitrogen (R-NH2+), could be crucial for activity. Interestingly, like the PDT variant, the as-isolated state of this [FeFe] hydrogenase appears to be the Hred state, whereas, under the same conditions, the native enzyme was in a mixture of Hox, Hred, HredH+ and HsredH+ states. This indicates that the N-methylamine group has a lower pKa and is unprotonated under these conditions (pH 8). This is counterintuitive as protonated tertiary amines are expected to be stabilised relative to protonated primary amines due to the inductive effect of the additional methyl carbon. However, the interaction with the protein matrix may play an additional role (discussed below). Another question regarding this variant is whether the methyl group points away from the open coordination site or toward it. The free complex is more stable with the methyl group pointing down toward the metals due to the anomeric effect and there will be reduced steric repulsion if the methyl group occupies space in the open coordination site. However, this would block the binding of H2, possibly providing another explanation for the lack of activity. Ultimately, crystal structures could reveal why this variant is so inactive.
The monocyanide form of the N-methyl-ADT variant was also tested and found to have 0.05% of the H2 production activity and 0.001% of the H2 oxidation activity of the native-like enzyme. This was surprising for two reasons. Firstly, N-methyl-ADT and monocyanide substitutions independently have little effect on the catalytic bias, but together shift the bias much more in favor of H2 production. Secondly, the monocyanide substitution of the ADT variant only halves the activity whereas this substitution on the N-methyl-ADT variant decreases the activity over 20-fold. Clearly, the factors affecting the enzyme activity are not well understood with multiple factors at play, so further investigation of cofactor variants using spectroscopic and electrochemical approaches could provide crucial insight.
The active site of an [FeFe] hydrogenase is intimately influenced by its interactions with the protein matrix. These interactions include the covalent attachment of [4Fe–4S]H and [2Fe]Hvia a cysteine residue, hydrogen-bonding interactions to the bridging thiolate, CO, and CN− ligands, and longer-range electrostatic interactions. The study by Knözer and coworkers was the first to try and understand these effects by mutating the protein matrix surrounding the H-cluster in CrHydA1 and CpI.26 They found that several surrounding amino acids play important roles in catalysis. A cysteine residue (Cys299) located within hydrogen bonding distance of the ADT ligand (Fig. 4) and thought to make up part of the proton transfer pathway was mutated to serine giving a completely inactive enzyme with unusual spectroscopic properties. A methionine (Met497), whose thioether group is positioned close to the ADT bridging ligand was mutated to leucine and gave a slightly less active enzyme. Spectroscopic analysis attributed this to a loss of the [2Fe]H subcluster from the active site. Another methionine (Met353) whose thioether group is positioned close to the bridging CO ligand was mutated to leucine giving a less active enzyme. Spectroscopic analysis was consistent with a high level of intact H-cluster, indicating that the lowered activity was related to the loss of an interaction between the thioether of the methionine and the bridging CO ligand. Lastly, mutation of an arginine residue (whose guanidinium group is positioned very close to the distal CN− ligand), when mutated to asparagine, gave an inactive enzyme which lacked both IR and EPR signals from the [2Fe]H site. From this, it was established that some amino acids surrounding the H-cluster are catalytically relevant while others play a role in stabilising the [2Fe]H subcluster.
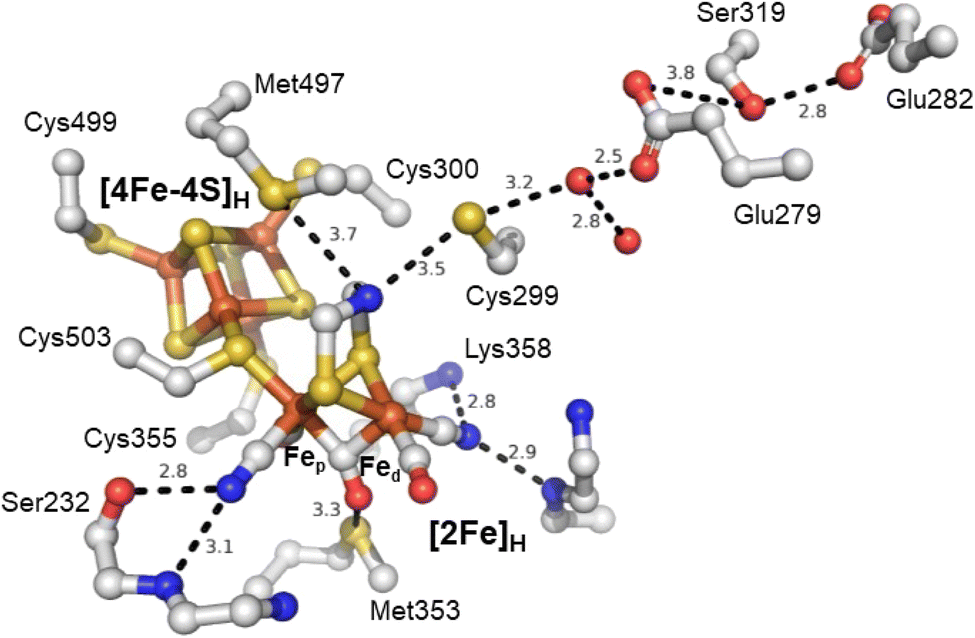 | ||
| Fig. 4 Structure of the active site H-cluster of [FeFe] hydrogenase showing the interacting amino acid residues and proton transfer pathway. Distances shown are in Å. Residue numbering is from the C. pasteurianum enzyme. The image was prepared using PDB ID 4XDC.24 | ||
Subsequent studies have further investigated the role of the cysteine in the proton transfer pathway, mutating it to serine,62–65 alanine59,63,66 and aspartic acid.62,63,66,67 The cysteine likely forms a hydrogen bond with the ADT ligand. It is generally accepted that this amino acid is responsible for directly exchanging protons between the H-cluster and the proton transfer pathway. Recent studies have also suggested that it directly influences the H-cluster's electronic structure.59,62 The IR spectrum of the as-isolated C169A mutant of CrHydA1 is essentially identical to that of the Hox state of wild-type CrHydA1,59 but EPR spectroscopy shows that the state has one more electron than Hox, and is Hred-like with IR bands shifted to higher energy. Oxidation of the C169A mutant gives an IR spectrum with higher energy IR bands and an EPR spectrum typical of Hox. This suggests that the C169A mutant has a more electron-deficient [2Fe]H core. Similar results have been reported for the C169S mutant65 but interpreted differently.
A further interesting feature of the C169A and C169S mutants is that they both stabilise a terminal hydride intermediate (Hhyd).39,64,65 This state is extremely persistent in these mutants and is observed during spectroelectrochemical titrations,64 confounding the initial kinetic proposal that a deficient proton transfer pathway slows the rate of transfer and traps the Hhyd state. By acting as a hydrogen bond acceptor C169 of the wild-type enzyme may increase the negative charge on the nitrogen of the ADT ligand, increasing its pKa and strengthening its basic properties. This would affect the thermodynamic hydricity at the H-cluster68,69 stabilising the protonated amine (HredH+ and HsredH+ states) over the protonated Fe (Hhyd).
A third argument in support of this idea is that the C169A mutants of CrHydA1, and the equivalent C178A mutant of DdHydAB, can tolerate the binding of CN− to the apical coordination site of the H-cluster much more readily than wild-type enzymes, whose H-clusters cannot tolerate the additional electron density and partially decompose on addition of CN−.59 The C169A and C178A mutants have no hydrogen bonding interaction with ADT, removing additional negative charge on the nitrogen. As the ADT ligand is now more electron-withdrawing the H-cluster can accept the extra electron density introduced by the additional CN− ligand; the free NH groups of their ADT ligands may hydrogen bond to the exogenous CN− ligand, further increasing the stability of the H-cluster.
Overall, the hypothesised hydrogen bond from the ADT ligand to the cysteine thiol helps explain several observations but is far from being conclusively demonstrated. Further experiments such as investigations of the C169A mutant containing the PDT or ODT cofactor are needed to provide additional insight.
The electron relay serves as a pathway for electrons to reach the active site but the properties of these clusters are thought to also influence catalytic activity, catalytic bias, and oxygen sensitivity. Removing the F-domain was achieved for the Clostridium acetobutylicum70 and Megasphaera elsdenii71 [FeFe] hydrogenases, leading to loss of activity. For the M. elsdenii enzyme, the catalytic bias was also affected. In the D. desulfuricans [FeFe] hydrogenase, it was observed that reduction of the F-clusters influenced the redox state of the H-cluster through electrostatic repulsion.34 This effect destabilised states in which the [4Fe–4S]H subcluster was reduced such as Hred and HsredH+. These states are still likely to form transiently during catalysis even if they are not observed during thermodynamic titrations.
Thus, it has been made clear that the H-cluster cannot be considered an isolated entity as it is intimately connected to the protein scaffold. This has an enormous influence on the electronic and geometric structure through various types of interactions. These interactions are only now beginning to be understood for a select group of model enzymes. It is very likely that studying the diversity of [FeFe] hydrogenases in nature will reveal additional insight and possibly specific evolutionary adaptations that modify the H-cluster for specific functions.
The diversity of [FeFe] hydrogenases: prototypical, ancestral and sensory type
In recent years it has become clear that there is huge diversity of [FeFe] hydrogenases in nature and so far, only a tiny fraction of this diversity has been studied. In 2001, Vignais, Billoud and Jacques reviewed the classification and phylogeny of hydrogenases based on the available data.72 They presented examples of [FeFe] hydrogenases containing only the catalytic H-domain (e.g. from the green algae C. reinhardtii and Chlorella fusca), containing accessory iron–sulfur cluster domains (e.g. from M. elsdenii and C. pasteurianum), possessing additional subunits (e.g. from Desulfovibrio fructosovorans and Thermotoga maritima) and even an enzyme with a similar composition to the D. fructosovorans and T. maritima enzymes but where the enzyme's subunits are fused into one polypeptide. Then, in 2007, Meyer classified [FeFe] hydrogenases into the more familiar M1–M5 subgroups, where M stands for monomeric.73 M1 indicates the presence of the H-domain only and M2–M5 indicate the presence of additional iron–sulfur cluster domains (Fig. 5). In this article, Meyer performed extensive phylogenetic analysis with a subset of the [FeFe] hydrogenase sequences available and found that the majority of sequences cluster together and include sequences from the M1–M5 groups.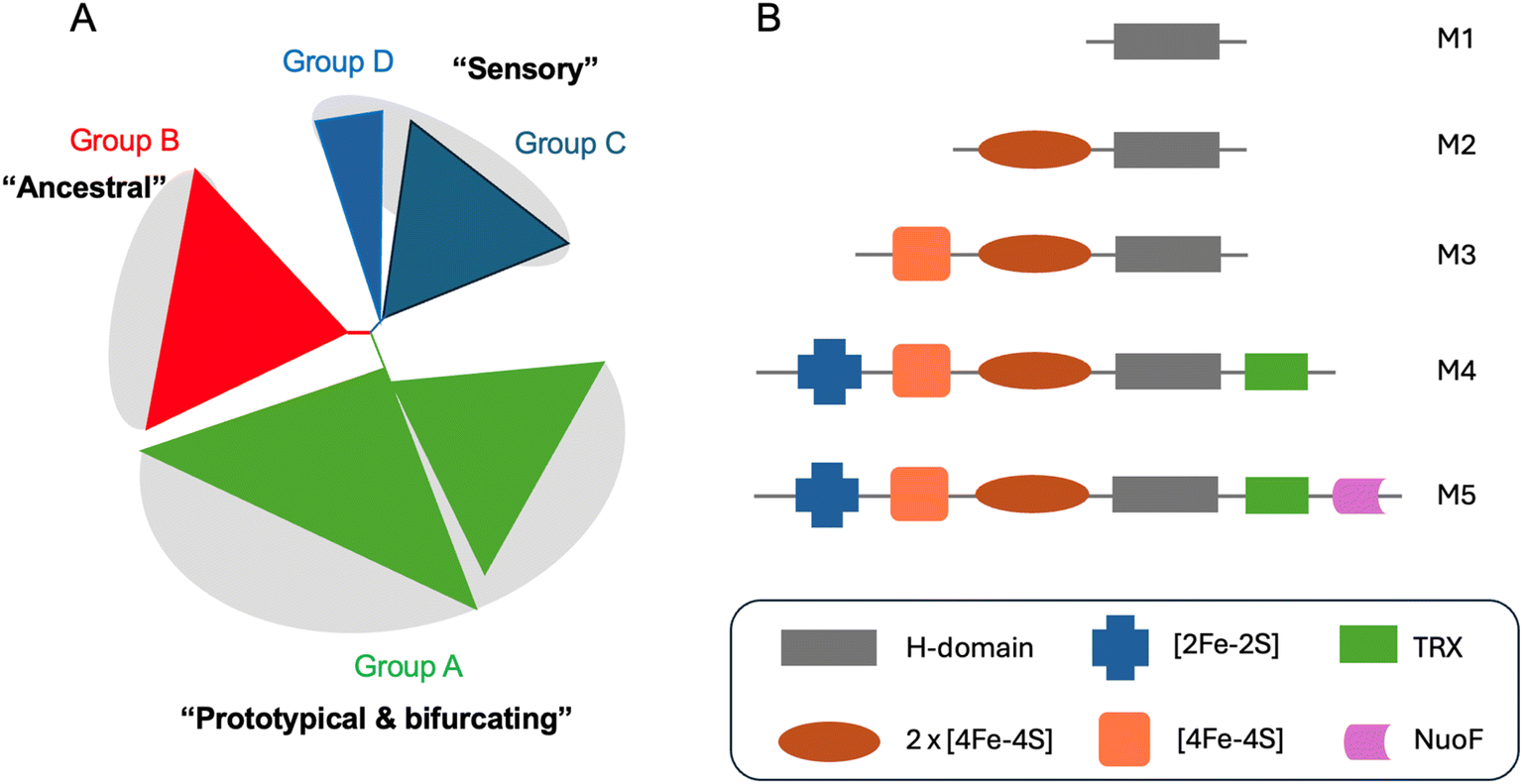 | ||
| Fig. 5 (A) Simplified view of the [FeFe] hydrogenases phylogeny diagram proposed by Greening et al. (2016).74 (B) Biodiversity of [FeFe] hydrogenases represented by five main subclasses named as M1–5. M stands for monomeric and number indicates the size. Legend: TRX – thioredoxin, NuoF – NADH: ubiquinone oxidoreductase chain F. | ||
Later, Calusinska et al. performed a phylogenetic analysis of hydrogenases with a focus on clostridia and found that [FeFe] hydrogenases could be divided into several groups (A, B, C and D), of which the first two groups were further subdivided (A1–8 and B1–3).75 Each of these groups corresponded well to phylogenetic groups identified previously by Meyer. Group C was remarked as consisting of dimeric PAS/PAC sensory domain containing hydrogenases and group D as consisting of putative hydrogenases. In 2016, Greening et al. carried out an even more extensive phylogenetic analysis on 1223 [FeFe] hydrogenase sequences and classified them similarly to Calusinka where group A was subdivided into four groups and group D was integrated into group C (Fig. 5).74 Overall, these studies show an enormous diversity of [FeFe] hydrogenase sequences of which very few have been investigated.
Group A, also known as the prototypical group, includes the vast majority of [FeFe] hydrogenases studied to date, and the only group for which structures are available. It includes CpI14,76 from C. pasteurianum, DdHydAB15,47 from D. desulfuricans and CrHydA177 from C. reinhardtii. Also included is the class of electron-bifurcating [FeFe] hydrogenases78,79 that reversibly reduce protons using NADH and ferredoxin synergistically, as well as a class of hydrogen uptake [FeFe] hydrogenases, A3, which includes CpII80,81 from C. pasteurianum, and the uniquely oxygen-stable A5 [FeFe] hydrogenase, CbA5H, from Clostridium beijerinckii.82
Group B, also known as the ancestral group, is much more poorly studied, with only a few characterised examples including the functionally and spectroscopically characterised CpIII from C. pasteurianum and the less well-characterised HydA2 from Clostridium acetobutylicum.
Group C, also known as the sensory group, is also poorly studied but two examples, HydS from Thermotoga maritima (TmHydS)44,83 and HydS from Thermoanaerobacter mathranii (TamHydS)84–86 have recently been produced and characterised both functionally and spectroscopically. These enzyme have very low activity and, at least in the case of TamHydS, show extremely irreversible electrochemical behavior.84 They are theorised to have a sensing function, presumably hydrogen, but, as yet, the mechanism of signal transduction is unknown.
To provide one example of how different [FeFe] hydrogenases from different groups can be, TmHydS and TamHydS both lack the crucial amino acids of the proton-transfer pathway identified in group A [FeFe] hydrogenases including a cysteine that directly interacts with the ADT ligand and is thought to be the direct proton donor/acceptor of the H-cluster. In the case of TamHydS an alternative proton transfer pathway has been identified.87 A proton transfer pathway is yet to be identified for TmHydS despite intense investigation.83
There is still much to learn about the wider diversity of [FeFe] hydrogenases. A crucial part of future research will be to characterise enzymes from groups B, C and the more diverse branches of group A. Understanding their unique physiological functions and how these influence their protein sequences and thus the structure and properties of the active site and accessory clusters is crucial to understanding how enzyme efficiency is maximised. By understanding these structure–function relationships we will learn more about designing optimal H2 oxidation and H2 production catalysts based on iron.
Future outlook: the missing pieces of the catalytic cycle
The hydride state, Hhyd
As discussed above, the first steps of the catalytic cycle in the H2 production direction, namely reduction, protonation and further reduction have been comprehensively studied, and we are reaching a point where these stages of the catalytic cycle are understood. It has been proposed that another form of the two-electron reduced, protonated state exists, called Hhyd, in which there is a terminal hydride bound to the apical position on Fed. How exactly this state forms is not clear but the following observations have been made:(1) Hhyd can be formed at low pH in the presence of high concentrations of sodium dithionite (Na2S2O4).39 This was taken as evidence that this state is transiently formed under conditions of high turnover.
(2) Hhyd can be formed under equilibrium conditions in enzymes where there have been mutations made to the terminal cysteine in the proton-transfer pathway65 or where the ADT bridging ligand is substituted with oxadithiolate (ODT).39 It has been assumed that these modifications restrict proton transfer and so the terminal hydride that is formed as an intermediate cannot be protonated to make H2.
(3) Alternative forms of Hhyd named Hhyd:ox and Hhyd:red can be formed upon illumination of HredH+ and HsredH+, respectively, under cryogenic conditions.43 These results may indicate that the Hhyd states are less stable, tautomeric forms of HredH+ and HsredH+, and so energy must be supplied to enable proton transfer from the nitrogen base to Fed.
(4) Electrochemical titrations of CpI crystals have shown to populate hydride species.88
Another explanation for the formation of Hhyd in the disrupted proton-transfer mutants and ODT enzyme stands. Both have less basic bridgeheads; mutation of the cysteine may influence the basicity of the ADT amine through disruption of hydrogen bonding and the ether bridgehead in ODT is clearly less basic than the native amine. A follow-up question then is why the PDT enzyme, in which the amine is substituted with methylene, does not behave in the same way as the ODT enzyme. This may be explained by differences in the redox potential of the diiron clusters between PDT and ODT, where ODT ought to be more positive than PDT. One might then expect that PDT can indeed form Hhyd but at more negative potentials than ODT, so far not accessed experimentally.
But what is the difference between Hhyd:ox and Hhyd:red and the Hhyd states observed at low pH and in mutants? Table 2 compares the IR bands and EPR g-values of the Hhyd states so far identified in CrHydA1.
| Name | IR | EPR | Ref. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CNp | CNd | COp | COd | COb | Average | Difference to Hhyd | g 1 | g 2 | g 3 | ||
| a Values reported in ref. 39 deviate substantially (>6 cm−1) from other ref. 40, 59, 66 and 89. b The same EPR spectrum has been assigned to a “Htrans-like” state in ref. 90 and a similar EPR spectrum was reported for the CN−-bound “Htrans-like” state in the C169A mutant of CrHydA1 (g = 2.068, 1.977, 1.916) in ref. 59. | |||||||||||
| Hhyd | 2082a (2088) | 2068a (2075) | 1978 (1979) | 1960 (1961) | 1860 (1861) | 1990 (1993) | — | 2.077 (2.080) | 1.935 (1.941) | 1.880 (1.884) | 39 (89) |
| +3 | |||||||||||
| Hhyd:ox | 2092 | 2086 | 1983 | 1954 | 1865 | 1996 | +6 (+3) | n.a. | n.a. | n.a. | 43 |
| Hhyd:red | 2087 | 2078 | 1972 | 1954 | 1851 | 1988 | −2 (−5) | 2.069 | 1.938 | 1.880 | 43 |
| Hhyd(ODT) | 2081a (2090) | 2076 (2075) | 1980 (1980) | 1962 (1963) | 1868 (1869) | 1993 (1995) | +3 (0) | (2.069) | (1.941) | (1.879) | 39 (40) |
| +5 (+2) | |||||||||||
| Hhyd(C169A) | 2082a (2089) | 2068a (2076) | 1978 (1980) | 1962 (1962) | 1862 (1864) | 1990 | 0 (−3) | (2.075) | (1.942) | (1.884) | 39 (59) |
| 1994 | +4 (+1) | ||||||||||
| Hhyd(C169S) | n.r. | n.r. | 1977 | 1960 | 1860 | 2.068 | 1.943 | 1.881 | 65 | ||
| Hhyd(E279A) | 2082 | 2068 | 1984 | 1970 | 1858 | 1992 | +2 (−1) | n.r. | n.r. | n.r. | 39 |
| HhydH+(C169S) | n.r. | n.r. | 1987 | 1967 | 1874 | 2.065b | 1.969b | 1.906b | 64 | ||
The first thing to note is some discrepancies between the IR band frequencies measured for what should be identical Hhyd states in different studies. In most cases these discrepancies are small (>3 cm−1) but for the CN− bands they appear to be larger. In general Hhyd:red appears to have the most red-shifted IR bands indicating the most electron-rich [2Fe]H site. Meanwhile, Hhyd:ox has more blue-shifted IR bands. The differences between these two states are thought to be due to the difference in the redox state of [4Fe–4S]H, which is oxidised in Hhyd:ox and reduced in Hhyd:red. The Hhyd state formed at low pH, on the other hand, possesses IR bands that are blue-shifted relative to Hhyd:red but red-shifted relative to Hhyd:ox, yet is also proposed to have a reduced [4Fe–4S]H like Hhyd:red. Clearly, there is something about the structure around the H-cluster in Hhyd that is different. Stripp and coworkers proposed that Hhyd has a protonated [4Fe–4S]H,36,66 which would shift some of the electron density from [2Fe]H back onto [4Fe–4S]H. This idea agrees with the observation of this state at low pH,39 although the protonation of the [4Fe–4S]H cluster in other catalytic states is still disputed.37,38 The Hhyd states formed in the ODT enzyme and a C169A mutant have similar IR bands to the Hhyd state formed at low pH.39 Again, these could represent states with protonated [4Fe–4S]H clusters, however, these Hhyd states are stable at neutral pH. Since neither of these changes (substitution of ADT with ODT or mutation of Cys169 to Ala) should influence the pKa of [4Fe–4S]H it seems unlikely that these states would represent protonated [4Fe–4S]H states. Instead, an alternative explanation could be that the removal of the hydrogen-bonding interaction between the ADT amine and the cysteine thiol group influences the electronic structure of the H-cluster in such a way that disfavours protonation of the bridging ADT and favours terminal hydride formation on Fed. If this is the case, then could this also explain the stabilisation of Hhyd at low pH and the differences in the IR signatures of Hhydvs. Hhyd:red? Evidence in this direction comes from comparison of the Hox and Hred states in the ODT enzyme and the C169A mutant (Table 3).
| Name | IR | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|
| CNp | CNd | COp | COd | COb | Average | Difference to Hox | ||
| a This spectrum was actually assigned to HoxH in ref. 66. However, in ref. 59 it was observed that this state formed under conditions typical for forming Hox and was associated with an Hox-like EPR spectrum. b This state was assigned to Hred in ref. 66. However, in ref. 59 it was observed that this state formed under conditions typical for forming Hred and was found to be an EPR-silent state, untypical for Hox | ||||||||
| Hox (WT/ADT) | 2088 | 2072 | 1964 | 1940 | 1804 | 1974 | — | 91 |
| Hox (WT/ODT) | 2092 | 2076 | 1970 | 1947 | 1811 | 1979 | +5 | 91 |
| Hox (C169A/ADT) | 2092a (n.r.) | 2075a (n.r.) | 1972a (1972) | 1946a (1948) | 1813a (1815) | 1980 | +6 | 66 (59) |
| Hred (WT/ADT) | 2084 | 2066 | 1962 | 1933 | 1792 | 1967 | −7 | 91 |
| Hred (WT/ODT) | 2083 | 2070 | 1964 | 1943 | 1804 | 1973 | −1 | 91 |
| Hred (C169A/ADT) | 2089b | 2068b | 1971b | 1939b | 1804b | 1974 | 0 | 59 |
From Table 3, the Hox spectra for ODT and C169A are blue-shifted compared to Hox in WT ADT, while the Hred spectra for ODT and C169A are like Hox in WT ADT but blue-shifted compared to Hred in WT ADT. This indicates that IR spectra for the ODT and C169A enzymes are blue-shifted compared to their WT counterparts.
Have we observed HhydH+ and/or Hox(H2)?
While there have been some reports of HhydH+ in the literature, these are yet to be substantiated with direct evidence and mostly rely on observed IR and EPR spectra that are somewhat similar to Hhyd but blue-shifted (in the case of the IR spectra). In 2014, Mulder observed IR bands at 1987, 1967 and 1874 cm−1 in the C169S mutant of CrHydA1, where the bridging CO shifted to 1964 cm−1 in D2O and was associated with an EPR spectrum with g-values of 2.065, 1.969 and 1.906.65 DFT calculations supported an assignment to HhydH+, with a protonated ADT and a reduced [4Fe–4S]H cluster. In 2021, Mészáros observed a similar IR spectrum with bands at 1988, 1959 and 1975 cm−1 for WT CrHydA1, associated with an EPR spectrum with g-values of 2.073, 1.935 and 1.881 and assigned this to the same HhydH+ state.92 While there are some similarities between the spectroscopic properties of these two putative HhydH+ states, they are clearly very different to the Hhyd states of WT and C169S CrHydA1, whose IR and EPR spectra are nearly identical. Thus, the assignment of these two new signals to HhydH+ seems premature. Furthermore, it would be surprising if the H-cluster of HhydH+ retained the same overall electronic structure as that of Hhyd. One might expect the protonation of the ADT ligand to trigger electron transfer yielding a [4Fe–4S]2+-[Fe(I)Fe(II)H−] electronic structure in which the reducing equivalent is on [2Fe]H. Such a state would be primed for H2 formation yielding the Hox state.While HhydH+ stabilisation in mutants may be achievable, it seems unlikely that it can be stabilised in WT enzymes due to their exceptionally fast turnover rates. The same is probable for any potential Hox(H2) intermediate. A possible way of capturing these states would be to use ultra-fast spectroscopic techniques such as pump-probe IR, which has been coupled to some kind of method for rapidly triggering catalysis. The Dyer group have pioneered methods for using CdS/CdSe dot-in-rod photosensitiser systems coupled to redox mediators for light-triggered reduction of [FeFe] hydrogenases among other enzymes.48,52 Work with CrHydA1 showed that the enzyme converted from Hox to Hred on a timescale of 10 to 100 μs; over the same time-period, HredH+ and HsredH+ were also formed. Hhyd was formed between 50 and 500 μs. All states eventually decayed reforming Hox on timescales similar to those expected based on the known catalytic rates for H2 production by CrHydA1 (≈1000 s−1 or 1 μs−1). A caveat of this study was that the starting state of the enzyme was a mixture of Hox, Hred and HredH+, which complicated the kinetic analysis as single electron reduction will convert Hox to Hred/HredH+ and reduce Hred/HredH+ to HsredH+/Hhyd, and so the kinetics of Hred/HredH+ formation are a convolution of both a formation and decay rate. Furthermore, in a subsequent study it was found that increasing the protein concentration led to faster kinetics indicating a diffusion limitation.51 Accordingly, this method could be enhanced by engineering photosensitisers directly onto [FeFe] hydrogenases, as has been previously shown.55–57,93,94
Have we already observed an H2-bound, Hox(H2), intermediate?
As we are unsure of what we are looking for, that is entirely possible. What do we expect it to look like? Here, some clues can be acquired from investigation of synthetic η2-H2 metal complexes.95 Many η2-H2 mono-metallic complexes have been studied over the years but examples of bimetallic complexes like [2Fe]H that stably bind η2-H2 are rarer.96–101 Generally, dihydrogen complexes are identified through structural characterization (X-ray or neutron crystallography), vibrational spectroscopy (IR, Raman, inelastic neutron scattering) and NMR spectroscopy. Free H2 in the gas phase has a vibrational frequency of around 4100 cm−1 (ref. 102) and the transition is IR invisible due to the absence of a change in dipole moment. Binding of H2 to metals gives IR active modes due to the dipolar character of the metal–hydrogen bonds and the coupling of the H–H stretch to other vibrations. A range of complexes show vibrational modes in the 2200–3200, 1100–1700 and 400–1000 cm−1 regions.95 Due to the low intensity of these peaks and their potential overlap with other bands from the protein matrix, the possibility of observing a dihydrogen bound species by IR is low. In NMR, metal-bound dihydrogen presents with chemical shifts in the 2.5 to −31 ppm region, which overlaps with the chemical shifts of metal-hydrides.95 For reference, the [FeFe] hydrogenase Hhyd state gives a peak at −9.6 ppm.103 However, observation of JHD coupling or short T1 relaxation times can provide support for a dihydrogen species. However, in reality, this can only be achieved with S = 0 systems as unpaired electrons tend to produce NMR line-broadening, as would be expected for a putative Hox(H2) intermediate.Specifically, for [FeFe] hydrogenases, EPR spectroscopy may provide a valuable tool for identifying and characterizing H2-aducts. The Peters and Hoffmann groups measured EPR and ENDOR on an H2-bound Fe(I) complex, which revealed a single electron–nuclear hyperfine interaction with a coupling tensor of [2.3, −40.6, −37.8] with both isotropic and anisotropic components.104
Returning to IR, it might be possible to observe the H2-bound intermediate indirectly through changes to the CO and CN− ligand vibrations upon H2 binding. Side-on binding of H2 has both a σ-donation and π-backdonation contribution. Binding of hydrogen to monometallic carbonyl complexes causes a small blue shift in the vibrational frequency of the carbonyl IR bands, likely reflecting the decreased backdonation due to some of this electron density being donated into the H2 σ*.95 A similar effect is seen in CO binding to the [FeFe] hydrogenase H-cluster forming the Hox–CO state; the IR band of the bridging CO is blue-shifted by around 10 cm−1.13 So, it can be postulated that an Hox(H2) state may show similar blue shifted IR bands.
NRVS has been used extensively to probe the Fe–H vibrations of the Hhyd states but could also be a tool to explore potential Hox(H2) states. For an Fe-η2-H2-hydride complex an intense NRVS symmetric Fe–H2 stretching mode was observed at 1052 cm−1 that shifts to 781 cm−1 upon D2 substitution.105 In addition, the weaker antisymmetric stretch mode was observed at 1774 cm−1 and bending modes were observed at 558, 584, 733 and 823 cm−1. These results suggest that in principle, an H2 bound intermediate of an [FeFe] hydrogenase could be probed with NRVS provided it is sufficiently enriched.
Lastly, X-ray crystallography is generally not considered as an effective method for observing protons due to their low electron density and bond libration effects that give anomalous bond lengths. Despite this, it has been used to provide evidence for the location of hydrides and protons in [NiFe] hydrogenase enzymes due to sub-angstrom resolution quality of the data.106 Alternatively, neutron diffraction could be employed, as neutrons are scattered predominantly by the nucleus and the coherent neutron scattering length of hydrogen is similar to heavy atoms. However, due to the negative sign of the scattering length of 1H and the large amount of incoherent scattering it is much easier to obtain information on the location of deuterium. Regardless, due to the weak intensity of neutron sources very large crystals are needed and data collection times are extremely long. Recently, a neutron structure was published for an oxidised form of [NiFe] hydrogenase,107 however, this technique is yet to reveal the locations of hydrides and protons from hydrogen splitting. The size of the crystals needed (at least 1 mm3) on top of the requirement for deuteration continues to cause problems for neutron diffraction experiments. Nevertheless, it is likely that the next few years will see the publication of neutron and subangstrom resolution X-ray structures of [FeFe] hydrogenases, revealing the locations of protons. If these can be carried out on the Hhyd states or potential HhydH+ and Hox(H2) intermediates, these structures will provide unprecedented insight into our understanding of biological H2 activation.
New tools: CryoEM and its potential in hydrogenase research
Electron imaging visualises electrostatic potential, which depends on the positions of both nuclei and electrons.108 Thus, hydrogens can be observed more clearly in cryoEM than in X-ray crystallography. Another advantage of cryoEM is that samples do not need to be crystallised. However, there is currently a size limit of around 50 kDa, below which obtaining cryoEM structures is exceptionally difficult, but still possible. In recent years, several cryoEM structures of hydrogenases have been published including various electron-bifucating [FeFe] hydrogenases.109–113 One structure of a [NiFe] hydrogenase reached 1.52 Å resolution, potentially paving the way toward resolving hydrogen atoms.113A multiple catalytic pathways scenario?
Another intriguing idea is that of multiple catalytic pathways. Earlier in this article, two hypotheses for the catalytic cycle of [FeFe] hydrogenase were presented, both with some evidence in support of them. However, it is possible that, to a certain extent, elements of both cycles are true, and that alternative pathways are taken by [FeFe] hydrogenases depending on (i) the conditions and (ii) which enzyme is being studied. It has already been demonstrated for CrHydA1 that at least three different forms of the Hhyd state exist including the ‘classical’ Hhyd observed at low pH and Hhyd:ox and Hhyd:red observed at high pH, mainly under cryogenic conditions and illumination but also thought to contribute at room temperature. During proton reduction, from the HredH+ state there are multiple potential pathways that can be taken (Fig. 6): if electron transfer is slow then tautomerization to Hhyd:ox may be faster than reduction to HsredH+. Hhyd:ox could then be reduced to Hhyd:red or at low pH become protonated to a putative Hhyd:oxH+ state, which could either become further reduced to a putative Hhyd:redH+ state or, again if electron transfer is slow, directly form H2. Formation of H2 from a putative Hhyd:oxH+ state would leave the [4Fe–4S]H cluster oxidised and the [2Fe]H cluster in an overoxidised Fe(II)Fe(II) oxidation state, a putative “Hsox” state, which may not be stable. Thus, it can already be seen that depending on the electron supply, pH, and enzyme properties such as redox potentials and pKa values, several pathways from HredH+ may exist. Since HsredH+ cannot be further reduced or protonated, it can only tautomerise to Hhyd:red. Hhyd:red is unlikely to be reducible but will become protonated to give Hhyd:redH+. Hhyd:redH+ formation is likely to involve PCET where [4Fe–4S]H transfers an electron to [2Fe]H. In principle, Hhyd:redH+ could be further reduced to give Hhyd:sredH+ where both the [4Fe–4S]H and [2Fe]H clusters are reduced, which would in turn form H2 bound to the Hred state in a Hred(H2) state. Likewise, it would also be possible to reduce the Hox(H2) state before releasing H2 giving Hred(H2). Such states could potentially be observed under conditions where there is a very fast electron supply. One could also envisage additional states such as the protonated Hox state (HoxH+) or the deprotonated HsredH+ (Hsred) state that could follow additional pathways. Whether any of these states are populated during turnover, even under extreme conditions, is not known but it is speculated to be the case based on electrochemical studies of amino acid variants.46 It may also be possible that accessing some of these alternative pathways is responsible for phenomena observed under extreme conditions during electrochemistry such as high potential inactivation114 and low potential inactivation.45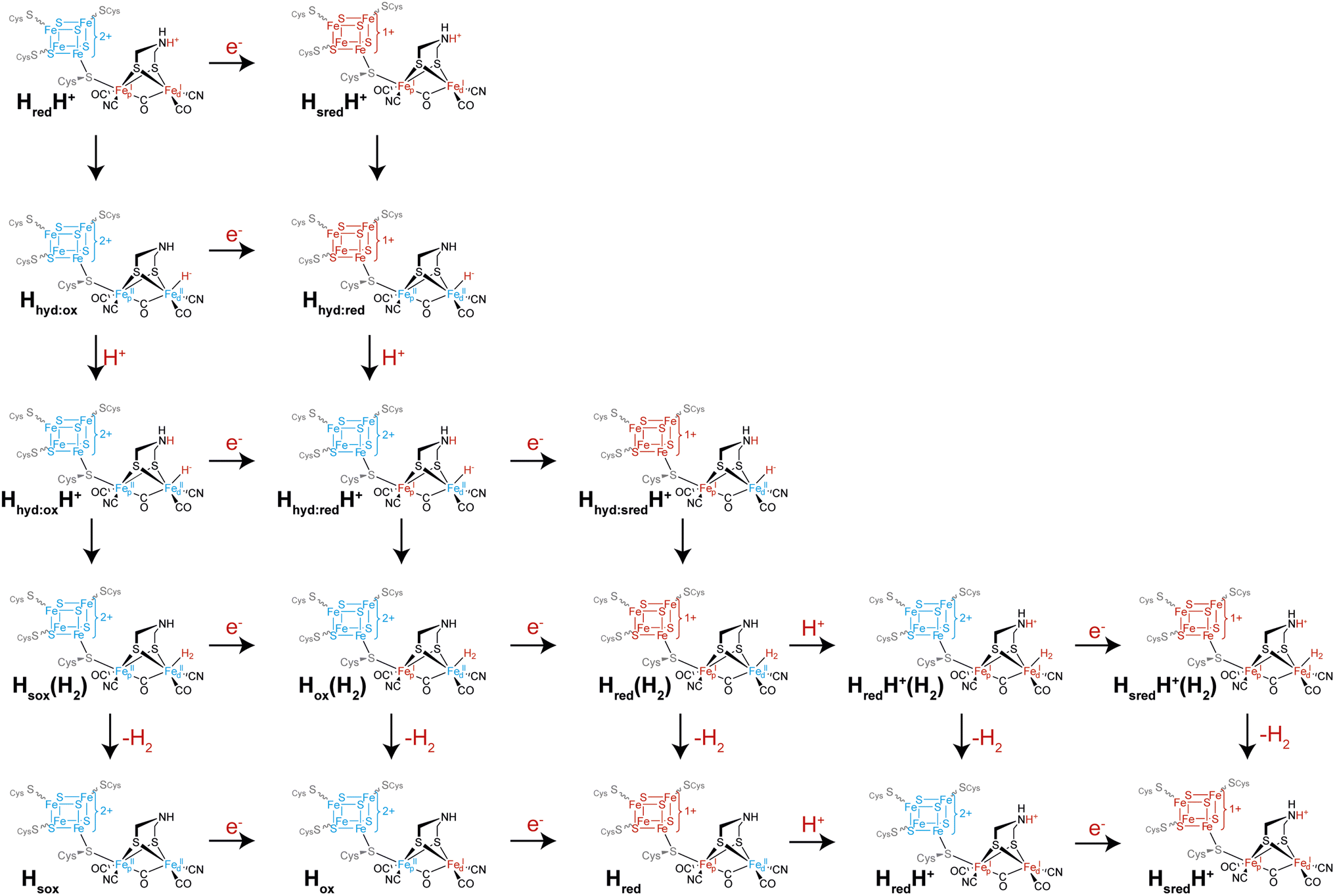 | ||
| Fig. 6 Possible alternative pathways taken by the H-cluster from the HredH+ state (top left), which can proceed by reduction to the HsredH+ state (horizontal arrow) or tautomerism to the Hhyd:ox state (vertical arrow). The HsredH+ state can also tautomerise (vertical arrow), yielding the Hhyd:red state. Both forms of the Hhyd state can then be protonated (vertical arrows) and the two possible forms of HhydH+ can then undergo reduction (horizontal arrows) or tautomerism (vertical arrows), and so on. | ||
It could also be possible that different pathways are followed depending on the direction of catalysis. For example, if interconversion of the HredH+ and HsredH+ states with their Hhyd tautomers is slow compared with electron transfer, then during proton reduction the pathway HredH+ → HsredH+ → Hhyd:red would be followed while the Hhyd:red → Hhyd:ox → HredH+ pathway would be followed during H2 oxidation. Determining whether [FeFe] hydrogenases do indeed follow multiple pathways depending on conditions requires the ability to carefully control the rate of electron supply while monitoring the composition of states using spectroscopy. This is very difficult using standard spectroscopic approaches where electrons are exchanged with the enzyme using mediators. Instead, an approach is needed where the enzyme is directly attached to an electrode surface. The techniques of surface enhanced IR spectroscopy (SEIRAS) and protein film infrared electrochemistry (PFIRE) are designed to do this.115 While [NiFe] hydrogenases have been quite extensively studied with SEIRAS116–119 and also by PFIRE,120,121 there are only two examples of SEIRAS being employed with an [FeFe] hydrogenase, specifically CrHydA1,122,123 and no examples of PFIRE applied to [FeFe] hydrogenases. In both SEIRAS studies only the spectra of the protein backbone amides could be observed and there was no evidence of the active site CO and CN− ligands, even though catalytic activity was observed. Thus, there is potential for this technique and for PFIRE to be incredibly useful in future studies, to particularly allow operando conditions.
Conclusion
[FeFe] hydrogenases are incredibly fascinating yet complex enzymes capable of extremely high turnover frequencies with exceptional efficiency and reversibility. How this is achieved is still not completely understood despite decades of research. In this perspective we have provided a summary of where we are now in terms of our understanding, how we have gotten here and where we think the field is moving and how it is going to get there, particularly highlighting the main challenges. The catalytic cycle of [FeFe] hydrogenases has been a difficult puzzle to build, but we believe that enough pieces are in place to provide a good overview of the picture we are putting together. However, the next pieces of the puzzle will be the most challenging to assemble, requiring new techniques and approaches or combinations of existing techniques. The promise of being able to design new catalytic materials motivates scientists to keep working on this challenging puzzle and we expect the next few years will see substantial progress toward this goal.Author contributions
All authors contributed to the conceptualisation, literature search, writing and figure making of the perspective and were involved in revising, editing, and proofreading the initial and revised versions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PRM thanks the Royal Society (grant no. RGS\R1\231433), the Royal Society of Chemistry (grant no. R23-6753486967) and the University of Leicester for funding. MTL is financially supported by a Future 50 Scholarship from the University of Leicester. JAB acknowledges funding from the Deutsche Forschungsgemeinschaft (DFG) Priority Programme “Iron–Sulfur for Life: Cooperative Function of Iron–Sulfur Centers in Assembly, Biosynthesis, Catalysis and Disease” (SPP 1927) Project BI 2198/1-1, the Royal Society (grant no. RG\R2\232336), the Royal Society of Chemistry (grant no. R22-2594924113), and the University of Essex for funding.References
- M. Yue, H. Lambert, E. Pahon, R. Roche, S. Jemei and D. Hissel, Hydrogen energy systems: A critical review of technologies, applications, trends and challenges, Renewable Sustainable Energy Rev., 2021, 146, 111180, DOI:10.1016/j.rser.2021.111180.
- K. T. Møller, T. R. Jensen, E. Akiba and H.-w. Li, Hydrogen - A sustainable energy carrier, Prog. Nat. Sci.: Mater., 2017, 27(1), 34–40, DOI:10.1016/j.pnsc.2016.12.014.
- IEA, The Future of Hydrogen, IEA, Paris, 2019, https://www.iea.org/reports/the-future-of-hydrogen Search PubMed.
- S. Shiva Kumar and V. Himabindu, Hydrogen production by PEM water electrolysis – A review, Mater. Sci. Energy Technol., 2019, 2(3), 442–454, DOI:10.1016/j.mset.2019.03.002.
- L. Bhatnagar, J. A. Krzycki and J. G. Zeikus, Analysis of hydrogen metabolism in Methanosarcina barkeri: Regulation of hydrogenase and role of CO-dehydrogenase in H2 production, FEMS Microbiol. Lett., 1987, 41(3), 337–343 CrossRef.
- S. Menon and S. W. Ragsdale, Unleashing hydrogenase activity in carbon monoxide dehydrogenase/acetyl-CoA synthase and pyruvate:ferredoxin oxidoreductase, Biochemistry, 1996, 35(49), 15814–15821, DOI:10.1021/bi9615598.
- B. Santiago and O. Meyer, Characterization of hydrogenase activities associated with the molybdenum CO dehydrogenase from Oligotropha carboxidovorans, FEMS Microbiol. Lett., 1996, 136(2), 157–162, DOI:10.1016/0378-1097(95)00498-X.
- J. Wilcoxen and R. Hille, The hydrogenase activity of the molybdenum/copper-containing carbon monoxide dehydrogenase of Oligotropha carboxidovorans, J. Biol. Chem., 2013, 288(50), 36052–36060, DOI:10.1074/jbc.M113.522441.
- K. Yang and W. W. Metcalf, A new activity for an old enzyme: Escherichia coli bacterial alkaline phosphatase is a phosphite-dependent hydrogenase, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(21), 7919–7924, DOI:10.1073/pnas.0400664101.
- J. Heo, M. T. Wolfe, C. R. Staples and P. W. Ludden, Converting the NiFeS carbon monoxide dehydrogenase to a hydrogenase and a hydroxylamine reductase, J. Bacteriol., 2002, 184(21), 5894–5897, DOI:10.1128/jb.184.21.5894-5897.2002.
- A. Winiarska, D. Hege, Y. Gemmecker, J. Kryściak-Czerwenka, A. Seubert, J. Heider and M. Szaleniec, Tungsten enzyme using hydrogen as an electron donor to reduce carboxylic acids and NAD+, ACS Catal., 2022, 12(14), 8707–8717, DOI:10.1021/acscatal.2c02147.
- T. M. Van Der Spek, A. F. Arendsen, R. P. Happe, S. Yun, K. A. Bagley, D. J. Stufkens, W. R. Hagen and S. P. J. Albracht, Similarities in the architecture of the active sites of Ni-hydrogenases and Fe-hydrogenases detected by means of infrared spectroscopy, Eur. J. Biochem., 1996, 237(3), 629–634, DOI:10.1111/j.1432-1033.1996.0629p.x.
- A. J. Pierik, M. Hulstein, W. R. Hagen and S. P. J. Albracht, A low-spin iron with CN and CO as intrinsic ligands forms the core of the active site in [Fe]-hydrogenases, Eur. J. Biochem., 1998, 258(2), 572–578, DOI:10.1046/j.1432-1327.1998.2580572.x.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, X-ray Crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 Angstrom resolution, Science, 1998, 282(5395), 1853–1858, DOI:10.1126/science.282.5395.1853.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Desulfovibrio desulfuricans iron hydrogenase: the structure shows unusual coordination to an active site Fe binuclear center, Structure, 1999, 7(1), 13–23, DOI:10.1016/S0969-2126(99)80005-7.
- Y. Nicolet, A. L. de Lacey, X. Vernède, V. M. Fernandez, E. C. Hatchikian and J. C. Fontecilla-Camps, Crystallographic and FTIR spectroscopic evidence of changes in Fe coordination upon reduction of the active site of the Fe-only hydrogenase from Desulfovibrio desulfuricans, J. Am. Chem. Soc., 2001, 123(8), 1596–1601, DOI:10.1021/ja0020963.
- H.-J. Fan and M. B. Hall, A capable bridging ligand for Fe-only hydrogenase: density functional calculations of a low-energy route for heterolytic cleavage and formation of dihydrogen, J. Am. Chem. Soc., 2001, 123(16), 3828–3829, DOI:10.1021/ja004120i.
- A. S. Pandey, T. V. Harris, L. J. Giles, J. W. Peters and R. K. Szilagyi, Dithiomethylether as a ligand in the hydrogenase H-cluster, J. Am. Chem. Soc., 2008, 130(13), 4533–4540, DOI:10.1021/ja711187e.
- A. Silakov, B. Wenk, E. Reijerse and W. Lubitz, 14N HYSCORE investigation of the H-cluster of [FeFe] hydrogenase: evidence for a nitrogen in the dithiol bridge, Phys. Chem. Chem. Phys., 2009, 11(31), 6592–6599, 10.1039/B905841A.
- A. Adamska-Venkatesh, S. Roy, J. F. Siebel, T. R. Simmons, M. Fontecave, V. Artero, E. Reijerse and W. Lubitz, Spectroscopic characterization of the bridging amine in the active site of [FeFe] hydrogenase using isotopologues of the H-cluster, J. Am. Chem. Soc., 2015, 137(40), 12744–12747, DOI:10.1021/jacs.5b06240.
- G. Rao, L. Tao and R. D. Britt, Serine is the molecular source of the NH(CH2)2 bridgehead moiety of the in vitro assembled [FeFe] hydrogenase H-cluster, Chem. Sci., 2020, 11(5), 1241–1247, 10.1039/C9SC05900H.
- A. Pagnier, B. Balci, E. M. Shepard, H. Yang, D. M. Warui, S. Impano, S. J. Booker, B. M. Hoffman, W. E. Broderick and J. B. Broderick, [FeFe]-hydrogenase: defined lysate-free maturation reveals a key role for lipoyl-H-protein in DTMA ligand biosynthesis, Angew. Chem., Int. Ed., 2022, 61(22), e202203413, DOI:10.1002/anie.202203413.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse and W. Lubitz, et al., Biomimetic assembly and activation of [FeFe]-hydrogenases, Nature, 2013, 499(7456), 66–69, DOI:10.1038/nature12239.
- J. Esselborn, N. Muraki, K. Klein, V. Engelbrecht, N. Metzler-Nolte, U. P. Apfel, E. Hofmann, G. Kurisu and T. Happe, A structural view of synthetic cofactor integration into [FeFe]-hydrogenases, Chem. Sci., 2016, 7(2), 959–968, 10.1039/C5SC03397G.
- J. Noth, J. Esselborn, J. Güldenhaupt, A. Brünje, A. Sawyer, U.-P. Apfel, K. Gerwert, E. Hofmann, M. Winkler and T. Happe, [FeFe]-hydrogenase with chalcogenide substitutions at the H-cluster maintains full H2 evolution activity, Angew. Chem., Int. Ed., 2016, 55(29), 8396–8400, DOI:10.1002/anie.201511896.
- P. Knörzer, A. Silakov, C. E. Foster, F. A. Armstrong, W. Lubitz and T. Happe, Importance of the protein framework for catalytic activity of [FeFe]-hydrogenases, J. Biol. Chem., 2012, 287(2), 1489–1499, DOI:10.1074/jbc.M111.305797.
- L. Kertess, A. Adamska-Venkatesh, P. Rodríguez-Maciá, O. Rüdiger, W. Lubitz and T. Happe, Influence of the [4Fe–4S] cluster coordinating cysteines on active site maturation and catalytic properties of C. reinhardtii [FeFe]-hydrogenase, Chem. Sci., 2017, 8(12), 8127–8137, 10.1039/C7SC03444J.
- P. Rodríguez-Maciá, L. Kertess, J. Burnik, J. A. Birrell, E. Hofmann, W. Lubitz, T. Happe and O. Rüdiger, His-ligation to the [4Fe–4S] subcluster tunes the catalytic bias of [FeFe] hydrogenase, J. Am. Chem. Soc., 2019, 141(1), 472–481, DOI:10.1021/jacs.8b11149.
- W. Roseboom, A. L. De Lacey, V. M. Fernandez, E. C. Hatchikian and S. P. J. Albracht, The active site of the [FeFe]-hydrogenase from Desulfovibrio desulfuricans. II. Redox properties, light sensitivity and CO-ligand exchange as observed by infrared spectroscopy, J. Biol. Inorg. Chem., 2006, 11(1), 102–118, DOI:10.1007/s00775-005-0040-2.
- A. Silakov, C. Kamp, E. Reijerse, T. Happe and W. Lubitz, Spectroelectrochemical characterization of the active site of the [FeFe] hydrogenase HydA1 from Chlamydomonas reinhardtii, Biochemistry, 2009, 48(33), 7780–7786, DOI:10.1021/bi9009105.
- C. Sommer, A. Adamska-Venkatesh, K. Pawlak, J. A. Birrell, O. Rüdiger, E. J. Reijerse and W. Lubitz, Proton coupled electronic rearrangement within the H-cluster as an essential step in the catalytic cycle of [FeFe] hydrogenases, J. Am. Chem. Soc., 2017, 139(4), 1440–1443, DOI:10.1021/jacs.6b12636.
- A. Adamska-Venkatesh, D. Krawietz, J. Siebel, K. Weber, T. Happe, E. Reijerse and W. Lubitz, New redox states observed in [FeFe] hydrogenases reveal redox coupling within the H-cluster, J. Am. Chem. Soc., 2014, 136(32), 11339–11346, DOI:10.1021/ja503390c.
- S. Katz, J. Noth, M. Horch, H. S. Shafaat, T. Happe, P. Hildebrandt and I. Zebger, Vibrational spectroscopy reveals the initial steps of biological hydrogen evolution, Chem. Sci., 2016, 7(11), 6746–6752, 10.1039/C6SC01098A.
- P. Rodriguez-Macia, K. Pawlak, O. Rudiger, E. J. Reijerse, W. Lubitz and J. A. Birrell, Intercluster redox coupling influences protonation at the h-cluster in [FeFe] hydrogenases, J. Am. Chem. Soc., 2017, 139(42), 15122–15134, DOI:10.1021/jacs.7b08193.
- M. Haumann and S. T. Stripp, The molecular proceedings of biological hydrogen turnover, Acc. Chem. Res., 2018, 51(8), 1755–1763, DOI:10.1021/acs.accounts.8b00109.
- M. Senger, K. Laun, F. Wittkamp, J. Duan, M. Haumann, T. Happe, M. Winkler, U.-P. Apfel and S. T. Stripp, Proton-coupled reduction of the catalytic [4Fe-4S] cluster in [FeFe]-hydrogenases, Angew. Chem., Int. Ed., 2017, 56(52), 16503–16506, DOI:10.1002/anie.201709910.
- P. Rodríguez-Maciá, N. Breuer, S. DeBeer and J. A. Birrell, Insight into the redox behavior of the [4Fe–4S] subcluster in [FeFe] hydrogenases, ACS Catal., 2020, 10(21), 13084–13095, DOI:10.1021/acscatal.0c02771.
- M. A. Martini, O. Rüdiger, N. Breuer, B. Nöring, S. DeBeer, P. Rodríguez-Maciá and J. A. Birrell, The nonphysiological reductant sodium dithionite and [FeFe] hydrogenase: influence on the enzyme mechanism, J. Am. Chem. Soc., 2021, 143(43), 18159–18171, DOI:10.1021/jacs.1c07322.
- M. Winkler, M. Senger, J. Duan, J. Esselborn, F. Wittkamp, E. Hofmann, U.-P. Apfel, S. T. Stripp and T. Happe, Accumulating the hydride state in the catalytic cycle of [FeFe]-hydrogenases, Nat. Commun., 2017, 8(1), 16115, DOI:10.1038/ncomms16115.
- E. J. Reijerse, C. C. Pham, V. Pelmenschikov, R. Gilbert-Wilson, A. Adamska-Venkatesh, J. F. Siebel, L. B. Gee, Y. Yoda, K. Tamasaku and W. Lubitz, et al., Direct observation of an iron-bound terminal hydride in [FeFe]-hydrogenase by nuclear resonance vibrational spectroscopy, J. Am. Chem. Soc., 2017, 139(12), 4306–4309, DOI:10.1021/jacs.7b00686.
- M. W. Ratzloff, J. H. Artz, D. W. Mulder, R. T. Collins, T. E. Furtak and P. W. King, CO-bridged H-cluster intermediates in the catalytic mechanism of [FeFe]-hydrogenase CaI, J. Am. Chem. Soc., 2018, 140(24), 7623–7628, DOI:10.1021/jacs.8b03072.
- J. A. Birrell, V. Pelmenschikov, N. Mishra, H. Wang, Y. Yoda, K. Tamasaku, T. B. Rauchfuss, S. P. Cramer, W. Lubitz and S. DeBeer, Spectroscopic and computational evidence that [FeFe] hydrogenases operate exclusively with CO-bridged intermediates, J. Am. Chem. Soc., 2020, 142(1), 222–232, DOI:10.1021/jacs.9b09745.
- C. Lorent, S. Katz, J. Duan, C. J. Kulka, G. Caserta, C. Teutloff, S. Yadav, U.-P. Apfel, M. Winkler and T. Happe, et al., Shedding light on proton and electron dynamics in [FeFe] hydrogenases, J. Am. Chem. Soc., 2020, 142(12), 5493–5497, DOI:10.1021/jacs.9b13075.
- N. Chongdar, J. A. Birrell, K. Pawlak, C. Sommer, E. J. Reijerse, O. Rüdiger, W. Lubitz and H. Ogata, Unique spectroscopic properties of the h-cluster in a putative sensory [FeFe] hydrogenase, J. Am. Chem. Soc., 2018, 140(3), 1057–1068, DOI:10.1021/jacs.7b11287.
- V. Hajj, C. Baffert, K. Sybirna, I. Meynial-Salles, P. Soucaille, H. Bottin, V. Fourmond and C. Léger, FeFe hydrogenase reductive inactivation and implication for catalysis, Energy Environ. Sci., 2014, 7(2), 715–719, 10.1039/C3EE42075B.
- O. Lampret, J. Duan, E. Hofmann, M. Winkler, F. A. Armstrong and T. Happe, The roles of long-range proton-coupled electron transfer in the directionality and efficiency of [FeFe]-hydrogenases, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(34), 20520–20529, DOI:10.1073/pnas.2007090117.
- B. R. Glick, W. G. Martin and S. M. Martin, Purification and properties of the periplasmic hydrogenase from Desulfovibrio desulfuricans, Can. J. Microbiol., 1980, 26(10), 1214–1223, DOI:10.1139/m80-203.
- B. L. Greene, G. J. Schut, M. W. W. Adams and R. B. Dyer, Pre-steady-state kinetics of catalytic intermediates of an [FeFe]-hydrogenase, ACS Catal., 2017, 7(3), 2145–2150, DOI:10.1021/acscatal.6b03276.
- C. Knight and G. A. Voth, The curious case of the hydrated proton, Acc. Chem. Res., 2012, 45(1), 101–109, DOI:10.1021/ar200140h.
- M. Mirmohades, A. Adamska-Venkatesh, C. Sommer, E. Reijerse, R. Lomoth, W. Lubitz and L. Hammarström, Following [FeFe] hydrogenase active site intermediates by time-resolved mid-IR spectroscopy, J. Phys. Chem. Lett., 2016, 7(16), 3290–3293, DOI:10.1021/acs.jpclett.6b01316.
- M. L. K. Sanchez, S. E. Konecny, S. M. Narehood, E. J. Reijerse, W. Lubitz, J. A. Birrell and R. B. Dyer, The laser-induced potential jump: a method for rapid electron injection into oxidoreductase enzymes, J. Phys. Chem. B, 2020, 124(40), 8750–8760, DOI:10.1021/acs.jpcb.0c05718.
- M. L. K. Sanchez, C. Sommer, E. Reijerse, J. A. Birrell, W. Lubitz and R. B. Dyer, Investigating the kinetic competency of CrHydA1 [FeFe] hydrogenase intermediate states via time-resolved infrared spectroscopy, J. Am. Chem. Soc., 2019, 141(40), 16064–16070, DOI:10.1021/jacs.9b08348.
- M. L. K. Sanchez, S. Wiley, E. Reijerse, W. Lubitz, J. A. Birrell and R. B. Dyer, Time-resolved infrared spectroscopy reveals the pH-independence of the first electron transfer step in the [FeFe] hydrogenase catalytic cycle, J. Phys. Chem. Lett., 2022, 13(25), 5986–5990, DOI:10.1021/acs.jpclett.2c01467.
- M. W. Ratzloff, M. B. Wilker, D. W. Mulder, C. E. Lubner, H. Hamby, K. A. Brown, G. Dukovic and P. W. King, Activation thermodynamics and H/D kinetic isotope effect of the Hox to HredH+ transition in [FeFe] hydrogenase, J. Am. Chem. Soc., 2017, 139(37), 12879–12882, DOI:10.1021/jacs.7b04216.
- M. B. Wilker, K. E. Shinopoulos, K. A. Brown, D. W. Mulder, P. W. King and G. Dukovic, Electron transfer kinetics in CdS nanorod–[FeFe]-hydrogenase complexes and implications for photochemical H2 generation, J. Am. Chem. Soc., 2014, 136(11), 4316–4324, DOI:10.1021/ja413001p.
- C. E. Lubner, A. M. Applegate, P. Knörzer, A. Ganago, D. A. Bryant, T. Happe and J. H. Golbeck, Solar hydrogen-producing bionanodevice outperforms natural photosynthesis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(52), 20988–20991, DOI:10.1073/pnas.1114660108.
- C. E. Lubner, P. Knörzer, P. J. N. Silva, K. A. Vincent, T. Happe, D. A. Bryant and J. H. Golbeck, Wiring an [FeFe]-hydrogenase with photosystem I for light-induced hydrogen production, Biochemistry, 2010, 49(48), 10264–10266, DOI:10.1021/bi1016167.
- J. Duan, A. Hemschemeier, D. J. Burr, S. T. Stripp, E. Hofmann and T. Happe, Cyanide binding to [FeFe]-hydrogenase stabilizes the alternative configuration of the proton transfer pathway, Angew. Chem., Int. Ed., 2023, 62(7), e202216903, DOI:10.1002/anie.202216903.
- M. A. Martini, K. Bikbaev, Y. Pang, C. Lorent, C. Wiemann, N. Breuer, I. Zebger, S. DeBeer, I. Span and R. Bjornsson, et al., Binding of exogenous cyanide reveals new active-site states in [FeFe] hydrogenases, Chem. Sci., 2023, 14(11), 2826–2838, 10.1039/D2SC06098A.
- M. Lorenzi, J. Gellett, A. Zamader, M. Senger, Z. Duan, P. Rodríguez-Maciá and G. Berggren, Investigating the role of the strong field ligands in [FeFe] hydrogenase: spectroscopic and functional characterization of a semi-synthetic mono-cyanide active site, Chem. Sci., 2022, 13(37), 11058–11064, 10.1039/D2SC02271K.
- J. F. Siebel, A. Adamska-Venkatesh, K. Weber, S. Rumpel, E. Reijerse and W. Lubitz, Hybrid [FeFe]-hydrogenases with modified active sites show remarkable residual enzymatic activity, Biochemistry, 2015, 54(7), 1474–1483, DOI:10.1021/bi501391d.
- E. C. Kisgeropoulos, V. S. Bharadwaj, D. W. Mulder and P. W. King, The contribution of proton-donor pKa on reactivity profiles of [FeFe]-hydrogenases, Front. Microbiol., 2022, 13, 903951, DOI:10.3389/fmicb.2022.903951.
- J. Duan, M. Senger, J. Esselborn, V. Engelbrecht, F. Wittkamp, U.-P. Apfel, E. Hofmann, S. T. Stripp, T. Happe and M. Winkler, Crystallographic and spectroscopic assignment of the proton transfer pathway in [FeFe]-hydrogenases, Nat. Commun., 2018, 9(1), 4726, DOI:10.1038/s41467-018-07140-x.
- D. W. Mulder, Y. Guo, M. W. Ratzloff and P. W. King, Identification of a catalytic iron-hydride at the H-cluster of [FeFe]-hydrogenase, J. Am. Chem. Soc., 2017, 139(1), 83–86, DOI:10.1021/jacs.6b11409.
- D. W. Mulder, M. W. Ratzloff, M. Bruschi, C. Greco, E. Koonce, J. W. Peters and P. W. King, Investigations on the role of proton-coupled electron transfer in hydrogen activation by [FeFe]-hydrogenase, J. Am. Chem. Soc., 2014, 136(43), 15394–15402, DOI:10.1021/ja508629m.
- M. Senger, S. Mebs, J. Duan, O. Shulenina, K. Laun, L. Kertess, F. Wittkamp, U.-P. Apfel, T. Happe and M. Winkler, et al., Protonation/reduction dynamics at the [4Fe–4S] cluster of the hydrogen-forming cofactor in [FeFe]-hydrogenases, Phys. Chem. Chem. Phys., 2018, 20(5), 3128–3140, 10.1039/C7CP04757F.
- S. Morra, S. Maurelli, M. Chiesa, D. W. Mulder, M. W. Ratzloff, E. Giamello, P. W. King, G. Gilardi and F. Valetti, The effect of a C298D mutation in CaHydA [FeFe]-hydrogenase: Insights into the protein-metal cluster interaction by EPR and FTIR spectroscopic investigation, Biochim. Biophys. Acta, Bioenerg., 2016, 1857(1), 98–106, DOI:10.1016/j.bbabio.2015.10.005.
- E. S. Wiedner, Thermodynamic hydricity of [FeFe]-hydrogenases, J. Am. Chem. Soc., 2019, 141(18), 7212–7222, DOI:10.1021/jacs.8b13084.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Thermodynamic Hydricity of Transition Metal Hydrides, Chem. Rev., 2016, 116(15), 8655–8692, DOI:10.1021/acs.chemrev.6b00168.
- C. Gauquelin, C. Baffert, P. Richaud, E. Kamionka, E. Etienne, D. Guieysse, L. Girbal, V. Fourmond, I. André and B. Guigliarelli, et al., Roles of the F-domain in [FeFe] hydrogenase, Biochim. Biophys. Acta, Bioenerg., 2018, 1859(2), 69–77, DOI:10.1016/j.bbabio.2017.08.010.
- G. Caserta, C. Papini, A. Adamska-Venkatesh, L. Pecqueur, C. Sommer, E. Reijerse, W. Lubitz, C. Gauquelin, I. Meynial-Salles and D. Pramanik, et al., Engineering an [FeFe]-hydrogenase: do accessory clusters influence O2 resistance and catalytic bias?, J. Am. Chem. Soc., 2018, 140(16), 5516–5526, DOI:10.1021/jacs.8b01689.
- P. M. Vignais, B. Billoud and J. Meyer, Classification and phylogeny of hydrogenases, FEMS Microbiol. Rev., 2001, 25(4), 455–501, DOI:10.1111/j.1574-6976.2001.tb00587.x.
- J. Meyer, [FeFe] hydrogenases and their evolution: a genomic perspective, Cell. Mol. Life Sci., 2007, 64(9), 1063, DOI:10.1007/s00018-007-6477-4.
- C. Greening, A. Biswas, C. R. Carere, C. J. Jackson, M. C. Taylor, M. B. Stott, G. M. Cook and S. E. Morales, Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival, ISME J., 2016, 10(3), 761–777, DOI:10.1038/ismej.2015.153.
- M. Calusinska, T. Happe, B. Joris and A. Wilmotte, The surprising diversity of clostridial hydrogenases: a comparative genomic perspective, Microbiology, 2010, 156(6), 1575–1588, DOI:10.1099/mic.0.032771-0.
- J.-S. Chen and L. E. Mortenson, Purification and properties of hydrogenase from Clostridium pasteurianum W5, Biochim. Biophys. Acta, Protein Struct., 1974, 371(2), 283–298, DOI:10.1016/0005-2795(74)90025-7.
- T. Happe and J. D. Naber, Isolation, characterization and N-terminal amino acid sequence of hydrogenase from the green alga Chlamydomonas reinhardtii, Eur. J. Biochem., 1993, 214(2), 475–481, DOI:10.1111/j.1432-1033.1993.tb17944.x.
- M. F. J. M. Verhagen, T. O'Rourke and M. W. W. Adams, The hyperthermophilic bacterium, Thermotoga maritima, contains an unusually complex iron-hydrogenase: amino acid sequence analyses versus biochemical characterization, Biochim. Biophys. Acta, Bioenerg., 1999, 1412(3), 212–229, DOI:10.1016/S0005-2728(99)00062-6.
- G. J. Schut and M. W. W. Adams, The iron-hydrogenase of Thermotoga maritima utilizes ferredoxin and NADH synergistically: a new perspective on anaerobic hydrogen production, J. Bacteriol., 2009, 191(13), 4451–4457, DOI:10.1128/jb.01582-08.
- M. W. W. Adams and L. E. Mortenson, The purification of hydrogenase II (uptake hydrogenase) from the anaerobic N2-fixing bacterium Clostridium pasteurianum, Biochim. Biophys. Acta, Bioenerg., 1984, 766(1), 51–61, DOI:10.1016/0005-2728(84)90216-0.
- J. B. Therien, J. H. Artz, S. Poudel, T. L. Hamilton, Z. Liu, S. M. Noone, M. W. W. Adams, P. W. King, D. A. Bryant and E. S. Boyd, et al., The physiological functions and structural determinants of catalytic bias in the [FeFe]-hydrogenases CpI and CpII of Clostridium pasteurianum strain W5, Front. Microbiol., 2017, 8, 1305, DOI:10.3389/fmicb.2017.01305.
- S. Morra, M. Arizzi, F. Valetti and G. Gilardi, Oxygen stability in the new [FeFe]-hydrogenase from Clostridium beijerinckii SM10 (CbA5H), Biochemistry, 2016, 55(42), 5897–5900, DOI:10.1021/acs.biochem.6b00780.
- N. Chongdar, P. Rodríguez-Maciá, E. J. Reijerse, W. Lubitz, H. Ogata and J. A. Birrell, Redox tuning of the H-cluster by second coordination sphere amino acids in the sensory [FeFe] hydrogenase from Thermotoga maritima, Chem. Sci., 2023, 14(13), 3682–3692, 10.1039/D2SC06432D.
- A. Fasano, H. Land, V. Fourmond, G. Berggren and C. Léger, Reversible or irreversible catalysis of H+/H2 conversion by FeFe hydrogenases, J. Am. Chem. Soc., 2021, 143(48), 20320–20325, DOI:10.1021/jacs.1c09554.
- H. Land, P. Ceccaldi, L. S. Mészáros, M. Lorenzi, H. J. Redman, M. Senger, S. T. Stripp and G. Berggren, Discovery of novel [FeFe]-hydrogenases for biocatalytic H2-production, Chem. Sci., 2019, 10(43), 9941–9948, 10.1039/C9SC03717A.
- H. Land, A. Sekretareva, P. Huang, H. J. Redman, B. Németh, N. Polidori, L. S. Mészáros, M. Senger, S. T. Stripp and G. Berggren, Characterization of a putative sensory [FeFe]-hydrogenase provides new insight into the role of the active site architecture, Chem. Sci., 2020, 11(47), 12789–12801, 10.1039/D0SC03319G.
- P. R. Cabotaje, K. Walter, A. Zamader, P. Huang, F. Ho, H. Land, M. Senger and G. Berggren, Probing substrate transport effects on enzymatic hydrogen catalysis: an alternative proton transfer pathway in putatively sensory [FeFe] hydrogenase, ACS Catal., 2023, 13(15), 10435–10446, DOI:10.1021/acscatal.3c02314.
- S. Morra, J. Duan, M. Winkler, P. A. Ash, T. Happe and K. A. Vincent, Electrochemical control of [FeFe]-hydrogenase single crystals reveals complex redox populations at the catalytic site, Dalton Trans., 2021, 50(36), 12655–12663, 10.1039/D1DT02219A.
- V. Pelmenschikov, J. A. Birrell, C. C. Pham, N. Mishra, H. Wang, C. Sommer, E. Reijerse, C. P. Richers, K. Tamasaku and Y. Yoda, et al., Reaction coordinate leading to H2 production in [FeFe]-hydrogenase identified by nuclear resonance vibrational spectroscopy and density functional theory, J. Am. Chem. Soc., 2017, 139(46), 16894–16902, DOI:10.1021/jacs.7b09751.
- M. Lorenzi, P. Ceccaldi, P. Rodríguez-Maciá, H. J. Redman, A. Zamader, J. A. Birrell, L. S. Mészáros and G. Berggren, Stability of the H-cluster under whole-cell conditions—formation of an Htrans-like state and its reactivity towards oxygen, J. Biol. Inorg. Chem., 2022, 27(3), 345–355, DOI:10.1007/s00775-022-01928-5.
- J. Duan, S. Mebs, K. Laun, F. Wittkamp, J. Heberle, T. Happe, E. Hofmann, U.-P. Apfel, M. Winkler and M. Senger, et al., Geometry of the catalytic active site in [FeFe]-hydrogenase is determined by hydrogen bonding and proton transfer, ACS Catal., 2019, 9(10), 9140–9149, DOI:10.1021/acscatal.9b02203.
- L. S. Mészáros, P. Ceccaldi, M. Lorenzi, H. J. Redman, E. Pfitzner, J. Heberle, M. Senger, S. T. Stripp and G. Berggren, Spectroscopic investigations under whole-cell conditions provide new insight into the metal hydride chemistry of [FeFe]-hydrogenase, Chem. Sci., 2020, 11(18), 4608–4617, 10.1039/D0SC00512F.
- K. A. Brown, M. B. Wilker, M. Boehm, G. Dukovic and P. W. King, Characterization of photochemical processes for H2 production by CdS nanorod–[FeFe] hydrogenase complexes, J. Am. Chem. Soc., 2012, 134(12), 5627–5636, DOI:10.1021/ja2116348.
- M. B. Wilker, J. K. Utterback, S. Greene, K. A. Brown, D. W. Mulder, P. W. King and G. Dukovic, Role of surface-capping ligands in photoexcited electron transfer between CdS nanorods and [FeFe] hydrogenase and the subsequent H2 gGeneration, J. Phys. Chem. C, 2018, 122(1), 741–750, DOI:10.1021/acs.jpcc.7b07229.
- G. J. Kubas, Fundamentals of H2 binding and reactivity on transition metals underlying hydrogenase function and H2 production and storage, Chem. Rev., 2007, 107(10), 4152–4205, DOI:10.1021/cr050197j.
- A. K. Justice, R. C. Linck, T. B. Rauchfuss and S. R. Wilson, Dihydrogen activation by a diruthenium analogue of the Fe-only hydrogenase active site, J. Am. Chem. Soc., 2004, 126(41), 13214–13215, DOI:10.1021/ja0455594.
- C. R. S. M. Hampton, I. R. Butler, W. R. Cullen, B. R. James, J. P. Charland and J. Simpson, Molecular dihydrogen and hydrido derivatives of ruthenium(II) complexes containing chelating ferrocenyl-based tertiary phosphine amine ligands and/or monodentate tertiary phosphine ligands, Inorg. Chem., 1992, 31(26), 5509–5520, DOI:10.1021/ic00052a029.
- C. Hampton, W. R. Cullen, B. R. James and J. P. Charland, The preparation and structure of a dinuclear η2-H2 complex (P-N)(η2-H2)Ru(μ-Cl)2(μ-H)Ru(H)(PPh3)2, P-N = Fe[η-C5H3(CHMeNMe2){P(i-Pr)2}-1,2](η-C5H5), J. Am. Chem. Soc., 1988, 110(20), 6918–6919, DOI:10.1021/ja00228a069.
- K. Abdur-Rashid, D. G. Gusev, A. J. Lough and R. H. Morris, Synthesis and characterization of RuH2(H2)2(PiPr3)2 and related chemistry. Evidence for a bis(dihydrogen) structure, Organometallics, 2000, 19(9), 1652–1660, DOI:10.1021/om990669i.
- T. Arliguie, B. Chaudret, R. H. Morris and A. Sella, Monomeric and dimeric ruthenium(II) η2-dihydrogen complexes with tricyclohexylphosphine co-ligands, Inorg. Chem., 1988, 27(4), 598–599, DOI:10.1021/ic00277a006.
- A. S. Weller and J. S. McIndoe, Reversible binding of dihydrogen in multimetallic complexes, Eur. J. Inorg. Chem., 2007, 2007(28), 4411–4423, DOI:10.1002/ejic.200700661.
- D. Veirs and G. M. Rosenblatt, Raman line positions in molecular hydrogen: H2, HD, HT, D2, DT, and T2, J. Mol. Spectrosc., 1987, 121(2), 401–419, DOI:10.1016/0022-2852(87)90058-0.
- S. Rumpel, C. Sommer, E. Reijerse, C. Farès and W. Lubitz, Direct detection of the terminal hydride intermediate in [FeFe] hydrogenase by NMR spectroscopy, J. Am. Chem. Soc., 2018, 140(11), 3863–3866, DOI:10.1021/jacs.8b00459.
- Y. Lee, R. A. Kinney, B. M. Hoffman and J. C. Peters, A nonclassical dihydrogen adduct of S = 1/2 Fe(I), J. Am. Chem. Soc., 2011, 133(41), 16366–16369, DOI:10.1021/ja207003m.
- M.-H. Chiang, V. Pelmenschikov, L. B. Gee, Y.-C. Liu, C.-C. Hsieh, H. Wang, Y. Yoda, H. Matsuura, L. Li and S. P. Cramer, High-frequency Fe–H and Fe–H2 modes in a trans-Fe(η2-H2)(H) complex: a speed record for nuclear resonance vibrational spectroscopy, Inorg. Chem., 2021, 60(2), 555–559, DOI:10.1021/acs.inorgchem.0c03006.
- H. Ogata, K. Nishikawa and W. Lubitz, Hydrogens detected by subatomic resolution protein crystallography in a [NiFe] hydrogenase, Nature, 2015, 520(7548), 571–574, DOI:10.1038/nature14110.
- T. Hiromoto, K. Nishikawa, S. Inoue, H. Ogata, Y. Hori, K. Kusaka, Y. Hirano, K. Kurihara, Y. Shigeta and T. Tamada, et al., New insights into the oxidation process from neutron and X-ray crystal structures of an O2-sensitive [NiFe]-hydrogenase, Chem. Sci., 2023, 14(35), 9306–9315, 10.1039/D3SC02156D.
- T. Nakane, A. Kotecha, A. Sente, G. McMullan, S. Masiulis, P. M. G. E. Brown, I. T. Grigoras, L. Malinauskaite, T. Malinauskas and J. Miehling, et al., Single-particle cryo-EM at atomic resolution, Nature, 2020, 587(7832), 152–156, DOI:10.1038/s41586-020-2829-0.
- H. M. Dietrich, R. D. Righetto, A. Kumar, W. Wietrzynski, R. Trischler, S. K. Schuller, J. Wagner, F. M. Schwarz, B. D. Engel and V. Müller, et al., Membrane-anchored HDCR nanowires drive hydrogen-powered CO2 fixation, Nature, 2022, 607(7920), 823–830, DOI:10.1038/s41586-022-04971-z.
- A. Katsyv, A. Kumar, P. Saura, M. C. Pöverlein, S. A. Freibert, S. T. Stripp, S. Jain, A. P. Gamiz-Hernandez, V. R. I. Kaila and V. Müller, et al., Molecular Basis of the Electron Bifurcation Mechanism in the [FeFe]-hydrogenase complex HydABC, J. Am. Chem. Soc., 2023, 145(10), 5696–5709, DOI:10.1021/jacs.2c11683.
- C. Furlan, N. Chongdar, P. Gupta, W. Lubitz, H. Ogata, J. N. Blaza and J. A. Birrell, Structural insight on the mechanism of an electron-bifurcating [FeFe] hydrogenase, eLife, 2022, 11, e79361, DOI:10.7554/eLife.79361.
- X. Feng, G. J. Schut, D. K. Haja, M. W. W. Adams and H. Li, Structure and electron transfer pathways of an electron-bifurcating NiFe-hydrogenase, Sci. Adv., 8(8), eabm7546, DOI:10.1126/sciadv.abm7546.
- R. Grinter, A. Kropp, H. Venugopal, M. Senger, J. Badley, P. R. Cabotaje, R. Jia, Z. Duan, P. Huang and S. T. Stripp, et al., Structural basis for bacterial energy extraction from atmospheric hydrogen, Nature, 2023, 615(7952), 541–547, DOI:10.1038/s41586-023-05781-7.
- M. del Barrio, M. Sensi, L. Fradale, M. Bruschi, C. Greco, L. de Gioia, L. Bertini, V. Fourmond and C. Léger, Interaction of the H-cluster of FeFe hydrogenase with halides, J. Am. Chem. Soc., 2018, 140(16), 5485–5492, DOI:10.1021/jacs.8b01414.
- J. Kozuch, K. Ataka and J. Heberle, Surface-enhanced infrared absorption spectroscopy, Nat. Rev. Methods Primers, 2023, 3(1), 70, DOI:10.1038/s43586-023-00253-8.
- D. Millo, P. Hildebrandt, M.-E. Pandelia, W. Lubitz and I. Zebger, SEIRA spectroscopy of the electrochemical activation of an immobilized [NiFe] hydrogenase under turnover and non-turnover conditions, Angew. Chem., Int. Ed., 2011, 50(11), 2632–2634, DOI:10.1002/anie.201006646.
- D. Millo, M.-E. Pandelia, T. Utesch, N. Wisitruangsakul, M. A. Mroginski, W. Lubitz, P. Hildebrandt and I. Zebger, Spectroelectrochemical study of the [NiFe] hydrogenase from Desulfovibrio vulgaris Miyazaki F in solution and immobilized on biocompatible gold surfaces, J. Phys. Chem. B, 2009, 113(46), 15344–15351, DOI:10.1021/jp906575r.
- O. Gutiérrez-Sanz, M. Marques, I. A. C. Pereira, A. L. De Lacey, W. Lubitz and O. Rüdiger, Orientation and function of a membrane-bound enzyme monitored by electrochemical surface-enhanced infrared absorption spectroscopy, J. Phys. Chem. Lett., 2013, 4(17), 2794–2798, DOI:10.1021/jz4013678.
- H. Krassen, A. Schwarze, B. Friedrich, K. Ataka, O. Lenz and J. Heberle, Photosynthetic hydrogen production by a hybrid complex of photosystem I and [NiFe]-hydrogenase, ACS Nano, 2009, 3(12), 4055–4061, DOI:10.1021/nn900748j.
- R. Hidalgo, P. A. Ash, A. J. Healy and K. A. Vincent, Infrared spectroscopy during electrocatalytic turnover reveals the Ni-L active site state during H2 oxidation by a NiFe hydrogenase, Angew. Chem., Int. Ed., 2015, 54(24), 7110–7113, DOI:10.1002/anie.201502338.
- P. A. Ash, S. E. T. Kendall-Price and K. A. Vincent, Unifying activity, structure, and spectroscopy of [NiFe] hydrogenases: combining techniques to clarify mechanistic understanding, Acc. Chem. Res., 2019, 52(11), 3120–3131, DOI:10.1021/acs.accounts.9b00293.
- H. Krassen, S. Stripp, G. von Abendroth, K. Ataka, T. Happe and J. Heberle, Immobilization of the [FeFe]-hydrogenase CrHydA1 on a gold electrode: Design of a catalytic surface for the production of molecular hydrogen, J. Biotechnol., 2009, 142(1), 3–9, DOI:10.1016/j.jbiotec.2009.01.018.
- H. Krassen, S. T. Stripp, N. Böhm, A. Berkessel, T. Happe, K. Ataka and J. Heberle, Tailor-made modification of a gold surface for the chemical binding of a high-activity [FeFe] hydrogenase, Eur. J. Inorg. Chem., 2011, 2011(7), 1138–1146, DOI:10.1002/ejic.201001190.
| This journal is © The Royal Society of Chemistry 2024 |