
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrategically designed metal-free deep-ultraviolet birefringent crystals with superior optical properties†
Yang
Li
 a,
Xinglong
Chen
*b and
Kang Min
Ok
*a
a,
Xinglong
Chen
*b and
Kang Min
Ok
*a
aDepartment of Chemistry, Sogang University, Seoul 04107, Republic of Korea. E-mail: kmok@sogang.ac.kr
bMaterials Science Division, Argonne National Laboratory, Lemont, Illinois 60439, USA
First published on 16th August 2024
Abstract
Finding new birefringent materials with deep-ultraviolet (DUV, λ < 200 nm) transparency is urgent, as current commercial materials cannot meet the rapidly growing demands in related application fields. Herein, three guanidinium-based compounds, C(NH2)3CH3SO3, β-C(NH2)3Cl, and γ-C(NH2)3Cl, all featuring [C(NH2)3·X]∞ (X = CH3SO3 and Cl) pseudo layers, were designed through structural motif tailoring. Theoretical calculations indicate that these metal-free compounds all possess broad bandgaps (6.49–6.71 eV, HSE06) and remarkable birefringence (cal. 0.166–0.211 @ 1064 nm). Centimeter-sized C(NH2)3CH3SO3 crystals have been grown using a feasible aqua-solution method. Subsequently, to further optimize the properties, β/γ-C(NH2)3Cl was remolded by further tailoring the [C(NH2)3]+ cationic unit and the acceptor Cl− anion, and then the fourth compound NH2COF was theoretically constructed. Interestingly, NH2COF exhibits the desired coexistence of a wider bandgap (7.87 eV, HSE06) and giant birefringence (cal. 0.241 @ 1064 nm) attributed to its higher density of well-aligned birefringence-active groups (BAGs). Furthermore, among these four designed compounds, C(NH2)3CH3SO3 has been experimentally synthesized and exhibits a short UV cutoff edge. Centimeter-sized crystals have been grown using a feasible aqueous solution method. This study provides an effective strategy to optimize the density of BAGs for large birefringence and offers valuable insights into the strategic design of metal-free DUV birefringent crystals.
Introduction
Birefringent materials can monitor and modulate the polarization of light and are widely used to fabricate polarizing devices such as polarizing prisms and fiber optic isolators, which are vital components in various advanced optical fields.1–4 Currently, in the DUV wavelength region, only two birefringent materials, namely, MgF2 and α-BaB2O4 (α-BBO), have been commercialized and used for DUV coherent light modulation.5,6 However, the materials still face some shortcomings, such as the small birefringence of MgF2 and the restricted bandgap of α-BBO. These limitations have stimulated researchers to investigate new DUV birefringent materials that balance large birefringence with short UV cutoff edges.7,8 An excellent DUV birefringent material must satisfy two imperative functional requirements simultaneously: (1) a birefringence larger than 0.10 at 1064 nm, and (2) a cutoff edge below 200 nm (bandgap Eg > 6.2 eV).9 To achieve these essential properties, the functional building blocks (FBBs) used to construct a DUV birefringent-active crystal structure should exhibit significant HOMO–LUMO gaps, large anisotropy polarizability (Δα), high spatial density, and optimal arrangements.10–13 Alkali metal and alkaline earth metal cations are widely used to investigate DUV compounds.14–16 However, commercial or promising DUV birefringent materials such as LiBO2, α-BBO, and Ca3(BO3)2 are usually grown at high temperatures with complex crystallization and growth conditions. Additionally, the inevitable high energy costs and the soaring prices of metal elements, such as lithium in LiBO2, further increase expenses. In contrast, metal-free compounds composed of earth-abundant elements, such as C, N, O, S, and H, could provide cheaper raw materials. Furthermore, these environmentally friendly metal-free compounds can typically be synthesized using a low-cost aqueous solution method, offering an alternative and promising system for researching new DUV birefringent materials.17–23In general, common BAGs can be planar π-conjugated groups or cations susceptible to second-order Jahn–Teller (SOJT) distortions. Planar π-conjugated groups include [NO3]−, [CO3]2−, [BO3]3−, [B3O6]3−, [C3N6H7]+, and [C3N3O3]3−.24–26 The cations susceptible to SOJT distortions include octahedrally coordinated d0 early transition metals (e.g., Mo6+, Nb5+, Ti4+), and cations with a stereochemically active lone pair (e.g., Pb2+, Bi3+, I5+).27–30 However, SOJT-distorted polyhedra usually red-shift the materials' cutoff edges, which negatively affects their applications in short-wavelength regions. Therefore, the design and syntheses of DUV birefringent materials (Δn > 0.1 @ 1064 nm) have mainly focused on the inorganic π-conjugated system, such as α-BBO ([B3O6]3−, 189 nm, 0.122 @ 1064 nm), LiBO2 ([BO2]∞−, 164 nm, 0.136 @ 1064 nm), and KSrCO3F ([CO3]2−, 195 nm, 0.1049 @ 1064 nm).6,8,31 In addition, among these inorganic π-conjugated groups, theoretical calculations indicate that isolated [BO3]3− and polymerized [BO2]∞− chains offer a better balance of the HOMO–LUMO gap (8.08 eV) and Δα (8.49) compared to the [CO3]2− and [NO3]− (Fig. 1). Therefore, borate compounds with the [BO3]3− group have played a dominant role in the development of DUV birefringent materials. In recent years, the organic π-conjugated [C(NH2)3]+ has garnered increasing attention because it not only has a wide HOMO–LUMO gap (8.36 eV) but also a considerable Δα (23.71), which is almost three times that of [BO3]3− (8.08) (Fig. 1).32–34 In other words, [C(NH2)3]+ can achieve a more favorable balance between bandgap and optical anisotropy. Moreover, the three [–NH2] groups in [C(NH2)3]+ can potentially serve as hydrogen bonding donors, providing more flexibility in structural configuration. Therefore, the [C(NH2)3]+ unit has a significant advantage in building new advanced DUV birefringent materials. To date, the guanidium-based compound system is still underdeveloped, with only two DUV compounds simultaneously achieving large birefringence (>0.15 @ 1064 nm) and a large bandgap: [C(NH2)3]3B3O3F2(OH)2 and [C(NH2)3]2B3O3F4(OH) (6.53–6.36 eV, 0.164–0.171 @ 1064 nm).21
 | ||
| Fig. 1 Calculated HOMO–LUMO gaps and polarizability (Δα) for inorganic and organic coplanar BAGs. Inorganic: [BO3]3−, [CO3]2−, [NO3]−, and [B3O6]3−. Organic: [C(NH2)3]+, [C3N6H7]+ (Me+), and [C3N3O3]3− (CY3−). | ||
To maximize the contribution of [C(NH2)3]+ to birefringence, the π-conjugated [C(NH2)3]+ needs to be arranged in a coplanar mode. In addition, it should have the highest possible density in the unit cell, which can be achieved by reducing the volume fraction of non-BAGs in the unit cell. Moreover, the substitution strategy based on a suitable template has been widely applied as a highly efficient approach for new compound design. In this context, the well-studied borate family provides various template options. Among these, KBe2BO3F2 (KBBF) and Sr2Be2B2O7 (SBBO) are widely used as structural templates to investigate new π-conjugated compounds with outstanding optical properties because they feature parallel arrangements of BO3–BeO3F/BeO4 layers.35,36 In a previous study, we successfully designed a KBBF-type compound, KF·B(OH)3, which shows a wide bandgap and moderate birefringence (7.63 eV and 0.114 @ 1064 nm).37 Given the above insights and experience, we see the prospect of exploring new guanidinium-based compounds with superior birefringence (≥0.20 @ 1064 nm) by remolding template compounds such as SBBO and KF·B(OH)3.
Herein, four metal-free compounds, C(NH2)3CH3SO3, β-C(NH2)3Cl, γ-C(NH2)3Cl, and NH2COF, have been rationally designed, among which β-C(NH2)3Cl and γ-C(NH2)3Cl are hypothetical compounds, and NH2COF was theoretically screened using DFT calculations. The bandgaps and birefringence of these compounds were predicted by DFT calculations. C(NH2)3CH3SO3 and β/γ-C(NH2)3Cl possess large bandgaps (6.49–6.71 eV, HSE06) and large birefringence (cal. 0.166–0.211 @ 1064 nm). C(NH2)3CH3SO3 displays a good growth habit, and centimeter-sized crystals have been obtained. Remarkably, designed using the “All-Three-in-One” strategy, NH2COF achieves the highest ρBAGs (16.18 × 10−3 Å−3), a remarkably wide bandgap (7.87 eV, HSE06), and the large birefringence (cal. 0.241 @ 1064 nm) among these four compounds, which are superior to those of the commercial α-BBO (6.56 eV and 0.122 @ 1064 nm). These findings can provide a new pathway to investigate novel DUV birefringent materials with superior birefringence and wide bandgaps.
Experimental section
CH3SO3H (Thermo Scientific, 98%) and [C(NH2)3]2CO3·H2O (TCI, 98%) were used as received.Single crystals of C(NH2)3CH3SO3 were easily grown via the slow evaporation method. [C(NH2)3]2CO3·H2O (30 mmol), CH3SO3H (2 mL), and 100 mL deionized water were mixed in a glass beaker and stirred for 3 hours. The beaker was put in a dust-free environment and evaporated at room temperature for one week. Transparent block-shaped crystals were washed with distilled water with a high yield of 90% based on [C(NH2)3]2CO3·H2O. According to our preliminary trials, centimeter-level crystals with a thickness of up to 0.3 cm can be easily grown in a short growth cycle, manifesting a good crystal growth habit.
The Rigaku MiniFlex 600 diffractometer equipped with a Cu Kα radiation (λ = 1.5418 Å) was used for collecting the PXRD data from 5° to 70° with a scan step width of 0.02° and a scan speed of 1° min−1.
Bruker D8 QUEST diffractometer with the monochromatic Mo Kα radiation (λ = 0.71073 Å) was used for determining the single crystal structure of C(NH2)3CH3SO3. Collected data were integrated with the SAINT program, and the program SCALE for the area detector was used to apply the multi-scan absorption correction. OLEX2 program with XT method and ShelXL software were applied to solve the structure.38 Non-hydrogen atoms were refined using full-matrix least-squares techniques, and the final least-squares refinement was performed on Fo2 with data having Fo2 ≥ 2σ(Fo2). The location of the hydrogen atoms was confirmed by the different Fourier maps and optimized further using the first-principles calculations. The PLATON checked the final structure for no missing symmetry.39 Crystallographic data and structure refinement information are listed in Table S1.† The atomic coordinates, equivalent isotropic displacement parameters, selected interatomic lengths and angles, and hydrogen bond distances are given in Tables S2–S6.†
IR spectrum was measured on a Bruker TENSOR 27 ATR-FT-IR spectrometer in the 500–4000 cm−1 range (Fig. S7†). The ground sample was contacted on the diamond attenuated-total-reflectance crystal.
The optical diffuse reflectance spectrum was measured at room temperature with a Lambda 1050 scan UV-vis spectrophotometer in the wavelength range from 190 to 800 nm. A polished crystal of C(NH2)3CH3SO3 was used to measure the UV-vis-NIR transmittance spectrum (190–1600 nm) with a Shimadzu SolidSpec-3700 DUV spectrophotometer at room temperature (Fig. S8†).
The birefringence (at 546 nm) was measured using the polarizing microscope method and calculated using the following equation: R = Δn × d. Here, R represents the optical path difference, which was recorded with a ZEISS Axiolab 5 polarizing microscope equipped with a Berek compensator. The thickness (d) and crystal plane of the examined crystal were determined using a Bruker D8 QUEST diffractometer.
The TGA of C(NH2)3CH3SO3 was investigated on a SCINCO TGA-N 1000 thermal analyzer. Ground polycrystalline samples were put into an alumina crucible and heated to 900 °C under flowing Ar at 10 °C min−1 (Fig. S9†).
The CASTEP package was used for the first-principles calculations via the plane-wave pseudo-potential method.40 The band structure and the optical properties were calculated based on the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof functional and TS method for DFT-D correction.41–44 Plane-wave cut-off energy of 830 eV was chosen, and the dense k-point sampling less than 0.04 Å−1 was adopted. The HSE06 functional provides bandgap calculations that are closely aligned with experimental values (relative error < 5%). Therefore, the HSE06 bandgap is used to evaluate the UV cutoff edge.45 The corresponding plane-wave cutoff energy is 750 eV, with a dense k-point sampling of less than 0.07 Å−1. The linear optical properties were examined based on the dielectric function ε(ω) = ε1(ω) + iε2(ω). The imaginary part of the dielectric function ε2 can be calculated based on the electronic structures. The real part is obtained by the Kramers–Kronig transformation; accordingly, the refractive indices and the birefringence (Δn) can be calculated. The OptaDOS was used to analyze the calculated results. Gaussian 09 was used to calculate the HOMO–LUMO gap and polarizability with a 6-31G (D3-BJ) basis set.46 CP2K, Multiwfn, VMD, and Vesta were utilized for the electron localized functional (ELF) map diagram, unit sphere representations of polarizability and independent gradient model based on Hirshfeld partition (IGMH) analyses.47
Results and discussion
C(NH2)3CH3SO3 crystallizes in the monoclinic space group, I2/m (no. 12).48 With the aid of the hydrogen bonds, each [C(NH2)3]+ is connected with three [CH3SO3]− and extends infinitely to form the neutral single layer of [C(NH2)3·CH3SO3]∞ (Fig. S1a–c†). Two adjacent [C(NH2)3·CH3SO3]∞ layers are anti-oriented to form the 2[C(NH2)3·CH3SO3]∞ double layer. These double layers connect with each other via hydrogen bonds to build the whole structure of C(NH2)3CH3SO3 (Fig. S1d†).Hypothetical β-C(NH2)3Cl and γ-C(NH2)3Cl crystallize in the space groups of trigonal P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 (no. 72) and monoclinic C2/m (no. 12), respectively. The phonon dispersion of β/γ-C(NH2)3Cl was performed and confirmed that they are dynamically stable (Fig. S3†). In their structures, Cl− is coordinated with three bidentate [–NH2] groups from [C(NH2)3]+ by NH2⋯Cl interactions, forming the single layer of [C(NH2)3·Cl]∞. In addition, this single layer adopts the same assembly style as C(NH2)3CH3SO3 to generate the 2[C(NH2)3·Cl]∞ double layer and the whole structure of β/γ-C(NH2)3Cl (Fig. S4 and S5†).
m1 (no. 72) and monoclinic C2/m (no. 12), respectively. The phonon dispersion of β/γ-C(NH2)3Cl was performed and confirmed that they are dynamically stable (Fig. S3†). In their structures, Cl− is coordinated with three bidentate [–NH2] groups from [C(NH2)3]+ by NH2⋯Cl interactions, forming the single layer of [C(NH2)3·Cl]∞. In addition, this single layer adopts the same assembly style as C(NH2)3CH3SO3 to generate the 2[C(NH2)3·Cl]∞ double layer and the whole structure of β/γ-C(NH2)3Cl (Fig. S4 and S5†).
NH2COF crystallizes in the orthorhombic space group of Ibam (no. 72).49 The hydrogen bonding of N–H⋯O connects the neutral [NH2COF] unit, generating the [NH2COF]∞ layer in the ab plane (Fig. S6†). Connected by van der Waals interactions, all these layers are parallelly distributed in the structure, with an interlayer distance of 3.06 Å, endowing NH2COF with a large optical anisotropy between the c and a/b axes. The evolutionary path from SBBO to C(NH2)3CH3SO3 and β/γ-C(NH2)3Cl, and the concordant arrangement of [C(NH2)3]+, are shown in Fig. 2. It is known that the π-conjugated [BO3]3− groups dominate the attractive optical properties of SBBO. The tetrahedral [BeO4]6− governs the arrangements of [BO3]3− in the structure, also the source of the structural polymorphism problem (stacking faults) in SBBO.50 During our design process, we proceeded based on the framework of SBBO, proposing the substitution of 2Sr2+ + 2Be2+ + O2− + 2[BO3]3− by 2[C(NH2)3]+ + 2[CH3SO3]− (Fig. 2a–c). As expected, C(NH2)3CH3SO3 maintains the structural gene of SBBO and features pseudo-double layers of 2[(C(NH2)3·CH3SO3)2]∞. The [C(NH2)3]+ units are well-aligned in C(NH2)3CH3SO3, benefiting the generation of large birefringence (0.166 @ 1064 nm) (Fig. 2c and d). Meanwhile, the “stacking faults” and toxic raw reagent BeO issues of SBBO were overcome. In the obtained C(NH2)3CH3SO3, the ρBAGs of π-conjugated [C(NH2)3]+ is 5.32 × 10−3 Å−3. Further increasing ρBAGs of [C(NH2)3]+ is anticipated to enhance the birefringence of compounds.
 | ||
| Fig. 2 Ball-and-stick representations of (a) SBBO, (b) [Be2B2O7]4− framework, (c) and (d) C(NH2)3CH3SO3 (GSOH), (e) KF·B(OH)3, (f) hydrogen bonding around F−, (g) hydrogen bonding around Cl−, (h) C(NH2)3Cl, (i) [C(NH2)3]+ and Cl− dimer, (j) [NH2COF]0 molecule, (k) hydrogen bonding around [NH2COF]0 molecule, and (l) structure of NH2COF (Sr: blue; K: purple; Be: orange; S: yellow; O: red; B: green; F and Cl: light green; C: black; N: brown; H: white). | ||
Thus, the larger birefringence-inert [CH3SO3]− groups in C(NH2)3CH3SO3 were substituted by smaller Cl− atoms to increase the ρBAGs. Subsequently, the β-/γ-C(NH2)3Cl compounds were designed. As expected, the density of [C(NH2)3]+ increased to 9.01 × 10−3 Å−3 in β-/γ-C(NH2)3Cl, and their birefringence reached 0.201–0.211 @ 1064 nm (Fig. 2h, S4 and S5†). Fig. 2e–h depict the tailoring strategy from KF·B(OH)3 to β-/γ-C(NH2)3Cl and then to NH2COF. KF·B(OH)3 features the [F·B(OH)3]∞ pseudo-layers with K+ atoms located in the interlayer space. These [F·B(OH)3]∞ pseudo-layers are perfectly replaced by neutral [C(NH2)3·Cl]∞ pseudo-layers, leading to the construction of β-/γ-C(NH2)3Cl compounds. Due to the larger Δα of [C(NH2)3]+, the birefringence of β-/γ-C(NH2)3Cl (0.201–0.211(@1064 nm)) is nearly twice that of KF·B(OH)3 (0.114(@1064 nm)). To further improve the birefringence, the [C(NH2)3·Cl]0 dimer was further modified: two [–NH2] were replaced by the hydrogen bonding acceptors (O2−/F−), resulting in a neutral π-conjugated [NH2COF]0. Consequently, the NH2COF compound was designed, with a very high ρBAGs value of 16.18 × 10−3 Å−3, larger than KBBF, SBBO, KF·B(OH)3, C(NH2)3CH3SO3, and β/γ-C(NH2)3Cl (Fig. 2l). The calculated birefringence is 0.241 @ 1064 nm. The experimental PXRD pattern of C(NH2)3CH3SO3 matches well with the simulated one (Fig. 3a). The IR spectrum confirms the presence of [C(NH2)3]+ and CH3SO3− groups (Fig. S7†). The UV-vis diffuse reflectance spectrum of C(NH2)3CH3SO3 indicates its UV cutoff edge is below 190 nm (Fig. 3b). To better estimate the UV cutoff edge, the UV-vis-NIR transmittance spectrum was measured. C(NH2)3CH3SO3 exhibits a short cutoff edge of 194 nm (Fig. S8†), consistent with the calculated bandgap of 6.71 eV using the HSE06 functional. The thermal stability curve indicates that it remains stable up to 300 °C (Fig. S9†). The polycrystalline samples of C(NH2)3CH3SO3 were immersed in methyl alcohol (MeOH), ethyl alcohol (EtOH), acetone, n-hexane, and isopropanol (IPA) for over 2 days for the chemical stability test. The measured PXRD patterns indicate that C(NH2)3CH3SO3 remains stable in various solvents, which is beneficial for its practical application. In addition, a sample of C(NH2)3CH3SO3 powder (0.330 g) was exposure to air for seven days to test its air stability, which is also essential for practical applications. The ambient temperature was maintained at 30 ± 5 °C and the humidity at 35 ± 5%. After one week, the mass (0.330 g) and the PXRD pattern showed almost no change, indicating that C(NH2)3CH3SO3 has good air stability under these conditions (Fig. S10†).
 | ||
| Fig. 3 (a) PXRD pattern and (b) UV-vis diffuse reflectance spectrum of C(NH2)3CH3SO3. Inset: as grown crystals of C(NH2)3CH3SO3. | ||
The calculated bandgaps by the GGA and HSE06 hybrid functional methods of C(NH2)3CH3SO3, β/γ-C(NH2)3Cl, and NH2COF are listed in Table S7† and illustrated in Fig. S11–S14.† The HSE06 (GGA) results are as follows: 6.71 (5.21) eV for C(NH2)3CH3SO3, 6.49 (4.96) eV for β-C(NH2)3Cl, 6.51 (4.93) eV for γ-C(NH2)3Cl, and 7.87 (6.15) eV for NH2COF.
The total/partial density of states for C(NH2)3CH3SO3, β/γ-C(NH2)3Cl, and NH2COF are depicted in Fig. S15–S17.† For C(NH2)3CH3SO3 and β/γ-C(NH2)3Cl, the upper regions of the valence band are dominated by the N-2p, C-2p, O-2p states (C(NH2)3CH3SO3), and Cl-3p states (β-/γ-C(NH2)3Cl). The conduction band's bottom areas contain C-2p, N-2p, and small amounts of H-1s states. Thus, the [C(NH2)3]+ moiety mainly determines the optical properties of C(NH2)3CH3SO3 and β/γ-C(NH2)3Cl, as expected. For NH2COF, the top of the valence band is mainly occupied by O-2p and N-2p states, with smaller amounts of F-2p and H-1s states. Meanwhile, the bottom of the conduction band is primarily composed of O-2p, C-2p, and N-2p states, with small quantities of F-2p and H-1s states.
The electron localized function maps indicate the π-conjugated electrons from [C(NH2)3]+ and NH2COF (Fig. S18 and S19†), which are expected sources of the large birefringence. The unit sphere and vector representations method visually display the polarizability, thus, it was applied to [C(NH2)3]+ and CH3SO3−, with results shown in Fig. S20.† Clearly, the larger polarizability tensorial components of [C(NH2)3]+ are mainly distributed in the xy-plane. However, for CH3SO3−, it is along the C–S bond in the z-axis, negatively contributing to the birefringence.
The HOMO–LOMO gaps (EH–L) and polarizability anisotropy (Δα) for [BO3]3−, [B(OH)3], [C(NH2)3]+, and NH2COF were displayed in Fig. 4a. The results indicate that NH2COF is an attractive BAG that maintains a good balance of EH–L and Δα. Although the Δα (NH2COF) is smaller than that of [C(NH2)3]+, the largest density still contributes to achieving the represented birefringence of 0.241 @ 1064 nm (Fig. 4b).
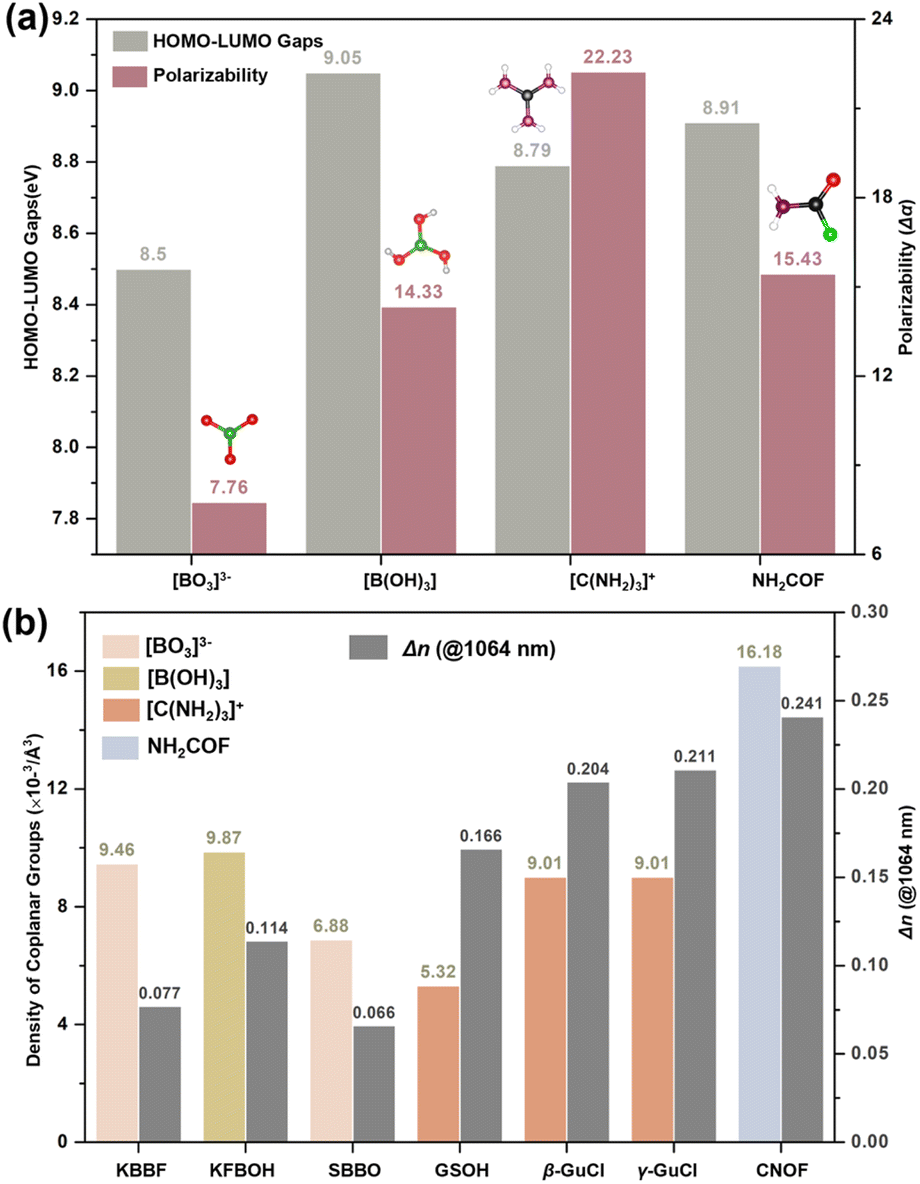 | ||
| Fig. 4 (a) Calculated HOMO–LUMO gaps and polarizability for coplanar groups. (b) Density of coplanar groups and calculated birefringence (@1064 nm) in KBBF, KFBOH, SBBO, C(NH2)3CH3SO3 (GSOH), β/γ-C(NH2)3Cl (GuCl), and NH2COF (CNOF). | ||
The selected DUV metal-free and metal-containing optical crystals are listed in Fig. 5. C(NH2)3CH3SO3, β/γ-C(NH2)3Cl, and NH2COF exhibit wide bandgaps (6.71–7.87 eV, HSE06) and large birefringence (cal. 0.166–0.241 @ 1064 nm), satisfying the fundamental requirements for DUV birefringent materials. Notably, the birefringence of γ-C(NH2)3Cl (cal. 0.211 @ 1064 nm) is significantly larger than that of other reported DUV metal-free compounds, such as [C(NH2)3][B3O3F2(OH)2] (GBF1, 0.171 @ 1064 nm) and (NH4)4[B12O16F4(OH)4] (ABFH, 0.114 @ 546 nm).21,51 More importantly, NH2COF exhibits a UV λcutoff (∼159 nm), which is 30 nm shorter than that of α-BBO (189 nm), and a birefringence (cal. 0.241 @ 1064 nm) nearly twice that of the α-BBO (0.122 @ 1064 nm), suggesting its potential as a promising DUV birefringent crystal. The experimental birefringence of C(NH2)3CH3SO3 was measured and compared with the calculated result. The natural crystal plane of C(NH2)3CH3SO3 for birefringence measurement is the (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) plane, where the [C(NH2)3]+ is located. The thickness is 16 μm and the optical path difference is 61.27 nm. Therefore, the measured birefringence is 0.004 @ 546 nm (Fig. S21†). This result fits the calculated birefringence of 0.002 @ 546 nm for this crystal plane, indicating that our measurements and calculations are reliable. Such small birefringence on this crystal plane is reasonable. First, C(NH2)3CH3SO3 belongs to the monoclinic crystal system, making it a biaxial crystal. According to the crystallographic symmetry, the principal optical axis Z is parallel to the crystal b direction. Second, the ideal [C(NH2)3]+ has C3 symmetry, and in C(NH2)3CH3SO3, the [C(NH2)3]+ has a weak distortion. Thus, the values of in-plane nx and ny are nearly the same and far beyond the out-plane nz. That is to say, nx ≈ ny ≫ nz. Third, in the structure of C(NH2)3CH3SO3, [C(NH2)3]+ is located at the (10) crystal plane; therefore, the birefringence on the (10) plane is equal to |nx − ny| and is reasonably close to zero.
) plane, where the [C(NH2)3]+ is located. The thickness is 16 μm and the optical path difference is 61.27 nm. Therefore, the measured birefringence is 0.004 @ 546 nm (Fig. S21†). This result fits the calculated birefringence of 0.002 @ 546 nm for this crystal plane, indicating that our measurements and calculations are reliable. Such small birefringence on this crystal plane is reasonable. First, C(NH2)3CH3SO3 belongs to the monoclinic crystal system, making it a biaxial crystal. According to the crystallographic symmetry, the principal optical axis Z is parallel to the crystal b direction. Second, the ideal [C(NH2)3]+ has C3 symmetry, and in C(NH2)3CH3SO3, the [C(NH2)3]+ has a weak distortion. Thus, the values of in-plane nx and ny are nearly the same and far beyond the out-plane nz. That is to say, nx ≈ ny ≫ nz. Third, in the structure of C(NH2)3CH3SO3, [C(NH2)3]+ is located at the (10) crystal plane; therefore, the birefringence on the (10) plane is equal to |nx − ny| and is reasonably close to zero.
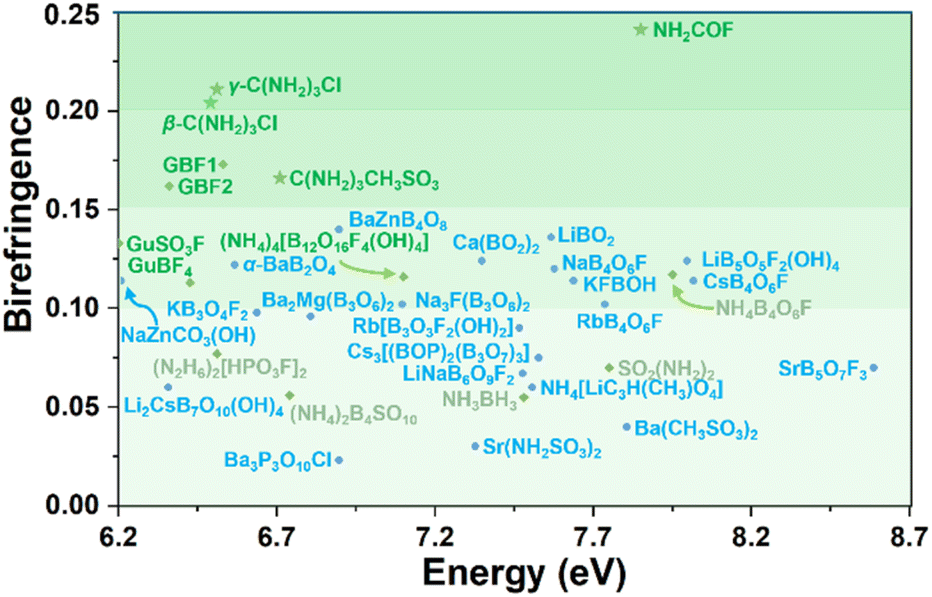 | ||
| Fig. 5 Birefringence of selected DUV metal-free (green), metal-containing optical crystals (blue), along with results from this work (star). Details are listed in Table S8.† | ||
In summary, three guanidium-based DUV birefringent materials, C(NH2)3CH3SO3, β-C(NH2)3Cl, and γ-C(NH2)3Cl, were rationally designed with wide bandgaps (6.49–6.71 eV, HSE06) and remarkable birefringence of 0.166–0.211 @ 1064 nm (cal.). Among them, C(NH2)3CH3SO3 has been experimentally realized, and centimeter-sized crystals were successfully grown via the mild aqua-solution method, indicating good growth habits. In addition, the fourth compound, NH2COF, was obtained via the “All-Three-in-One” strategy. Theoretical calculations indicate that NH2COF possesses a wide bandgap of 7.87 eV (HSE06) and a large birefringence of 0.241 @ 1064 nm (cal.), which are significantly enhanced compared to C(NH2)3CH3SO3, β-/γ-C(NH2)3Cl, and the commercial α-BBO (6.56 eV and 0.122 @ 1064 nm). This work demonstrates that [C(NH2)3]+ and NH2COF units can be promising DUV BAGs, shedding light on the tremendous potential of metal-free compounds as advanced DUV birefringent materials. Our further work is to try to grow a large crystal of NH2COF to experimentally evaluate its potential as a promising DUV birefringent material.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for C(NH2)3CH3SO3 has been deposited at the CCDC under 2357582 and can be obtained from https://www.ccdc.cam.ac.uk/structures.Author contributions
K. M. O. conceived the project. Y. L. and X. C. conducted the experimental studies. All authors contributed to the writing and editing of the manuscript and agreed on the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (Grant No. 2018R1A5A1025208 and 2019R1A2C3005530). The authors thank Dr Hao Li for helpful discussions and technical assistance.Notes and references
- A. Tagaya, H. Ohkita, M. Mukoh, R. Sakaguchi and Y. Koike, Science, 2003, 301, 812–814 CrossRef CAS PubMed.
- L. H. Nicholls, F. J. Rodríguez-Fortuño, M. E. Nasir, R. M. Córdova-Castro, N. Olivier, G. A. Wurtz and A. V. Zayats, Nat. Photonics, 2017, 11, 628–633 CrossRef CAS.
- S. Niu, G. Joe, H. Zhao, Y. Zhou, T. Orvis, H. Huyan, J. Salman, K. Mahalingam, B. Urwin, J. Wu, Y. Liu, T. E. Tiwald, S. B. Cronin, B. M. Howe, M. Mecklenburg, R. Haiges, D. J. Singh, H. Wang, M. A. Kats and J. Ravichandran, Nat. Photonics, 2018, 12, 392–396 CrossRef CAS.
- D. Brida, C. Manzoni and G. Cerullo, Opt. Lett., 2012, 37, 3027 CrossRef PubMed.
- M. J. Dodge, Appl. Opt., 1984, 23, 1980 CrossRef CAS.
- G. Zhou, J. Xu, X. Chen, H. Zhong, S. Wang, K. Xu, P. Deng and F. Gan, J. Cryst. Growth, 1998, 191, 517–519 CrossRef CAS.
- F. Zhang, X. Chen, M. Zhang, W. Jin, S. Han, Z. Yang and S. Pan, Light: Sci. Appl., 2022, 11, 252 CrossRef CAS PubMed.
- X. Chen, B. Zhang, F. Zhang, Y. Wang, M. Zhang, Z. Yang, K. R. Poeppelmeier and S. Pan, J. Am. Chem. Soc., 2018, 140, 16311–16319 CrossRef CAS PubMed.
- A. Tudi, S. Han, Z. Yang and S. Pan, Coord. Chem. Rev., 2022, 459, 214380 CrossRef CAS.
- X. Hao, M. Luo, C. Lin, G. Peng, F. Xu and N. Ye, Angew. Chem., Int. Ed., 2021, 60, 7621–7625 CrossRef CAS PubMed.
- J. Lu, J. N. Yue, L. Xiong, W. K. Zhang, L. Chen and L. M. Wu, J. Am. Chem. Soc., 2019, 141, 8093–8097 CrossRef CAS PubMed.
- G. Shi, Y. Wang, F. Zhang, B. Zhang, Z. Yang, X. Hou, S. Pan and K. R. Poeppelmeier, J. Am. Chem. Soc., 2017, 139, 10645–10648 CrossRef CAS PubMed.
- C. Huang, M. Mutailipu, F. Zhang, K. J. Griffith, C. Hu, Z. Yang, J. M. Griffin, K. R. Poeppelmeier and S. Pan, Nat. Commun., 2021, 12, 2597 CrossRef CAS PubMed.
- P. Gong, L. Kang and Z. Lin, J. Am. Chem. Soc., 2020, 142, 15157–15163 CrossRef CAS PubMed.
- Q. Ding, X. Liu, S. Zhao, Y. Wang, Y. Li, L. Li, S. Liu, Z. Lin, M. Hong and J. Luo, J. Am. Chem. Soc., 2020, 142, 6472–6476 CrossRef CAS PubMed.
- H. Liu, H. Wu, Z. Hu, J. Wang, Y. Wu and H. Yu, J. Am. Chem. Soc., 2023, 145, 12691–12700 CrossRef CAS PubMed.
- S. Zhang, X. Wu, Y. Song, D. Ni, B. Hu and T. Zhou, J. Cryst. Growth, 2003, 252, 246–250 CrossRef CAS.
- T. W. Kasel, Z. Deng, A. M. Mroz, C. H. Hendon, K. T. Butler and P. Canepa, Chem. Sci., 2019, 10, 8187–8194 RSC.
- M. Luo, C. Lin, D. Lin and N. Ye, Angew. Chem., Int. Ed., 2020, 59, 15978–15981 CrossRef CAS PubMed.
- H. Qiu, F. Li, C. Jin, Z. Yang, J. Li, S. Pan and M. Mutailipu, Angew. Chem., Int. Ed., 2024, 63, e202316194 CrossRef CAS PubMed.
- C. Jin, H. Zeng, F. Zhang, H. Qiu, Z. Yang, M. Mutailipu and S. Pan, Chem. Mater., 2022, 34, 440–450 CrossRef CAS.
- C. Hu, Q. Chen, F. Kong and J. Mao, Inorg. Chem. Front., 2024, 11, 3150–3158 RSC.
- M. Mutailipu, J. Han, Z. Li, F. Li, J. Li, F. Zhang, X. Long, Z. Yang and S. Pan, Nat. Photonics, 2023, 17, 694–701 CrossRef CAS.
- H. Wu, Z. Wei, Z. Hu, J. Wang, Y. Wu and H. Yu, Angew. Chem., Int. Ed., 2024, 63, e202406318 CrossRef CAS PubMed.
- Z. Bai and K. M. Ok, Angew. Chem., Int. Ed., 2024, 63, e202315311 CrossRef CAS PubMed.
- J. Lu, Y. Lian, L. Xiong, Q. Wu, M. Zhao, K. Shi, L. Chen and L. Wu, J. Am. Chem. Soc., 2019, 141, 16151–16159 CrossRef CAS.
- L. Qi, X. Jiang, K. Duanmu, C. Wu, Z. Lin, Z. Huang, M. G. Humphrey and C. Zhang, J. Am. Chem. Soc., 2024, 146, 9975–9983 CrossRef CAS.
- P. Li, C. Hu, Y. Li, J. Mao and F. Kong, J. Am. Chem. Soc., 2024, 57, 7868–7874 CrossRef.
- M. Liang, Y. Zhang, E. Izvarin, M. J. Waters, J. M. Rondinelli and P. S. Halasyamani, Chem. Mater., 2024, 36, 2113–2123 CrossRef CAS.
- W. Zeng, Y. Tian, X. Dong, L. Huang, H. Zeng, Z. Lin and G. Zou, Chem. Mater., 2024, 36, 2138–2146 CrossRef CAS.
- W. Zhang and P. S. Halasyamani, CrystEngComm, 2017, 19, 4742–4748 RSC.
- Y. Shang, H. Sha, Z. Wang, R. Su, C. He, X. Yang and X. Long, Adv. Opt. Mater., 2024, 2302844 CrossRef CAS.
- X. Liu, L. Kang, R. Guo and Z. Lin, Dalton Trans., 2021, 50, 17495–17498 RSC.
- J. X. Wang, S. F. Li, M. M. Ren, M. H. Lv, R. L. Tang, J. Chen, H. Huang, B. Zhang and D. Yan, Inorg. Chem., 2024, 63, 4487–4491 CrossRef CAS PubMed.
- B. Wu, D. Tang, N. Ye and C. Chen, Opt. Mater., 1996, 5, 105–109 CrossRef CAS.
- C. Chen, Y. Wang, B. Wu, K. Wu, W. Zeng and L. Yu, Nature, 1995, 373, 322–324 CrossRef CAS.
- Y. Li, X. Chen and K. M. Ok, Chem. Commun., 2022, 58, 8770–8773 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr. - Cryst. Mater., 2005, 220, 567–570 CrossRef CAS.
- W. Wang, H. Fan and Y. Ye, Polymer, 2010, 51, 3575–3581 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- K. Liu, H. Fan, P. Ren and C. Yang, J. Alloys Compd., 2011, 509, 1901–1905 CrossRef CAS.
- J. Lin, A. Qteish, M. C. Payne and V. Heine, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 4174 CrossRef PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- V. A. Russell, M. C. Etter and M. D. Ward, J. Am. Chem. Soc., 1994, 116, 1941–1952 CrossRef CAS.
- A. F. Baxter, K. O. Christe and R. Haiges, Struct. Chem., 2017, 28, 303–307 CrossRef CAS.
- H. Huang, J. Yao, Z. Lin, X. Wang, R. He, W. Yao, N. Zhai and C. Chen, Chem. Mater., 2011, 23, 5457–5463 CrossRef CAS.
- C. Jin, F. Li, B. Cheng, H. Qiu, Z. Yang, S. Pan and M. Mutailipu, Angew. Chem., Int. Ed., 2022, 61, e202203984 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data, birefringence of selected DUV metal-free and metal-containing compounds, IR spectrum, UV-vis-NIR transmittance spectrum, TGA diagram, chemical and air stability test, phonon dispersion, independent gradient model based on Hirshfeld partition (IGMH) analyses, band structures, density of states, ELF diagrams, unit sphere representations of polarizability under a static electric field, birefringence measurement and HOMO and LUMO energy gaps. CCDC 2357582 contains the supplementary crystallographic data for C(NH2)3CH3SO3. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04155k |
| This journal is © The Royal Society of Chemistry 2024 |