
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
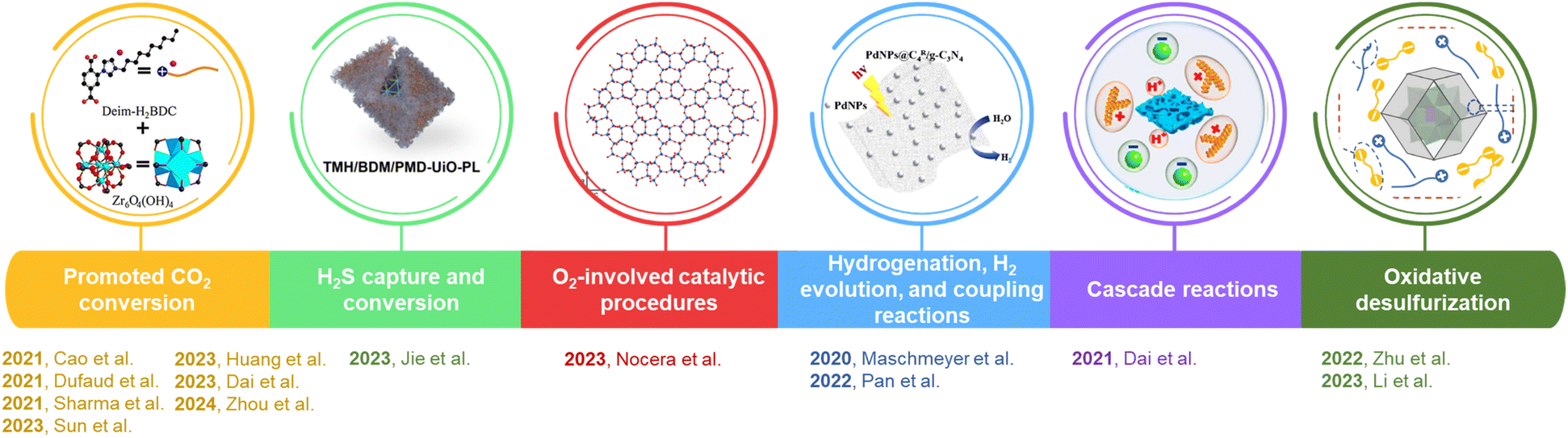
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePorous liquids: an integrated platform for gas storage and catalysis
Errui
Li
a,
Kevin M.
Siniard
a,
Zhenzhen
Yang
*b and
Sheng
Dai
 *a
*a
aDepartment of Chemistry, Institute for Advanced Materials and Manufacturing, University of Tennessee, Knoxville, TN 37996, USA
bChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: yangz3@ornl.gov; dais@ornl.gov
First published on 18th October 2024
Abstract
Porous liquids (PLs) represent a new frontier in materials design, combining the unique features of fluidity in liquids and permanent porosity in solids. By engineering well-defined pores into liquids via designed structure modification techniques, the greatly improved free volume significantly enhances the gas transport and storage capability of PL sorbents. Triggered by the promising applications of PLs in gas separation, PLs are further explored in catalysis particularly to integrate the gas storage and catalytic transformation procedure. This emerging field has demonstrated promising progress to advance catalytic procedures using PLs as catalysts, with performance surpassing that of the pure liquid and porous host counterparts. In this perspective article, the recent discoveries and progress in the field of integrated gas storage and catalysis by leveraging the PL platforms will be summarized, particularly compared with the traditional homogeneous or heterogeneous catalytic procedures. The unique features of PLs endow them with combined merits from liquid and solid catalysts and beyond which will be illustrated first. This will be followed by the unique techniques being utilized to probe the porosity and active sites in PLs and the structural evolution during the catalytic procedures. The catalytic application of PLs will be divided by the reaction categories, including CO2-involving transformation, O2-involving reaction, H2S conversion, hydrogenation reaction, and non-gas involving cascade reactions. In each reaction type, the synthesis approaches and structure engineering techniques of PLs, structure characterization, catalytic performance evaluation, and reaction mechanism exploration will be discussed, highlighting the structure–performance relationship and the advancement benefiting from the unique features of PLs.
1. Introduction
Porous liquids (PLs) represent a promising category of systems capable of combining the merits of flowing liquids and porous solids.1–8 In PLs, well-defined pores ranging from micro- to macro-scales are engineered into liquids to afford greatly improved free volumes within the dense liquid phase and tunable physicochemical properties. Diverse synthesis approaches have been developed to produce diverse PL categories, which have been obtained by liquifying molecules with stiff interior voids (type I PL),9,10 dissolving rigid porous hosts in flowing liquids (type II PL),11,12 dispersing porous frameworks in high steric hindrance solvents (type III PL),13 and employing neat meltable extended frameworks (type IV PL),14 respectively. Porous hosts with rigid scaffolds, such as metal–organic frameworks (MOFs),15,16 covalent organic frameworks (COFs),17 zeolites,18,19 hollow silica spheres,20–22 silicalite,23,24 hollow carbon spheres,10,25 porous cages,9,11,26–28 and macromolecules,29 could be liquified by surface modification, physical dispersion, or surface deposition. Accordingly, the liquid counterparts could adopt ionic liquids (ILs),18,30–32 bulky organic solvents,11,33–35 water,23,36 oil,37,38 and polyfluoroalkyl substances (PFAS).39 To provide high-quality PLs, the scaffolds of the porous host should be rigid, and exhibit no collapse, no degradation, and no phase segregation after integrating with the liquid component. The liquid phase should possess low viscosity, good stability, and capability to stabilize the involved porous host solute, nanoparticles, or clusters.The unique features and large structure/property tunability of PLs render them promising applications in gas separation and storage. The benefits are twofold: (1) the cavities in PLs provide more free volume for gas storage than the dense liquid phase; and (2) the chemical properties and textural structure of the porous host, liquid counterpart, and solid–liquid interphase can be tuned to facilitate the sorption of targeted gas molecules via a molecular sieving effect or thermodynamic differentiation. For example, the gas uptake selectivity of PLs can be improved by tuning the cavity size of the porous host and creating a gating effect, facilitating the separation of gas molecules with similar sizes, such as N2, CO2, CH4, and Xe, and leading to highly selective gas sorption.34,40 The application of PLs has been further extended to separate small organic molecules and solvents. For example, the enantio-resolution of racemic nucleosides41 and separation of isomeric alcohols and chlorofluorocarbons9 have been achieved by type I cyclodextrin- and coordination cage-derived PLs, respectively. The large free volume in PLs could also significantly enhance membrane-based gas separation performance, which exhibited much higher gas solubility and gas transport efficiency than the dense liquid phase.20
Triggered by the promising performance of PLs in gas separation and storage, the application of PLs in catalysis—particularly to integrate the gas storage and catalytic transformation procedure—has been further explored. Structure engineering over flowing liquids, the porous host, and the solid–liquid interphase has been leveraged to advance the catalytic procedures. Progress has been made by using PLs to overcome the poor gas solubility in pure liquids, the mass transfer limitation in gas–solid phase reaction, and the difficulty of integrating multiple active sites possessing different or even incompatible properties in a single phase. The designed PL engineering approach can afford efficient catalytic systems combining the merits of homogeneous and heterogeneous pathways with improved catalytic efficiency and long-term durability.
In this perspective article, the recent progress in the discovery and development of PLs for catalysis applications will be summarized and compared with traditional catalytic systems. The unique features of PLs will be introduced first, particularly the efforts that cannot be achieved by using traditional liquid or solid catalyst-promoted pathways, which make PLs a new frontier in catalysis. Next, advanced characterization techniques deployed to accommodate the PL features will be demonstrated, providing insights into the chemical/textural structure evolution and interactions on the surface/interface, as well as exploring the reaction pathways. Third, the catalytic applications of PLs will be introduced and divided by the reaction categories, which include CO2-involving transformations, O2-involving reactions, H2S conversion, hydrogenation reactions, and non-gas involving cascade reactions (Fig. 1). In these reaction procedures, the design of PLs, synthesis approaches, structure engineering techniques, detailed characterization, catalysis performance evaluation, and reaction mechanism exploration will be discussed, highlighting the structure–performance relationship and the advancements benefiting from the unique features of PLs. Finally, the existing challenges and upcoming opportunities in this emerging and promising research area will be addressed. The insights provided herein could generate opportunities for the discovery and development of high-performance catalytic systems to combine the merits of homogeneous and heterogeneous approaches. This fundamental understanding will provide guidance on how to leverage the unique features of PLs to solve the existing challenges in current catalytic procedures and open up new avenues in this integrated field of gas storage and catalysis.
 | ||
| Fig. 1 Chronology of the development of PLs as a platform to integrate gas storage and catalysis application. | ||
2. Unique features of PLs to integrate gas storage and catalysis
Although the exploration of PLs in catalysis fields is still in its infancy, their unique features and attractive performance surpassing the single liquid or solid components have been demonstrated in diverse catalytic procedures. Compared with the porous host integrating the active sites in the solid state, upon PL formation via liquification, the catalytic procedure will be transformed from heterogeneous to homogeneous, leading to enhanced mass transfer and reaction kinetics. In addition, the properties of the liquid phase, such as ILs, organic solvents, and H2O could be further harnessed to tune the reactivity and selectivity. The potential advantages and unique opportunities provided by PL engineering to enhance the catalytic performance include the following aspects (Fig. 2):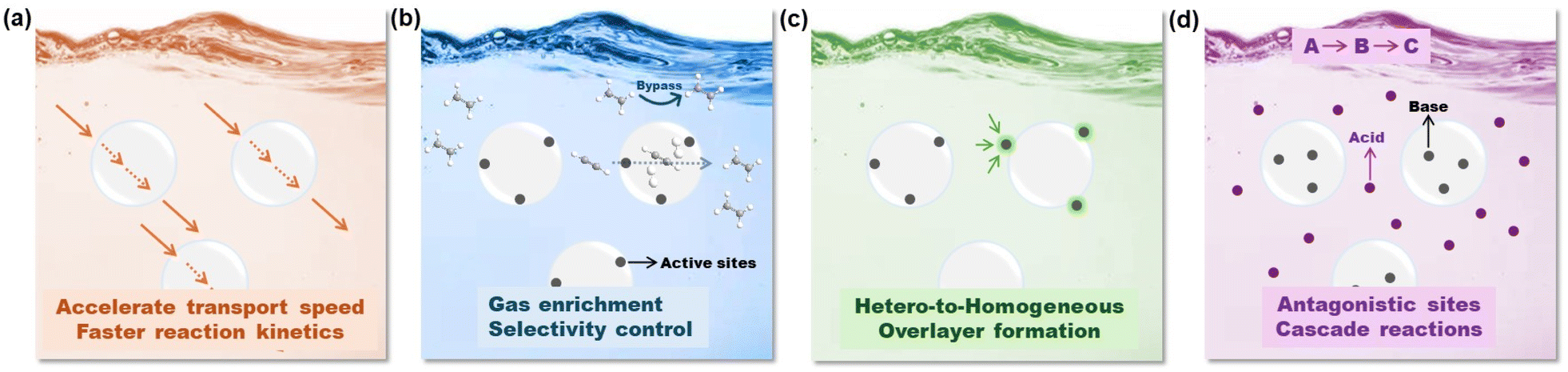 | ||
| Fig. 2 Potential advantages and unique opportunities provided by PL engineering to enhance the catalytic performance. (a) Accelerating the reaction kinetics by providing relatively large free volume. (b) Improving the gas concentration around the catalytic actives sites and tuning reaction selectivity via fine-tuning over the porosity of the porous host. (c) Transforming a heterogeneous catalytic system into a homogeneous one and creating an overlayer on top of the active metal sites to improve their stability. (d) Providing the opportunity to include active sites with antagonistic properties in one system to facilitate cascade reactions. | ||
(1) Tuning the chemical structures of the flowing liquids to stabilize the PL phase especially to enhance gas diffusion (Fig. 2a). The transport of substrates/products in the reaction media plays a critical role in accelerating the reaction kinetics. Compared with the dense liquid phase, by engineering permanent porosity/cavities within the PL media, the free volume will be greatly improved, and correspondingly the diffusion rate, particularly for gas molecules, will be accelerated, leading to improved reaction efficiency. The improved diffusion rate of gas molecules in type I PLs composed of an IL fluid phase and hollow silica sphere host has been demonstrated in membrane-based CO2/N2 separation.42
(2) Enhancing the gas solubility in PLs and stabilizing the catalytic sites by harnessing chemical/textural structure engineering over the porous hosts (Fig. 2b). For gas-involving reaction procedures, i.e., CO2 conversion, hydrogenation reactions (H2), and oxidative reaction processes (O2), the solubility of the gas components in the reaction media will directly affect catalytic efficiency, particularly the concentration of the gas components in the local environment of the active sites.
The construction of PLs provides an alternative solution to enrich the gas substrates around the conversion centres, which in turn could enhance the gas-involving catalytic procedure. In addition, the gating effect of the porous host by porosity size or surface property engineering could be leveraged to sieve the location of different gas molecules and allow selective gas transformation. For example, a porous host with controllable porosity may achieve selective acetylene semihydrogenation within the pores in the presence of a large amount of ethene,43 with the prerequisite of precise catalytic site design.44 The versatile structural engineering of PLs provides the opportunity to achieve enhanced selectivity control in liquid-phase reaction procedures, which are currently mainly feasible in gas-phase reactions, including the semihydrogenation of acetylene to provide high-purity ethene resources.
(3) Leveraging the interface of the porous host/flowing liquid to anchor and stabilize active metal sites via bottom-up or in situ deposition (Fig. 2c). Besides activity and selectivity control, the long-term durability of the catalysts is one of the issues frequently encountered in diverse catalytic procedures. For homogeneous catalysts, the deactivation can be caused by the catalyst separation procedure via a solvent washing or high temperature-involving distillation process to remove the substrates/products. For heterogeneous catalysts, sintering or coking-deduced catalytic performance decay is widely observed in supported noble or transition metal-promoted gas-phase reactions particularly at high temperatures under a reducing or oxidative atmosphere.45 The emergence of PLs provides the opportunity to transform supported catalysts into homogeneous systems. By engineering active metal nanoparticles (NPs) or single atoms (SAs) on the surface or within the porous channels of the host, the integrated porous host components can be liquified via PL construction. The liquid can assist in the dispersion and stabilization of the active metal sites. Beyond this, the liquid phase can act as a protective overlayer and enhance the sintering resistance of the metal sites, via a behaviour similar to strong metal–support interaction (SMSI) in heterogeneous catalyst design.46,47 The extra benefits will provide the possibility to tune the electron density on the metal sites via the metal–liquid phase interaction, leading to altered adsorption strength of the substrate, intermediate, and product as well as whole reaction selectivity variation.
(4) Involving multiple active sites within the liquid and solid compositions in PLs separately towards cascade reaction procedures (Fig. 2d). As PLs have the capability to integrate two separated components in the same medium, this may provide the possibility to include catalytic sites possessing antagonistic properties in the same reaction mixture, i.e., acidic and basic sites. This combination is rarely reported in traditional homogeneous reaction media, as rapid neutralization will take place, leading to complete deactivation. Anchoring acidic and basic sites in the same catalyst can be achieved in heterogeneous catalyst design via protection/deprotection steps, and the density of the active sites is generally low. PLs provide a platform to include high density active sites with antagonistic nature in the same media, for example, by confining the acid sites within the porous host channels and excluding the base sites in the liquid phase. This efficient integration will allow PLs to catalyse cascade reactions particularly those requiring catalysts with diverse properties in each step.
3. Techniques for characterizing the structures of PLs and probing the catalytic procedures
The unique features of PLs with combined solid and liquid features make it challenging to characterize the chemical/textural structures, monitor the structural evolution during the catalysis procedure, and probe the reaction pathways, which requires unique characterization techniques, particularly operando spectroscopy, and X-ray- and neutron-based scattering techniques, assisted by computational simulation.One of the attractive features of PLs is the incorporation of pores in the dense liquid phase. While porosity analysis of porous solid materials can be provided and calculated by gas uptake isotherms being collected at low temperatures (e.g., N2 isotherms at 77 K and CO2 isotherms at 195 K), this approach cannot be used for PLs. Particularly at low temperatures, the liquid phase of PLs will freeze, preventing gas molecules from penetrating the cavities and providing porosity information. Instead, characterization techniques capable of providing porosity information via evaluation under ambient conditions are preferred. Positron annihilation lifetime spectroscopy (PALS) could be deployed to provide information on the inherent cavities, particularly via the comparison with the pure liquid phase. PALS is a non-destructive, in situ pore/void characterization technique used to probe the microstructure of materials by measuring the lifetime of a positron, which can be correlated to the average size and concentration of the void space in materials determined by the relationship of the positron lifetime and vacancies, voids and defects in the material (Fig. 3a).10,52–54
 | ||
Fig. 3 (a) Positron annihilation lifetime of PEGS, PILs-PEGS and HCS-liquid collected at 298 K. Reproduced with permission.10 Copyright 2017, Wiley-VCH. (b) 129Xe NMR spectra for Xe gas in CC33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :133-R and CC15-R PLs compared to those in neat hexachloropropene. Reproduced with permission.40 Copyright 2020, Wiley-VCH. (c) SAXS scattering patterns of PS-OS@SiNR liquid samples prepared from SiNRs with aspect ratios. Reproduced with permission.48 Copyright 2019, The Royal Society of Chemistry. (d) WAXS scattering patterns of the linear-chained ionic liquids. Reproduced with permission.49 Copyright 2023, The Royal Society of Chemistry. (e) Neutron pair distribution function (NPDF) of deuterated sample NIL n–4 at 300 K. Reproduced with permission.50 Copyright 2016, American Chemical Society. (f) SANS profiles of dry MOF-919 and MOF-919 in the contrast matching point of a 50% D2O and 50% H2O mixture (wet). Reproduced with permission.51 Copyright 2023, The Authors, published by Springer Nature. :133-R and CC15-R PLs compared to those in neat hexachloropropene. Reproduced with permission.40 Copyright 2020, Wiley-VCH. (c) SAXS scattering patterns of PS-OS@SiNR liquid samples prepared from SiNRs with aspect ratios. Reproduced with permission.48 Copyright 2019, The Royal Society of Chemistry. (d) WAXS scattering patterns of the linear-chained ionic liquids. Reproduced with permission.49 Copyright 2023, The Royal Society of Chemistry. (e) Neutron pair distribution function (NPDF) of deuterated sample NIL n–4 at 300 K. Reproduced with permission.50 Copyright 2016, American Chemical Society. (f) SANS profiles of dry MOF-919 and MOF-919 in the contrast matching point of a 50% D2O and 50% H2O mixture (wet). Reproduced with permission.51 Copyright 2023, The Authors, published by Springer Nature. | ||
Although gas uptake at ultra-low temperatures cannot be deployed for porosity structure analysis of PLs, the gas uptake evaluation under ambient conditions can provide information on the capacity and selectivity of the targeted gas sorption performance by PLs. In addition, the pressure-swing and temperature-swing gas uptake isotherms can be utilized to probe the retention of the free volume in PLs by comparison with the porous host and the liquid phase. Further, the interaction strength between gas molecules and the PL phase can be predicted via thermodynamic calculations. For example, the gas uptake evaluation using CO2,18,34,55,56 CH4,37,57 O2,23 N2 (ref. 20) etc. as the probe molecules could demonstrate the benefits of PLs in gas storage in terms of capacity and selectivity, providing guidance on the structure–performance relationship in gas-involving reactions.
129Xe operando nuclear magnetic resonance (NMR) spectroscopy is another technique capable of providing information on the relative free volume of PLs. Xe nuclei are highly sensitive to the local chemical environment. Compared with the Xe in the dense liquid phase, upon PL formation and the introduction of extra cavities, the Xe species are transformed from a confined (in pure liquid) state to a more free environment (in PL), leading to chemical shift variations. The chemical shift difference can be utilized as an indicator of free volume in PLs (Fig. 3b).40
For PLs containing porous hosts with crystalline and ordered structures, X-ray scattering techniques, including small-angle X-ray scattering (SAXS) (Fig. 3c),48 wide-angle X-ray scattering (WAXS) (Fig. 3d),49 and pair distribution function (PDF) (Fig. 3e)50 techniques can be deployed to provide structural information about the porous host and liquid canopy in PLs, focusing on the short-, intermediate-, and long-range order associated with adjacency correlations between neighbouring atoms, charge alternation and polar/non-polar domains. Correspondingly, neutron-based techniques58 such as small angle neutron scattering (SANS) (Fig. 3f)51 and neutron-PDF are highly sensitive to light elements, which is particularly helpful for PLs composed of organic porous hosts such as organic cages, COFs, and carbon spheres. Besides the ex situ analysis, the scattering curves collected in situ as a function of pressure and temperature under specific gas atmospheres, such as CO2, N2, CO and CH4, can provide information on the structural evolution of PLs upon exposure to the gas molecules. Neutron spin echo (NSE)59 could be used to examine the dynamics of the liquid matrix at the interface, measuring slow dynamical processes such as molecular motion on the nanoscale, and is particularly suited to measure the motion of the liquid as a function of gas uptake and liquid composition.
Furthermore, operando characterization techniques, such as in situ Fourier-transform infrared spectroscopy (FTIR) under temperature and pressure-swing procedures in the presence of a determined gas atmosphere,60,61operando NMR particularly by deploying isotope-labeled PLs, the dosing gas molecules, other reaction substrates,62 and in situ X-ray photoelectron spectroscopy (XPS)63–65 under a determined gaseous atmosphere are powerful tools to study the active sites, reactive intermediates, reaction pathways and reaction kinetics.
Notably, theoretical simulations34,56,57,66 could provide information on the assembly of liquid and solid counterparts, functionality interaction at the solid–liquid interface, guest molecule interaction, reaction pathway prediction, and thermodynamic/kinetic simulation. Together with the experimental characterization, computational chemistry can be leveraged to understand the structure-catalytic performance relationship, particularly in comparison with the pure solid or liquid counterpart.
4. Applications of PLs in catalysis
The combination of permanent porosity, multiple active sites, and liquification of the scaffolds makes PLs unique systems in catalysis applications. In addition, the large tuning space in both the porous host and the liquid component provides diverse opportunities to improve the catalytic performance of PLs in certain reaction procedures. On the premise that porosity ensures good mass transfer, the retentive liquid fluidity of PLs improves the contact between precursors and active sites, even transforming heterogeneous reactions into homogeneous procedures. These unique properties drive researchers to explore the capability of PLs in the field of catalysis and to extend the merits of PLs in gas storage to gas molecule-involving transformations.The intrinsic pores of PLs could serve as soft cylinders well suited for gas storage, and subsequent chemical conversion catalysed by multiple active sites on the host leads to the production of value-added chemicals. PLs being synthesized via facile physical mixing of porous materials with ILs, surface ionization, surface rigidity tuning, surface deposition, etc. have been deployed to improve gas capture and storage and facilitate mass transfer during the catalytic procedure. These synthesis methodologies can not only ensure good solubility of porous materials in the liquid phase but also fully expose the catalytic sites (Table 1).
| PLs | Type | Synthesis approach | Porous hosts | Liquid phase | Reaction process | Ref. |
|---|---|---|---|---|---|---|
| Im-UiO-PL | Type I | Surface ionization | MOF | — |
|
67 |
| PIL-CF3-Py-RhMOP, PIL-diz-RhMOP | Type II | Surface rigidity modification | MOP | IL | 68 | |
| PoIL | Type III | Physical mixing | MOF | IL |
|
69 |
| Im-PL-Cage | Type I | In situ coordination | MOC | — |
|
63 |
| MCM22-Ag in [M2070H]Br | Type III | Surface deposition | Zeolite | IL |
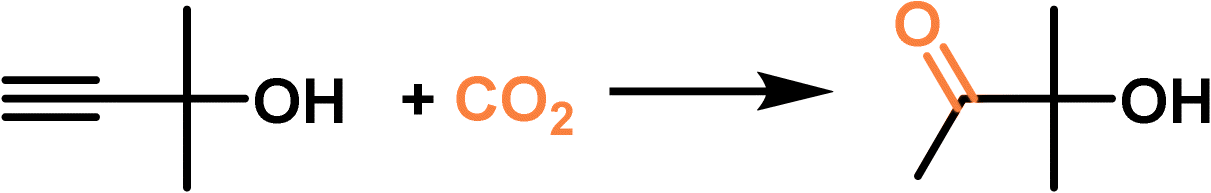
|
70 |
| CdS/NH2-UIO-66 PL | Type III | Surface modification | MOF | IL |
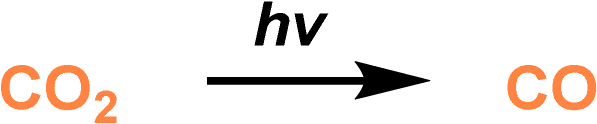
|
71 |
| PoLi | Type I | Surface modification | Hollow silica | — |

|
72 |
| TMH/BDM/PMD-UiO-PL | Type I | Surface modification | MOF | — |
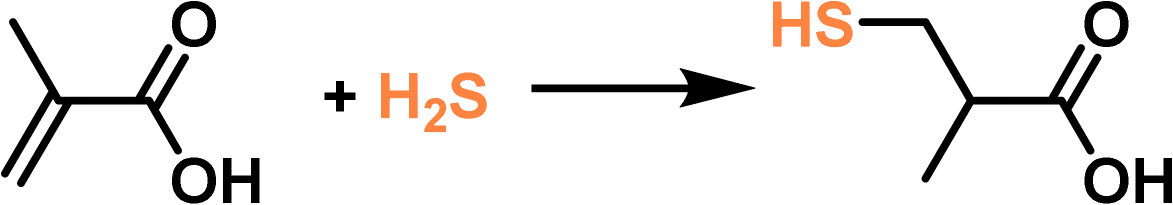
|
73 |
| Silicalite-1 aqueous | Type III | Physical mixing | Silicalite | Water |
|
24 |
| Pt@HS-SiO2 PL | Type I | Surface modification | Hollow silica | — |
|
74 |
| Pd NPs@COC/g-C3N4 | Type III | Solubilization | COC | Water |

|
75 |
| NHC–Pd@SiO2 PL | Type I | Surface modification | Hollow silica | — |
|
76 |
| MCM-22-PL | Type III | Physical mixing | Zeolite | IL |
|
61 |
| HPMo@ZIF-8-PIL | Type III | Physical mixing | MOF | IL |
|
77 |
| UiO-66-T3PILs | Type III | Physical mixing | MOF | IL | 78 | |
| UiO-66 | Type III | Physical mixing | MOF | IL | 79 |
4.1 PL-promoted CO2 conversion
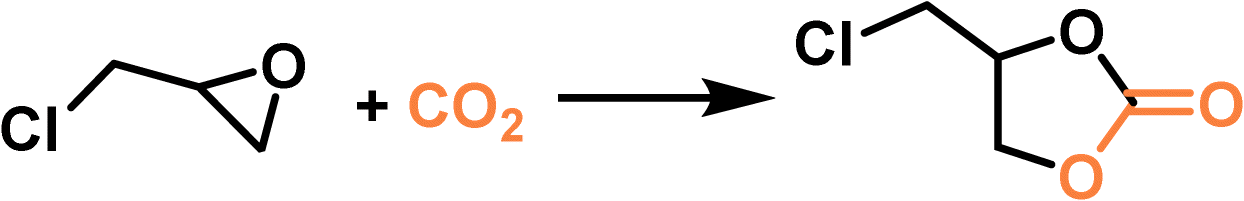
PLs with engineered structures have the potential to act as soft cylinders, that is, they can store gas in a frozen state (at low temperature) and release gas in the liquid state (at temperatures higher than the melting point). As a proof of concept, a MOF-derived PL (denoted as Im-UiO-PL) has been designed and synthesized via a surface ionization method for this purpose with the cycloaddition reaction of CO2 with epoxides as a probe reaction.67 A cationic MOF (Deim-UiO-66) was constructed by introducing imidazolium cations with long alkyl chains coupled with Cl− anions into the ligands of the MOF architecture. Then the anionic poly(ethylene glycol)-tailed sulfonate (PEGS) canopy was modified on the MOF surface through ion exchange with Cl− to liquify the cationic MOF porous host (Fig. 4a). The uniform distribution of the MOF core in the liquid phase was confirmed by energy-dispersive X-ray spectroscopy (EDS) of Zr element (on the MOF host) and S element (on the PEGS anion). Through density functional theory (DFT) calculations, it was determined that the size of PEGS is large enough to prevent its penetration through the cavities of Deim-UiO-66 possessing a surface area of 103 m2 g−1 and pore size of 8 and 11 Å being calculated using the Horvath–Kawazoe method. Gravimetric gas solubility measurements demonstrated that the obtained Im-UiO-PL showed excellent adsorption capability for CO2 (5.93 mmol g−1 at 9 bar), which was approximately 14 times larger than that of pure PEGS (0.43 mmol g−1) (Fig. 4b). The enhanced CO2 adsorption capacity confirms the presence of permanent porosity in this MOF-based PL. Differential scanning calorimetry (DSC) analysis revealed the melting temperature of 28 °C and crystallization temperature of −6 °C for Im-UiO-PL. The phase changing behaviour was then leveraged to make the Im-UiO-PL a soft cylinder for CO2 storage and conversion. The CO2 sorption procedure was conducted under 20 bar CO2 pressure and then slowly released back to atmospheric pressure after saturation. Then epichlorohydrin (substrate) and tetrabutyl-ammonium bromide (TBAB, catalyst) were added, and the mixture was heated to 120 °C to allow the cycloaddition reaction between epoxide and CO2 released from the Im-UiO-PL. During the reaction procedure, the stored CO2 in the PL was slowly released and efficiently utilized to synthesize cyclic carbonate without introducing an extra CO2 source (Fig. 4c). It turned out that in an identical gas storage and conversion procedure, 79.6% yield of chloropropene carbonate was achieved by the Im-UiO-PL sorbent, which was much higher than that achieved by PEGS (liquid phase, 20.9%), Deim-UiO-66 (porous host, 19.7%), and their physical mixture (25.9%). The enhanced catalytic performance in PL was provided by the inherent porosity and fluidity of the system ensuring high CO2 storage capacity and effective mass and heat transfer.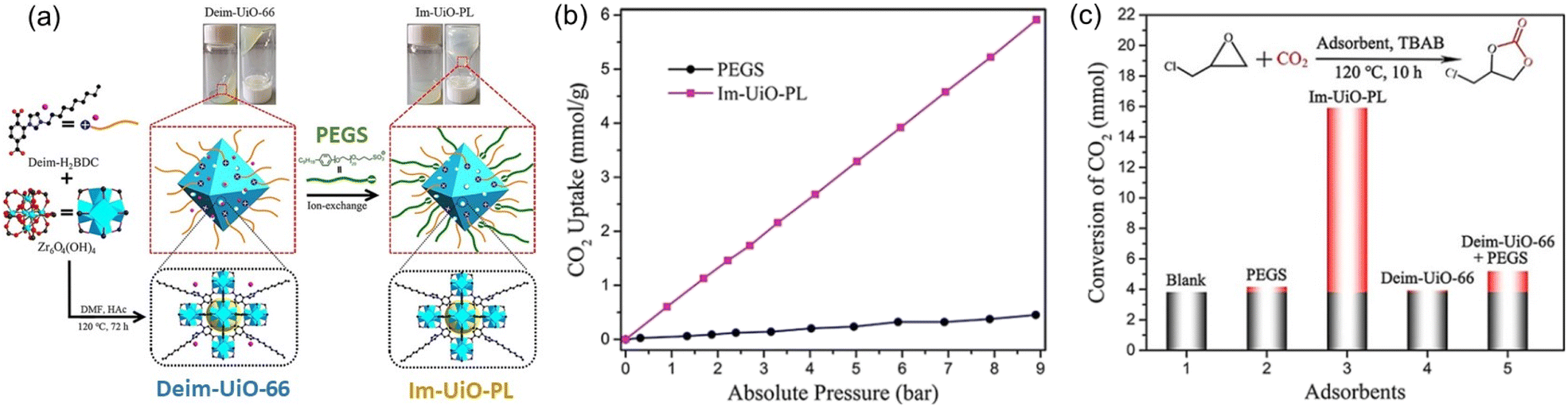 | ||
| Fig. 4 (a) The synthesis approach of Im-UiO-PL via surface modification and ionic exchange. (b) The CO2 uptake isotherms of PEGS and Im-UiO-PL at 298 K with CO2 pressure up to 9 bar. (c) Cycloaddition reaction of epichlorohydrin with CO2 stored in Im-UiO-PL and the separated liquid or solid counterparts in the presence of TBAB. (a–c) Reproduced with permission.67 Copyright 2021, Wiley-VCH. | ||
By integrating the catalytic sites within the porous host or liquid counterpart of PL, CO2 conversion can be performed without adding extra catalysts. A structure engineering approach was developed to convert PL composed of rhodium(II)-based metal–organic polyhedra (RhMOP) and imidazolium IL (IL-Br) from type III to type II (Fig. 5a).68 By introducing additional coordinating reagent, 1-dodecylimidazole (diz) or 4-(trifluoromethyl)pyridine (CF3-Py), to RhMOP, the surface rigidity of the porous host was improved, transforming insoluble RhMOP into a surface modified MOP that can be dissolved in the size-excluded IL-Br. The type III nature of the initial PL composed of RhMOP and IL-Br was confirmed by dynamic light scattering (DLS) and scanning electron microscopy (SEM), demonstrating the presence of MOP particles around 15–30 nm in size. Molecular dynamics (MD) simulations indicated that the bulky IL-Br cannot enter and block the inner cavity of RhMOP, and the interaction energy between IL-Br and RhMOP was weak (ΔE = 68.2 kcal mol−1). Comparatively, after grafting N-donor molecules (diz and CF3-Py) onto the open metal sites of RhMOP (Fig. 5a), the as-obtained MOPs became soluble in the bulky IL-Br phase, leading to the formation of type II PLs. MD simulation revealed the increased interaction energy between IL-Br and the CF3-Py-RhMOP (ΔE = 113.9 kcal mol−1), which was further illustrated by the chemical shift of the characteristic peaks of IL-Br and CF3-Py-RhMOP in NMR spectra.
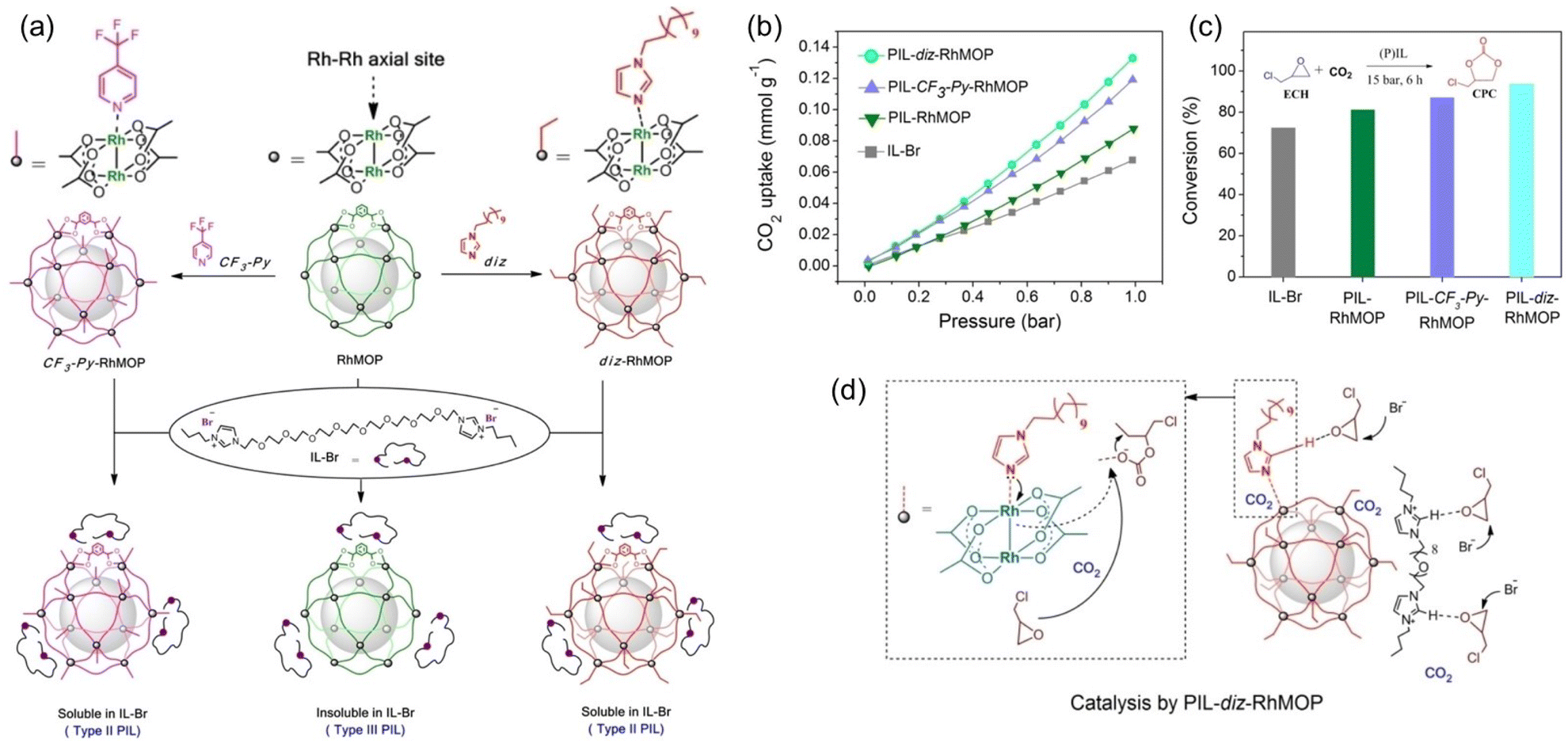 | ||
| Fig. 5 (a) The strategy to transform a type II PL (PIL-RhMOP) into a type III PL via surface rigidity modification by introducing an N-donor (PIL-CF3-Py-RhMOP and PIL-diz-RhMOP). (b) The CO2 uptake isotherms (at 273 K) and (c) catalytic performance of IL-Br, PIL-RhMOP, PIL-CF3-Py-RhMOP, and PIL-diz-RhMOP in the cycloaddition reaction of CO2 with epichlorohydrin (75 °C, CO2 pressure 15 bar, 6 h). (d) The proposed reaction mechanism catalysed by PIL-diz-RhMOP. (a-d) Reproduced with permission.68 Copyright 2023, Wiley-VCH. | ||
The CO2 uptake evaluation revealed that a higher CO2 sorption capacity was achieved by the type II PL, PIL-diz- (0.133 mmol g−1) and PIL-CF3-Py-RhMOP (0.121 mmol g−1), and not by the type III PIL-RhMOP (0.088 mmol g−1) and neat IL-Br (0.068 mmol g−1) at 273 K and CO2 pressure of 1 bar (Fig. 5b). The cycloaddition reaction of CO2 with epichlorohydrin was performed to demonstrate the benefits of using PLs in gas–liquid phase reactions. As the IL-Br can catalyse cyclic carbonate formation, no additional catalysts were required. At 75 °C and CO2 pressure of 15 bar within 6 h, the catalytic activity was decreased in the order of IL-Br < PIL-RhMOP < PIL-CF3-Py-RhMOP < PIL-diz-RhMOP, which was the same trend as the changes in CO2 uptake capacity (Fig. 5c), emphasizing the critical role of high free volume in promoting the CO2 transformation.
As shown in the plausible catalytic mechanism (Fig. 5d), the imidazolium cations can activate the epoxide via hydrogen-bond formation, and the Br-anions promote the ring opening via nucleophilic attack. In PLs, the Rh–Rh unit in the coordination cage can further activate the epoxide substrate and the additional cavities endow the systems with improved CO2 solubility. In addition, the post-grafted N-donor molecules contain additional sites (imidazole group) to stabilize the intermediates, further promoting catalytic performance.
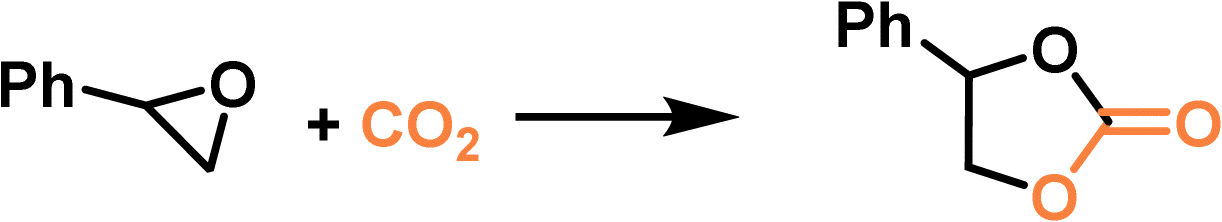
The type III PL composed of a MOF and IL with halide anions formed by physical mixing also exhibited catalytic activity and selectivity improvement for the cycloaddition reaction of CO2 with styrene oxide.69 By combining alkyl-phosphonium ILs with ZIF-8, different PLs were constructed, and the existence of porosity in PLs was demonstrated by the increased CO2 adsorption capacity compared with that of the pure IL. Under identical reaction conditions, PLs exhibited higher catalytic activity and selectivity towards cyclic carbonate production against pure ILs. Dispersing MOFs in ILs synergistically increased gas solubility and improved mass transport efficiency to facilitate CO2 transformation.
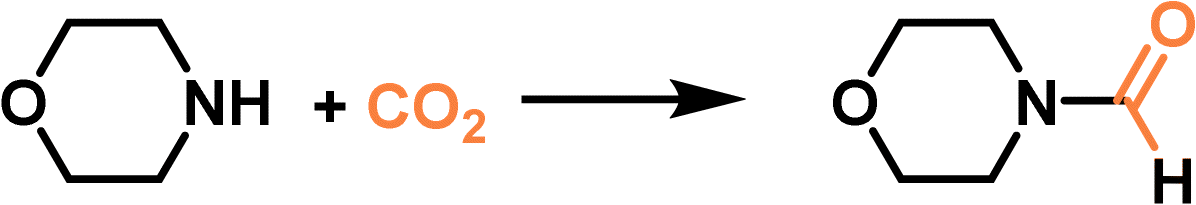
Compared with type II and III PLs, type I PLs have no boundary between the solid host and the external liquid phase, leading to the formation of a single fluid phase. An in situ coordination approach was developed to afford a type I PL from a PEG-imidazolium chain functional benzene dicarboxylic acid linker, zinc salts, and sulfonylcalix[4]arene (Im-PL-Cage). The as-formed rigid metal–organic cage (MOC) architecture afforded permanent porosity in the PL system (Fig. 6a).63 The successful construction of the as-designed structure was confirmed by spectroscopy- and X-ray-based techniques. For example, both Im-PL-Cage and the Zn-cage being obtained in the solid state (without the long PEG chain on the carboxylic acid linker) exhibited the presence of imidazolium N+ species (by XPS) and Zn2+ sites (by Synchrotron-based X-ray absorption spectroscopy (XAS)). MD simulations revealed the existence of 4–6 Å pores in Im-PL-Cage, which can be easily penetrated by CO2 and effectively block bulky PEG-imidazolium chains. PALS analysis over Im-PL-Cage derived a void diameter of around 9.34 Å. Benefiting from the high free volume in Im-PL-Cage, enhanced CO2 uptake capacity of 1.78 mmol g−1 at 298 K and a CO2 pressure of 10 bar were achieved (Fig. 6b), compared with the pure liquid PEG-Im-H2BDC component (0.31 mmol g−1). The formylation reaction of amine in the presence of hydrosilane was selected as a probe reaction using the stored CO2 as a gas resource. The Im-PL-Cage and other control samples were saturated with CO2 first at high pressure (20 bar), then external CO2 was released. After adding morpholine (substrate) and solvent, the reaction mixture was heated to 60 °C allowing the release of the stored CO2 and occurrence of the formylation reaction. Notably, under identical conditions, a much higher yield of the N-formylmorpholine product was obtained by Im-PL-Cage (96.7%) than that of the pure liquid component PEG-Im-H2BDC (21.5%), the porous Zn-Cage solid (24.5%), and the physical mixture of Zn-Cage and PEG-Im-H2BDC without strong surface interaction (31.9%) (Fig. 6c). The major reason for this difference lies in the presence of permanent pores in Im-PL-Cage, leading to much higher CO2 storage capacity during the saturation procedure. For the solid Zn-Cage, most of the CO2 adsorbed in the cavities could be quickly released during the desorption process due to the lack of the dense liquid wall. It is worth noting that Im-PL-Cage not only had the advantages of homogeneous catalytic systems such as good solubility and mass transfer, but also showed good recyclability. Stable assembly was well maintained and an N-formylmorpholine yield of 93% was obtained after cycling for five runs.
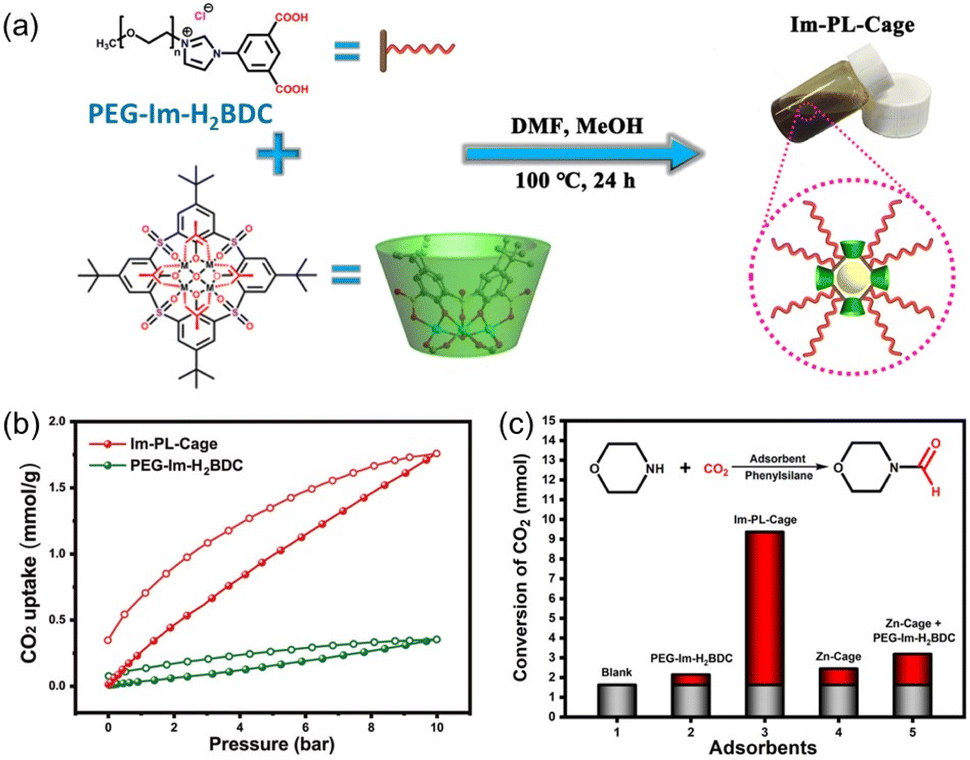 | ||
| Fig. 6 (a) Schematic diagram for the preparation of Im-PL-Cage via coordination between the PEG-Im-H2BDC linker, zinc salts, and calixarene. (b) The CO2 uptake isotherms of Im-PL-Cage and the pure liquid PEG-Im-H2BDC linker at 298 K. (c) The formylation reaction of morpholine in the presence of phenylsilane using stored CO2 as the resource at 60 °C. (a–c) Reproduced with permission.63 Copyright 2023, The Authors, published by Springer Nature. | ||
The catalytic active sites can be homogeneously coated on the porous host via in situ surface deposition during the PL synthesis procedure.70 A zeolite with nanosheet morphology (MCM-22) and Na+ cations within the scaffolds was used as the precursor, which was treated by cation exchange to afford MCM22-Ag containing Ag+ cations. Then, an IL composed of ammonium cations and bromide anions ([M2070]Br) was deployed as the liquid phase (Fig. 7a and b). Upon mixing MCM22-Ag and [M2070]Br, ultra-small AgBr species were formed and uniformly distributed on the surface of the porous host, leading to the formation of an ultra-stable type III PL. The crystallinity of the MCM-22-Ag was well maintained and confirmed by X-ray-based techniques. The dynamic rheological analysis within a wide range of oscillation strain, angular frequency, and temperature revealed the good fluidity of the as-synthesized PLs. Compared with the pure IL [M2020H]Br having a CO2 uptake capacity of 1.94 wt% at 298 K and CO2 pressure of 10 bar, the PL containing 40% zeolite achieved a CO2 uptake capacity of 3.48 wt%, indicating a much higher free volume existed in the PL system. The 129Xe NMR technique was further deployed to characterize the existence of cavities in the PL. The Xe peak was shifted from −5165.3 ppm in the pure IL phase [M2070H]Br to −5180.9 ppm in the PL containing zeolite, indicating that the local environment around the Xe gas molecules is less confined in PL (Fig. 7c). The surface deposition approach can be extended to other ILs containing bromide anions (e.g., tetradecyltrihexylphosphonium bromide, [P66614]Br). By exchanging the Na+ in MCM-22 with Ba2+, a PL can be constructed by mixing with sulfonate-derived ILs ([P66614]2[SO4]) and BaSO4 was uniformly coated on the surface of the zeolite host, leading to the formation of a stable type III PL.
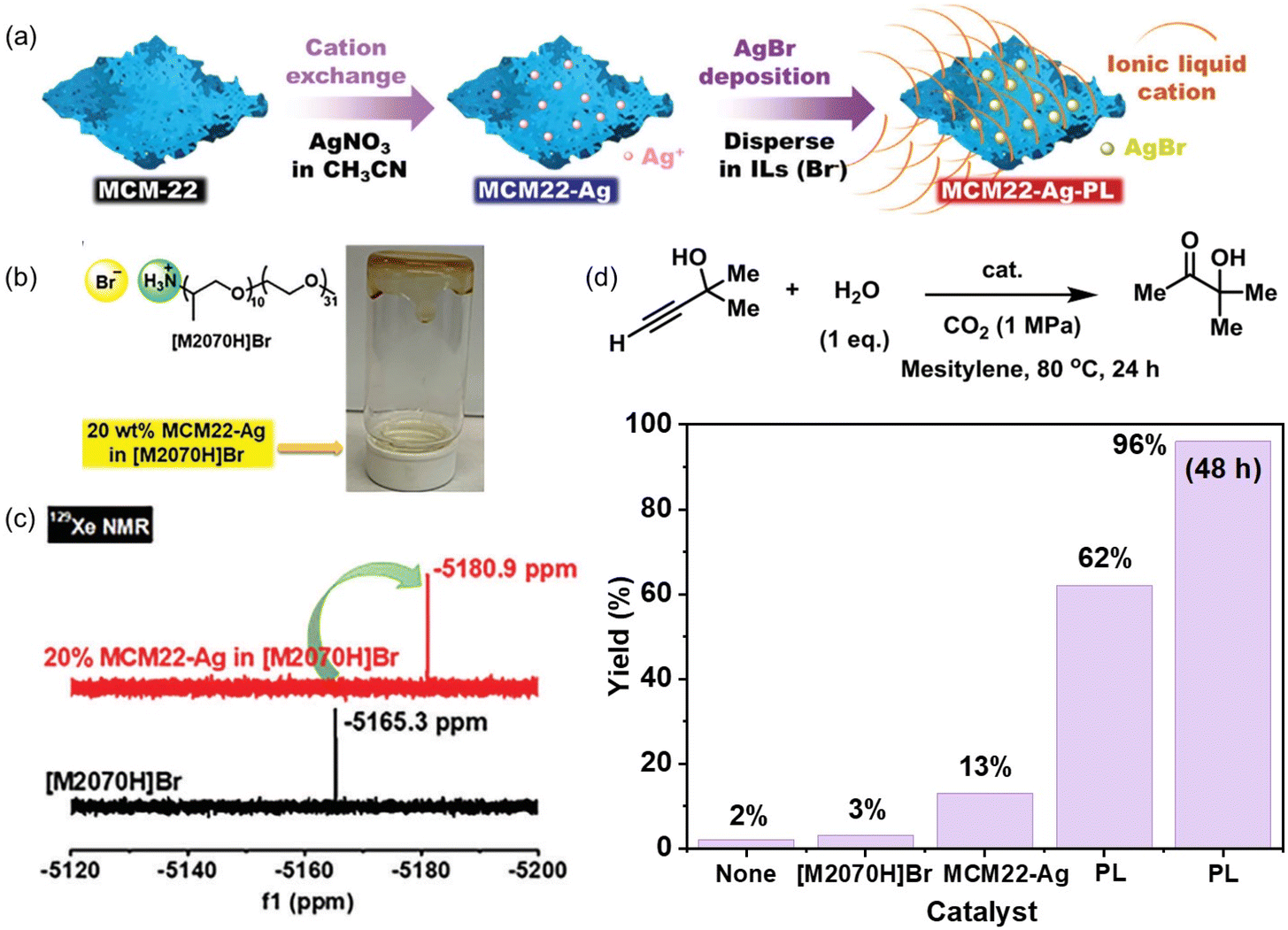 | ||
| Fig. 7 (a) Synthesis of type III PLs via surface deposition using Ag+ containing zeolite nanosheets and ILs containing bromide anions. (b) Structure of the IL [M2070H]Br and image of the as-obtained PL. (c) 129Xe NMR spectra of the pure liquid phase [M2070H]Br and the PL composed of 20% MCM22-Ag in [M2070H]Br. (d) Hydration reaction of 2-methyl-3-butyn-2-ol in the presence of CO2 and H2O catalysed by [M2070H]Br, MCM22-Ag, and the as-obtained PL composed of 20%MCM-Ag in [M2070H]Br. (a–d) Reproduced with permission.70 Copyright 2023, Wiley-VCH. | ||
The as-derived PL was leveraged to catalyse the propargylic alcohol hydration reaction taking 2-methyl-3-butyn-2-ol as the substrate (Fig. 7d), which involved a cascade procedure including a cycloaddition reaction of CO2 with the substrate and the hydrolysis of the formed cyclic carbonate intermediate.80 In the as-formed type III PL, the surface deposited Ag+ species can activate the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond via coordination, and the bromide anion can promote the CO2 insertion and hydrolysis step, making them promising candidates in the hydration reaction of propargylic alcohol. The catalytic performance evaluation was performed under a CO2 pressure of 1 MPa at 80 °C within 24 h, in which 62% yield of the 3-hydroxy-3-methyl-2-butanone product was obtained by PL being synthesized from MCM22-Ag and [M2070H][Br], while the zeolite MCM22-Ag only afforded a yield of 13% under identical conditions. A quantitative yield of the hydration product was obtained by PL within 48 h. The higher catalytic activity of the PL was facilitated by the homogeneous nature, high CO2 solubility, and improved gas–liquid–solid mass transport efficiency.
C bond via coordination, and the bromide anion can promote the CO2 insertion and hydrolysis step, making them promising candidates in the hydration reaction of propargylic alcohol. The catalytic performance evaluation was performed under a CO2 pressure of 1 MPa at 80 °C within 24 h, in which 62% yield of the 3-hydroxy-3-methyl-2-butanone product was obtained by PL being synthesized from MCM22-Ag and [M2070H][Br], while the zeolite MCM22-Ag only afforded a yield of 13% under identical conditions. A quantitative yield of the hydration product was obtained by PL within 48 h. The higher catalytic activity of the PL was facilitated by the homogeneous nature, high CO2 solubility, and improved gas–liquid–solid mass transport efficiency.
CO2 photoreduction represents an attractive approach to fulfill CO2 transformation to value-added chemicals using clean solar energy.81 A PL design strategy was demonstrated to facilitate the CO2-to-CO conversion via photoirradiation via engineering the active sites on the surface of the porous host and tuning the electronic properties of the active sites via amide bond formation.71 A heterostructure composed of CdS NPs on the surface of a MOF containing –NH2 on the linker was deployed as the porous host (CdS/NH2-UIO-66), and an IL containing ammonium cations and carboxylate anions was used as the fluid phase ([M2070][IPA]) (Fig. 8a). The PL was obtained upon mixing and thermal treatment of the solid and liquid counterparts and the ionic pairs were modified on the surface of the MOF core via amide bond formation, as revealed by the FTIR and XPS spectra. Theoretical calculation demonstrated that the IL molecule with dimensions of 24, 18, and 20 Å cannot enter the micropores of the MOF with the size of around 6 Å (Fig. 8b). The well-maintained cavity in the CdS/NH2-UIO-66 PL was further illustrated by the improved CO2 uptake capacity up to 4 bar compared with the pure IL ([M2070][IPA]). Control experiments revealed that coating the IL moieties on the surface of CdS/NH2-UIO-66 improved the hydrophobicity via water-contact angle analysis and stability in acetonitrile (Fig. 8c).
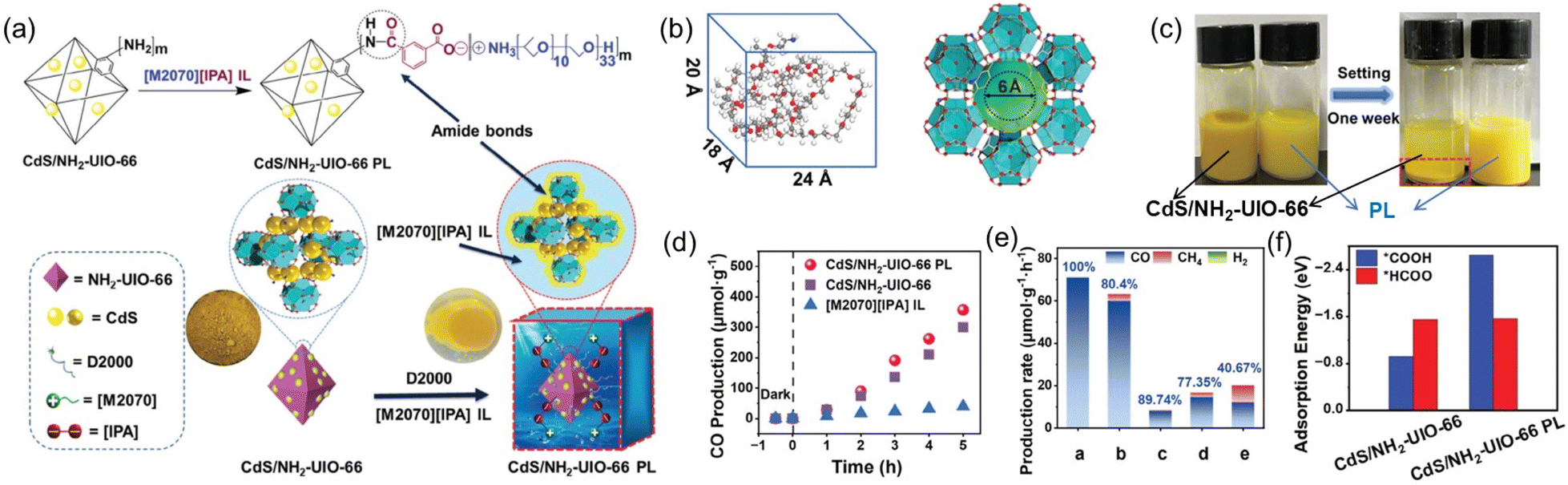 | ||
| Fig. 8 (a) Synthesis of CdS/NH2-UIO-66 PL via surface modification and amide bond formation between CdS/NH2-UIO-66 and IL [M2070][IPA]. (b) Size of the IL [M2070][IPA] (left) and the porous host (right). (c) Comparison of the stability of CdS/NH2-UIO-66 and the PL in acetonitrile. (d) The CO2 photoreduction performance within 5 h. (e) The CO evolution rate achieved by different catalysts. a: PL; b: CdS/NH2-UIO-66; c: IL; d: physical mixture of b and c; e: PL from IL and CdS/UIO-66 without amide bond formation. (f) Adsorption energy of different intermediates from theoretical simulation. (a–f) Reproduced with permission.71 Copyright 2024, Wiley-VCH. | ||
CO2 photoreduction analysis was conducted using triethanolamine (TEOA) as the sacrificial reagent and acetonitrile as the solvent. The CdS/NH2-UIO-66 PL exhibited much higher CO evolution amount and selectivity (356.86 μmol g−1 and 100%) than the pure IL (39.53 μmol g−1, 89.74%) and the porous host CdS/NH2-UIO-66 (300.04 μmol g−1, 80.4%) (Fig. 8d and e). Detailed studies via in situ FT-IR combined with theoretical calculations revealed that the reaction pathway was altered via surface modification over the porous host. For the MOF with active sites (CdS/NH2-UIO-66), the *HCOO intermediate-involving reaction pathway dominated, and besides CO, the intermediate was beneficial to the production of other reducing products such as CH4. Meanwhile, in the presence of amide bonds on the surface of CdS/NH2-UIO-66 in PL, the Zr4+ are more electron-rich, leading to enhanced adsorption strength of the *COOH intermediate, and the *COOH-involving pathway was more conducive to CO generation (Fig. 8f).
Composite PLs containing enzyme components could catalyse CO2 conversion into utility chemicals.72 The as-developed composite possessed two core–shell conjugates, hollow silica nanorod-based PL (PoLi) and carbonic anhydrase enzyme (bCA), both of which were modified by a polymer surfactant on the surface (Fig. 9a). The PL was formed by treating phosphate buffer, lyophilization, and heating. The cavity generated from the hollow silica host can store CO2 with a capacity of 5.5 cm3 g−1 at 273 K and a CO2 pressure of 1 bar, while the carbonic anhydrase enzyme can promote the in situ enzymatic hydration of CO2 to form HCO3− ions. By involving Ca2+ species into the PL system, CaCO3 precipitation was detected, which was revealed by spectroscopy, microscopy, and X-ray diffraction monitoring (Fig. 9b). The CO2-to-utility chemical transformation was facilitated by the large free volume of the PL, ensuring high CO2 solubility and mass transfer during the reaction procedure. The enzyme sites were uniformly distributed and stabilized in the PL phase to ensure stable and continuous CO2-to-CaCO3 transformation.
 | ||
| Fig. 9 (a) Synthesis procedure of PL composed of hollow silica nanorods and carbonic anhydrase enzyme. (b) Reaction mechanism of CO2 sequestration, storage, releasing, conversion, and transformation to CaCO3 in PL PoLi-bCA/CaCl2. (a and b) Reproduced with permission.72 Copyright 2021, Wiley-VCH. | ||
4.2 PLs in H2S capture and conversion
PLs have shown the capability to remove trace amounts of gas impurities such as H2S for natural gas (e.g., CH4) purification and convert the captured H2S into value-added chemicals.73 The key lies in introducing H2S-philic functionalities (e.g., tertiary amines) on the surface of the porous host. Current existing technologies for the purification of natural gas, removal of trace amounts of CO2 and H2S from large amounts of CH4, rely on liquid sorbents such as amine solutions, possessing the inherent limitations of volatility, corrosivity, and inferior thermal/chemical stability. Deploying porous solid sorbents (e.g., zeolites, MOFs, COFs, etc.) modified by targeted gas-philic moieties could improve the stability but cannot accommodate existing continuous flow separation infrastructures and involve tedious synthesis procedures.82–85 In addition, how to deal with the captured waste gases such as the highly hazardous H2S remains an arduous challenge.With these considerations, a type I PL was developed to integrate the gas impurity removal and its catalytic transformation. UiO-66-OH with abundant hydroxyl groups on the surface and a surface area of 1044 m2 g−1 was deployed as the porous host. Then an IL containing siloxane and tertiary amine groups was grafted onto the surface of UiO-66-OH via O–Si bond formation. Anion exchange with PEGS led to the formation of the targeted type I PLs (Fig. 10a). The crystallinity of the porous host was well-maintained in the PL phase, as confirmed by the PXRD patterns. The particle size of PLs being dispersed in methanol was around 260 nm, as obtained by dynamic light scattering (DLS) measurement.
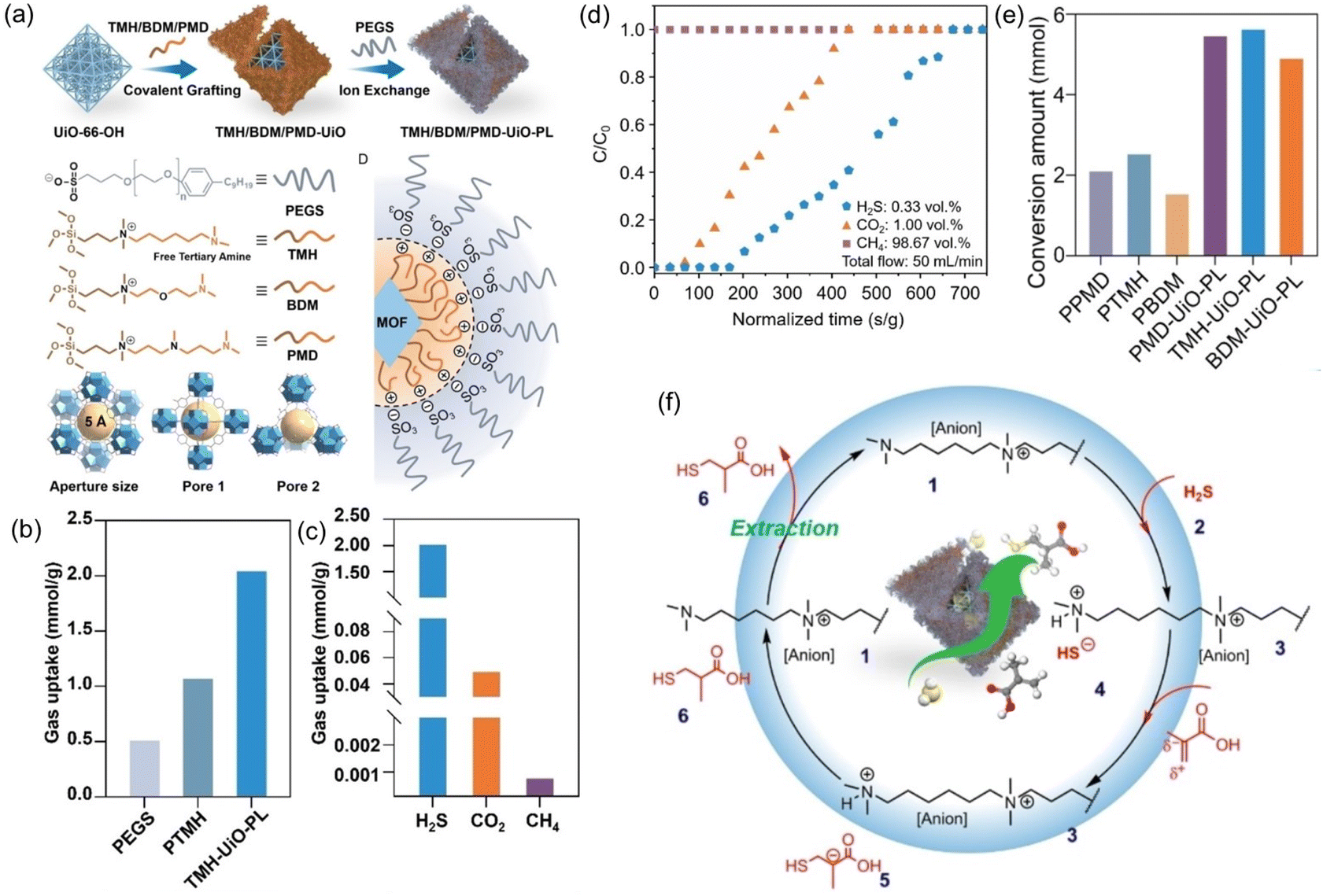 | ||
| Fig. 10 (a) Synthesis procedure of type I PL with a tertiary amine-modified MOF as the porous host and PEGS and anions. (b) The H2S uptake capacity of PL (TMH-UiO-PL), IL (PTMH), and PEGS at 303 K. (c) Comparison of the H2S, CO2, and CH4 uptake capacity obtained by TMH-UiO-PL. (d) Breakthrough evaluation of TMH-UiO-PL using H2S:CO2:CH4 (volume ratio of 0.33:1.00:98.67) as the feed gas at 313 K and 1 bar. (e) Catalytic performance of PLs and the corresponding ILs in the reaction of H2S with α-methacrylic acid to produce 3-mercaptoisobutyric acid. (f) The proposed reaction mechanism based on theoretical simulation. (a–f) Reproduced with permission.73 Copyright 2023, Wiley-VCH. | ||
The H2S adsorption was evaluated at 303 K and 1 bar pressure, in which the uptake capacity of the PL (2.04 mmol g−1) was much higher than that of the IL (1.07 mmol g−1) and the anion component PEGS (0.51 mmol g−1) (Fig. 10b). In addition, the PL exhibited much higher H2S uptake capacity (2.04 mmol g−1) than CO2 (0.05 mmol g−1) and CH4 (0.0008 mmol g−1) (Fig. 10c). The high selectivity of the PL for H2S was benefited by the tertiary group on the liquid–solid interface, possessing more negative reaction enthalpy with H2S (−54.91 kJ mol−1) than CO2 (−14.14 kJ mol−1) and CH4 (−2.28 kJ mol−1). The breakthrough test with H2S (0.33 vol%), CO2 (1.00 vol%), and CH4 (98.67%) as the feed gas revealed that pure CH4 can be obtained initially, followed by the appearance of CO2 and then H2S (Fig. 10d).
The high uptake capacity and selectivity of the tertiary amine-functionalized PL were then leveraged to promote the reaction of the captured H2S with α-methacrylic acid to produce 3-mercaptoisobutyric acid. With PL as the catalyst, >80% yield was achieved, while the separated porous host and liquid counterparts delivered no or low yields (<43%) (Fig. 10e). The reaction mechanism study demonstrated an anti-Markovnikov addition pathway, including deprotonation of H2S by the N sites, nucleophilic attack of the SH- group on the positively charged β-C, and H transfer (Fig. 10f). The as-developed strategy showcases the merits of PLs in integrating gas storage and conversion via sophisticated structure engineering.
4.3 PLs in O2-involving catalytic procedures
Water is a ubiquitous solvent for diverse applications, possessing extensive hydrogen bonding networks capable of promoting the solvation of polar compounds but exhibiting low solubility to most gases. The low solubility of gases, especially oxygen, in water is a critical challenge for many energy and biomedical technologies that require high concentrations of gas molecules to be rapidly transported through an aqueous environment.86 A thermodynamic approach was leveraged to create aqueous solutions with permanent pores, that is, PL with H2O as the liquid phase. The critical factor is to deploy nanocrystals with abundant micropores, a hydrophobic inner surface, and hydrophilic outer surface, which can be stably dispersed in water and prohibit the water filling into the pores.23,36Silicalite-1 with terminal silanol groups on the external surface (hydrophilic) and SiO4 tetrahedra on the pore surface (hydrophobic) (Fig. 11a), with a surface area of 457 m2 g−1 and the average diameter around 59 or 90 nm, was used to create pores in water.23 Density analysis was first deployed to determine the porous nature of aqueous silicalite-1 colloidal solution and compared with theoretical calculations (Fig. 11b). Notably, the measured density value of the aqueous solution of silicalite-1 with different concentrations confirmed the maintenance of dry micropores in liquid water. The gas solubility was then evaluated to detect the existence of pores in water. Compared with the pure water, an O2 solubility of 1.1 mmol L−1 (at 0.84 bar) and a CO2 solubility of 23 mmol L−1 (at 0.67 bar) was obtained, and comparatively, improved performance was achieved by the solution composed of 20 wt% silicalite-1 in water, with an O2 solubility of 26 mmol L−1 and a CO2 solubility of 284 mmol L−1 (Fig. 11c). Additionally, the gas sorption procedures were reversible. The porous water creation was further extended to ZIF-67 with the outer surface modified by water soluble proteins (bovine serum albumin). The O2 solubility of the aqueous protein BSA/ZIF-67 solution achieved 80% of the theoretical value assuming all the pores of the ZIF-67 host are dry. In addition, surface modification of hydrophobic ZIF-8 via chemical reaction with methoxy polyethylene epoxide through N–C bond formation can create a promising porous host for porous water formation. ZSM-5 with a Si/Al ratio of 64 and average diameter of 193 nm can form a porous water solution with a concentration of 40%, leading to a high O2 solubility of 187 mL dL−1. All these systems demonstrated the possibility of creating porous water guided by thermodynamic principles.
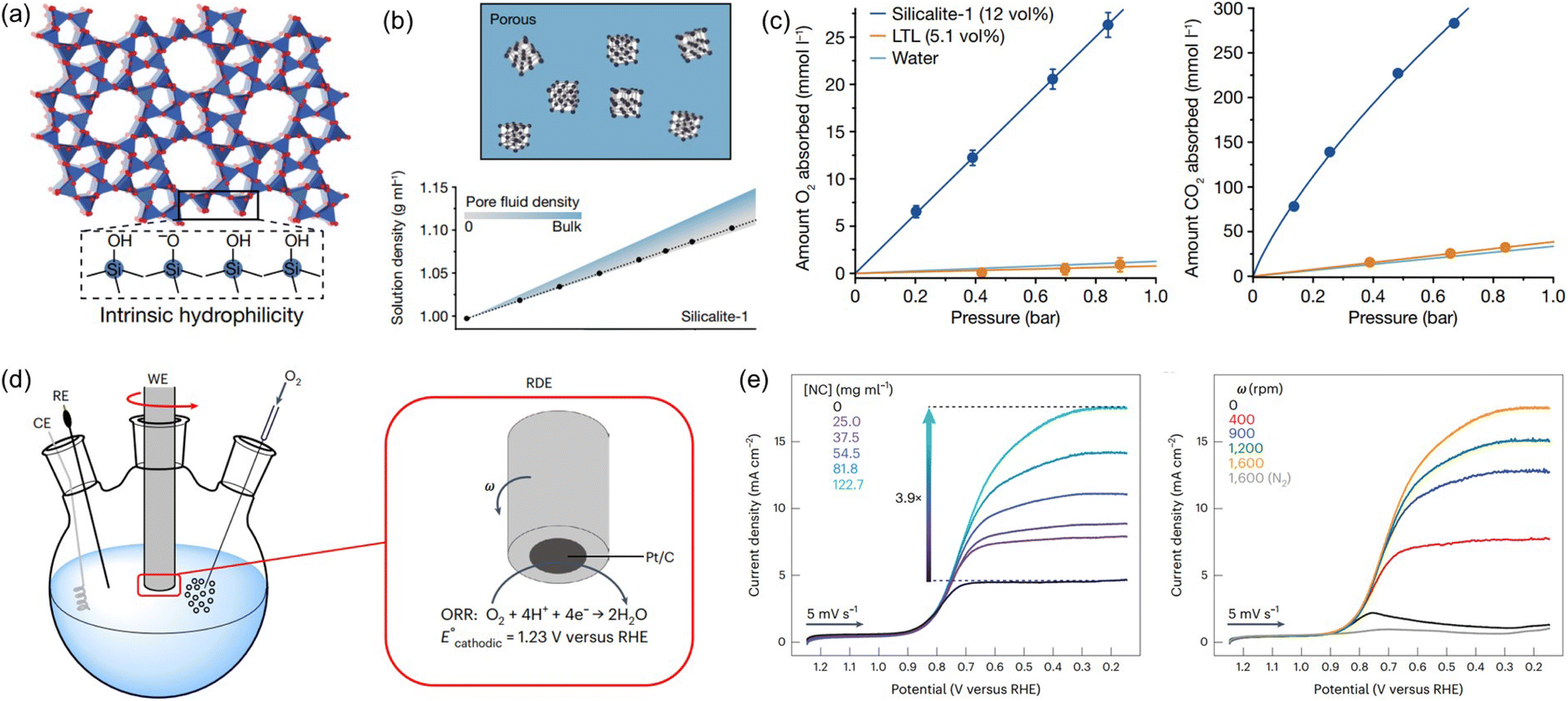 | ||
| Fig. 11 (a) The crystal structure of porous host silicalite-1. (b) The density of aqueous silicalite-1 solution being obtained by experimental measurement and theoretical calculation. (c) The uptake capacity of O2 and CO2 by aqueous solution with and without silicalite-1. (d) The ORR electrocatalysis performance evaluation using the RDE setup. (a–c) Reproduced with permission.23 Copyright 2022, Springer Nature. (e) The RDE voltammograms being collected in O2-saturated buffer with different concentrations of silicalite porous host (rotation rate = 1600 rpm) (left) and different rotation rates (silicalite concentration of 122.7 mg mL−1) (right). (d and e) Reproduced with permission.24 Copyright 2023, Springer Nature. | ||
By creating permanent micropores in water, the benefits of high gas solubility and fast mass transport can be leveraged to promote the gas–liquid phase reaction using water as the solvent. Taking the oxygen reduction reaction (ORR) electrocatalysis as a probe reaction, the porous water composed of A-silicalite-1 nanocrystals with an average diameter of 197 nm stably dispersed in water was used as the electrolyte.24 The initial rotating disk electrode (RDE) voltammetry evaluation was performed (Fig. 11d). Using phosphate-buffered water at pH 7.0, a plateau current density of only 4.5 mA cm−2 was obtained, which increased to 17.5 mA cm−2 with the added silicalite porous host (122.7 mg mL−1) (Fig. 11e). In addition, the current density in the ORR increased with the rotation rate. Notably, the enhanced catalytic activity endowed by the deployment of silicalite in water was further illustrated by the ORR performance in 0.5 M H2SO4 with the pH of 0.3; at 0.25 V versus RHE, the current density of 21.1 mA cm−2 was achieved, which was 3.8 times higher than the value obtained by the electrolyte without pores. Control experiments using a water-filled or nonporous silicalite host revealed that the major reason for the improved ORR current density was the much higher O2 concentration around the electrocatalyst in the porous water electrolyte.
4.4 PL-promoted hydrogenation reaction, H2 evolution reaction, and coupling reaction
Hydrogenation reactions are of great significance in both organic synthesis and industrial production.87,88 In the liquid phase, the utilization of PLs as the reaction media has the potential to increase the H2 gas concentration around the active sites, leading to enhanced reaction activity. The type I PL composed of hollow silica spheres with the surface modified by corona–canopy ionic pairs (ammonium cations and sulfonate anions) was well established20 and Pt NPs (around 1.2 nm) can be introduced within the spheres via a thiol group-assisted procedure (Fig. 12a).74,89 The hydrogenation reaction of 1-decene was selected as the probe reaction, which was conducted at 40 °C under ambient H2 pressure using ethanol as the solvent.90 Initial reaction performance evaluation revealed that the reaction followed first-order kinetics and was controlled by diffusion when Pt@HS-SiO2 was used as the catalyst. The catalytic activity of different catalysts, as indicated by the apparent turnover frequency, increased in the order of Pt@HS-SiO2 PL (using KPEGS as the solvent, 768 h−1) < OS-(Pt@HS-SiO2) (without PEGS anion exchange, 930 h−1) < Pt@HS-SiO2 (without corona–canopy modification, 2006 h−1) < Pt@HS-SiO2 PL (2062 h−1) (Fig. 12b). The Pt@HS-SiO2 PL system can be recycled for at least three times with well-maintained catalytic activity. In addition, this Pt NP-involving PL can be utilized in the hydrogenation reaction of diverse substrates such as aliphatic linear and cyclic alkenes and nitrobenzene derivatives.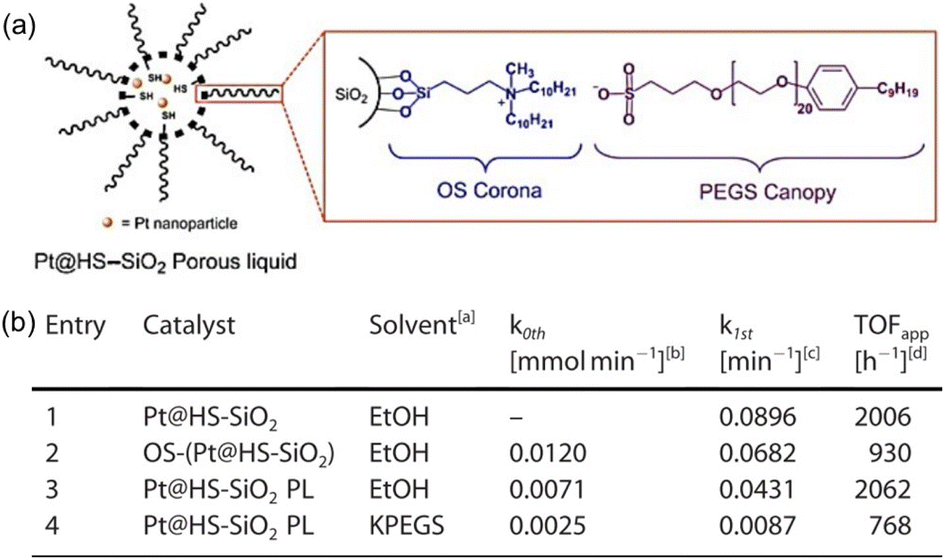 | ||
| Fig. 12 (a) Schematic diagram of the Pt NP-involving type I PL. (b) Catalytic activity and reaction kinetics of different catalytic systems in the hydrogenation reaction of 1-decene to decane. (a and b) Reproduced with permission.74 Copyright 2020, Wiley-VCH. | ||
In order to enhance the complexation of the porous host with noble metal species, the covalent organic cages (COCs) were synthesized via the Schiff base formation reaction using tris(2-aminoethyl)amine (Tren) and aromatic aldehydes as the precursors, followed by hydrogenation using NaBH4 as the reducing reagent.75 The as-afforded COCs with bipyridine linkers exhibited the capability to bind metal species (e.g., Pd(OAc)2), which were then reduced to Pd NPs by NaBH4 with good dispersibility and an average size of 2.6 nm (Fig. 13a). In addition, most of the Pd NPs are located on the surface of bipyridine-functionalized COCs. Then, the COC-stabilized Pd NPs were loaded onto carbon nitride nanosheets to promote the photo-catalytic H2 evolution from water. Under identical reaction conditions with triethanolamine as the sacrificial agent, the H2 evolution rate of the Pd NPs@COC/g-C3N4 reached 5487 μmol g−1 h−1, which was 2.5 times higher than that obtained by Pd NPs @g-C3N4 (2040 μmol g−1 h−1) (Fig. 13b). Good recyclability of the Pd NPs@COC/g-C3N4 was obtained, with no significant activity loss or Pd NP agglomeration over at least four cycles (Fig. 13c). The photoluminescence and decay lifetime evaluation revealed that the presence of COCs can suppress the recombination of photoinduced electrons and facilitate electron transport, leading to enhanced photocatalytic performance. This co-catalyst system combines the advantages of homogeneous and heterogeneous catalysis, ensuring high catalytic activity and long-term durability for hydrogen evolution.
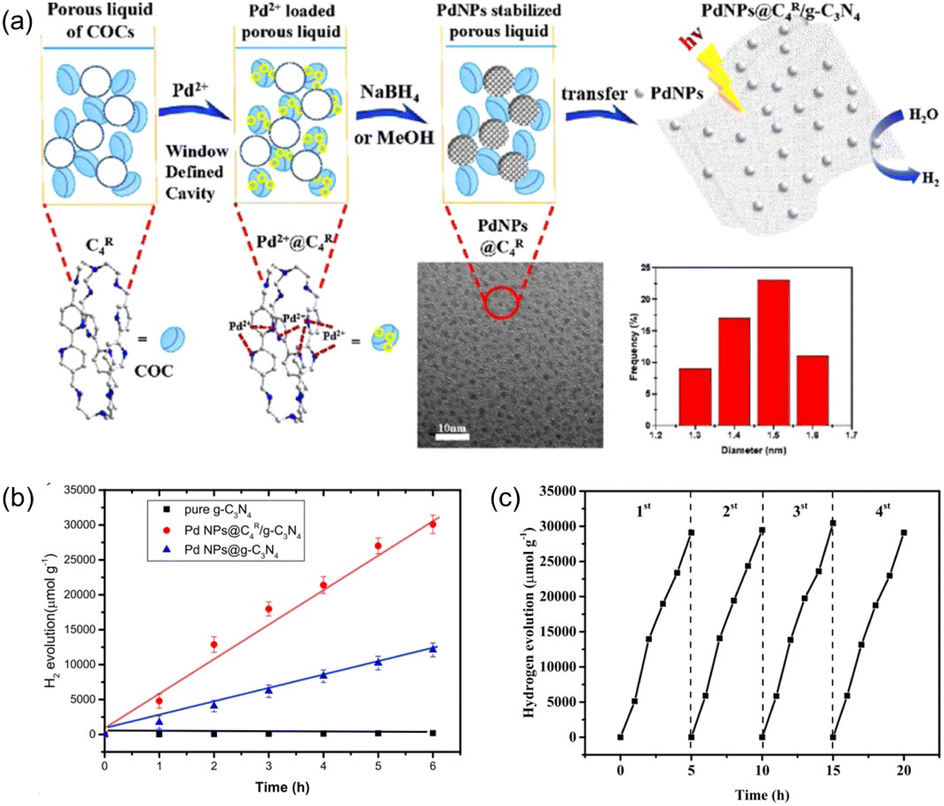 | ||
| Fig. 13 (a) Schematic illustration of the formation and stabilization of Pd NPs in the porous liquid of C4R COCs and then transfer onto g-C3N4 nanosheets for photocatalytic hydrogen evolution. (b) The performance of different catalytic systems in photocatalytic hydrogen evolution reaction. (c) Recyclability performance of the Pd NPs@C4R COCs/g-C3N4 catalyst within four cycles. (a–c) Reproduced with permission.75 Copyright 2020, American Chemical Society. | ||
Not only can metal NPs, but also homogeneous noble metal species be immobilized within a type I PL via complexation.76 First, the inner surface of the hollow silica nanospheres was chemically modified with N-heterocyclic carbenes (NHCs), which then acted as anchoring sites to complex with Pd2+ species (Fig. 14a). Finally, the outer surface of the silica spheres was modified via the corona–canopy strategy to afford Type I PL (NHC–Pd@SiO2 PL). The Heck coupling was selected as the probe reaction to determine the catalytic activity of the Pd-complexed PL.91,92 The results showed that this PL system exhibited a significant catalytic effect on the coupling reaction between aryl iodides and butyl acrylate despite the low Pd loading (Fig. 14b). Notably, for diverse aryl iodide substrates containing electron-donating or electron-withdrawing groups, the Heck reactions could be accomplished successfully.
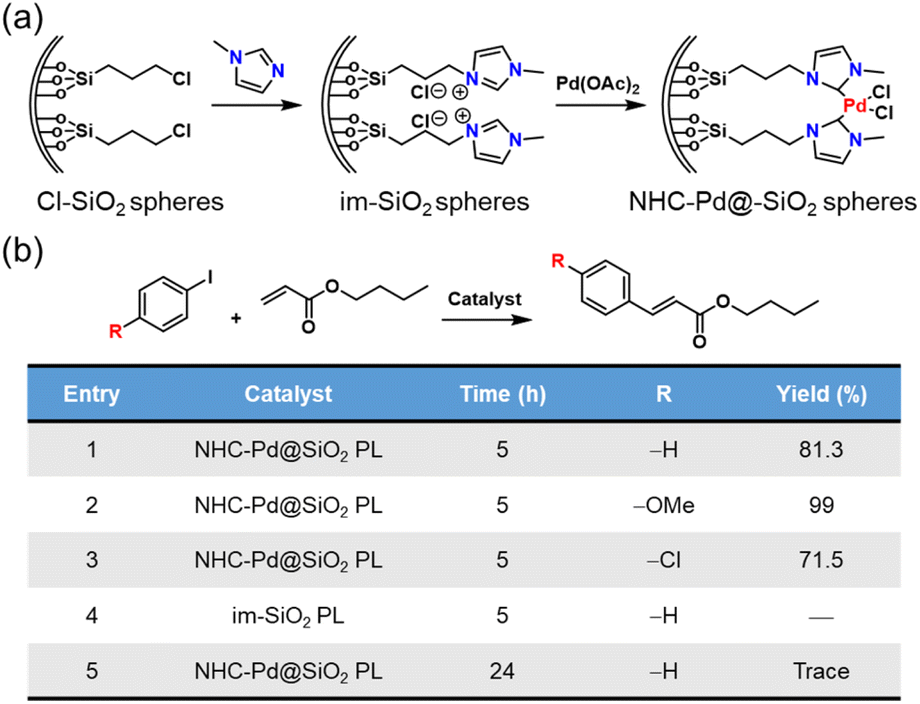 | ||
| Fig. 14 (a) Synthesis of NHC–Pd@SiO2 for PL construction. (b) Heck reaction between different aryl iodides and butyl acrylate catalysed by Pd2+-involving PLs. | ||
4.5 Application of PLs in cascade reactions
Catalytic systems capable of promoting cascade reactions, particularly those requiring active sites possessing antagonistic properties, are highly desired to fulfill multiple chemical transformations in a one-pot procedure without separation and purification of the reaction intermediates.93–97 The unique feature of PLs provides the opportunity to anchor and separate active sites with diverse or even incompatible properties in a single catalytic system, as the liquid phase and porous host in PLs are not tightly or completely combined with each other. Towards this purpose, a type III PL composed of zeolite and IL was developed (Fig. 15a).61 The zeolite MCM-22 with MWW crystalline structure and nanosheet morphology was deployed as the solid counterpart, possessing a surface area of 391 m2 g−1 and micropores around 0.5 and 1.4 nm. MCM-22 provided the acidic sites for the cascade reaction (Brønsted acid sites of 245.6 μmol g−1 and Lewis acid sites of 208 μmol g−1). The IL composed of ammonium cations with long alkyl chains and chloride anions (TOMAC) was deployed as the liquid phase and provided the basic sites (Cl−). The as-prepared MCM-22-PL has 26.0 wt% MCM-22 in TOMAC. The PALS analysis over MCM-22-PL exhibited the ortho-positronium lifetime tau3 of 2.139 ns, corresponding to the cavity diameter of 0.59 nm within the liquid phase, which was close to the porosity of the MCM-22 host. In the 129Xe NMR spectrum of the IL TOMAC, the peak was located at −5161.6 ppm, which was obviously upfield shifted to −5167.3 ppm in the MCM-22-PL, indicating the higher free volume in MCM-22-PL and the existence of permanent pores (Fig. 15b).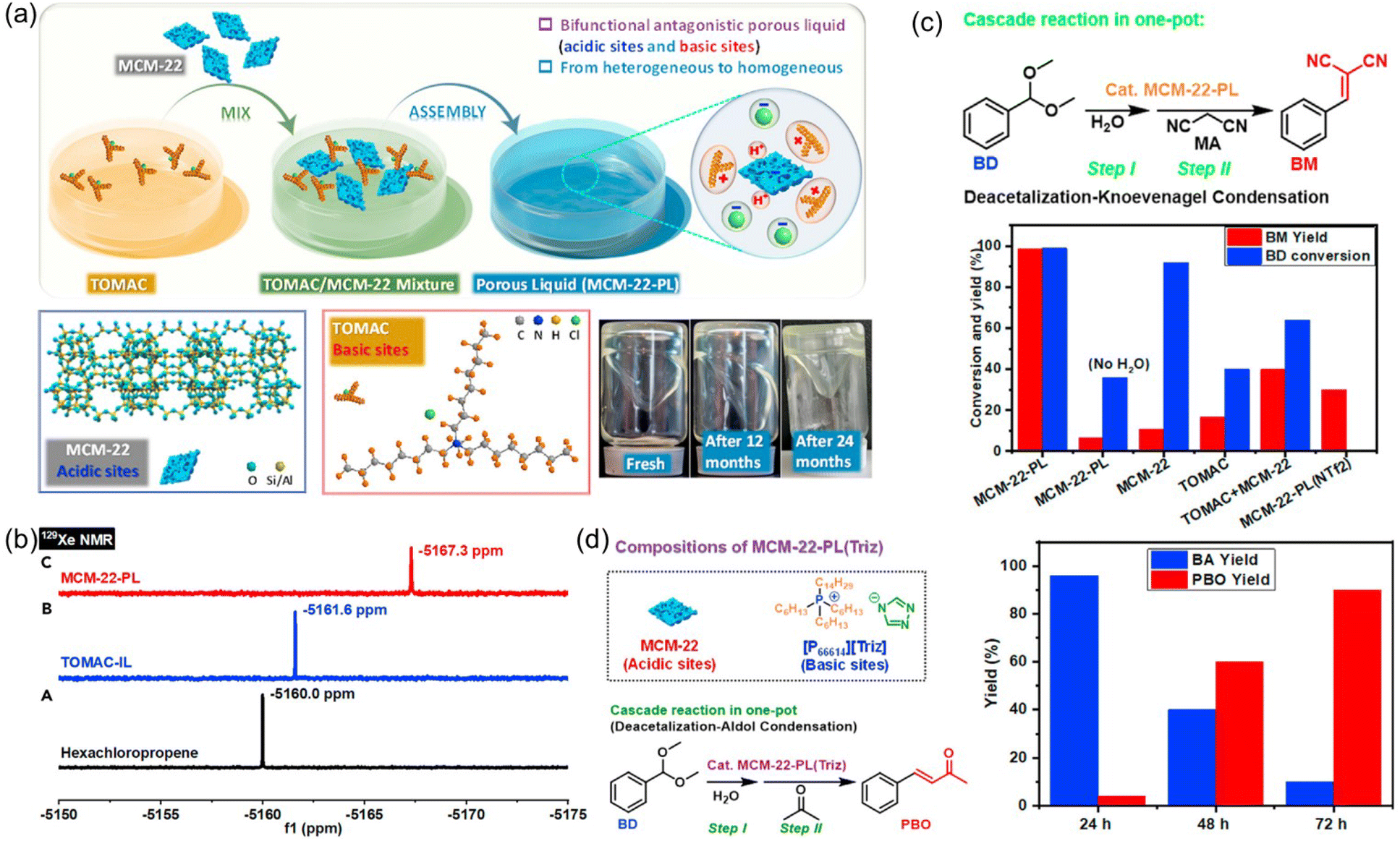 | ||
| Fig. 15 (a) Synthesis procedure of the MCM-22-PL, structures of the MCM-22 (solid) and IL TOMAC (liquid), and images of the as-synthesized PL. (b) 129Xe NMR spectra of the bulky solvent, IL solution, and PL solution. (c) The performance of the diverse catalytic systems including MCM-22-PL in the cascade deacetalization–Knoevenagel condensation reaction procedure. (d) The cascade deacetalization–aldol condensation catalysed by MCM-22-PL(Triz). (a–d) Reproduced with permission.61 Copyright 2021, Elsevier. | ||
Then the deacetalization–Knoevenagel condensation was used as a probe reaction, in which acidic and basic sites are required respectively for the first and second steps (Fig. 15c). The co-existence of acidic and basic sites in MCM-22-PL made it a promising candidate to promote this cascade procedure in a single batch. When benzaldehyde dimethylacetal (BD) and malononitrile (MA) were used as the substrates, complete BD conversion and >99% yield of the benzylidene malononitrile (BM) was achieved, catalysed by MCM-22-PL in toluene at 80 °C within 3 h. Control experiments revealed that MCM-22 was responsible for the BD-to-BA conversion and the TOMAC was required to promote the BA-to-BM transformation. Notably, by engineering the active sites in the PL, the MCM-22-PL allowed the reactions to take place in the liquid phase and the catalyst to be recycled after removing the solvents and products by thermal treatment under a vacuum. A BM yield of 94% can be achieved after recycling MCM-22-PL five times. The superior performance of MCM-22-PL was due to its unique features including possessing well-preserved acidic and basic catalytic sites in the solid and liquid counterparts respectively, transforming the process from heterogeneous to homogeneous in nature, and shortened distance between the acidic and basic sites via structural assembly. The reaction mechanism was studied in detail via in situ DRIFTS and theoretical simulation, revealing the existence of a semi-acetal as the active intermediate.
Then the PL composed of MCM-22 and IL with a triazolate anion was synthesized and used in the cascade deacetalization–aldol condensation, in which the second step needs active sites with stronger basicity (Fig. 15d). With BD and acetone as the reaction substrates, a 90% yield of the 4-phenyl-3-buten-2-one (PBO) product can be obtained within 72 h. The achievements made in this work demonstrated a promising way to facilitate complicated reaction procedures via structural engineering over the PL systems.
4.6 Application of PLs in oxidative desulfurization
Oxidative desulfurization of sulfides represents an efficient approach to remove impurities and upgrade the fuel oil and phosphomolybdic acid was demonstrated as one of the catalysts to oxidize the sulfur-containing compounds to sulfone, which can then be removed via extraction. The PL construct can facilitate the mass transfer, integrate with the extraction procedure, and stabilize the catalyst via the confinement effect.98,99 Towards this purpose, the porous host was obtained via in situ assembly of zinc salts, the 2-MeIm ligand, and phosphomolybdic acid (HPMo, as active sites) and denoted as HPMo@ZIF-8, in which HPMo was encapsulated within the cavity of the ZIF-8 framework.77 The structure of the porous host was characterized by PXRD, FT-IR, microscopy, and elemental mapping. The PL was then synthesized by dispersing the HPMo@ZIF-8 NPs in an IL [Emim][NTf2]. The SO2 sorption capacity was determined via the vapor–liquid equilibrium method, from which higher SO2 uptake capacity within the pressure range up to 1 bar was achieved by the HPMo@ZIF-8-PIL than the pure IL (Fig. 16a), indicating the presence of additional free volume in PL to accommodate more SO2 molecules in the PL. Molecular displacement experiments further provided evidence of porosity in the obtained PL. A smaller guest molecule (CHCl3) was chosen as the gas displacer because it can easily enter the cages of ZIF-8 and expel the gas in the cavity, leading to the formation of bubbles. Then, the HPMo@ZIF-8-PIL catalyst was deployed to catalyse the oxidation of dibenzothiophene (DBT) in the presence of H2O2 and 100% sulfur removal was achieved by HPMo@ZIF-8-PIL at 30 °C, which was much higher than that of the physical mixture of HPMo with IL (Fig. 16b). Notably, the PL-derived catalytic system can be further extended to other sulfur-containing substrates such as 4-methyldibenzothiophene (4-MDBT) and 4,6-dimethyldibenzothiophene (4,6-DMDBT). The cycling experiments revealed that the sulfur removal rate of 95% can be achieved at the sixth run, during which CCl4 extraction treatment was conducted at each cycle to remove the product combined with the active sites.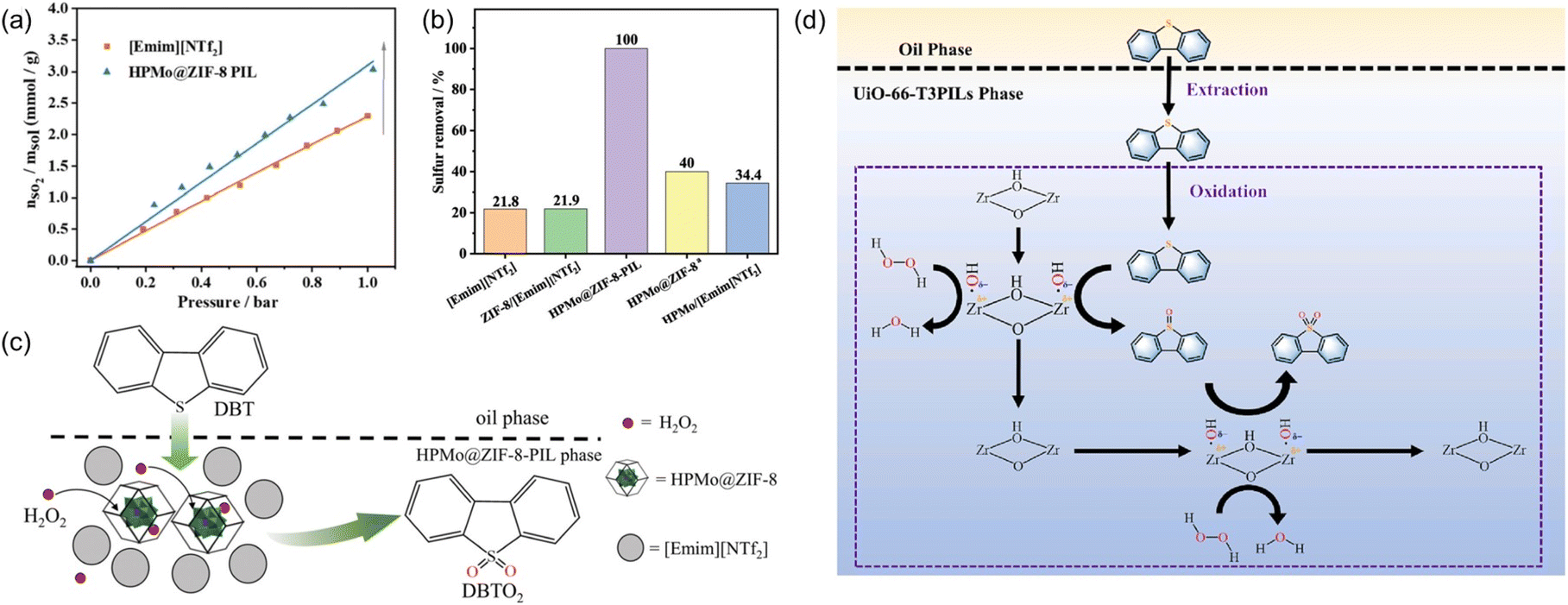 | ||
| Fig. 16 (a) The SO2 uptake isotherm of pure IL [Emim][NTf2] and the type III PL containing HPMo@ZIF-8 NPs. (b) The sulfur removal efficiency of different catalytic systems under identical conditions. (c) The proposed reaction mechanism of oxidative desulfurization of DBT promoted by PL. (a–c) Reproduced with permission.77 Copyright 2022, The Royal Society of Chemistry. (d) The integrated extraction and oxidation pathway in the UiO-66-T3PIL system. Reproduced with permission.78 Copyright 2023, Elsevier. | ||
The PL-promoted desulfurization strategy was then extended to the system composed of UiO-66(Zr) in IL [P66614][NTf2].78 The study revealed that the good catalytic desulfurization performance of PL (denoted as UiO-66-T3PILs) was generated by the extraction of the sulfur compounds from the oil phase into the IL phase, which was then oxidized by the ·OH radicals in the UiO-66 porous host in the presence of H2O2 (Fig. 16c). Recently, another type III PL (denoted UiO-66-BAPILs) was constructed by dispersing UiO-66 into a Brønsted acidic ionic liquid ([BSPy][CF3SO3]).79 Similarly, when H2O2 was used as the oxidant, the bifunctional UiO-66-BAPILs acted as both extractants and catalysts during the reaction, achieving a sulfur removal efficiency of 99.5%.
5. Conclusions and future perspectives
The unique properties of PLs and facile structure engineering approaches led to initial success in their catalysis applications in CO2, H2S, O2, and H2-involving reactions, as well as the cascade procedures. The merits by using PLs in catalysis have been demonstrated to combine the features of the porous solid and fluid liquid. Type I PLs have better stability than type II and III PLs since the liquid phase is chemically integrated with the porous host such as MOFs, hollow silica spheres, and hollow carbon spheres. As type II PLs are formed by solubilizing the porous host such as organic cages in solvents, it's difficult to integrate the catalytic sites within the porous host. In addition, the solvents (e.g., crown ether) being deployed in type II PLs have relatively low boiling points and inferior thermal stability. Type III PLs are most extensively studied and deployed in separation and catalysis-integrated procedures, benefiting from their large tunability in both liquid and porous host counterparts, which allows anchoring the catalytic active sites and integrating diverse sites in the liquid and solid components respectively. However, as type III PLs are formed via physically dispersing the porous host in the liquid phase, the stability of the dispersion, particularly in the presence of substrates and under the reaction conditions, should be well maintained to ensure the quality of the adopted PLs.Although related research is still at the preliminary stage, the unique features of PLs will provide alternative solutions and new opportunities in combined separation and catalysis procedures that cannot be fulfilled in traditional homogeneous or heterogeneous procedures. Further improvements can be made in the following aspects to further improve and extend the application of PLs in catalysis.
• Although in the gas-involving reactions the high solubility of the gas sources could benefit the reaction procedure, the currently deployed PLs still exhibit inferior gas capacity since only physisorption is involved. PLs possessing stronger interaction strength with the gas molecules, i.e., via chemisorption pathway, could benefit not only the gas uptake capacity, but also activate the gas molecules.
• Most of the current PL-involving catalytic procedures are still focused on those below 100 °C. In catalytic applications, particularly for gas-phase reactions, high temperatures are needed (e.g., up to 800 °C in dry reforming of methane to produce CO and H2 from CH4 and CO2, as well as in methane cleavage to generate H2).100,101 Potential application of PLs in these high temperature-involving procedures is more challenging and rarely explored. Increased gas solubility and fast mass transport at high temperatures are more difficult to be achieved, while PLs based on molten salts combined with stable and suitable porous hosts may play such roles.102
• Only limited systems could engineer the active sites within the PLs, and most of them still require additional catalysts, which caused problems in mass transfer and catalyst recycling. PLs capable of integrating the gas molecule concentration and catalytic site involvement are preferred in terms of proximity of the substrate-active sites, sustainability, and facile catalyst recycling.
• PLs have exhibited the capability of tuning the gas selectivity by the molecular sieving effect or interaction strength in gas separation applications, but this has not been utilized in catalysis, which could benefit selective catalysis procedures.
• The preliminary results of PL-catalysed cascade reactions have demonstrated the great potential of PLs in simplifying reaction procedures without protection, separation, and purification. This will rely on the design and selection of the porous host and the liquid component to engineer active sites with different properties together without sacrificing their activity. For example, PLs composed of strong base and Lewis acid sites can be used to catalyze aldol condensation and dehydration to form conjugated enones in a single sequence, and cascade Michael addition and cyclization to synthesize cyclic ketones, lactams, or lactones.103,104
• Sophisticated characterization techniques are still required to perform the structural evaluation during the reaction procedure, particularly on the solid–liquid interface, and the case will be more complicated if the active sites are on the interphase location. Operando techniques are preferred to monitor the reaction procedures and understand the structure–performance relationship.
Progress in the above aspects will contribute to the wider application of PLs in catalysis and beyond.
Data availability
No new data were generated or analysed during the writing of this perspective.Author contributions
All authors contributed to the outline discussion, participated in the writing of the original draft, and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported financially by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, US Department of Energy.Notes and references
- A. Bavykina, A. Cadiau and J. Gascon, Coord. Chem. Rev., 2019, 386, 85–95 CrossRef CAS.
- P. F. Fulvio and S. Dai, Chem, 2020, 6, 3263–3287 CAS.
- K. Jie, Y. Zhou, H. P. Ryan, S. Dai and J. R. Nitschke, Adv. Mater., 2021, 33, 2005745 CrossRef CAS PubMed.
- T. D. Bennett, F.-X. Coudert, S. L. James and A. I. Cooper, Nat. Mater., 2021, 20, 1179–1187 CrossRef CAS.
- D. Wang, Y. Xin, D. Yao, X. Li, H. Ning, H. Zhang, Y. Wang, X. Ju, Z. He and Z. Yang, Adv. Funct. Mater., 2022, 32, 2104162 CrossRef CAS.
- B. D. Egleston, A. Mroz, K. E. Jelfs and R. L. Greenaway, Chem. Sci., 2022, 13, 5042–5054 RSC.
- N. O'Reilly, N. Giri and S. L. James, Chem.–Eur. J., 2007, 13, 3020–3025 CrossRef PubMed.
- D. Wang, Y. Ying, Y. Xin, P. Li, Z. Yang and Y. Zheng, Acc. Mater. Res., 2023, 4, 854–866 CrossRef CAS.
- L. Ma, C. J. E. Haynes, A. B. Grommet, A. Walczak, C. C. Parkins, C. M. Doherty, L. Longley, A. Tron, A. R. Stefankiewicz, T. D. Bennett and J. R. Nitschke, Nat. Chem., 2020, 12, 270–275 CrossRef CAS PubMed.
- P. Li, J. A. Schott, J. Zhang, S. M. Mahurin, Y. Sheng, Z. A. Qiao, X. Hu, G. Cui, D. Yao, S. Brown, Y. Zheng and S. Dai, Angew. Chem., Int. Ed., 2017, 56, 14958–14962 CrossRef CAS PubMed.
- R. J. Kearsey, B. M. Alston, M. E. Briggs, R. L. Greenaway and A. I. Cooper, Chem. Sci., 2019, 10, 9454–9465 RSC.
- Z. Zhang, B. Yang, B. Zhang, M. Cui, J. Tang and X. Qiao, Nat. Commun., 2022, 13, 2353 CrossRef CAS PubMed.
- S. He, L. Chen, J. Cui, B. Yuan, H. Wang, F. Wang, Y. Yu, Y. Lee and T. Li, J. Am. Chem. Soc., 2019, 141, 19708–19714 CrossRef CAS PubMed.
- R. Gaillac, P. Pullumbi, K. A. Beyer, K. W. Chapman, D. A. Keen, T. D. Bennett and F.-X. Coudert, Nat. Mater., 2017, 16, 1149–1154 CrossRef CAS PubMed.
- A. Koutsianos, R. Pallach, L. Frentzel-Beyme, C. Das, M. Paulus, C. Sternemann and S. Henke, Nat. Commun., 2023, 14, 4200 CrossRef CAS PubMed.
- L. Qiu, L. Peng, D. Moitra, H. Liu, Y. Fu, Z. Dong, W. Hu, M. Lei, D. e. Jiang, H. Lin, J. Hu, K. A. McGarry, I. Popovs, M. Li, A. S. Ivanov, Z. Yang and S. Dai, Small, 2023, 19, 2302708 CrossRef CAS PubMed.
- R. E. Mow, A. S. Lipton, S. Shulda, E. A. Gaulding, T. Gennett and W. A. Braunecker, J. Mater. Chem. A, 2020, 8, 23455–23462 RSC.
- W. Shan, P. F. Fulvio, L. Kong, J. A. Schott, C.-L. Do-Thanh, T. Tian, X. Hu, S. M. Mahurin, H. Xing and S. Dai, ACS Appl. Mater. Interfaces, 2018, 10, 32–36 CrossRef CAS PubMed.
- P. Li, H. Chen, J. A. Schott, B. Li, Y. Zheng, S. M. Mahurin, D.-e. Jiang, G. Cui, X. Hu and Y. Wang, Nanoscale, 2019, 11, 1515–1519 RSC.
- J. Zhang, S. H. Chai, Z. A. Qiao, S. M. Mahurin, J. Chen, Y. Fang, S. Wan, K. Nelson, P. Zhang and S. Dai, Angew. Chem., Int. Ed., 2015, 54, 932–936 CrossRef CAS PubMed.
- S. Liu, L. Meng and J. Fan, ChemistrySelect, 2021, 6, 5027–5033 CrossRef CAS.
- T. Shi, Y. Zheng, T. Wang, P. Li, Y. Wang and D. Yao, ChemPhysChem, 2018, 19, 130–137 CrossRef CAS PubMed.
- D. P. Erdosy, M. B. Wenny, J. Cho, C. DelRe, M. V. Walter, F. Jimenez-Angeles, B. Qiao, R. Sanchez, Y. Peng, B. D. Polizzotti, M. O. de la Cruz and J. A. Mason, Nature, 2022, 608, 712–718 CrossRef CAS.
- A. E. Thorarinsdottir, D. P. Erdosy, C. Costentin, J. A. Mason and D. G. Nocera, Nat. Catal., 2023, 6, 425–434 CrossRef CAS.
- F. Su, X. Li, Y. Wang, Z. He, L. Fan, H. Wang, J. Xie, Y. Zheng and D. Yao, Sep. Purif. Technol., 2021, 277, 119410 CrossRef CAS.
- K. Jie, N. Onishi, J. A. Schott, I. Popovs, D. E. Jiang, S. Mahurin and S. Dai, Angew. Chem., Int. Ed., 2020, 59, 2268–2272 CrossRef CAS PubMed.
- Z. Deng, W. Ying, K. Gong, Y. J. Zeng, Y. Yan and X. Peng, Small, 2020, 16, 1907016 CrossRef CAS.
- M. Atilhan, A. Cincotti and S. Aparicio, J. Mol. Liq., 2021, 330, 115660 CrossRef CAS.
- Y. Wang, Y. Sun, H. Bian, L. Zhu, D. Xia and H. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 45916–45928 CrossRef CAS.
- X. Zhao, Y. Yuan, P. Li, Z. Song, C. Ma, D. Pan, S. Wu, T. Ding, Z. Guo and N. Wang, Chem. Commun., 2019, 55, 13179–13182 RSC.
- X. Li, D. Wang, Z. He, F. Su, N. Zhang, Y. Xin, H. Wang, X. Tian, Y. Zheng and D. Yao, Chem. Eng. J., 2021, 417, 129239 CrossRef CAS.
- J. Wu, X. Wu, P. Zhao, Z. Wang, L. Zhang, D. Xu and J. Gao, Fuel, 2021, 300, 121013 CrossRef CAS.
- P. Li, D. Wang, L. Zhang, C. Liu, F. Wu, Y. Wang, Z. Wang, Z. Zhao, W. Wu and Y. Liang, Small, 2021, 17, 2006687 CrossRef CAS PubMed.
- R. L. Greenaway, D. Holden, E. G. Eden, A. Stephenson, C. W. Yong, M. J. Bennison, T. Hasell, M. E. Briggs, S. L. James and A. I. Cooper, Chem. Sci., 2017, 8, 2640–2651 RSC.
- F. Zhang, F. Yang, J. Huang, B. G. Sumpter and R. Qiao, J. Phys. Chem. B, 2016, 120, 7195–7200 CrossRef CAS PubMed.
- C. DelRe, H. Hong, M. B. Wenny, D. P. Erdosy, J. Cho, B. Lee and J. A. Mason, J. Am. Chem. Soc., 2023, 145, 19982–19988 CrossRef CAS.
- J. Cahir, M. Y. Tsang, B. Lai, D. Hughes, M. A. Alam, J. Jacquemin, D. Rooney and S. L. James, Chem. Sci., 2020, 11, 2077–2084 RSC.
- B. Lai, J. Cahir, M. Y. Tsang, J. Jacquemin, D. Rooney, B. Murrer and S. L. James, ACS Appl. Mater. Interfaces, 2020, 13, 932–936 CrossRef PubMed.
- S. Van Glubt and M. L. Brusseau, Environ. Sci. Technol., 2021, 55, 3706–3715 CrossRef CAS PubMed.
- B. D. Egleston, K. V. Luzyanin, M. C. Brand, R. Clowes, M. E. Briggs, R. L. Greenaway and A. I. Cooper, Angew. Chem., Int. Ed., 2020, 59, 7362–7366 CrossRef CAS PubMed.
- Y. Wang, Y. Sun, H. Bian, L. Zhu, D. Xia and H. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 45916–45928 CrossRef CAS PubMed.
- J. Zhang, S.-H. Chai, Z.-A. Qiao, S. M. Mahurin, J. Chen, Y. Fang, S. Wan, K. Nelson, P. Zhang and S. Dai, Angew. Chem., Int. Ed., 2015, 54, 932–936 CrossRef CAS PubMed.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS.
- Z.-A. Qiao, P. Zhang, S.-H. Chai, M. Chi, G. M. Veith, N. C. Gallego, M. Kidder and S. Dai, J. Am. Chem. Soc., 2014, 136, 11260–11263 CrossRef CAS PubMed.
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Chem. Rev., 2020, 120, 623–682 CrossRef CAS PubMed.
- H. Chen, S. Z. Yang, Z. Yang, W. Lin, H. Xu, Q. Wan, X. Suo, T. Wang, D. E. Jiang, J. Fu and S. Dai, ACS Cent. Sci., 2020, 6, 1617–1627 CrossRef CAS PubMed.
- Y. Sun, Z. Yang and S. Dai, J. Phys. Chem. Lett., 2023, 14, 2364–2377 CrossRef CAS.
- R. Kumar, P. Dhasaiyan, P. M. Naveenkumar and K. P. Sharma, Nanoscale Adv., 2019, 1, 4067–4075 RSC.
- S. Koutsoukos, J. Avila, N. J. Brooks, M. C. Gomes and T. Welton, Phys. Chem. Chem. Phys., 2023, 25, 6316–6325 RSC.
- K. Yang, Z. Cai, M. Tyagi, M. Feygenson, J. C. Neuefeind, J. S. Moore and Y. Zhang, Chem. Mater., 2016, 28, 3227–3233 CrossRef CAS.
- X. Wang, L. He, J. Sumner, S. Qian, Q. Zhang, H. O'Neill, Y. Mao, C. Chen, A. M. Al-Enizi, A. Nafady and S. Ma, Nat. Commun., 2023, 14, 973 CrossRef CAS PubMed.
- Y. Jean, J. D. Van Horn, W.-S. Hung and K.-R. Lee, Macromolecules, 2013, 46, 7133–7145 CrossRef CAS.
- S. Liu, J. Liu, X. Hou, T. Xu, J. Tong, J. Zhang, B. Ye and B. Liu, Langmuir, 2018, 34, 3654–3660 CrossRef CAS PubMed.
- Y. Liu, Y. Bai and T. Tian, Materials, 2019, 12, 3984 CrossRef CAS PubMed.
- J. Avila, L. F. Lepre, C. C. Santini, M. Tiano, S. Denis-Quanquin, K. Chung Szeto, A. A. Padua and M. Costa Gomes, Angew. Chem., Int. Ed., 2021, 60, 12876–12882 CrossRef CAS PubMed.
- A. Kai, B. D. Egleston, A. Tarzia, R. Clowes, M. E. Briggs, K. E. Jelfs, A. I. Cooper and R. L. Greenaway, Adv. Funct. Mater., 2021, 31, 2106116 CrossRef CAS.
- M. Costa Gomes, L. Pison, C. Červinka and A. Padua, Angew. Chem., Int. Ed., 2018, 57, 11909–11912 CrossRef CAS PubMed.
- J. Zhao, Z. Jin, Q. Hu, Z. Jin, T. J. Barber, Y. Zhang and M. Bleuel, Sci. Rep., 2017, 7, 15413 CrossRef PubMed.
- J. L. Bañuelos, G. Feng, P. F. Fulvio, S. Li, G. Rother, N. Arend, A. Faraone, S. Dai, P. T. Cummings and D. J. Wesolowski, Carbon, 2014, 78, 415–427 CrossRef.
- E. J. Kim, R. L. Siegelman, H. Z. Jiang, A. C. Forse, J.-H. Lee, J. D. Martell, P. J. Milner, J. M. Falkowski, J. B. Neaton and J. A. Reimer, Science, 2020, 369, 392–396 CrossRef CAS PubMed.
- H. Chen, Z. Z. Yang, H. G. Peng, K. C. Jie, P. P. Li, S. M. Ding, W. Guo, X. Suo, J. X. Liu, R. Yan, W. M. Liu, C. L. Do-Thanh, H. M. Wang, Z. D. Wang, L. Han, W. M. Yang and S. Dai, Chem, 2021, 7, 3340–3358 CAS.
- N. J. Williams, C. A. Seipp, F. M. Brethomé, Y.-Z. Ma, A. S. Ivanov, V. S. Bryantsev, M. K. Kidder, H. J. Martin, E. Holguin and K. A. Garrabrant, Chem, 2019, 5, 719–730 CAS.
- C. He, Y. H. Zou, D. H. Si, Z. A. Chen, T. F. Liu, R. Cao and Y. B. Huang, Nat. Commun., 2023, 14, 3317 CrossRef CAS PubMed.
- J. Cole, Z. Henderson, A. G. Thomas, C. Castle, A. J. Greer, C. Hardacre, M. Scardamaglia, A. Shavorskiy and K. L. Syres, JPhys Mater., 2023, 6, 045012 CrossRef CAS.
- I. Niedermaier, C. Kolbeck, N. Taccardi, P. S. Schulz, J. Li, T. Drewello, P. Wasserscheid, H. P. Steinrück and F. Maier, ChemPhysChem, 2012, 13, 1725–1735 CrossRef CAS.
- L. Sheng, Z. Chen and Y. Wang, Appl. Surf. Sci., 2021, 536, 147951 CrossRef CAS.
- Y. H. Zou, Y. B. Huang, D. H. Si, Q. Yin, Q. J. Wu, Z. Weng and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 20915–20920 CrossRef CAS PubMed.
- M. K. Dinker, M. M. Li, K. Zhao, M. Zuo, L. Ding, X. Q. Liu and L. B. Sun, Angew. Chem., Int. Ed., 2023, 62, e202306495 CrossRef.
- Y. Zhou, J. Avila, N. Berthet, S. Legrand, C. C. Santini, M. Costa Gomes and V. Dufaud, Chem. Commun., 2021, 57, 7922–7925 RSC.
- L. Qiu, H. Peng, Z. Yang, J. Fan, M. Li, S. Yang, D. M. Driscoll, L. Ren, S. M. Mahurin, L. N. He and S. Dai, Adv. Mater., 2023, 35, e2302525 CrossRef PubMed.
- Y. Xu, Y. Ren, G. Zhou, S. Feng, Z. Yang, S. Dai, Z. Lu and T. Zhou, Adv. Funct. Mater., 2024, 34, 2313695 CrossRef CAS.
- A. Bhattacharjee, R. Kumar and K. P. Sharma, ChemSusChem, 2021, 14, 3303–3314 CrossRef CAS PubMed.
- H. Ning, M. Shi, Q. Yang, J. Huang, X. Zhang, Y. Wu and K. Jie, Angew. Chem., Int. Ed., 2023, 62, e202310741 CrossRef CAS PubMed.
- E. B. Hemming, A. F. Masters and T. Maschmeyer, Chem.–Eur. J., 2020, 26, 7059–7064 CrossRef CAS PubMed.
- J. H. Zhang, M. J. Wei, Y. L. Lu, Z. W. Wei, H. P. Wang and M. Pan, ACS Appl. Energy Mater., 2020, 3, 12108–12114 CrossRef CAS.
- E. B. Hemming, A. F. Masters and T. Maschmeyer, Aust. J. Chem., 2020, 73, 1296–1300 CrossRef CAS.
- N. Yang, L. Lu, L. Zhu, P. Wu, D. Tao, X. Li, J. Gong, L. Chen, Y. Chao and W. Zhu, Inorg. Chem. Front., 2022, 9, 165–178 RSC.
- J. Yin, W. D. Fu, J. R. Zhang, X. Y. Liu, X. M. Zhang, C. Wang, J. He, W. Jiang, H. P. Li and H. M. Li, Chem. Eng. J., 2023, 470, 144290 CrossRef CAS.
- J. Yin, W. Fu, J. Zhang, X. Zhang, W. Qiu, W. Jiang, L. Zhu, H. Li and H. Li, Chem. Eng. J., 2024, 492, 152349 CrossRef CAS.
- Y. Zhao, Z. Yang, B. Yu, H. Zhang, H. Xu, L. Hao, B. Han and Z. Liu, Chem. Sci., 2015, 6, 2297–2301 RSC.
- X. Jiao, K. Zheng, L. Liang, X. Li, Y. Sun and Y. Xie, Chem. Soc. Rev., 2020, 49, 6592–6604 RSC.
- Y. Belmabkhout, N. Heymans, G. De Weireld and A. Sayari, Energy Fuels, 2011, 25, 1310–1315 CrossRef CAS.
- Y. Belmabkhout, P. M. Bhatt, K. Adil, R. S. Pillai, A. Cadiau, A. Shkurenko, G. Maurin, G. Liu, W. J. Koros and M. Eddaoudi, Nat. Energy, 2018, 3, 1059–1066 CrossRef CAS.
- G. Liu, A. Cadiau, Y. Liu, K. Adil, V. Chernikova, I. D. Carja, Y. Belmabkhout, M. Karunakaran, O. Shekhah and C. Zhang, Angew. Chem., Int. Ed., 2018, 57, 14811–14816 CrossRef CAS PubMed.
- E. S. Grape, J. G. Flores, T. Hidalgo, E. Martínez-Ahumada, A. Gutiérrez-Alejandre, A. Hautier, D. R. Williams, M. O'Keeffe, L. Öhrström and T. Willhammar, J. Am. Chem. Soc., 2020, 142, 16795–16804 CrossRef CAS PubMed.
- Y. Peng, J. N. Kheir and B. D. Polizzotti, Chem.–Eur. J., 2018, 24, 18820–18829 CrossRef CAS PubMed.
- H. U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner and M. Studer, Adv. Synth. Catal., 2003, 345, 103–151 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- E. B. Hemming, A. F. Masters and T. Maschmeyer, Chem. Commun., 2019, 55, 11179–11182 RSC.
- S. J. Roseblade and A. Pfaltz, Acc. Chem. Res., 2007, 40, 1402–1411 CrossRef CAS PubMed.
- A. V. Astakhov, O. V. Khazipov, A. Y. Chernenko, D. V. Pasyukov, A. S. Kashin, E. G. Gordeev, V. N. Khrustalev, V. M. Chernyshev and V. P. Ananikov, Organometallics, 2017, 36, 1981–1992 CrossRef CAS.
- J. Sherwood, J. H. Clark, I. J. Fairlamb and J. M. Slattery, Green Chem., 2019, 21, 2164–2213 RSC.
- M. J. Climent, A. Corma and S. Iborra, Chem. Rev., 2011, 111, 1072–1133 CrossRef CAS PubMed.
- I. Wheeldon, S. D. Minteer, S. Banta, S. C. Barton, P. Atanassov and M. Sigman, Nat. Chem., 2016, 8, 299–309 CrossRef CAS PubMed.
- Z. Jia, K. Wang, B. Tan and Y. Gu, ACS Catal., 2017, 7, 3693–3702 CrossRef CAS.
- M. A. Isaacs, C. M. Parlett, N. Robinson, L. J. Durndell, J. C. Manayil, S. K. Beaumont, S. Jiang, N. S. Hondow, A. C. Lamb and D. Jampaiah, Nat. Catal., 2020, 3, 921–931 CrossRef CAS.
- Y. Zhao, H. Zhou, X. Zhu, Y. Qu, C. Xiong, Z. Xue, Q. Zhang, X. Liu, F. Zhou and X. Mou, Nat. Catal., 2021, 4, 134–143 CrossRef CAS.
- W. Jiang, H. Jia, H. Li, L. Zhu, R. Tao, W. Zhu, H. Li and S. Dai, Green Chem., 2019, 21, 3074–3080 RSC.
- W. Jiang, X. An, J. Xiao, Z. Yang, J. Liu, H. Chen, H. Li, W. Zhu, H. Li and S. Dai, ACS Catal., 2022, 12, 8623–8631 CrossRef CAS.
- Y. Song, E. Ozdemir, S. Ramesh, A. Adishev, S. Subramanian, A. Harale, M. Albuali, B. A. Fadhel, A. Jamal, D. Moon, S. H. Choi and C. T. Yavuz, Science, 2020, 367, 777–781 CrossRef CAS PubMed.
- L. Chen, Z. Song, S. Zhang, C.-K. Chang, Y.-C. Chuang, X. Peng, C. Dun, J. J. Urban, J. Guo, J.-L. Chen, D. Prendergast, M. Salmeron, G. A. Somorjai and J. Su, Science, 2023, 381, 857–861 CrossRef CAS PubMed.
- W. Xiao and D. Wang, Chem. Soc. Rev., 2014, 43, 3215–3228 RSC.
- M. Markert, M. Mulzer, B. Schetter and R. Mahrwald, J. Am. Chem. Soc., 2007, 129, 7258–7259 CrossRef CAS PubMed.
- C.-B. Miao, M.-L. Chen, X.-Q. Sun and H.-T. Yang, Synthesis, 2019, 51, 2945–2958 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |