
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pd-incorporated polyoxometalate catalysts for electrochemical CO2 reduction†
Kimitake
Kawakami
a,
Tomohiro
Yabe
 a,
Fumiaki
Amano
b,
Kazuya
Yamaguchi
a and
Kosuke
Suzuki
*a
a,
Fumiaki
Amano
b,
Kazuya
Yamaguchi
a and
Kosuke
Suzuki
*a
aDepartment of Applied Chemistry, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan. E-mail: ksuzuki@appchem.t.u-tokyo.ac.jp
bDepartment of Applied Chemistry for Environment, Graduate School of Urban Environmental Sciences, Tokyo Metropolitan University, 1-1 Minami-Osawa, Hachioji, Tokyo 192-0397, Japan
First published on 19th August 2024
Abstract
Polyoxometalates (POMs), representing anionic metal–oxo clusters, display diverse properties depending on their structures, constituent elements, and countercations. These characteristics position them as promising catalysts or catalyst precursors for electrochemical carbon dioxide reduction reaction (CO2RR). This study synthesized various salts—TBA+ (tetra-n-butylammonium), Cs+, Sr2+, and Ba2+—of a dipalladium-incorporated POM (Pd2, [γ-H2SiW10O36Pd2(OAc)2]4−) immobilized on a carbon support (Pd2/C). The synthesized catalysts—TBAPd2/C, CsPd2/C, SrPd2/C, and BaPd2/C—were deposited on a gas-diffusion carbon electrode, and the CO2RR performance was subsequently evaluated using a gas-diffusion flow electrolysis cell. Among the catalysts tested, BaPd2/C exhibited high selectivity toward carbon monoxide (CO) production (ca. 90%), while TBAPd2/C produced CO and hydrogen (H2) with moderate selectivity (ca. 40% for CO and ca. 60% for H2). Moreover, BaPd2/C exhibited high selectivity toward CO production over 12 h, while palladium acetate, a precursor of Pd2, showed a significant decline in CO selectivity during the CO2RR. Although both BaPd2/C and TBAPd2/C transformed into Pd nanoparticles and WOx nanospecies during the CO2RR, the influence of countercations on their product selectivity was significant. These results highlight that POMs and their countercations can effectively modulate the catalytic performance of POM-based electrocatalysts in CO2RR.
Introduction
Anthropogenic carbon dioxide (CO2) emissions are widely recognized as major contributors to recent global warming, requiring prompt and effective mitigation strategies.1 In this context, the electrochemical CO2 reduction reaction (CO2RR) stands out as a promising solution, attracting considerable attention from both academia and industry.2 Utilizing multiple electrons and protons, the CO2RR process electrochemically converts CO2 into numerous valuable chemicals and fuels, such as carbon monoxide (CO), methanol, and ethylene (C2H4).3 Among these products, CO holds particular importance as a component of syngas, a vital feedstock for hydrocarbon synthesis via methanol or Fischer–Tropsch processes.4 Notably, the selectivity of the CO2RR is significantly influenced by the metals used in the electrocatalysts, with Au, Ag, and Zn demonstrating high selectivity toward CO production.5 In addition to these, Pd can also produce CO with high selectivity; however, its efficiency is considerably influenced by the applied potential and the state of the Pd catalyst. For instance, with increasing overpotential, the primary CO2RR product of Pd changes from formate (HCOO−) to CO, and smaller Pd nanoparticles (NPs) produce CO with greater selectivity.6,7 However, given the widespread use of aqueous electrolytes for the CO2RR, the competing hydrogen evolution reaction (HER) is inevitable,8 and the resulting degradation in catalytic performance emerges as a significant concern.4 Hence, developing robust CO2RR catalysts capable of selectively producing specific products is critical.Previous studies have extensively investigated the effects of alkali metal cations in electrolytes on the CO2RR. Based on their findings, these cations have been widely recognized to play critical roles in driving the CO2RR,9 with larger cations such as K+ and Cs+ demonstrating greater productivity for CO, HCOO−, and C2H4 compared to smaller cations such as Li+.9–19 Conversely, studies conducted by Schizodimou and Kyriacou have demonstrated higher CO2RR activity for electrolytes containing multivalent cations, particularly at low overpotentials.20 Meanwhile, other reports have suggested that multivalent cations, such as alkaline earth metal cations, enhance the partial current density of CO.14 Notably, electrolytes with small amounts of Ba2+ have been observed to reduce HER activity while increasing CO2RR activity at high overpotentials.17 Conversely, Bhargava et al. have reported that electrolytes containing substantial amounts of CaCl2 and BaCl2 compromise the CO2RR performance of Ag.16 This diminished performance is attributed to the coverage of active sites by the deposited oxides, hydroxides, and other compounds of alkaline earth metals. Beyond these factors, the practical application of alkaline earth metal salts as electrolytes is further constrained by their lower solubility in water,12 often necessitating excessive quantities for effective use, and their higher cost compared to corresponding alkali metal salts. Consequently, developing systems that minimize the use of alkaline earth metal salts while demonstrating optimal CO2RR performance is essential.
Polyoxometalates (POMs), representing anionic metal–oxo clusters comprising multiple {MOx} units (M = W, Mo, V, etc.), exhibit diverse properties depending on their structures and constituent elements.21–32 These attributes position POMs as promising catalysts and catalyst precursors for various electrochemical and photochemical reactions including CO2RR.30,33–38 Among the known varieties of POMs, particularly noteworthy are lacunary POMs, which lack one or more {MOx} units and possess reactive oxygen atoms, thus functioning as inorganic multidentate ligands.39,40 Our research group has pioneered techniques for synthesizing well-defined multinuclear metal-incorporated POMs by reacting tetra-n-butylammonium (TBA+) salts of lacunary POMs with metal ions in organic solvents.40–43 These metal-incorporated POMs have been demonstrated to serve as promising catalysts44–48 or catalyst precursors49 for various reactions. For instance, we utilized a TBA+ salt of a diiron-incorporated POM (TBAFe2, TBA8[H4(SiW10O36)2Fe2O]) supported on silica (TBAFe2/SiO2) as a catalyst precursor for methane oxidation, selectively obtaining formaldehyde and CO.49 Interestingly, tungsten-oxide nanoclusters derived from TBAFe2 protected in situ-formed FeOx subnanoclusters, thereby avoiding catalyst deactivation and product oxidation. Thus, metal-incorporated POMs hold immense potential for developing robust CO2RR catalysts capable of synthesizing specific products with high selectivity.
Notably, the countercations of POMs not only balance their anionic charges but also substantially influence their solubility and physicochemical properties.50,51 Typically, W-, Mo-, and V-based POMs exhibit reduced solubility in water when larger countercations such as Cs+ and Ba2+ are incorporated. Leveraging this characteristic, Blasco-Ahicart et al. used Cs+ and Ba2+ salts of nonacobalt-incorporated POMs as heterogeneous electrocatalysts for the oxygen evolution reaction in a 1 M H2SO4 aqueous solution.52 Thus, we envisaged that selecting appropriate countercations of POMs holds significant promise for developing robust and selective catalysts for the CO2RR.
Building upon these insights, this study is the first to investigate the CO2RR performance of Pd-incorporated POMs and the effect of their countercations. Specifically, a TBA+ salt of a dipalladium-incorporated POM (Pd2, [γ-H2SiW10O36Pd2(OAc)2]4−, Fig. 1a)44 immobilized on a carbon support (TBAPd2/C) was first synthesized. Subsequently, to explore the influence of different countercations, various analogs—CsPd2/C, SrPd2/C, and BaPd2/C—were prepared by exchanging TBA+ with alkali metal or alkaline earth metal cations (Cs+, Sr2+, or Ba2+) in an organic solvent (Fig. 1b). These catalysts were then deposited on a gas-diffusion carbon electrode, and CO2RR was conducted using a gas-diffusion flow electrolysis cell containing a 1 M potassium bicarbonate (KHCO3) aqueous solution. Our results revealed that BaPd2/C exhibited the highest selectivity toward CO production (ca. 90%) and maintained its catalytic performance over 12 h. Conversely, TBAPd2/C produced CO and hydrogen (H2) with moderate selectivity (ca. 40% for CO and ca. 60% for H2) (Fig. 1b). Subsequent analyses of the electrodes after participating in the CO2RR revealed the transformation of both BaPd2/C and TBAPd2/C into Pd NPs and WOx nanospecies. Furthermore, Pd acetate (Pd(OAc)2), a precursor of Pd2, showed a significant decline in CO selectivity during the CO2RR. These results reveal the significant influence of POMs and their countercations on the performance of POM-based electrocatalysts for the CO2RR.
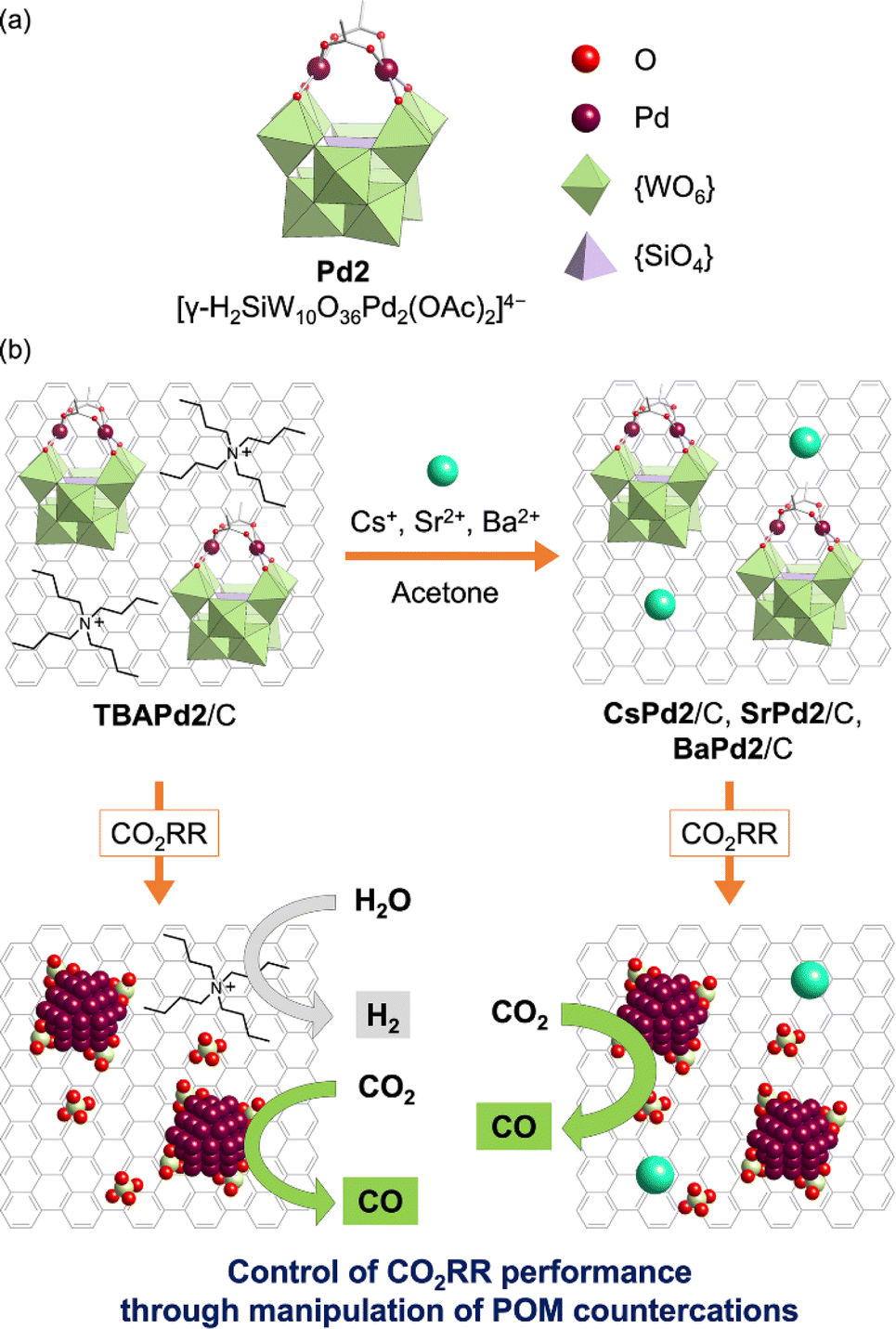 | ||
| Fig. 1 Preparation of POM-based electrocatalysts and their application in electrochemical CO2RR. (a) Structure of Pd2 ([γ-H2SiW10O36Pd2(OAc)2]4−). (b) Schematic depicting the preparation procedure of CsPd2/C, SrPd2/C, and BaPd2/C through manipulation of countercations of TBAPd2/C and their applications in electrochemical CO2RR. | ||
Results and discussion
Preparation and characterization of catalysts
Initially, TBAPd2 (Fig. 1a)44 was immobilized on a carbon support by mixing an acetone solution of TBAPd2 with an ethyl acetate suspension of the carbon support, yielding TBAPd2/C (Fig. 1b).53 Subsequently, cation exchange was performed by immersing TBAPd2/C in an acetone solution containing trifluoromethanesulfonate salts of Cs+, Sr2+, and Ba2+ to obtain CsPd2/C, SrPd2/C, and BaPd2/C, respectively (Fig. 1b, see ESI† for the detailed procedure).54 This approach of conducting cation exchange on a carbon support was preferred over direct immobilization of Cs+, Sr2+, and Ba2+ salts of Pd2 (i.e., CsPd2, SrPd2, and BaPd2) owing to the low solubility of the resulting POMs in water and organic solvents,50 which complicates their direct immobilization.Following synthesis, the prepared catalysts were characterized through X-ray absorption fine structure (XAFS) measurements. Notably, the Pd K-edge and W L3-edge extended X-ray absorption fine structure (EXAFS) oscillation patterns, as well as the Fourier-transformed EXAFS spectra, of TBAPd2/C closely resembled those of TBAPd2 (Fig. 2), suggesting that the anion structure of TBAPd2 remained intact upon immobilization on the carbon support. Subsequently, the EXAFS spectra of the cation-exchanged samples were similarly examined. As depicted in Fig. 2a, c, S1a and Table S1,† although the Fourier-transformed Pd K-edge EXAFS spectrum of BaPd2/C presented a lower intensity of scattering from the Pd–O bond compared to that of TBAPd2/C, the Pd K-edge EXAFS oscillation pattern of BaPd2/C closely resembled that of TBAPd2/C. Similarly, the W L3-edge EXAFS oscillation pattern of BaPd2/C closely matched that of TBAPd2/C (Fig. 2b). However, compared to the Fourier-transformed W L3-edge EXAFS spectrum of TBAPd2/C, that of BaPd2/C displayed increased scattering intensity from the terminal W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (peak around 1.3 Å)55 and decreased scattering intensity from the W–O(–W) bond (bonds between tungsten atoms and the bridging oxygen atom, with a peak around 1.7 Å)55 (Fig. 2d, S1b and Table S1†). These findings suggest that although the coordination environment of Pd and W partially changed during the cation exchange process, the fundamental structure of Pd2 was intrinsically maintained.
O bond (peak around 1.3 Å)55 and decreased scattering intensity from the W–O(–W) bond (bonds between tungsten atoms and the bridging oxygen atom, with a peak around 1.7 Å)55 (Fig. 2d, S1b and Table S1†). These findings suggest that although the coordination environment of Pd and W partially changed during the cation exchange process, the fundamental structure of Pd2 was intrinsically maintained.
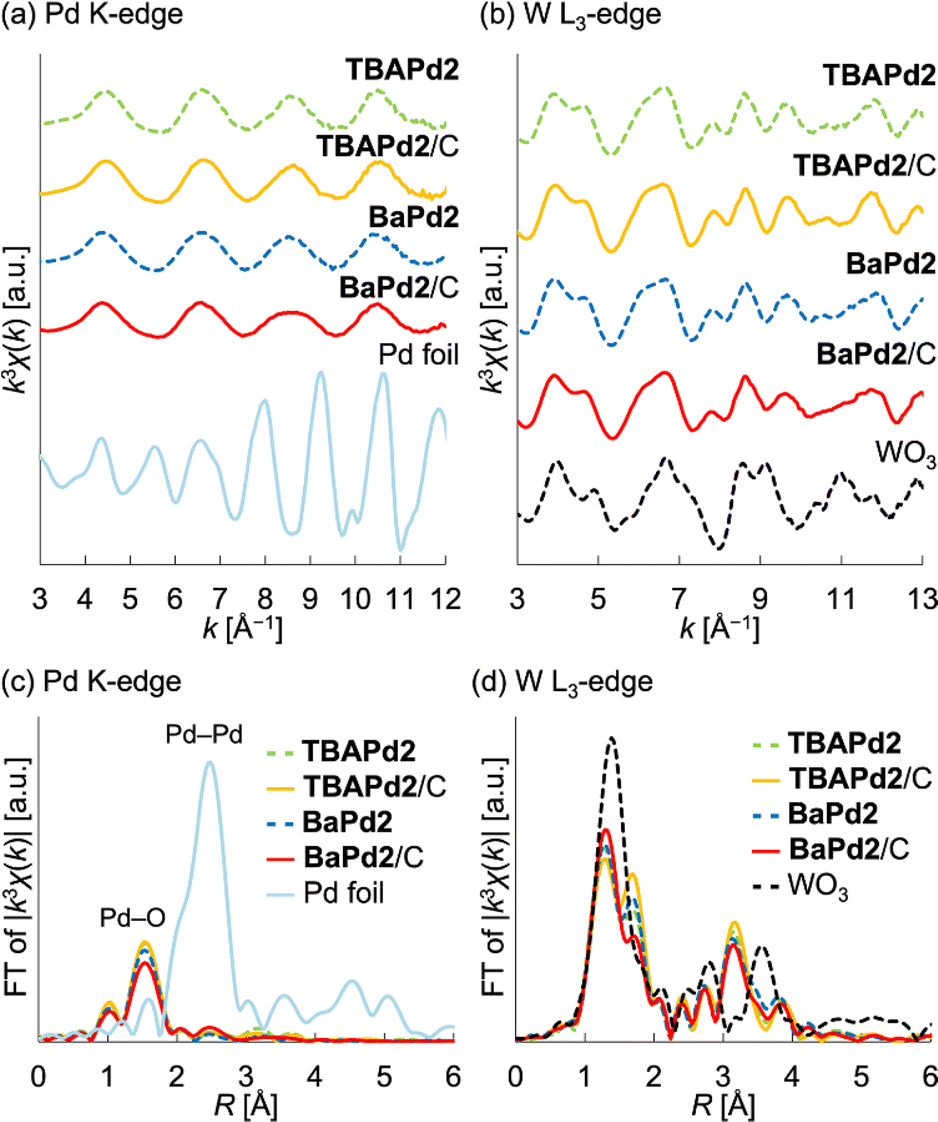 | ||
| Fig. 2 XAFS characterizations of TBAPd2/C and BaPd2/C. (a) k3-weighted Pd K-edge EXAFS oscillations (k = 3–12 Å−1). (b) k3-weighted W L3-edge EXAFS oscillations (k = 3–13 Å−1). (c) Fourier-transformed Pd K-edge EXAFS spectra. (d) Fourier-transformed W L3-edge EXAFS spectra. | ||
Based on these XAFS findings, we further investigated the structure of Pd2 during cation exchange by subjecting TBAPd2/C and BaPd2/C to infrared (IR) measurements. However, IR peaks originating from the POMs were difficult to discern owing to interference from the carbon support. To resolve this, we subsequently analyzed the IR spectra of TBAPd2 and BaPd2 without their carbon supports (Fig. S2†). Specifically, BaPd2 was synthesized through the cation exchange of TBAPd2 with Ba2+. Interestingly, the IR spectrum of BaPd2 displayed no peak corresponding to TBA+ in the range of 2800–3000 cm−1, indicating successful cation exchange. Furthermore, the IR spectrum of BaPd2 in the range of 300–1000 cm−1 closely resembled that of TBAPd2. Given that this region is characteristic of Keggin-type POMs,56 the cation exchange was deemed to not adversely affect the anion structure of Pd2.
Our next investigation focused on the elemental analysis of the prepared catalysts. As indicated in Table S2,† the ratio of Pd to W remained nearly constant at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 across all samples, consistent with the composition of TBAPd2, containing two Pd atoms and ten W atoms. Hence, this finding further corroborates the results of the EXAFS analysis, indicating that the structure of Pd2 is largely preserved during immobilization on the carbon support and cation exchange. Furthermore, our elemental analysis revealed that the numbers of Cs, Sr, and Ba species in CsPd2/C, SrPd2/C, and BaPd2/C were approximately four, two, and two, respectively. Given that Pd2 is a tetravalent anion ([γ-H2SiW10O36Pd2(OAc)2]4−) and Cs+, Sr2+, and Ba2+ are monovalent, divalent, and divalent, respectively, these results suggest that most of the TBA+ was exchanged with the corresponding alkali metal and alkaline earth metal cations.
:10 across all samples, consistent with the composition of TBAPd2, containing two Pd atoms and ten W atoms. Hence, this finding further corroborates the results of the EXAFS analysis, indicating that the structure of Pd2 is largely preserved during immobilization on the carbon support and cation exchange. Furthermore, our elemental analysis revealed that the numbers of Cs, Sr, and Ba species in CsPd2/C, SrPd2/C, and BaPd2/C were approximately four, two, and two, respectively. Given that Pd2 is a tetravalent anion ([γ-H2SiW10O36Pd2(OAc)2]4−) and Cs+, Sr2+, and Ba2+ are monovalent, divalent, and divalent, respectively, these results suggest that most of the TBA+ was exchanged with the corresponding alkali metal and alkaline earth metal cations.
CO2RR study
After characterizing the prepared catalysts, we evaluated their catalytic performance in the electrochemical CO2RR using a gas-diffusion flow electrolysis cell (Fig. S3†).57,58 The working electrodes for the CO2RR were fabricated by depositing TBAPd2/C, CsPd2/C, SrPd2/C, and BaPd2/C onto a gas-diffusion carbon electrode (Sigracet 39 BB, see ESI† for the detailed method). These electrodes were then put in the gas-diffusion flow electrolysis cell, and their catalytic performance was assessed via constant potential electrolysis at approximately −0.8 VRHE for 1 h, using a 1 M KHCO3 aqueous solution as the electrolyte. As illustrated in Fig. 3a and S4 and Table S3,†TBAPd2/C exhibited a faradaic efficiency for CO production (FECO) of 40.5 ± 7.7% and a total current density of 27.4 ± 1.1 mA cm−2. Conversely, CsPd2/C, SrPd2/C, and BaPd2/C displayed FECO values of 72.9 ± 0.1%, 78.9 ± 2.5%, and 92.9 ± 4.8%, respectively. Furthermore, the total current densities for CsPd2/C, SrPd2/C, and BaPd2/C were 26.8 ± 1.2, 27.2 ± 0.8, and 24.3 ± 0.6 mA cm−2, respectively. These findings indicate significant enhancements in CO2RR performance following cation exchange from TBA+ to Cs+, Sr2+, and Ba2+, with BaPd2/C demonstrating the highest efficiency. Collectively, these results underscore the critical significance of countercation identity in catalytic performance, as well as the superior efficiency of BaPd2/C for the CO2RR. Encouraged by these outcomes, we subsequently compared the catalytic performance of BaPd2/C with that of Pd(OAc)2, a precursor of Pd2, deposited on a gas-diffusion carbon electrode in a similar manner to the other catalysts, at the same Pd content (Fig. 3a). The results revealed that Pd(OAc)2 exhibited lower FECO (83.9 ± 3.6%) and lower total current density (11.66 ± 0.04 mA cm−2) compared to BaPd2/C (FECO 92.9 ± 4.8%, 24.3 ± 0.6 mA cm−2). Although various reports have addressed CO2RR using POM-containing electrodes in aqueous electrolytes, there have been no reports on CO2RR using Pd-incorporated POMs or the effect of the countercations of POMs on CO2RR (Table S8†).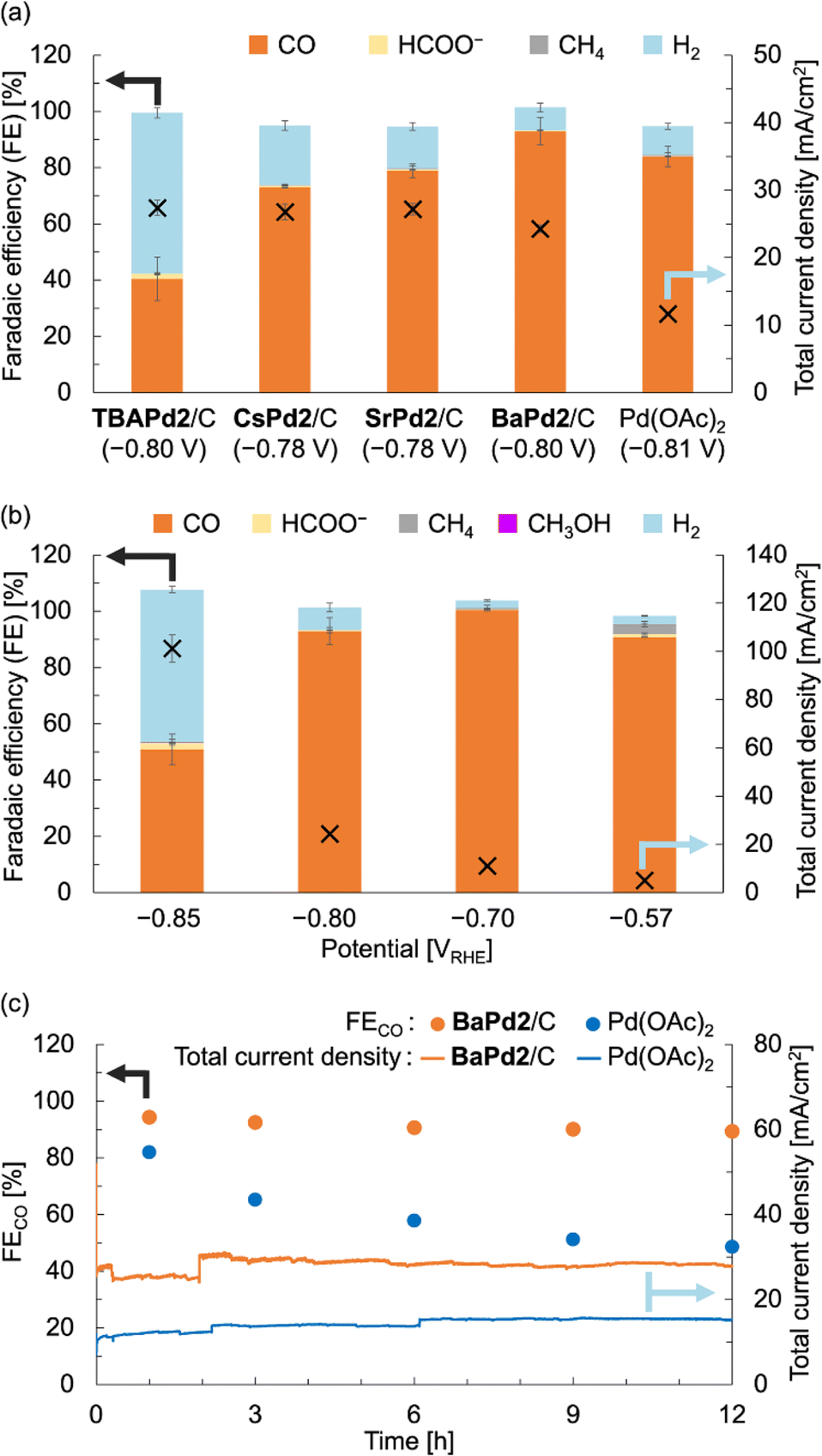 | ||
| Fig. 3 CO2RR performance of BaPd2/C-modified electrode and other electrodes in a 1 M KHCO3 aqueous solution. (a) FEs for all CO2RR products (left axis) and total current densities (right axis) of TBAPd2/C-, CsPd2/C-, SrPd2/C-, BaPd2/C-, and Pd(OAc)2-modified electrodes determined through constant potential electrolysis at −0.8 VRHE for 1 h. (b) FEs for all CO2RR products (left axis) and total current density (right axis) of BaPd2/C-modified electrode determined through constant potential electrolysis at different potentials for 1 h. (c) FECO (left axis) and total current density (right axis) of BaPd2/C-modified electrode (−0.75 VRHE, orange) and Pd(OAc)2-modified electrode (−0.73 VRHE, light blue) for 12 h. Error bars represent standard deviations calculated from two independent measurements. Symbols X in (a and b) denote the total current density (right vertical axes). All potentials were IR-corrected (see ESI†). | ||
Subsequently, CO2RR experiments were performed utilizing the BaPd2/C-modified electrode at various applied potentials to elucidate the potential dependence of its catalytic performance. Although the CO2RR experiments at −0.70 and −0.57 VRHE exhibited reduced total current densities compared to the reaction at −0.80 VRHE, CO was exclusively produced at each potential (Fig. 3b and Table S4†). However, reducing the potential to −0.85 VRHE resulted in a decrease in FECO and an increase in a faradaic efficiency for H2 production (FEH2) to nearly 50%. We also confirmed that CO and HCOO− were not produced in reactions using an electrode without Pd2 deposition or in reactions conducted in an Ar atmosphere (Table S5†).
To assess the long-term stability of the catalyst, a CO2RR experiment employing BaPd2/C as the catalyst was performed over 12 h at an applied potential of −0.75 VRHE (Fig. 3c and Table S6†). Note that due to the instability of Sigracet 39 BB in prolonged CO2RR, AvCarb P75T was used as a carbon electrode to prepare a BaPd2/C-modified electrode in this experiment. As depicted in Fig. 3c, although the total current density of the catalyst fluctuated during the reaction, its selectivity toward CO production was maintained consistently throughout the experiment. In contrast, a 12 h CO2RR experiment using Pd(OAc)2 as the catalyst exhibited a significant decline in FECO during the reaction, accompanied by an increase in FEH2 (Fig. 3c and Table S7†). The total current density in this case was stable around 14 mA cm−2 throughout the reaction, approximately half that of BaPd2/C. These results highlight the significant influence of POMs for the CO2RR performance.
Characterization of a BaPd2/C-modified electrode after the CO2RR
Based on the results of the CO2RR experiments, we performed XAFS measurements to probe the structure and electronic state of a BaPd2/C-modified electrode obtained after a 1 h CO2RR at −0.80 VRHE. To investigate the electronic state of the Pd species, we measured the Pd K-edge X-ray absorption near-edge structure (XANES) spectra of the electrode before and after the CO2RR (Fig. 4a). Notably, the XANES spectrum of the electrode before the reaction was intermediate to the corresponding spectra of a Pd foil and BaPd2/C. Conversely, the XANES spectrum of the electrode after the reaction closely resembled that of the Pd foil. These findings indicate that Pd2+ underwent partial reduction during electrode fabrication and complete reduction to Pd0 during the CO2RR. Subsequently, to glean insights into the structure of the POM before and after the reaction, we performed Pd K-edge and W L3-edge EXAFS analysis (Fig. 4b–e). Notably, the Pd K-edge EXAFS oscillation pattern of the electrode after the CO2RR resembled that of the Pd foil but not that of BaPd2/C (Fig. 4b). Furthermore, the Fourier-transformed Pd K-edge EXAFS spectra revealed that the electrode after the CO2RR exhibited reduced scattering intensity from the Pd–O bond and increased scattering intensity from the Pd–Pd bond compared to that before the reaction (Fig. 4d). These results suggest the structural transformation of BaPd2 to Pd NPs during the reaction. Conversely, the W L3-edge EXAFS analysis of the electrode after the reaction displayed a similar yet partially distinct oscillation pattern compared to that of WO3 (Fig. 4c). Furthermore, compared to the Fourier-transformed W L3-edge EXAFS spectra of WO3, that of the electrode after the CO2RR displayed differences around the second coordination sphere (Fig. 4e).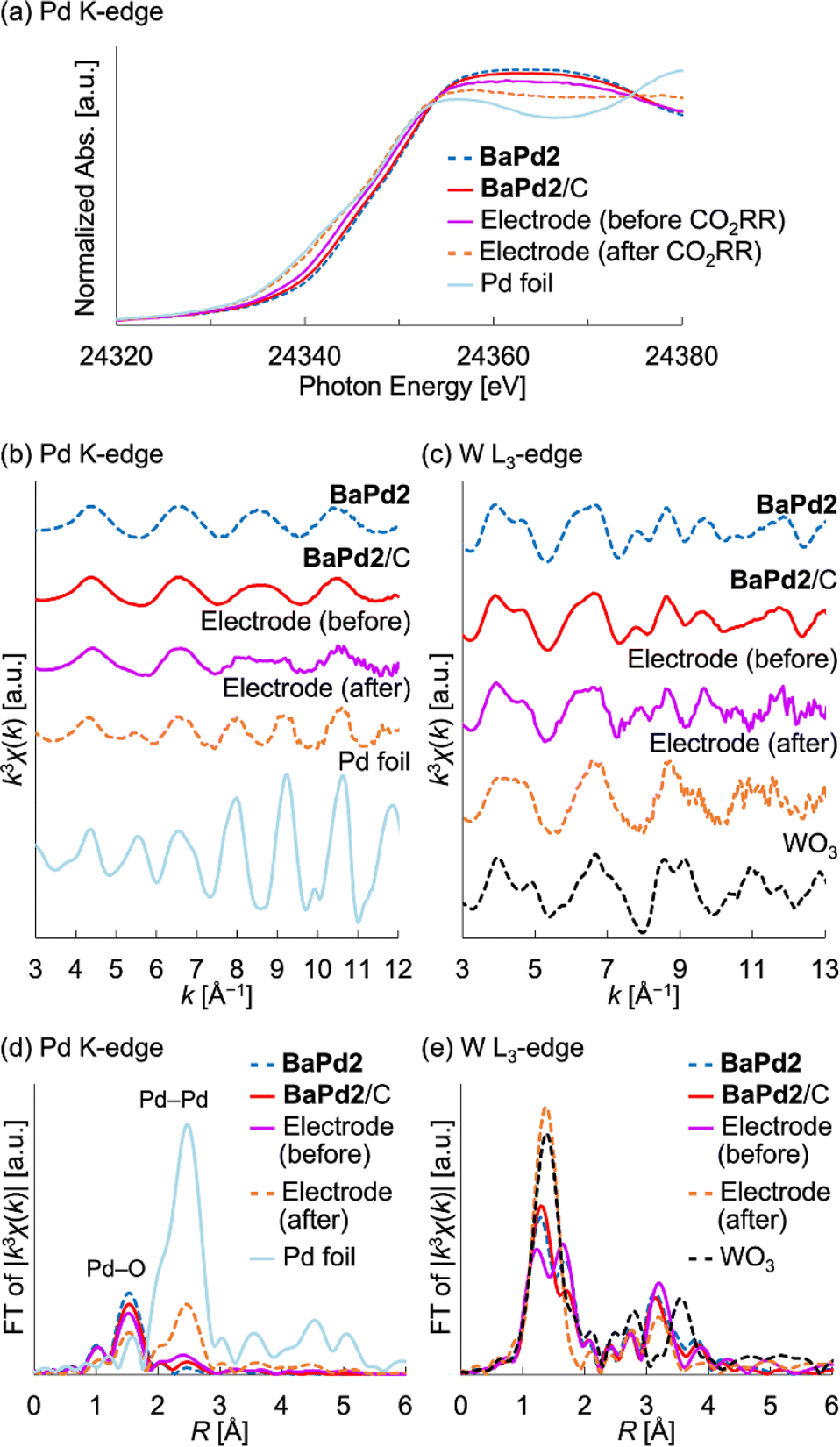 | ||
| Fig. 4 XAFS characterization of the BaPd2/C-modified electrode obtained after a 1 h CO2RR. (a) Pd K-edge XANES spectra. (b) k3-weighted Pd K-edge EXAFS oscillation pattern (k = 3–12 Å−1). (c) k3-weighted W L3-edge EXAFS oscillation pattern (k = 3–13 Å−1). (d) Fourier-transformed Pd K-edge EXAFS spectra. (e) Fourier-transformed W L3-edge EXAFS spectra. | ||
For comparison, we also conducted XAFS measurements on a Pd(OAc)2-modified electrode subjected to a 1 h CO2RR (Fig. S5 and S6†). Notably, the Pd K-edge XANES spectrum of the electrode before the reaction was intermediate to those of the Pd foil and Pd(OAc)2, whereas after the CO2RR, it closely resembled that of the Pd foil (Fig. S5†). Similarly, the Pd K-edge EXAFS oscillation pattern of the electrode after the CO2RR resembled that of the Pd foil but not that of Pd(OAc)2 (Fig. S5†). Furthermore, the Fourier-transformed Pd K-edge EXAFS spectrum of the Pd(OAc)2-modified electrode after the CO2RR exhibited a similar intensity of scattering from the Pd–O bond but a greater intensity of scattering from the Pd–Pd bond compared to the BaPd2/C-modified electrode after the reaction (Fig. S6†). These results suggest that the Pd NPs transformed from Pd(OAc)2 were larger than those formed from BaPd2/C after the CO2RR.
Based on these findings, we subjected the BaPd2/C-modified electrode obtained after the CO2RR to electron microscopy observations. Notably, transmission electron microscopy (TEM) images of the BaPd2/C after the CO2RR revealed the formation of NPs with an average particle size of 4.0 ± 1.3 nm on the carbon support (Fig. 5a and b). Conversely, larger NPs with a broader size distribution (an average particle size of 6.0 ± 3.1 nm) were formed on the Pd(OAc)2-modified electrode after the CO2RR (Fig. S7†), consistent with the XAFS results. Subsequently, high-angle annular dark-field scanning TEM (HAADF-STEM) images (Fig. 5c) and STEM-energy-dispersive spectroscopy (EDS) mappings (Fig. 5d–g) of the BaPd2/C after the reaction revealed that most Pd NPs were surrounded by W atoms, with some W species present in locations where Pd NPs were absent. This observation also confirms the retention of Ba on the electrode after the CO2RR. Collectively, these results indicate that BaPd2 transformed into WOx nanospecies and Pd NPs surrounded by WOx nanospecies during the reaction. Adopting a similar approach, our research group previously developed various metal NPs stabilized by lacunary POMs.59–61 Furthermore, previous reports suggest that WOx nanoclusters derived from diiron-incorporated POMs hinder the aggregation of in situ-formed FeOx subnanoclusters.49 Consequently, it is plausible that WOx nanospecies prevented the aggregation of Pd NPs in our CO2RR system.
 | ||
| Fig. 5 TEM and HAADF-STEM images of the BaPd2/C-modified electrode obtained after a 1 h CO2RR. (a) TEM image and (b) the corresponding size-distribution histogram. (c) HAADF-STEM image and (d–g) the corresponding STEM-EDS mappings (Pd, pink; W, turquoise; Ba, yellow). | ||
Effect of countercations on the CO2RR
To examine the impact of countercations on CO2RR performance, we characterized the TBAPd2/C-modified electrode subjected to a 1 h CO2RR at −0.80 VRHE, which exhibited poor CO2RR performance compared to the BaPd2/C-modified electrode (Fig. 3a and Table S3†). Notably, the Pd K-edge and W L3-edge EXAFS oscillation and Fourier-transformed EXAFS spectra of the TBAPd2/C-modified electrode after the CO2RR closely resembled those of the BaPd2/C-modified electrode after the reaction (Fig. S6 and S8†). Furthermore, the TEM image of the TBAPd2/C after the CO2RR revealed the formation of NPs with an average particle size of 2.9 ± 1.1 nm on the carbon support (Fig. S9a and S9b†). Similarly, the HAADF-STEM image (Fig. S9c†) and STEM-EDS mappings (Fig. S9d–9f†) of the TBAPd2/C after the CO2RR were nearly identical to those of the BaPd2/C after the CO2RR (Fig. 5). These results indicate that similar species were formed after the CO2RR regardless of the countercation type.An explanation for the enhanced CO production could be related to the electronic state of the Pd species. For instance, electron-rich Pd species modified with N-heterocyclic carbene ligands have been reported to demonstrate superior CO2RR performance.62,63 To probe the electronic states of the Pd species, we performed CO stripping voltammetry experiments on the TBAPd2/C-, CsPd2/C-, SrPd2/C-, and BaPd2/C-modified electrodes after the CO2RR (Fig. S10, see ESI† for additional details). Notably, the CO stripping potential of the TBAPd2/C-modified electrode was more positive than those of the CsPd2/C-, SrPd2/C-, and BaPd2/C-modified electrodes, indicating the presence of more electron-rich Pd NPs in the TBAPd2/C-modified electrode.62 Considering that POMs can donate electrons to metal NPs depending on their anion charges, as evidenced by the findings of our previous studies,59–61 the observed varying electronic states of Pd NPs likely stem from the differential ability of WOx nanospecies to donate electrons to Pd NPs depending on the countercations. However, despite exhibiting a more electron-rich state, Pd NPs derived from the TBAPd2/C-modified electrode displayed poor CO2RR performance (Fig. 3a and Table S3†), indicating that the electronic state of Pd NPs does not critically influence the CO2RR performance of our system.
Another plausible explanation for the enhanced CO production could be related to the cationic effect. Generally, in the CO2RR, electrolytes containing larger cations often demonstrate higher productivity for various CO2RR products such as CO.9–19 Several theories have been proposed to elucidate the origin of this cationic effect, including (i) modulation of the electric field near the electrode surface, (ii) pH buffering at the electrode interface, and (iii) stabilization of reaction intermediates.9,64–67 For instance, Jiang et al. recently investigated the CO2RR performance of Pd NPs formed on a carbon support using electrolytes containing various alkali metal cations. As anticipated, they observed higher FECO values for electrolytes containing larger cations, and they attributed these findings to the intensified electric field near the electrode surface.19 Although the primary influencing factor in our system remains unidentified at this stage, we propose that the enhanced CO production observed for CsPd2/C, SrPd2/C, and BaPd2/C may be attributed to the abovementioned cationic effect caused by the POM countercations.
Conclusions
In conclusion, this study highlighted the significant impact of the countercations of POM-based electrocatalysts on their catalytic performance in the CO2RR. Specifically, TBAPd2/C, synthesized by immobilizing TBAPd2 on a carbon support, was compared with its analogs CsPd2/C, SrPd2/C, and BaPd2/C, synthesized by exchanging the TBA+ cations of TBAPd2/C with alkali metal and alkaline earth metal cations (Cs+, Sr2+, or Ba2+) in acetone. Assessments of their catalytic CO2RR performance in a 1 M KHCO3 aqueous solution using a flow electrolysis cell with gas-diffusion carbon electrodes modified with these catalysts revealed that the BaPd2/C-modified electrode exhibited selective CO production over 12 h, whereas the TBAPd2/C-modified electrode exhibited lower CO selectivity owing to inadequate suppression of the HER. Detailed analyses indicated that both TBAPd2 and BaPd2 transformed into Pd NPs and WOx nanospecies during the CO2RR, highlighting the significant influence of countercations on the reactivity of the electrocatalyst. Moreover, Pd(OAc)2 demonstrated a marked decline in the CO selectivity during the CO2RR, and the formation of larger Pd NPs with a broader size distribution than BaPd2 was observed after the CO2RR. These results highlight the critical role of POMs and their countercations in modulating the CO2RR performance of POM-based electrocatalysts. As POMs continue to demonstrate promise in CO2RR applications, we believe that our findings will contribute to the development of highly effective and durable POM-based electrocatalysts for future CO2RR applications.Data availability
The data supporting this manuscript is available in the ESI† and available on request.Author contributions
K. K. performed the synthesis, reaction, and characterizations. T. Y. performed the XAFS analysis. K. S. conceived and directed the project. All authors analyzed and discussed the results and co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by JSPS KAKENHI (Grant 24K01448, 24H00463, and 22H04971), JST FOREST (JPMJFR213M), UTEC-UTokyo FSI Research Grant Program, and Murata Science Foundation. A part of this work was supported by Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Grant Number JPMXP1223UT0308 and JPMXP1224UT0068. We thank Mr Hiroyuki Oshikawa and Ms Mari Morita (The University of Tokyo) for their assistance with TEM, HAADF-STEM, and EDS analyses. We thank Dr Hironori Ofuchi (Japan Synchrotron Radiation Research Institute, SPring-8) for the support of XAFS measurements at BL14B2 (Proposal Number 2023B1658 and 2024A1546).Notes and references
- V. Masson-Delmotte, P. Zhai, A. Pirani, S. L. Connors, C. Péan, S. Berger, N. Caud, Y. Chen, L. Goldfarb, M. I. Gomis, M. Huang, K. Leitzell, E. Lonnoy, J. B. R. Matthews, T. K. Maycock, T. Waterfield, O. Yelekçi, R. Yu and B. Zhou, IPCC, 2021, Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, 2021 Search PubMed.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112 RSC.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorf, Chem. Rev., 2019, 119, 7610 CrossRef CAS PubMed.
- S. Hernández, M. A. Farkhondehfal, F. Sastre, M. Makkee, G. Saracco and N. Russo, Green Chem., 2017, 19, 2326 RSC.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266 CrossRef CAS PubMed.
- D. Gao, H. Zhou, F. Cai, J. Wang, G. Wang and X. Bao, ACS Catal., 2018, 8, 1510 CrossRef CAS.
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137, 4288 CrossRef CAS PubMed.
- Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Chem. Soc. Rev., 2020, 49, 6632 RSC.
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654 CrossRef CAS.
- A. Murata and Y. Hori, Bull. Chem. Soc. Jpn., 1991, 64, 123 CrossRef CAS.
- M. R. Thorson, K. I. Siil and P. J. A. Kenis, J. Electrochem. Soc., 2012, 160, F69 CrossRef.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006 CrossRef CAS PubMed.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277 CrossRef CAS PubMed.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001 RSC.
- A. S. Malkani, J. Anibal and B. Xu, ACS Catal., 2020, 10, 14871 CrossRef CAS.
- S. S. Bhargava, E. R. Cofell, P. Chumble, D. Azmoodeh, S. Someshwar and P. J. A. Kenis, Electrochim. Acta, 2021, 394, 139055 CrossRef CAS.
- M. C. O. Monteiro, F. Dattila, N. López and M. T. M. Koper, J. Am. Chem. Soc., 2022, 144, 1589 CrossRef CAS PubMed.
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Nat. Catal., 2022, 5, 268 CrossRef CAS.
- T.-W. Jiang, S.-S. Wang, X. Qin, W.-Y. Zhang, H. Li, X.-Y. Ma, K. Jiang, S. Zou and W.-B. Cai, J. Catal., 2024, 434, 115520 CrossRef CAS.
- A. Schizodimou and G. Kyriacou, Electrochim. Acta, 2012, 78, 171 CrossRef CAS.
- M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer, Berlin, 1983 Search PubMed.
- C. L. Hill and C. M. Prosser-McCartha, Coord. Chem. Rev., 1995, 143, 407 CrossRef CAS.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171 CrossRef CAS PubMed.
- N. Mizuno and M. Misono, Chem. Rev., 1998, 98, 199 CrossRef CAS PubMed.
- A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893 CrossRef CAS PubMed.
- L. Vilà-Nadal and L. Cronin, Nat. Rev. Mater., 2017, 2, 17054 CrossRef.
- N. I. Gumerova and A. Rompel, Nat. Rev. Chem, 2018, 2, 0112 CrossRef CAS.
- K. Suzuki, N. Mizuno and K. Yamaguchi, ACS Catal., 2018, 8, 10809 CrossRef CAS.
- B. Fabre, C. Falaise and E. Cadot, ACS Catal., 2022, 12, 12055 CrossRef CAS.
- J.-H. Kruse, M. Langer, I. Romanenko, I. Trentin, D. Hernández-Castillo, L. González, F. H. Schacher and C. Streb, Adv. Funct. Mater., 2022, 32, 2208428 CrossRef CAS.
- J. M. Cameron, G. Guillemot, T. Galambos, S. S. Amin, E. Hampson, K. M. Haidaraly, G. N. Newton and G. Izzet, Chem. Soc. Rev., 2022, 51, 293 RSC.
- N. Li, J. Liu, B.-X. Dong and Y.-Q. Lan, Angew. Chem., Int. Ed., 2020, 59, 20779 CrossRef CAS PubMed.
- K. Li, T. Liu, J. Ying, A. Tian and X. Wang, J. Mater. Chem. A, 2024, 12, 13576 RSC.
- M. Girardi, S. Blanchard, S. Griveau, P. Simon, M. Fontecave, F. Bedioui and A. Proust, Eur. J. Inorg. Chem., 2015, 3642 CrossRef CAS.
- D. Azaiza-Dabbah, C. Vogt, F. Wang, A. Masip-Sánchez, C. de Graaf, J. M. Poblet, E. Haviv and R. Neumann, Angew. Chem., Int. Ed., 2022, 61, e202112915 CrossRef CAS PubMed.
- A. M. Khenkin, I. Efremenko, L. Weiner, J. M. L. Martin and R. Neumann, Chem.–Eur. J., 2010, 16, 1356 CrossRef CAS PubMed.
- J. Zhu, H. Zhou, X. Liang, P. Feng, S. Zhao, Y. Sun, B. Ma, Y. Ding and X. Han, Chem. Commun., 2024, 60, 6761 RSC.
- L.-L. Liu, L. Wang, W.-Y. Xiao, P. Yang, J. Zhao and U. Kortz, Coord. Chem. Rev., 2024, 506, 215687 CrossRef CAS.
- K. Suzuki, N. Mizuno and K. Yamaguchi, J. Jpn. Pet. Inst., 2020, 63, 258 CrossRef CAS.
- K. Yonesato, H. Ito, H. Itakura, D. Yokogawa, T. Kikuchi, N. Mizuno, K. Yamaguchi and K. Suzuki, J. Am. Chem. Soc., 2019, 141, 19550 CrossRef CAS PubMed.
- T. Minato, D. Salley, N. Mizuno, K. Yamaguchi, L. Cronin and K. Suzuki, J. Am. Chem. Soc., 2021, 143, 12809 CrossRef CAS PubMed.
- C. Li, A. Jimbo, K. Yamaguchi and K. Suzuki, Chem. Sci., 2021, 12, 1240 RSC.
- T. Hirano, K. Uehara, K. Kamata and N. Mizuno, J. Am. Chem. Soc., 2012, 134, 6425 CrossRef CAS PubMed.
- K. Suzuki, F. Tang, Y. Kikukawa, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2014, 53, 5356 CrossRef CAS PubMed.
- K. Sato, K. Yonesato, T. Yatabe, K. Yamaguchi and K. Suzuki, Chem.–Eur. J., 2022, 28, e202104051 CrossRef CAS PubMed.
- K. Yonesato, D. Yanai, S. Yamazoe, D. Yokogawa, T. Kikuchi, K. Yamaguchi and K. Suzuki, Nat. Chem., 2023, 15, 940 CrossRef CAS PubMed.
- Y. Koizumi, K. Yonesato, S. Kikkawa, S. Yamazoe, K. Yamaguchi and K. Suzuki, J. Am. Chem. Soc., 2024, 146, 14610 CrossRef CAS PubMed.
- K. Wachi, T. Yabe, T. Suzuki, K. Yonesato, K. Suzuki and K. Yamaguchi, Appl. Catal., B, 2022, 314, 121420 CrossRef CAS.
- A. Misra, K. Kozma, C. Streb and M. Nyman, Angew. Chem., Int. Ed., 2020, 59, 596 CrossRef CAS PubMed.
- Q. Hu, S. Chen, T. Wågberg, H. Zhou, S. Li, Y. Li, Y. Tan, W. Hu, Y. Ding and X. Han, Angew. Chem., Int. Ed., 2023, 62, e202303290 CrossRef CAS PubMed.
- M. Blasco-Ahicart, J. Soriano-López, J. J. Carbó, J. M. Poblet and J. R. Galan-Mascaros, Nat. Chem., 2017, 10, 24 CrossRef PubMed.
- H. Wang, N. Kawasaki, T. Yokoyama, H. Yoshikawa and K. Awaga, Dalton Trans., 2012, 41, 9863 RSC.
- T. Suzuki, T. Yabe, K. Wachi, K. Yonesato, K. Suzuki and K. Yamaguchi, ChemNanoMat, 2023, 9, e202200428 CrossRef CAS.
- J. Evans, M. Pillinger and J. M. Rummey, J. Chem. Soc., Dalton Trans., 1996, 2951 RSC.
- A. Tézé and G. Hervé, Inorg. Synth., 1990, 27, 85 CrossRef.
- K. Liu, W. A. Smith and T. Burdyny, ACS Energy Lett., 2019, 4, 639 CrossRef CAS PubMed.
- H.-P. Iglesias van Montfort, S. Subramanian, E. Irtem, M. Sassenburg, M. Li, J. Kok, J. Middelkoop and T. Burdyny, ACS Energy Lett., 2023, 8, 4156 CrossRef CAS.
- K. Xia, T. Yatabe, K. Yonesato, T. Yabe, S. Kikkawa, S. Yamazoe, A. Nakata, K. Yamaguchi and K. Suzuki, Angew. Chem., Int. Ed., 2022, 61, e202205873 CrossRef CAS PubMed.
- K. Xia, T. Yatabe, K. Yonesato, S. Kikkawa, S. Yamazoe, A. Nakata, R. Ishikawa, N. Shibata, Y. Ikuhara, K. Yamaguchi and K. Suzuki, Nat. Commun., 2024, 15, 851 CrossRef CAS PubMed.
- K. Xia, T. Yatabe, K. Yamaguchi and K. Suzuki, Dalton Trans., 2024, 53, 11088 RSC.
- L. Zhang, Z. Wei, S. Thanneeru, M. Meng, M. Kruzyk, G. Ung, B. Liu and J. He, Angew. Chem., Int. Ed., 2019, 58, 15834 CrossRef CAS PubMed.
- Z. Cao, J. S. Derrick, J. Xu, R. Gao, M. Gong, E. M. Nichols, P. T. Smith, X. Liu, X. Wen, C. Copéret and C. J. Chang, Angew. Chem., Int. Ed., 2018, 57, 4981 CrossRef CAS PubMed.
- M. Moura de Salles Pupo and R. Kortlever, ChemPhysChem, 2019, 20, 2926 CrossRef CAS PubMed.
- G. Marcandalli, M. C. O. Monteiro, A. Goyal and M. T. M. Koper, Acc. Chem. Res., 2022, 55, 1900 CrossRef CAS PubMed.
- B. Deng, M. Huang, X. Zhao, S. Mou and F. Dong, ACS Catal., 2022, 12, 331 CrossRef CAS.
- H. Khani, A. R. Puente Santiago and T. He, Angew. Chem., Int. Ed., 2023, 62, e202306103 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Fig. S1–S10 and Tables S1–S8. See DOI: https://doi.org/10.1039/d4sc04304a |
| This journal is © The Royal Society of Chemistry 2024 |