
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpin effect in dual-atom catalysts for electrocatalysis
Xiaoqin
Xu
and
Jingqi
Guan
 *
*
Institute of Physical Chemistry, College of Chemistry, Jilin University, Changchun 130021, PR China. E-mail: guanjq@jlu.edu.cn
First published on 19th August 2024
Abstract
The development of high-efficiency atomic-level catalysts for energy-conversion and -storage technologies is crucial to address energy shortages. The spin states of diatomic catalysts (DACs) are closely tied to their catalytic activity. Adjusting the spin states of DACs' active centers can directly modify the occupancy of d-orbitals, thereby influencing the bonding strength between metal sites and intermediates as well as the energy transfer during electro reactions. Herein, we discuss various techniques for characterizing the spin states of atomic catalysts and strategies for modulating their active center spin states. Next, we outline recent progress in the study of spin effects in DACs for the oxygen reduction reaction (ORR), oxygen evolution reaction (OER), hydrogen evolution reaction (HER), electrocatalytic nitrogen/nitrate reduction reaction (eNRR/NO3RR), and electrocatalytic carbon dioxide reduction reaction (eCO2RR) and provide a detailed explanation of the catalytic mechanisms influenced by the spin regulation of DACs. Finally, we offer insights into the future research directions in this critical field.
1. Introduction
The growing consumption of fossil fuels and serious climate change seriously hinder the sustainable development of human society, prompting researchers to seek effective strategies to deal with them.1,2 Renewable energy-conversion technologies involve various chemical transformation processes, providing a sustainable and green route to relieve these urgent issues.3,4 Among these technologies, electrochemical conversion processes, such as fuel cells and Zn–air batteries (involving the ORR and OER), water splitting (involving the OER and HER), and eCO2RRs and eNRRs/eNO3RRs, which can facilitate the transformation of H2O, CO2, and N2 in the atmosphere into high-value-added products through coupling with renewable energy, represent a promising avenue for the development of sustainable energy solutions.5–7 Catalysts play a crucial role in energy-conversion techniques, with more than 80% of industrial processes utilizing catalysts to improve the speed, efficiency, and selectivity of reactions.8 Currently, precious metal catalysts (such as Pt, Au, Ru, and Ir) generally perform better than non-noble metal materials in facilitating electrocatalytic reaction kinetics. However, the limited supply and high cost of precious metals have greatly prevented them from being used on a large scale commercially.9 Therefore, powerful and cost-effective metal-based (TM) catalysts are needed to help the chemical industry to move toward a more sustainable future.Most catalytic processes occur on the metal surface, resulting in low catalyst utilization and limiting the catalytic performance. An effective strategy to improve metal utilization is downsizing catalysts and reducing the size of the active metal in solid metal catalysts to the atomic level to form single-atom catalysts (SACs), as proposed by Zhang et al. in 2011,10 thus making full use of metal atoms.11–13 Apart from maximized atomic utilization, SACs possess the advantages of uniform active sites, and the way their electronic structure and unsaturated coordination environment work together can improve the performances of various catalytic reactions.14,15 Although SACs show great prospects in catalysis, there are various limitations in this class of single-atom structures, such as low metal loading, only one type of specific active sites, and relatively weak interaction, which hinder their development.16,17 The advent of bimetallic nanocatalysts has led to the development of DACs, which have been considered a promising extension of SACs that can address the aforementioned challenges. DACs comprise isolated pairs of metal atoms, which act as the active sites. In homonuclear DACs, the same metal is present, whereas in heteronuclear DACs, different metals are present.18 In the 1980s, Muetterties et al. documented the semi-hydrogenation of internal alkynes to trans- or (E)-isomers using di-nuclear rhodium organic compounds, which was one of the earliest instances of catalytic reduction utilizing a synthetic DAC.19 DACs have many advantages over SACs, including: (1) the dual metal sites exhibit greater flexibility, enabling them to optimize the adsorption energy of specific intermediates and break the linear progression relationship;20 (2) the tunable electronic structure and spin states of DACs facilitate an enhancement of the ORR/OER kinetics, thus improving the energy-conversion efficiency and the cycle life of the associated energy-storage devices; thus paving the way for future practical applications of DACs;21 (3) DACs have a greater potential to adjust the d-band center through interactions of the electronic orbits compared SACs; thus allowing tuning the binding energy of the intermediates at the catalytic sites.1,22
The initiation of chemical reactions depends on two essential factors: the energies involved (both free and activation energies) and the angular momentum (spin) of the reactants.23 Since water oxidation, fuel cell reactions, and CO2 conversion involve transitions between triplet and singlet states or the participation of radicals, the influence of electron spin dynamics in these reactions cannot be ignored. Recent research has shown that the spin states (low, medium, and high spins) or magnetic structures of the active centers in DACs can directly influence the reaction barriers of catalytic reactions by adjusting the bonding of the reaction intermediates species with catalytic sites, as the formation energies of singlet products are generally higher than those of triplet ground-state products.24–26 Furthermore, the majority of DACs with unpaired spin electrons invariably demonstrate spin-dependent magnetism, filtering effective spin electrons through the magnetic field and thus effectively accelerating spin-dependent electrocatalytic reactions.27 Besides the influence of magnetism, other factors can affect the spin of electrons, such as the crystal field, coordination environment, oxidation state, bonding characteristics, electron count, and conductivity.28
The spin effect has been applied to the whole catalytic system; however, there is currently no reports providing a systematic summary of the spin effect in DACs. Consequently, this review highlights recent progress in the discovery of the influence of electron spin in DACs on the electrocatalysis of the HER, OER, ORR, CO2RR, and eNRR/eNO3RR, with a primary development schedule shown in Fig. 1 highlighting the major steps in the progress in DACs research. The concept of the spin effect was first introduced by Compton et al. in 1921,29 marking the beginning of its development in the context of electrocatalysts. It was not until the 1980s that binuclear molecules were first proposed and diatomic catalysts began to emerge as a prominent area of study.30 Subsequently, the study of DACs advanced from their initial preparation and modulation to an examination of the synergistic impact of charge iteration and electron spin polarization, which broadened the potential applications of DACs.33 Despite remaining in its infancy, the findings emerging from the investigation of the spin effect of DACs have attracted significant interest across various catalytic domains. This examination focuses on a detailed analysis of the crucial roles of the electron properties, specifically charge and spin, in these reactions. Additionally, we summarize strategies for optimizing the catalytic efficiency by manipulating these properties. We systematically discuss the physical and electrochemical properties of electrons and spin, and their interactions with reactants and intermediates. This review addresses the challenges and opportunities in developing high-performance catalysts, concluding with a forward-looking exploration of the future of spin catalysis.
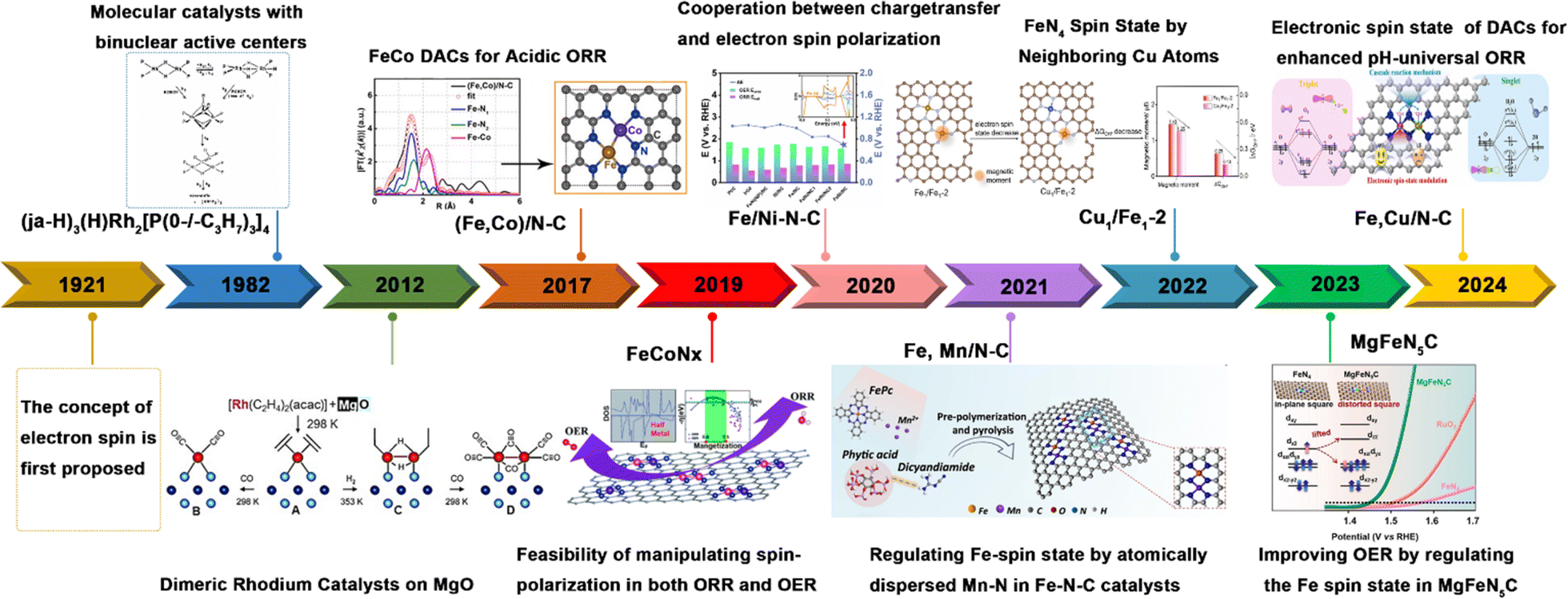 | ||
| Fig. 1 History of DACs with spin regulation for electrocatalysis, including the concept of electron spin. Reproduced with permission.29 Copyright 1921, Elsevier. Also shown are some representative DACs: (ja-H)3(H)Rh2[P(0-/-C3H7)3]4. Reproduced with permission.19 Copyright 1982, American Chemical Society. Dimeric Rh catalysts on MgO. Reproduced with permission.30 Copyright 2012, Wiley-VCH. (Fe, Co)/N–C. Reproduced with permission.31 Copyright 2017, American Chemical Society. FeCoNx. Reproduced with permission.32 Copyright 2019, Royal Society of Chemistry. Fe/Ni–N–C. Reproduced with permission.33 Copyright 2021, Elsevier. Fe, Mn/N–C. Reproduced with permission.34 Copyright 2021, Nature. Cu1/Fe1-2. Reproduced with permission.35 Copyright 2022, Wiley-VCH. MgFeN5C. Reproduced with permission.36 Copyright 2023, Wiley-VCH. and Fe, Cu/N–C. Reproduced with permission.37 Copyright 2024, Elsevier. | ||
2. Fundamentals of spin
The spin of the electron is an important fundamental property, which arises from its internal angular momentum, and plays a crucial role in manipulating various aspects of catalysis. In the context of quantum mechanics, the co-localization of two electrons in the same orbital is possible only if they possess opposite spins.38 The state in which an electron spins in an atomic or molecular orbital is called a spin state. Following the interaction, the five sub-d-orbitals split into a low-energy t2g bonding orbital that includes dxy, dyz, and dxz and a high-energy eg antibonding orbital that includes dx2−y2 and dz2, as demonstrated in Fig. 2.33,39 Fission energy (Δ0) is an important concept, which is the difference in energy levels between eg and t2g orbitals. Another important concept is the electron pairing energy (P), which is the energy needed to pair electrons with opposite spins within the same orbital. The degree of spin polarization is determined by the relative values of these two energies: when Δ0 exceeds P, electrons tend to preferentially occupy the triple degenerate t2g orbitals, which are associated with the low-spin state in a strong field. Conversely, the electrons occupy the orbits in accordance with the analogous Hunt rule, designated as the high-spin state in a weak field.39 Under specific conditions, the electron spin states in atoms or molecules align in a particular direction, a phenomenon known as spin polarization.40 Moreover, the radical-pair mechanism was initially proposed by Closs and independently identified by Lawler in 1969.41 In chemical reactions, due to the selection rules, reaction is prohibited if a change in spin is required.42 More and more scientists have realized that modulating the electron spin properties is a feasible means to heighten the intrinsic activity of catalysts, and thus the concept of spin catalysis has emerged.43 Since the specific mechanism of the spin effect in different electrocatalytic reactions varies, understanding the spin of metals is important for the design of DACs with enhanced electrocatalytic activities. This section begins with an introduction to the descriptors, the catalytic mechanism, and the regulation method of spin polarization for electrocatalytic reactions.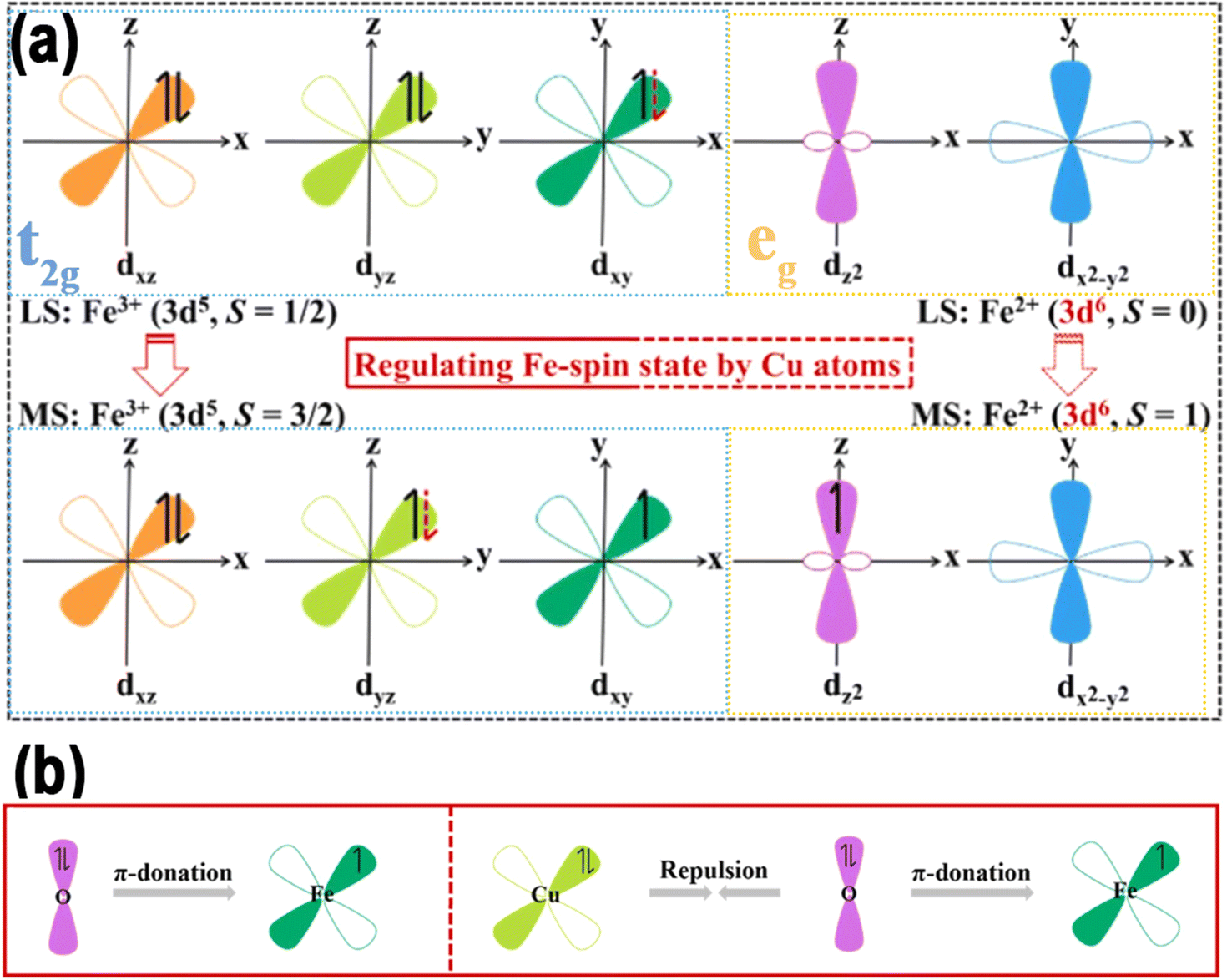 | ||
| Fig. 2 (a) Five d-orbitals in Fe, Cu/N–C. (b) Scheme of dxy electron interaction in Fe/N–C and Fe, Cu/N–C. Reproduced with permission.37 Copyright 2024, Elsevier. | ||
2.1. Catalytic descriptors based on spin polarization
In recent years, more and more researchers have combined experimental results with theoretical calculations to enhance the comprehension of electrocatalytic processes. Therefore, the descriptors derived from theoretical aspects play a significant role using the physical and chemical properties of an electrocatalyst as an indicator of its electrocatalytic activity in the electrocatalytic process.44 In this section, we discuss the development of descriptors that correlate the electrocatalytic activity of electrocatalysts, especially DACs, with spin polarization.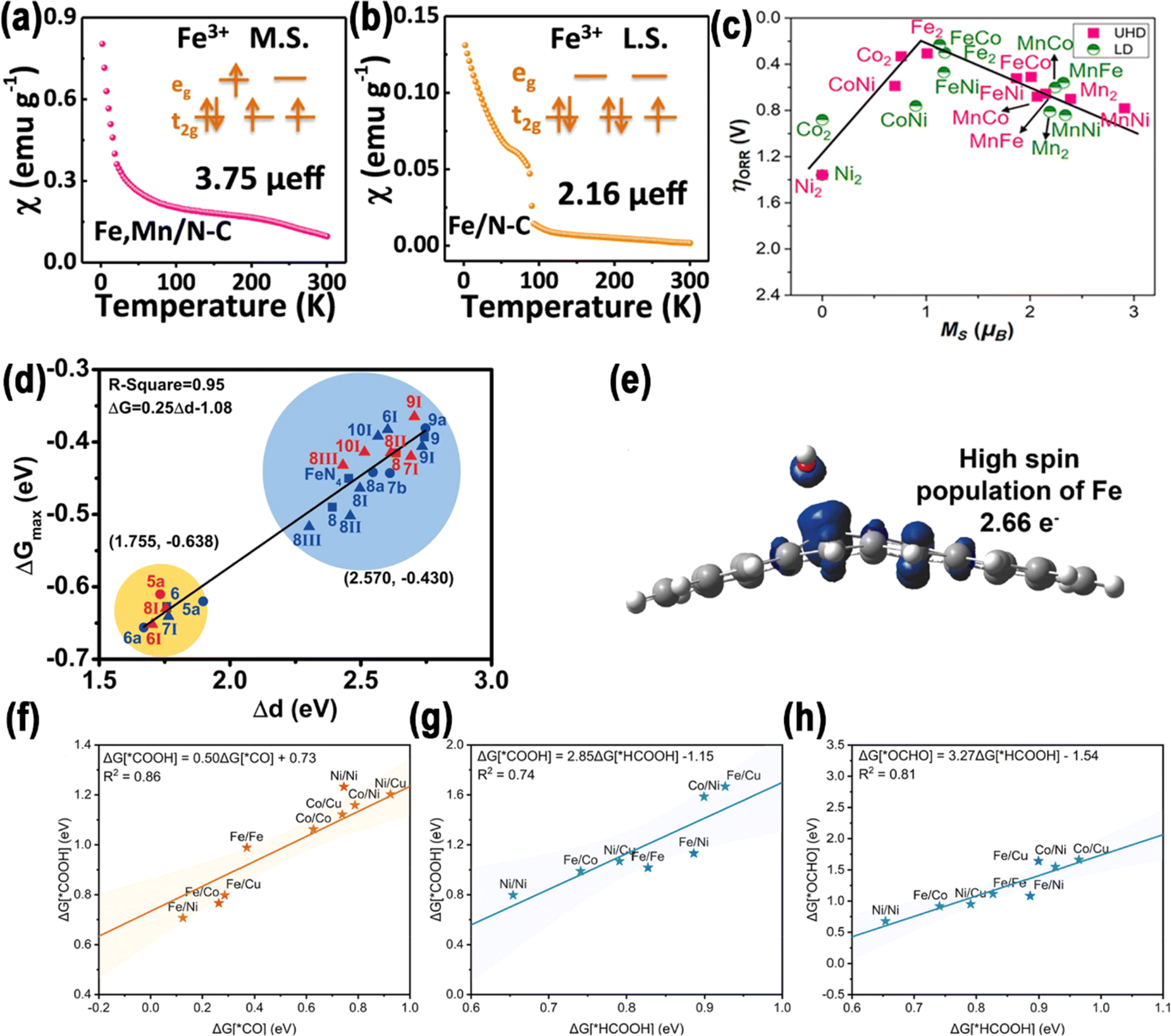 | ||
| Fig. 3 (a) Zero-field cooling temperature-dependent magnetic susceptibility of Fe, Mn/N–C, (b) Fe/N–C. Reproduced with permission.34 Copyright 2021, Nature. (c) Volcano plots illustrating the relationship between the local spin magnetic moments of metal atoms, with the optimal pathway for *OH anchoring presented. Reproduced with permission.52 Copyright 2023, American Chemical Society. (d) Correlation between Δd and ΔG. Reproduced with permission.53 Copyright 2022, Nature. (e) Charge transfer in FeNiN6-DAC. Reproduced with permission.54 Copyright 2021, American Chemical Society. Comparisons of the binding energies between (f) *COOH and *CO, (g) *OCHO and *HCOOH, as well as (h) *COOH and *HCOOH. Reproduced with permission.55 Copyright 2023, American Chemical Society. | ||
2.2. Spin state characterizations
An invaluable approach to the modulation of electronic structures is the induction of spin state transitions. This procedure demands different characterization techniques to measure the electron occupancy in t2g and eg orbitals. In this section, we detail the analytical methods used in the characterization techniques.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:5 to investigate the spin state of Fe(III).67 The M–T tests showed that PQD-Fe and o-MQFe-10:20:5 had μeff values of 1.69 μB and 3.65 μB, with unpaired electron numbers of 0.96 and 2.78, respectively (Fig. 4b).
:20:5 to investigate the spin state of Fe(III).67 The M–T tests showed that PQD-Fe and o-MQFe-10:20:5 had μeff values of 1.69 μB and 3.65 μB, with unpaired electron numbers of 0.96 and 2.78, respectively (Fig. 4b).
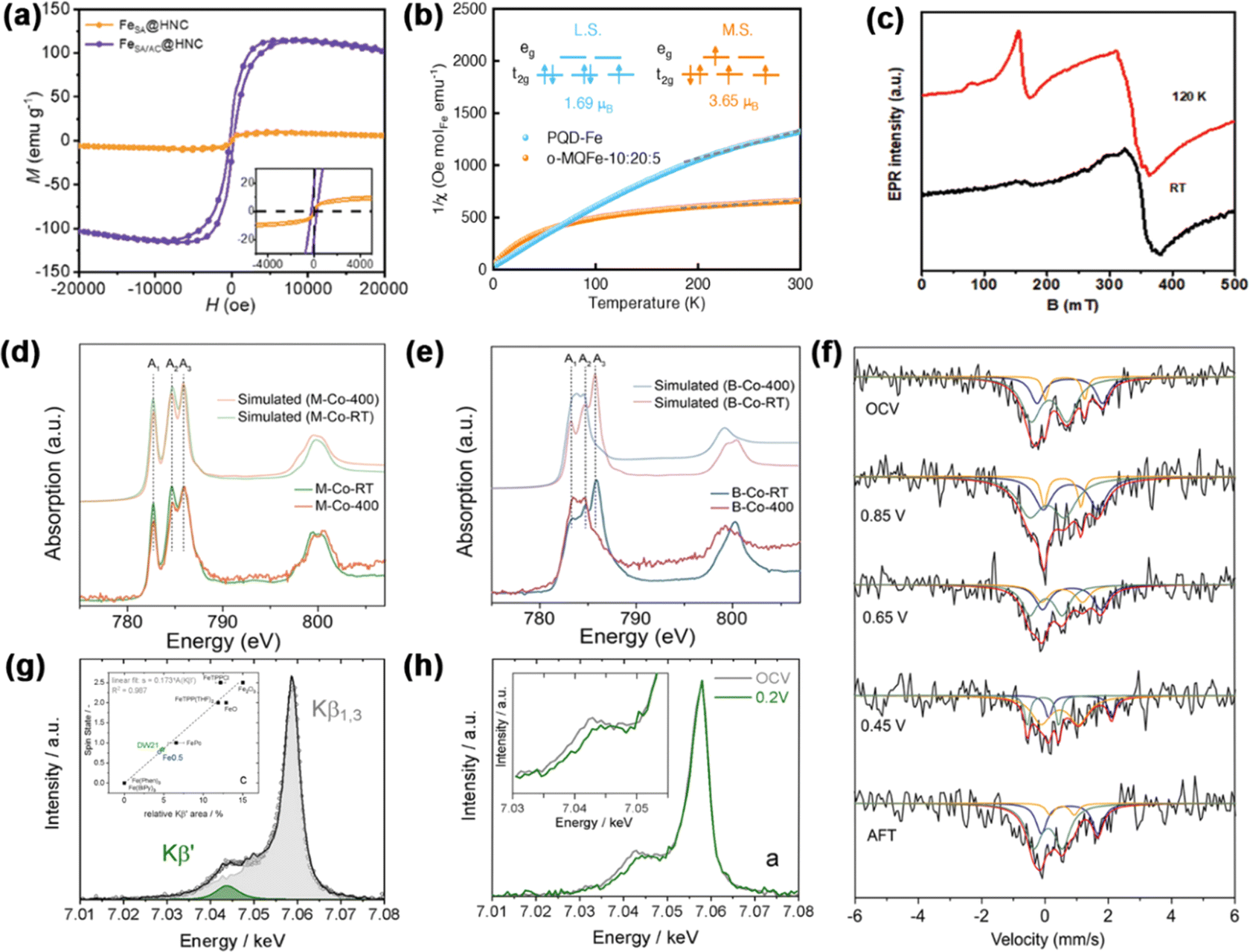 | ||
| Fig. 4 (a) Magnetic hysteresis loops of catalysts at room temperature. Reproduced with permission.66 Copyright 2024, Wiley-VCH. (b) Plots of the reciprocal of magnetic susceptibility (1/χm). Reproduced with permission.67 Copyright 2022, Wiley-VCH. (c) EPR spectra of FeSA-NSC-90. Reproduced with permission.68 Copyright 2022, Wiley-VCH. The experimental and simulated cobalt L2,3-edge XAS spectra of (d) M-CoPc-RT/400 and (e) B-CoPc-RT/400. Reproduced with permission.69 Copyright 2023, Nature. (f) Operando57Fe Mössbauer spectra of Fe1-NS1.3C at different voltage conditions. Reproduced with permission.70 Copyright 2021, Wiley-VCH. (g) Kβ mainline XES data. Inset of (g): relationship between the relative area of the Kβ′ peak compared to the total Kβ line and the spin state of the reference compounds. (h) In situ Kβ mainline; inset: amplification of the Kβ′ area. Reproduced with permission.71 Copyright 2021, Wiley-VCH. | ||
2.3. Regulation of the spin polarization of DACs
The electronic structure of DACs is an important reason for their synergistic effect. Three primary approaches have been devised to regulate the electronic structure of DACs by regulating the spin polarization: regulation of the central atoms, the distance between two metal sites, and the coordination environment.2.3.1.1. Coupled with a homonuclear metal. In homonuclear DACs, two identical metal atoms are situated adjacent to one another and may form a bond.1 Huang et al. developed a NPs-to-SAs-to-DAs atomization and sintering method via a N-removal and heat-induced migration process (Fig. 5a).82 To be specific, it was shown that it was possible to adjust the existing configurations of Co species at the atomic scale, thereby producing nanoparticles, CoN4 SAs, and Co2N5 DAs on hollow carbon spheres with N-doping. The difference in Gibbs free energy between ΔGOOH* and ΔGOH* (|ΔGOOH*–OH*|) is a significant descriptor for the ORR/OER, and ideally should be valued at 2.46 eV. As shown in Fig. 5b, the spin magnetic moment exhibited a positive, nearly linear correlation (R2 = 0.962) with |ΔGOOH*–OH*|. The Co2N5 dual atom's ΔGOOH* to ΔGOH* coordinate was near the ideal scaling relation with a |ΔGOOH*–OH*| value of 2.55 eV, near to the ideal 2.46 eV for ORR/OER catalysts (Fig. 5c). This suggests that Co2N5 has the most suitable O* intermediate adsorption/desorption energy. The likely reason for this is that the reduced spin magnetic moment of Co2N5 disrupts the universal ΔGOOH* to ΔGOH* scaling relation, leading to an optimal balance of O* adsorption.
 | ||
| Fig. 5 (a) Bader charge diagrams; (b) direct relationship between the magnetic moment and |ΔGOOH*–OH*|. (c) Comparison of the scaling of ΔGOOH* to ΔGOH* for all models to the universal and ideal scaling lines82 Copyright 2022, Nature. (d) Diagrammatic representation of M–N–C (where M = Fe or Ni), Fe/Ni–N–C (coordinated with 3 N atoms), and Fe/Ni–N–C (coordinated with 4 N atoms, with 2 N atoms shared by Ni and Fe). (e) Density of states for all the samples. Reproduced with permission.33 Copyright 2021, Elsevier. | ||
2.3.1.2. Coupled with a heteronuclear metal. In heteronuclear DACs, two distinct metal atoms are positioned next to each other, potentially forming a bond.1 Zhao et al. employed DFT to forecast that Fe–Ni dual-atom active centers would demonstrate moderate spin magnetization at the reaction sites and possess limited spin-polarized conduction electrons.33 Fe 3d, the active center of Fe–N–C, exhibits high spin polarization, with a spin magnetic moment value of 1.88 μB, and localized charge distribution. However, the metal of Ni–N–C is paramagnetic, and the spin magnetic moment value is almost zero. By constructing a heteronuclear Fe–Ni dual-atom pair, the spin coupling between the two metals weakened the spin polarization of the Fe 3d electron and reduced its spin magnetic moment to 1.48 μB (Fig. 5d and e). Based on the theoretical research results, an Fe/Ni–N–C catalyst with bifunctional ORR/OER performance was prepared by a wet chemical approach.
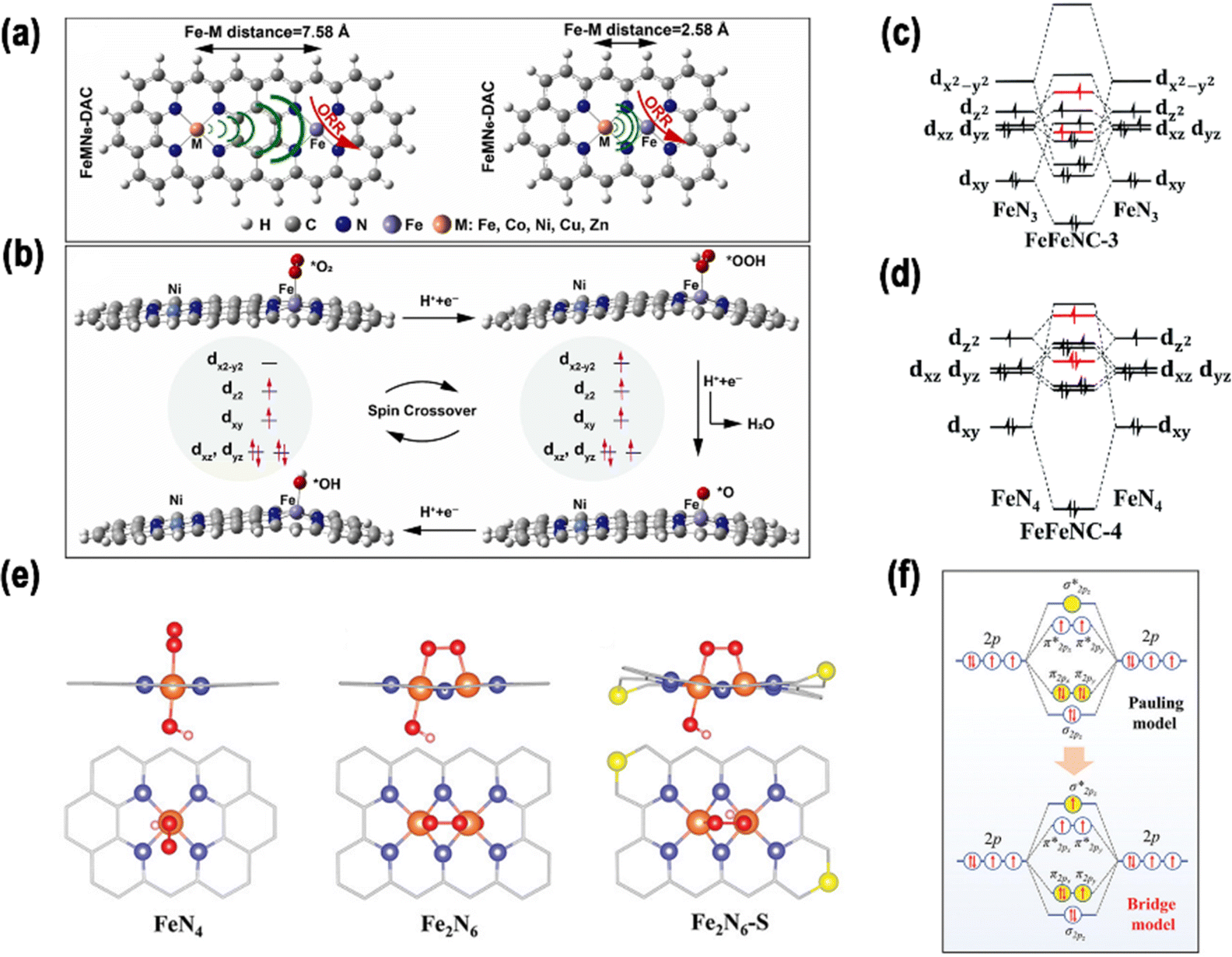 | ||
| Fig. 6 (a) Structures of FeMN6-DAC and FeMN8-DAC. (b) Side perspective of reaction intermediates of FeNiN8-DAC in the ORR. Reproduced with permission.60 Copyright 2023, Elsevier. Orbital interactions between Fe atoms in (c) FeFeNC-3 and (d) FeFeNC-4. Reproduced with permission.84 Copyright 2022, the Royal Society of Chemistry (e) geometric models of FeN4, Fe2N6, and Fe2N6-S with absorbed O2. (f) Schematic representation of the potential electron orbital configurations of O 2p orbitals in various oxygen-adsorption arrangements. Reproduced with permission.85 Copyright 2023, Wiley-VCH. | ||
Zhang et al. successfully synthesized Mn–Fe binuclear sites, anchored on a GO-like structure.86 The findings from aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) analysis revealed an approximate distance of 0.3–0.4 nm between the Mn and Fe atoms. This distance was consistent with the approximate sum of their van der Waals radii, and the proximity of two atomic sites may result in a small-distance electronic interaction. The small-distance electronic interaction between the Mn and Fe atom sites resulted in a transformation of the Fe spin state from a medium to a high-spin state. According to Sabatier's principle, the dz2 orbital of Fe in the high self-selection state is half-full, which is conducive to the absorption and desorption of oxygenated species during the ORR.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O dissociation and thus promoted the ORR kinetics. In addition, due to the variation of TMs in DACs on the left of the element periodic table, the dual-metal active sites demonstrated a higher spin polarization, giving rise to a more robust adsorption of *OH and a reduction in ORR activity.
O dissociation and thus promoted the ORR kinetics. In addition, due to the variation of TMs in DACs on the left of the element periodic table, the dual-metal active sites demonstrated a higher spin polarization, giving rise to a more robust adsorption of *OH and a reduction in ORR activity.
Liu et al. proposed an S-doped di-nuclear Fe-based ORR catalyst (Fe2N6-S) by simulating the catalytic center of ferredoxin, which was composed of Fe–Fe dual-metal sites and nonmetal S sites.85 In this process, the second Fe site and the heteroatom S act as “double modulators” in conjunction with one another to complete the accurate tuning of the electronic configuration. Additionally, spin-polarized DFT was predominantly utilized to anticipate the impact of the Fe–NC coordination structure on the O2 absorption characteristics. This is a critical determinant of the ORR activity. First, three kinds of carbon-based electrocatalyst models were constructed, as shown in Fig. 6e, in which four N atoms cooperated with Fe SA, Fe DA, and S-doped Fe DA, which were defined as FeN4, Fe2N6, and Fe2N6-S, respectively. Second, in a follow-up study, the authors modified the structures of FeN4, Fe2N6, and Fe2N6-S with OH*. The migration of electrons from the bonding orbital π2px or π2py to the antibonding orbital  in the bridge model weakened the O–O bond, thereby promoting the subsequent reaction process (Fig. 6f). These initial theoretical findings indicate that the dual Fe–Fe sites play a pivotal role in optimizing the oxygen-adsorption behavior, facilitating the efficient activation of O–O bonds, and consequently, enabling the acceleration of the ORR kinetics. Furthermore, the doping of S atoms around the Fe–Fe sites also exerts a modulating influence on the oxygen binding energy.
in the bridge model weakened the O–O bond, thereby promoting the subsequent reaction process (Fig. 6f). These initial theoretical findings indicate that the dual Fe–Fe sites play a pivotal role in optimizing the oxygen-adsorption behavior, facilitating the efficient activation of O–O bonds, and consequently, enabling the acceleration of the ORR kinetics. Furthermore, the doping of S atoms around the Fe–Fe sites also exerts a modulating influence on the oxygen binding energy.
3. Spin effect in dual-atom electrocatalysis
Recently, it has been increasingly acknowledged that the spin state of DACs plays a pivotal role in determining their catalytic activities for the HER,91 CO2RR,92 NRR,93 ORR,94 and OER.20 Modulation of the spin states offers the possibility to accurately design DACs with optimized electronic structures and good performances. In the following, we introduce the application of the spin-state regulation of DACs in various catalytic reactions in detail.3.1. Oxygen electrocatalysis
Clean and renewable energy sources and energy-conversion techniques have attracted extensive interest due to the current global energy and environmental challenges, and include metal–air batteries, fuel cells, and electrochemical water decomposition. Oxygen electrocatalytic reactions play a key role in these applications, determining the efficiency of hydrogen/oxygen-related reactions. Oxygen electrocatalytic reactions include the OER and ORR. From the perspective of orbital mechanics, oxygen molecules exist in two distinct spin states, the triplet state (3O2, ground state), which is the lowest energy configuration and represents the ground state, and the singlet state (1O2, metastable state), which is a metastable state.953O2 is generally considered to be the dominant form of oxygen in electrocatalysis due to its energy (0.00 eV) being lower than that of 1O2 (0.98 eV).95 As shown in Fig. 7, since the spin quantum number of oxygen species is not conserved between the OER and ORR, the high energy barrier of the two reactions is associated with spin-state transitions involving spin electrons, such as the singlet OH−/H2O to 3O2 spin transition.25 Therefore, DACs have been developed to reduce energy expenditure by reducing the energy barrier and promoting spin transitions.96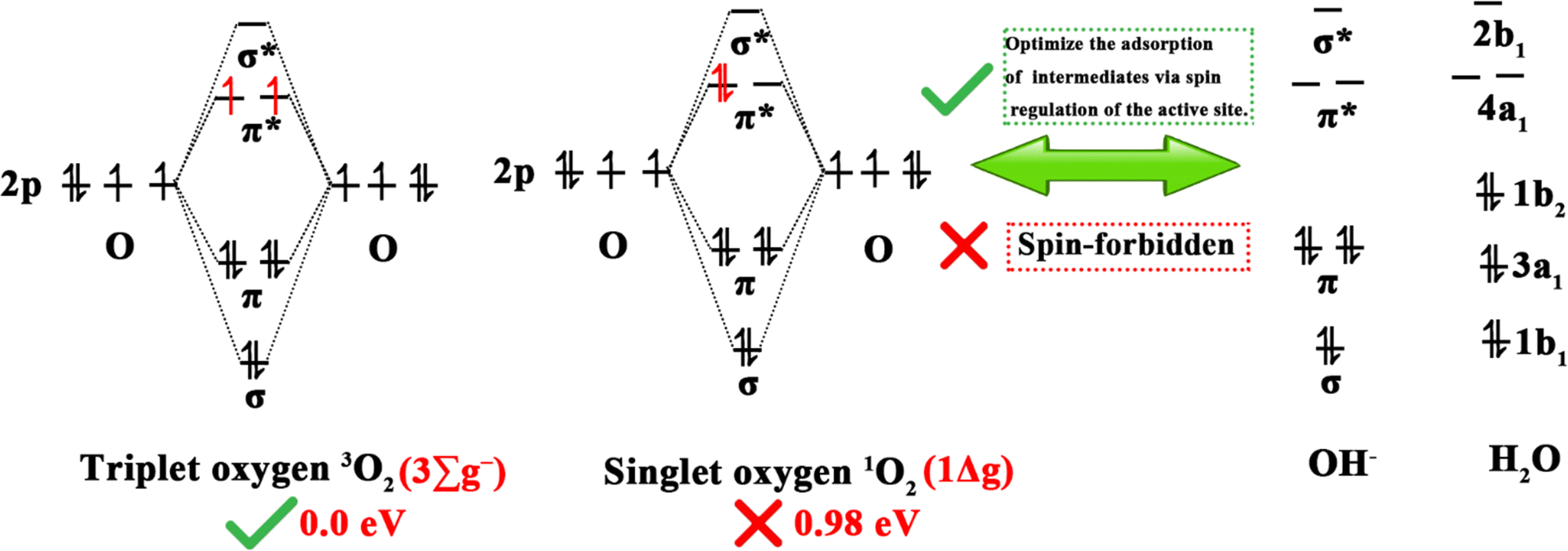 | ||
| Fig. 7 Electronic configurations of singlet OH−, H2O, 1O2 and 3O2. | ||
3.1.1.1. ORR mechanisms. As shown in Fig. 8, the ORR typically follows two reaction pathways on DACs: the reduction of O2 through OOH*, O*, and OH* intermediates, resulting in the formation of two H2O molecules via a 4e− pathway, or the formation of an H2O2 molecule via a 2e− transfer pathway. Depending on the reaction pathway, it can be further divided into the dissociation mechanism (DM) and the association mechanism (AM).97,98 The 4e− transfer of 3O2 and 1O2 in the DM (right of Fig. 8b) was taken as an illustration by Chen et al. to demonstrate the electron-transfer mechanism of the ORR.95 In the case of the 3O2RR, the π* orbital of 3O2 contains two unpaired electrons with parallel spins, requiring the first and second transferred electrons to have antiparallel spins to those already present. In accordance with the supposition that the electrons in the π* orbitals of 3O2 are in an up-spin configuration, the subsequent electrons should accordingly be in a down-spin configuration. Following the acceptance of these electrons by the two π* orbitals of the adsorbed O–O intermediate, the third electron is constrained to occupy only the higher-energy σ* orbital of the adsorbed OOH intermediate due to orbital occupancy constraints. The higher-energy antibonding orbital, which is filled by electrons, results in an instability of the O–O bond. This instability causes the adsorbed OOH to transform into adsorbed O and OH− intermediates. It could be postulated that the third electron occupies an orbital of the adsorbed O intermediate, with its spin likely antiparallel to the unpaired t2g electron. Finally, the last spin-down electron pairs with the third electron. For 1O2RR, the presence of an empty π* orbital indicates that the initial electron transfer is not impeded by Coulomb repulsion. It is highly likely that the spin orientation of the initial electron is opposite to that of the unpaired t2g electron. Subsequent electron transfers from the second to the last electron follow the same steps as in 3O2RR.
 | ||
| Fig. 8 Several reaction pathways for the ORR involving an (a) AM and (b) DM in both acidic and alkaline environments, alongside the mechanisms of the 3O2RR and 1O2RR, taking into account the spin of the transferred electrons.97,98 | ||
3.1.1.2. Spin effect in the ORR. Due to their maximum atomic utilization, adjustable electronic structures, and tunable coordination environment, SACs exhibit extraordinary activity and selectivity in the ORR.99,100 Despite the achievement and progress made by SACs, one of the most significant challenges in the exploitation of SACs is the scaling relationship between the adsorption/desorption toward a complicated multi-intermediate limit.101 Fe–N4 single-atom sites, characterized by a clearly defined structure, demonstrate a robust interaction with O species. This interaction presents a challenge during the ORR process, as it hinders the conversion of *O to *OH through protonation.102 In a study by Chen et al., an approach was taken to optimize the binding energy of O species.103 To this end, they constructed a catalyst featuring diatomic sites with FeCo molecular heterostructures. The FeCo-MHs were formed by overlapping CoPc molecules on FePc molecules, with the degree of overlapping dependent on the level of partial or complete overlap (Fig. 9a and b). As shown in Fig. 9c, the formation of the FePc/CoPc molecular heterostructure endowed the catalyst with higher performance in ORR/OER processes. In contrast to its Fe–N4 single-atom site counterpart, the heterostructure comprising molecular components was observed to influence the magnetic moments of the metal active centers, thereby increasing the proportion of the low-spin Fe(II)–N4 unit, which exhibited higher intrinsic activity compared to the intermediate-spin form. This optimized the intrinsic activity of the ORR (E1/2 = 0.95 V) at the summit of the volcano plot (Fig. 9d and e). It is worth noting that the arrangement of the FeCo molecular heterostructure could regulate the magnetic moments of both the Fe center (from 1.97 to 2.01) and the Co center (from 1.03 to 0.98) and thus change the binding strength for *O (Gibbs free energy from 4.95 to 1.42 eV), *OH (2.91 to 0.90 eV), and *OOH (5.35 to 4.06 eV) (Fig. 9f–h), which confirmed that the spin effect, achieved by building molecular heterostructures with dual-atom sites, is an effective strategy for regulating the ORR/OER catalytic activity.
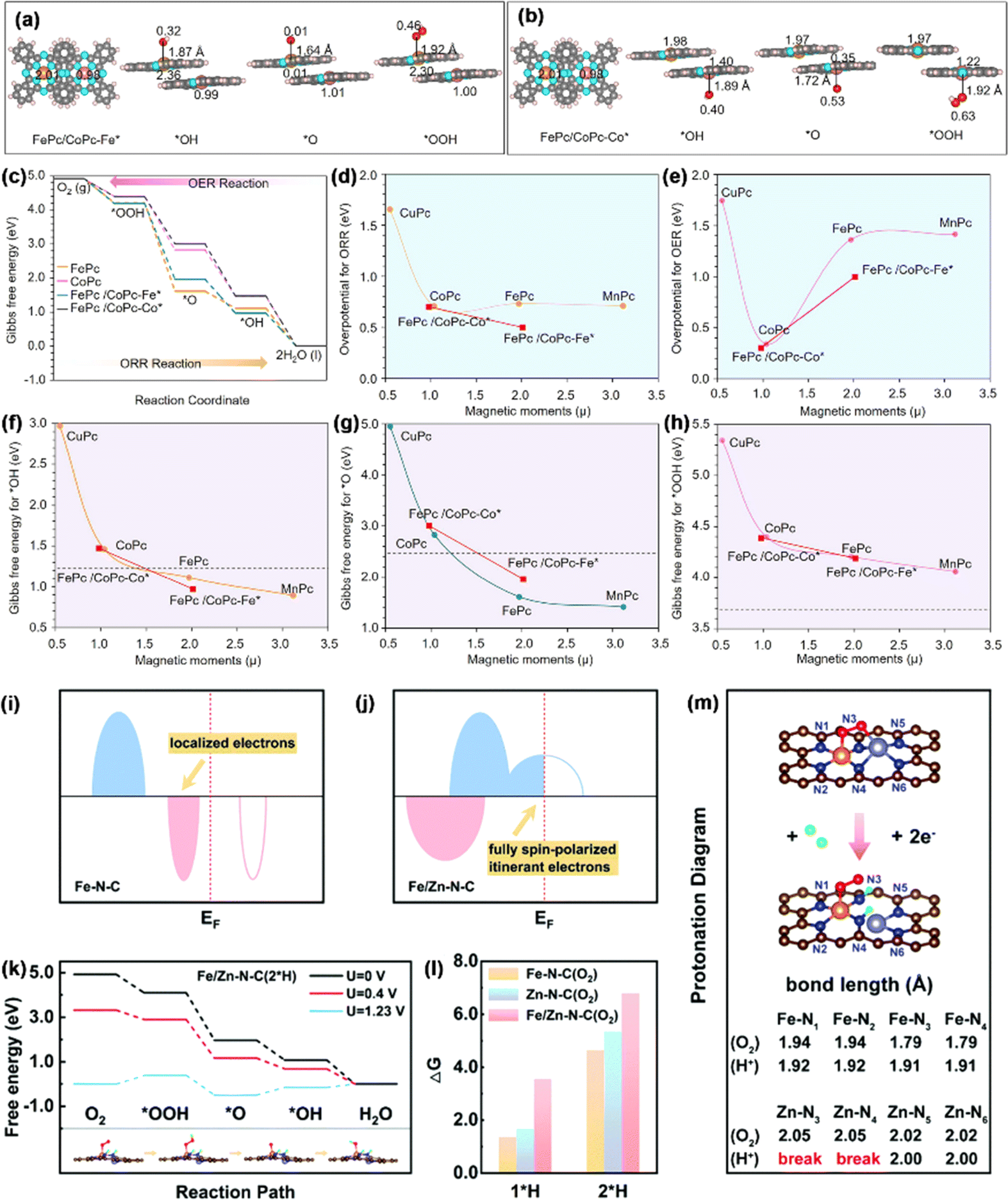 | ||
| Fig. 9 Optimized configurations of (a) FePc/CoPc-Fe* and (b) FePc/CoPc-Co* together with their respective intermediates. (c) Gibbs free energies for the ORR/OER. Graphs depicting overpotential versus magnetic moment (μ) for (d) the ORR and (e) OER. Magnetic moment analysis together with (f) *OH, (g) *O, and (h) *OOH. Reproduced with permission.103 Copyright 2023, American Chemical Society. (i and j) Diagrammatic representations of the electronic configurations of Fe–N–C and Fe/Zn–N–C. (k) Free energy of Fe/Zn–N–C. (l) ΔG1*H and ΔG2*H for catalysts exposed to O2. (m) Protonation map of Fe/Zn–N–C. Reproduced with permission.104 Copyright 2022, the Royal Society of Chemistry. | ||
Additionally, during the extended ORR process, the intermediate valence state of Fe2+ can initiate various side reactions, resulting in a degradation of the performance of Fe-based SACs. Li et al. reported a distinct electronic structure with half-metallic characteristics in a diatomic Fe/Zn–N–C catalyst, induced by the cooperative effects of Fe and Zn, as revealed through theoretical screening.104 This unique structure could generate spontaneous spin-polarized conduction electrons near the Fermi level (EF), thereby improving the capture and bonding of O2 molecules. Specifically, a conversion from a semiconductor to half-metal was noticed in the catalyst with the coordination of Zn (Fig. 9i and j), resulting in the emergence of spontaneous spin-polarized electrons at the EF. As explained above, in the stray field of a spin-polarized catalyst, the spins of two unpaired electrons in the antibonding π* orbits of paramagnetic O2 align in the same direction.
The full spin polarization of itinerant electrons promotes the formation of a bonded state, improving the capture of O2, which is the initial step in the ORR process. Binding energy calculations indicated that the adsorption of *O2 enhances the activity in Fe/Zn–N–C. On the other hand, DACs demonstrate greater stability during protonation. The protonation diagrams (Fig. 9k and l) showed a theoretical overpotential of 0.41 V for Fe/Zn–N–C after 2*H adsorption, matching the pre-protonation value, which confirmed that DACs could maintain their reactivity after post-protonation. This occurs because the protonation process leaves the electronic structure of the FeN4 active center unchanged (though ZnN4 is altered to ZnN2). As a result, the fully spin-polarized conduction electrons at the EF are preserved, maintaining the DACs' high reaction activity. Additionally, after protonation, the bond lengths of Fe–N3 and Fe–N4 were slightly increase from 1.79 to 1.91 Å (Fig. 9m). This change helps protect the FeN4 sites, ensuring a continuous and durable ORR process. The synthesized Fe/Zn–N–C showed exceptional ORR activity, with E1/2 values of 0.91 V in 0.1 M KOH and 0.81 V in 0.1 M HClO4, significantly surpassing those of Fe–N–C and Zn–N–C.
3.1.2.1. OER principles. Based on the established AEM and LOM routes, a potential mechanism for the generation of 3O2 and 1O2 was suggested by taking into account the spin orientation of the 4e− transfer during the OER (Fig. 10).95 LOM occurs when the oxygen p-band of lattice oxygen is elevated above the metal d-band center. O2 molecules produced via the LOM pathway originate from two sources (Fig. 10a): either both oxygen atoms originate from the oxide lattice, or one comes from the lattice oxygen while the other is sourced from the electrolyte (right in Fig. 10a). The AEM pathway includes two subcategories: Eley–Rideal (ER) (left in Fig. 10b) and Langmuir–Hinshelwood (LH) (right in Fig. 10b).
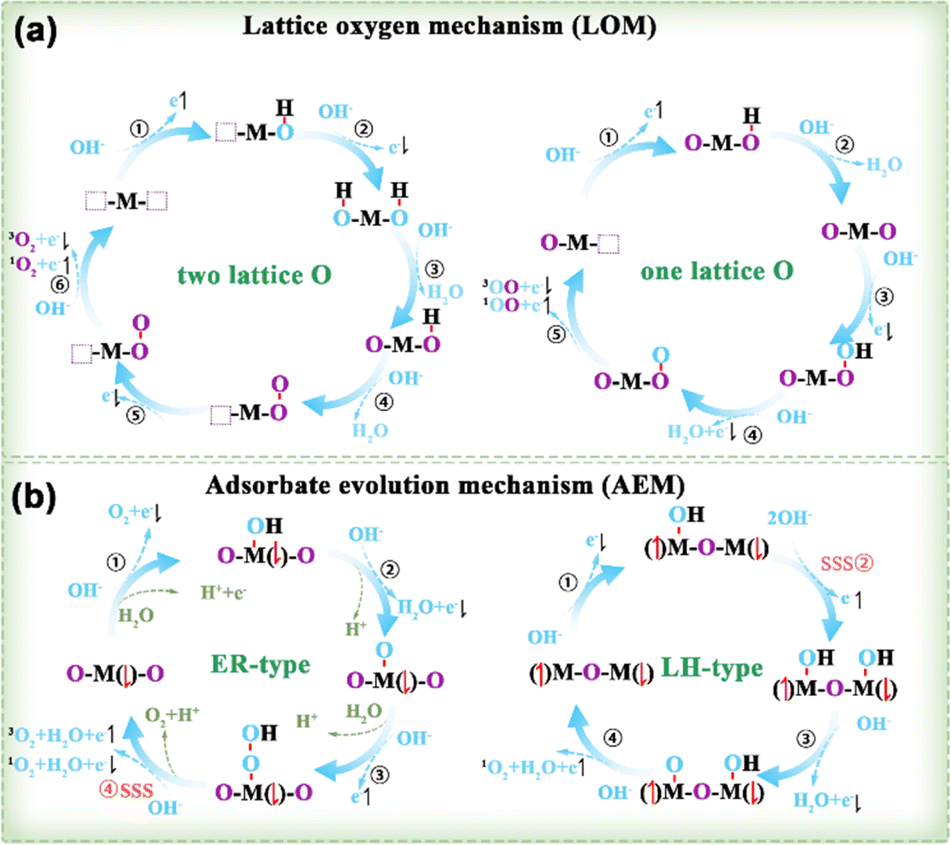 | ||
| Fig. 10 (a) LOM and (b) AEM of OER examined by taking into account the spin of the transferred electrons.95 M denotes the metal site, with the adjacent arrow indicating the spin direction of unpaired t2g electrons. Purple O and blue O represent the lattice and adsorbed O species, respectively. SSS stands for the spin selective step. | ||
3.1.2.2. Spin effect in the OER. The OER is essential for devices involved in energy-related fields, such as water splitting, fuel cells, and metal–air batteries (MABs).105–107 Basically, the OER process is thermodynamically challenging, involving multiple steps of proton-coupled electron transfer, and the inherently slow reaction dynamics requires highly efficient catalysts to lower the reaction overpotential for practical use.108 At present, many DACs have been effectively synthesized and implemented in applications pertaining to the OER.109–113 Nevertheless, the spin reaction mechanism at dual-metal active sites for improving the OER activity still needs further investigation.
Patzke et al. synthesized Ni, Co, and Fe single-atom sites, as well as Ni–Fe dual-atom sites on g-C3N4, denoted as Ni-CNG, Co-CNG, Fe-CNG, and NiFe-CNG.114 To gain insights into the adsorption behavior at Fe and Ni sites, they employed DFT calculations to assess the spin density of the catalysts. In all the samples (Fig. 11a–c), the results showed that Ni atoms exhibited a low-spin configuration, whereas Fe atoms demonstrated a high-spin configuration, revealing the presence of unpaired electrons in both the t2g and eg orbitals of Fe. In the in situ-formed Ni–O–Fe bonds, spin disparities between the Ni and Fe sites enhanced electron transfer during the OER process by establishing effective spin channels. Meanwhile, a new OER mechanism involving cooperative interaction between Ni and Fe sites in dual-site NiFe DACs was proposed (Fig. 11d). The OER initiation hinged on intricate interatomic electronic interactions at emerging Ni–O–Fe active sites. The local electronic structure was optimized to take advantage of unobstructed spin channels within the Ni–O–Fe bonds, thereby significantly augmenting the adsorption energies of the intermediates.
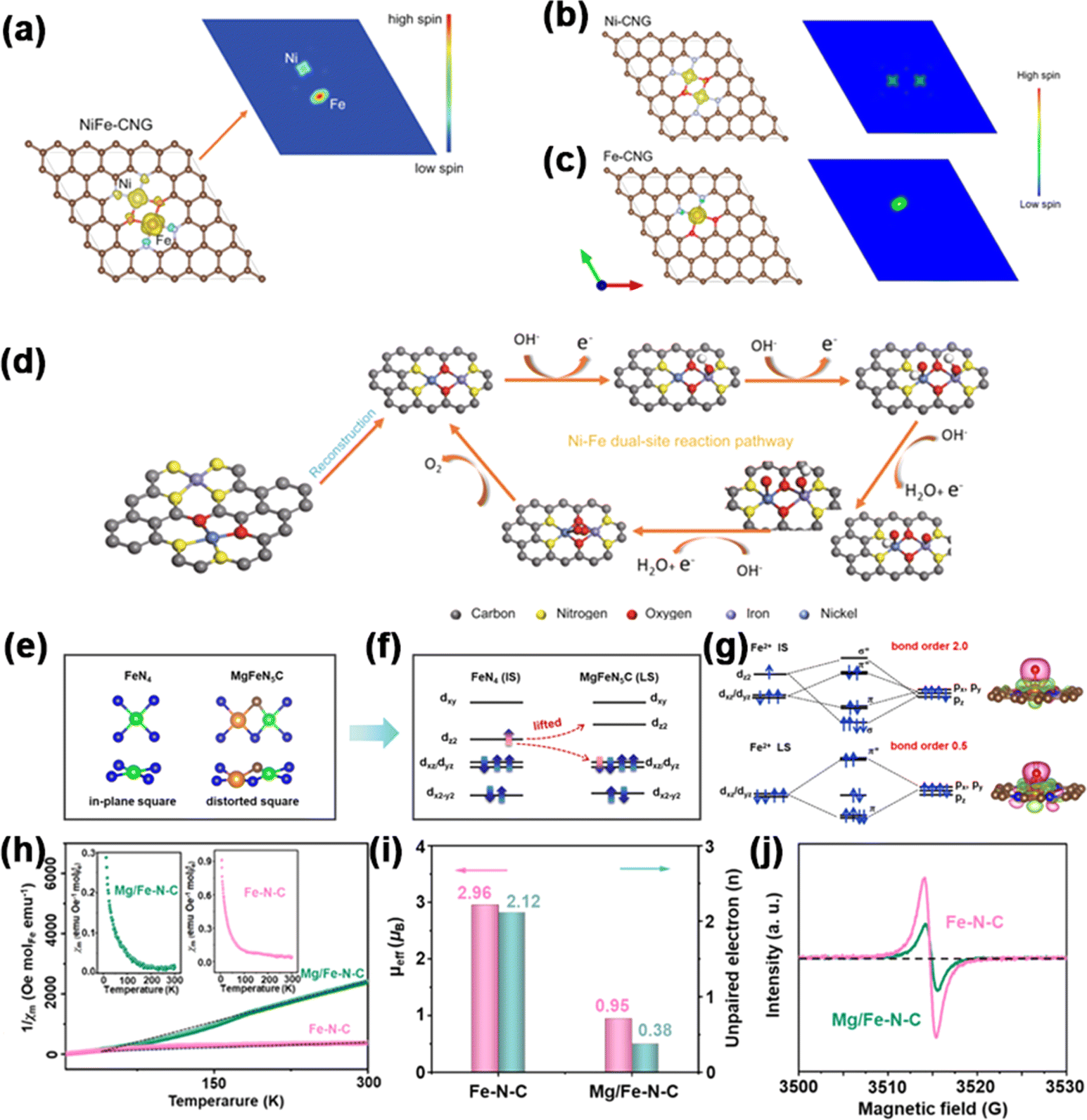 | ||
| Fig. 11 Spin density schema and spin channels of (a) NiFe-CNG, (b) Ni-CNG, and (c) Fe-CNG. (d) Proposed OER pathway diagram. Reproduced with permission.114 Copyright 2021, Nature. (e) Local geometry. (f) Spin state. (g) Charge density difference. (h) 1/χm–T curves; inset of (h) shows χm–T curves. (i) μeff and number of unpaired electrons (n) for Fe–N–C and Mg/Fe–N–C. (j) EPR spectra. Reproduced with permission.115 Copyright 2023, Wiley-VCH. | ||
Lu et al. reported a Mg–Fe dual-site catalyst (denoted as MgFeN5C) immobilized on a nitrogen-doped carbon substrate for the OER fabricated by an easy wet chemical method.115 The study found that introducing the Mg element into the Fe–N–C structure causes a significant mismatch in the ionic radii of Mg2+ and Fe2+ (rMg2+ = 7.2 Å, rFe2+ = 6.1 Å). This mismatch induces local crystal-field distortion in the FeN4 plane, elevating the energy level of Fe dz2. Consequently, Fe2+ undergoes a spin transition from an intermediate state (dxy2dxz2dyz1dz21, 2.96 μB) to a low state (dxy2dxz2dyz2, 0.95 μB). The low-spin (LS) state, characterized by smaller bond order, alleviates the effect of ΔG*O. As shown in Fig. 11e, in a typical FeN4 site, the central metal and circumambient N atoms exhibited an in-plane D4h symmetry, splitting the fivefold degenerate orbitals into dxy, dz2, dxz/dyz, and dx2−y2. Fe2+ attained the intermediate spin (IS) state through the partially occupied spin-up and spin-down channels. A strong interaction between Fe2+ and O intermediates arose from the unpaired dz2 and dxy/yz electrons, causing a maximal overlap of *O/*OH and *OOH with O2pz/2py and π* along the z-direction (Fig. 10f and g). However, in the dual-site Mg/Fe, the significant difference in ionic radii from Mg to Fe led to geometric distortion in the FeN4 moiety. DFT calculations revealed that within the MgFeN5C1 moiety, the three lowest orbitals—comprising dx2−y2 and the degenerate dxz/yz—were completely took up in the spin-up and spin-down channels. Conversely, the dz2 orbital was vacant, resulting in a low-spin (LS) state and consequently a reduced bond order with *O. To experimentally confirm the spin repartition, the authors conducted temperature-dependent magnetic susceptibility (χm) studies under ZFC-T conditions and employed EPR spectroscopy analysis. These analyses elucidated the electron spin allocations. The μeff values of Fe–N–C and Mg/Fe–N–C were calculated to be 2.96 μB and 0.95 μB, respectively (Fig. 10h and i). The d-electron count of Fe2+ declined from around 2.12 in Fe–N–C to about 0.38 in Mg/Fe–N–C, significantly affecting the geometry of the signal peaks (Fig. 11j), as acknowledged in the field for its impact on electron–environment interactions. Therefore, these results indicate that local field distortion can effectively adjust Fe spin configurations, and the LS state is more favorable for enhancing the intrinsic OER performance.
Recent studies have indicated that the high electronegativity of the coordinated N hinders intermediate adsorption at the metal center, raising the potential barrier and reducing the reaction kinetics.116 Therefore, Hu et al. utilized a P-doped metal—organic framework as a porous substrate to synthesize Co1-PNC/Ni1-PNC, with a P atom replacing a N atom in the M–N4 structure. This modification enhanced the electron spin density and altered the electronegativity, resulting in improved ORR/OER activities.117 The catalyst demonstrated high OER activity with η10 = 0.39 V, outperforming Co1–NC/Ni1–NC (η10 = 0.41 V) synthesized without P-doping.
3.2. Hydrogen evolution reaction
| H+(aq.) + e− → Hads | (1) |
Step (2) can proceed via two distinct pathways: when the surface concentration of H* is high, two H* species combine to form H2 in a process known as the Tafel step, which involves chemical desorption.
| Hads + Hads → H2(g) |
Otherwise, H* reacts with another H3O+ + e− (via proton-coupled electron transfer) to form H2 (Heyrovsky step, electrochemical desorption).
| Hads + H+(aq.) + e− → H2(g) |
This process is shown in Fig. 12a. Similarly, in alkaline electrolytes, the HER mechanism follows either the Volmer–Tafel or Volmer–Heyrovsky pathway, with water molecules acting as the reactant (Fig. 12b).
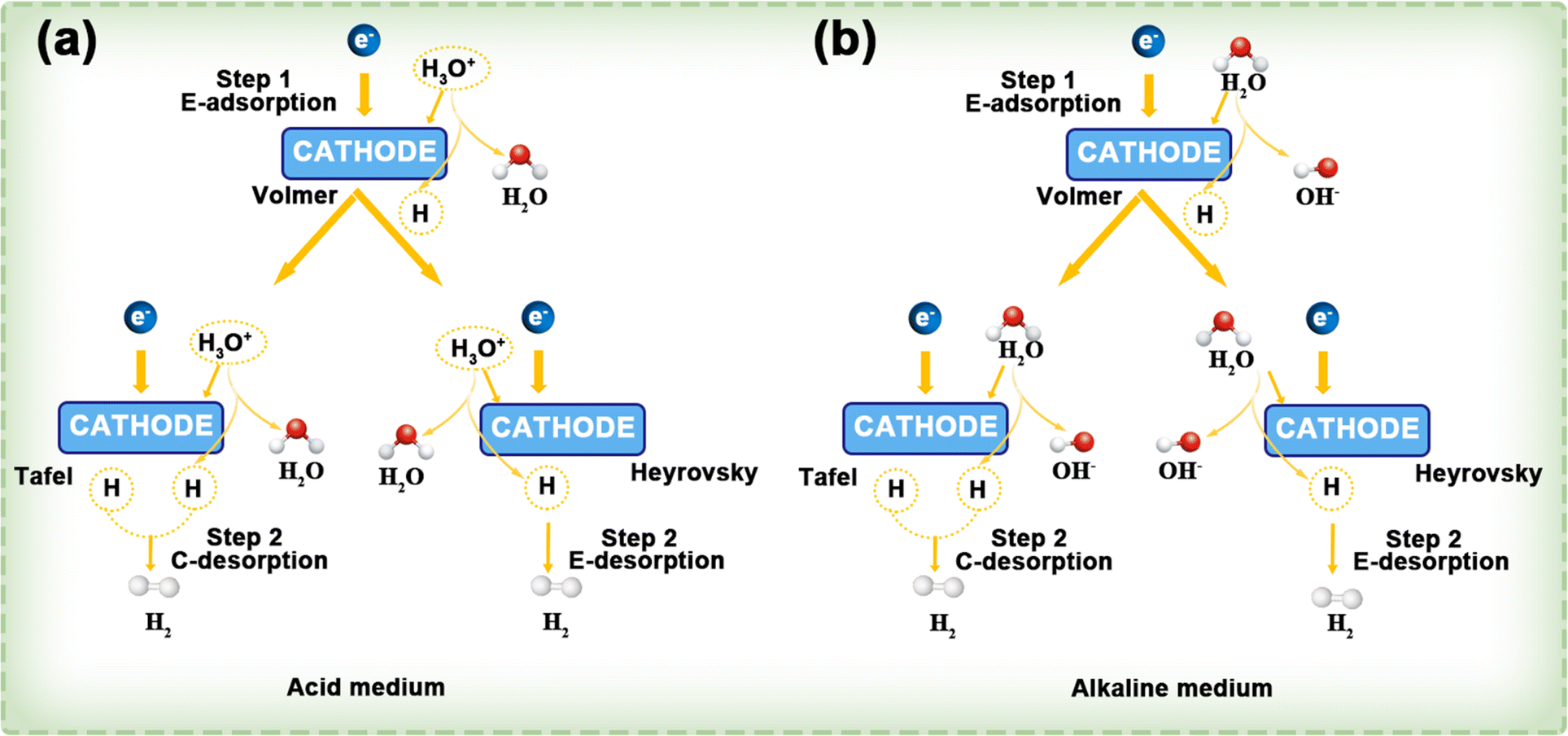 | ||
| Fig. 12 In both acidic (a) and alkaline (b) environments, the HER follows the Volmer–Tafel and Volmer–Heyrovsky mechanisms. The terms E- and C- indicate electrochemical and chemical processes, respectively. Reproduced with permission.119 Copyright 2022, American Chemical Society. | ||
Huang and colleagues developed a strategy for the atomization and sintering of nanoparticles, single atoms, and diatoms (NP–SA–DA).82 This approach allowed generating Co nanoparticles, CoN4 single atoms, and Co2N5 diatoms on N-doped hollow carbon spheres. In the HER, the spin magnetic moment is negatively linearly related to the Gibbs free energy of H* (ΔGH*). Further, ΔGH* decreases linearly with the increase in the spin magnetic moment. Additionally, the dual-atom Co2N5 exhibited the highest ΔGH*, suggesting that incorporating homonuclear Co atoms results in a low-spin state. This low-spin state is beneficial for the balanced adsorption and desorption of H* intermediates, thereby enhancing the HER performance. The Gibbs free energy of H* intermediates for HER in Co2N5 and CoN4 revealed that the Co2N5 dual-atom configuration had an H* adsorption energy of −0.04 eV. This value is nearer to 0 eV, signifying superior HER activity.
3.3. Electrocatalytic carbon dioxide reduction reaction
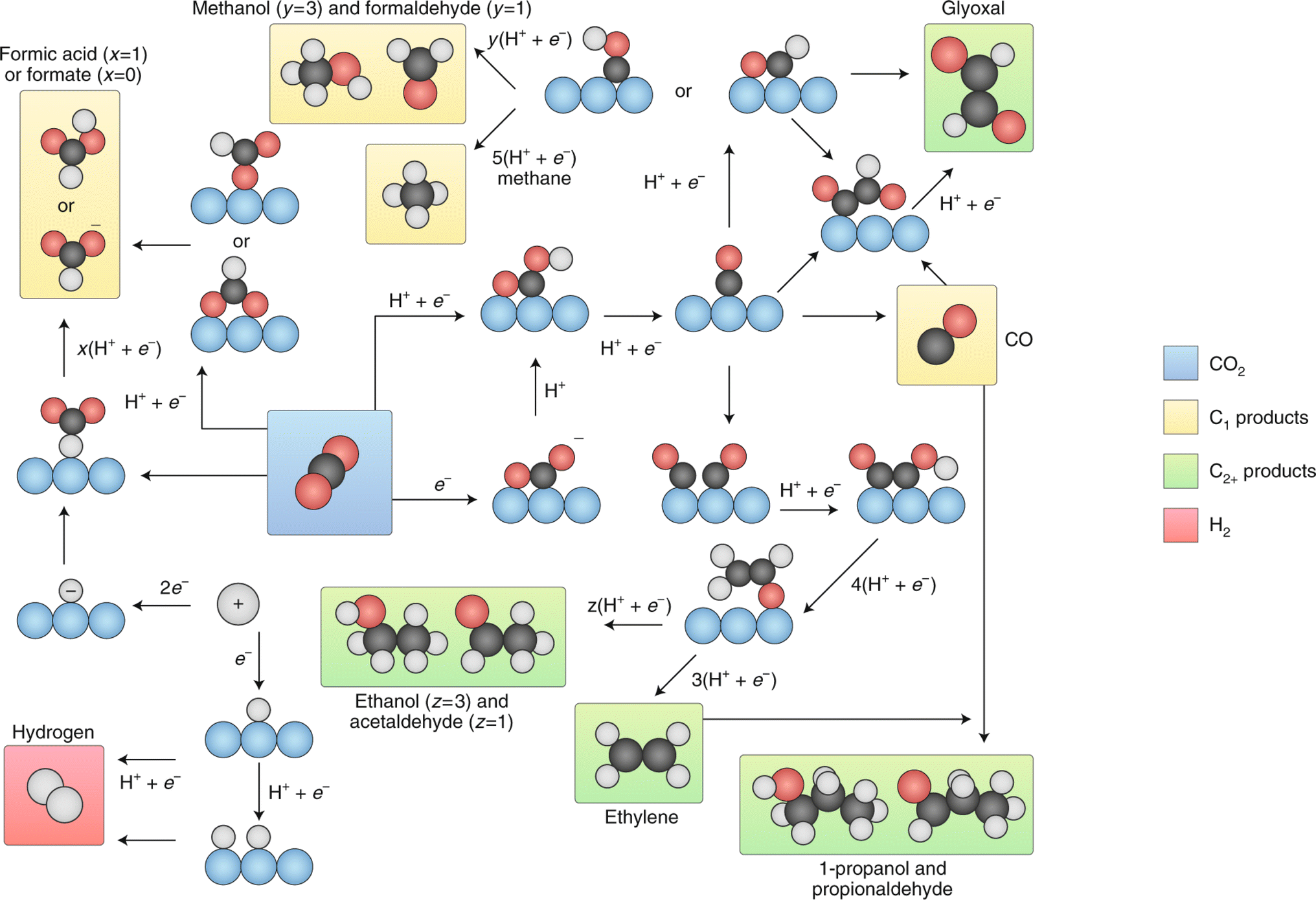 | ||
| Fig. 13 Summary of the reaction pathways for the ECO2RR leading to various products. Reproduced with permission.123 Copyright 2019, Nature. | ||
In work by Lu and co-workers, FeCo–NC and Fe–NC/Co–NC were adopted as probes to investigate the ECO2RR performance, aiming at the potential formation of C1 product.127 First, the models were decided on using spin-polarized DFT (Fig. 14a). It was shown that dual atoms of Fe and Co collaborate to provide bridge sites for stable CO2 adsorption and modify the electronic structure by analyzing CO2 adsorption configurations and energy (Fig. 14b–d). Also, electronic DOS analysis revealed the impact of the ηFe(C)–ηCo(O) configuration on FeCo–NC (Fig. 14e and f). These give FeCo–NC a significant advantage in CO2 adsorption and initial activation compared to the corresponding SACs. The energy levels of the Fe and Co diatomic center d-orbitals in FeCo–NC were found to align closely with the CO 2π* orbitals around −1.00 eV, as revealed by the DOS analysis of CO adsorption. The interplay between these orbitals leads to the constitution of a partly filled d–2π* orbital below the EF and an empty d–2π* orbital above it, which is essential for the stable adsorption of CO. Due to powerful spin polarization, the majority α-spin orbitals receive considerable exchange stabilization energy. The α-spin d-orbitals at the Fe and Co dual-atom centers exhibited an energy level of 2.00 eV, which is lower than the energy level of the CO 2π* orbitals. Additionally, the spin density diagram (Fig. 14g) showed there was significant spin polarization at the Fe and Co diatomic centers, facilitating CO adsorption and activation.
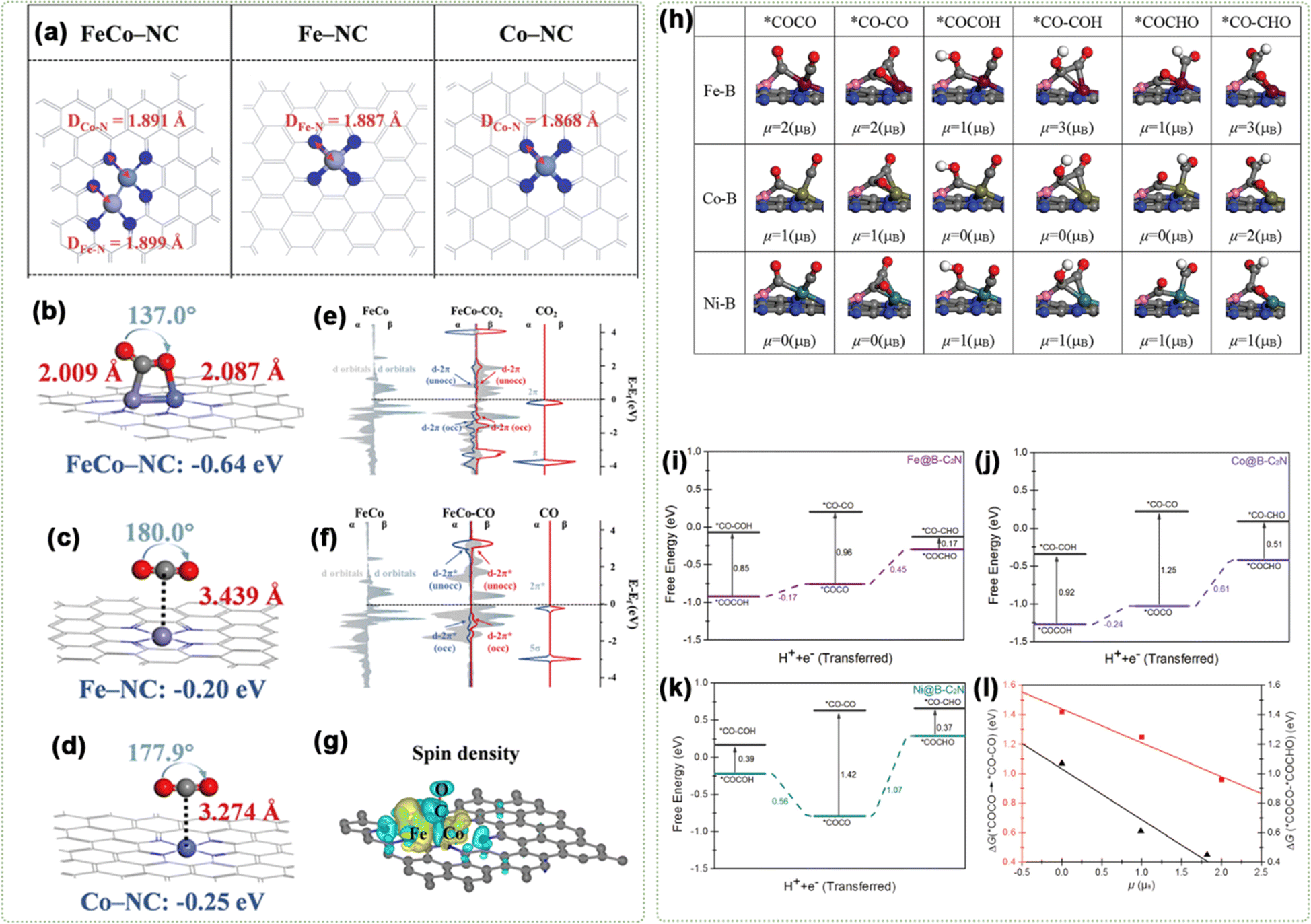 | ||
| Fig. 14 (a) Optimized structural models. Predominant configurations of CO2 adsorption for (b) FeCo–NC, (c) Fe–NC, and (d) Co–NC. Projected density of state (PDOS) analysis of (e) the 3d orbitals of Fe and Co dual-atom centers and the 2p orbitals of CO2; (f) the 3d orbitals of Fe, Co dual-atom centers, 2p orbitals of the CO gas molecule. (g) Spin density of CO-assisted FeCo–NC. Reproduced with permission.127 Copyright 2023, Wiley-VCH. (h) Optimized structure of C–C coupling. Free energy profiles for C–C coupling on (i) Fe@B-C2N, (j) Co@B-C2N, and (k) Ni@B-C2N at 0 V (vs. RHE). (l) Correlation of the reaction free energy as a function of the spin moment (μ) for adsorbed reactants. Reproduced with permission.130 Copyright 2021, Wiley-VCH. | ||
The μeff value can reveal the influence of different spin states on C–C coupling. For instance, through spin-polarized DFT, Liu's group showed that M@X-C2N (M = Fe, Co, Ni, and Cu; X = B) exhibited stronger functionality than single/double-M centers in promoting the CO2RR, with a particular enhancement observed in C–C coupling.130 Specifically, the energetics and spin moment of C–C coupling via from *COCHO to *CO–CHO, from *COCOH to *CO–COH, and from *COCO to *CO–CO were calculated, as displayed in Fig. 14h. As depicted in Fig. 14i–k, among M@B-C2N (M = Fe (μ = 2 μB), Co (μ = 1 μB), and Ni (μ = 0 μB)), Fe@B-C2N showed the lowest Gibbs free energy barrier of 0.17 eV. Among them, Ni@B-C2N (μ = 0 μB) primarily produces CH4, exhibiting the lowest energy barrier at 0.42 eV. On Fe–B or Co–B sites, the transition from *COCHO to *CO–CHO (Δμ = 2 μB) was more favorable than the transition from *COCOH to *CO–COH. On Ni–B sites, both transitions—*COCHO to *CO–CHO and *COCOH to *CO–COH were equally probable with Δμ = 0 μB. This indicates that C–C coupling feasibility depends on the initial spin state and intermediates. Fig. 14l illustrates that the high-spin state of the M–B dual-atom sites significantly enhances the energetics of both chemical and electrochemical coupling processes. Specifically, chemical C–C coupling (*COCO → *CO–CO) and the electrochemical coupling (*COCO + H+ + e− → *COCHO) are more actively beneficial under these conditions, indicating that the high spin moment of M in M–B dual-atom sites promotes C–C coupling and protonation in the ECO2RR under ambient conditions.
To elucidate why almost no DACs can efficiently produce substantial quantities of multi-carbon products, Li et al. conducted an in-depth examination of the surface states of representative homonuclear and heteronuclear DACs. They employed spin-polarized DFT calculations, incorporating van der Waals interactions, to explore the underlying mechanisms of the CO2RR.55 They discovered that the favorable surface states of DACs are covered by CO either unilaterally or bilaterally at the M–M bridge sites, making CO the primary product of DACs. However, C–C coupling predominantly occurs at the top sites of metal surfaces. However, CO tends to preferentially occupy the bridge sites between the two metal atoms. This preferential adsorption of CO at the bridge sites can hinder subsequent C–C coupling reactions. Furthermore, based on the energy changes, the occurrence of C–C coupling on the surface of DACs is both thermodynamically and kinetically unfeasible.
3.4. Electrochemical nitrogen reduction reaction
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond, the energy input is high before the hydrogenation reaction occurs, and the two separate nitrogen atoms are converted into each other by the hydrogenation process. Taking into account various sequences of H addition to N atoms, the hydrogenation process in the associative mechanism can follow three distinct pathways: distal, alternating, and enzymatic. As shown in Fig. 15b, in the distal pathway, one of the nitrogen atoms in a nitrogen molecule is connected to the active site of the catalyst, and the hydrogen process is carried out continuously from the N atom far away from the catalyst until the ammonia gas formed is separated, and then will continue to hydrogenate the nitrogen atom near the catalyst to generate a second ammonia molecule. In comparison, the alternating pathway also occurs in the end-on adsorption configuration, where a nitrogen atom is connected to the active site of the catalyst, but in the hydrogenation process, the two nitrogen atoms alternately bind hydrogen atoms until the final step of the pathway. In Fig. 15c, it can be seen that the enzymatic pathway occurs in a side-on adsorption configuration, in which the nitrogen molecule lies horizontally above the catalyst active site and two N atoms of N2 are bonded to the catalytic active site simultaneously, and they alternately bind with H atoms until they release two NH3 molecules. Additionally, a novel mechanism, entirely distinct from the dissociative and associative pathways, has been raised for the conversion of N2 to NH3, known as the MvK pathway. As illustrated in Fig. 15d, it is believed that NH3 formation on the surface of transition metal nitrides (TMNs) follows this MvK pathway (or a combination of pathways including the MvK pathway). In accordance with this mechanism, N atoms in the TMN are reduced to NH3, and the N2 from the environment fills the vacancies left by the extracted lattice N atoms, thereby completing the catalyst renewal and preparing for a new reaction cycle.
N bond, the energy input is high before the hydrogenation reaction occurs, and the two separate nitrogen atoms are converted into each other by the hydrogenation process. Taking into account various sequences of H addition to N atoms, the hydrogenation process in the associative mechanism can follow three distinct pathways: distal, alternating, and enzymatic. As shown in Fig. 15b, in the distal pathway, one of the nitrogen atoms in a nitrogen molecule is connected to the active site of the catalyst, and the hydrogen process is carried out continuously from the N atom far away from the catalyst until the ammonia gas formed is separated, and then will continue to hydrogenate the nitrogen atom near the catalyst to generate a second ammonia molecule. In comparison, the alternating pathway also occurs in the end-on adsorption configuration, where a nitrogen atom is connected to the active site of the catalyst, but in the hydrogenation process, the two nitrogen atoms alternately bind hydrogen atoms until the final step of the pathway. In Fig. 15c, it can be seen that the enzymatic pathway occurs in a side-on adsorption configuration, in which the nitrogen molecule lies horizontally above the catalyst active site and two N atoms of N2 are bonded to the catalytic active site simultaneously, and they alternately bind with H atoms until they release two NH3 molecules. Additionally, a novel mechanism, entirely distinct from the dissociative and associative pathways, has been raised for the conversion of N2 to NH3, known as the MvK pathway. As illustrated in Fig. 15d, it is believed that NH3 formation on the surface of transition metal nitrides (TMNs) follows this MvK pathway (or a combination of pathways including the MvK pathway). In accordance with this mechanism, N atoms in the TMN are reduced to NH3, and the N2 from the environment fills the vacancies left by the extracted lattice N atoms, thereby completing the catalyst renewal and preparing for a new reaction cycle.
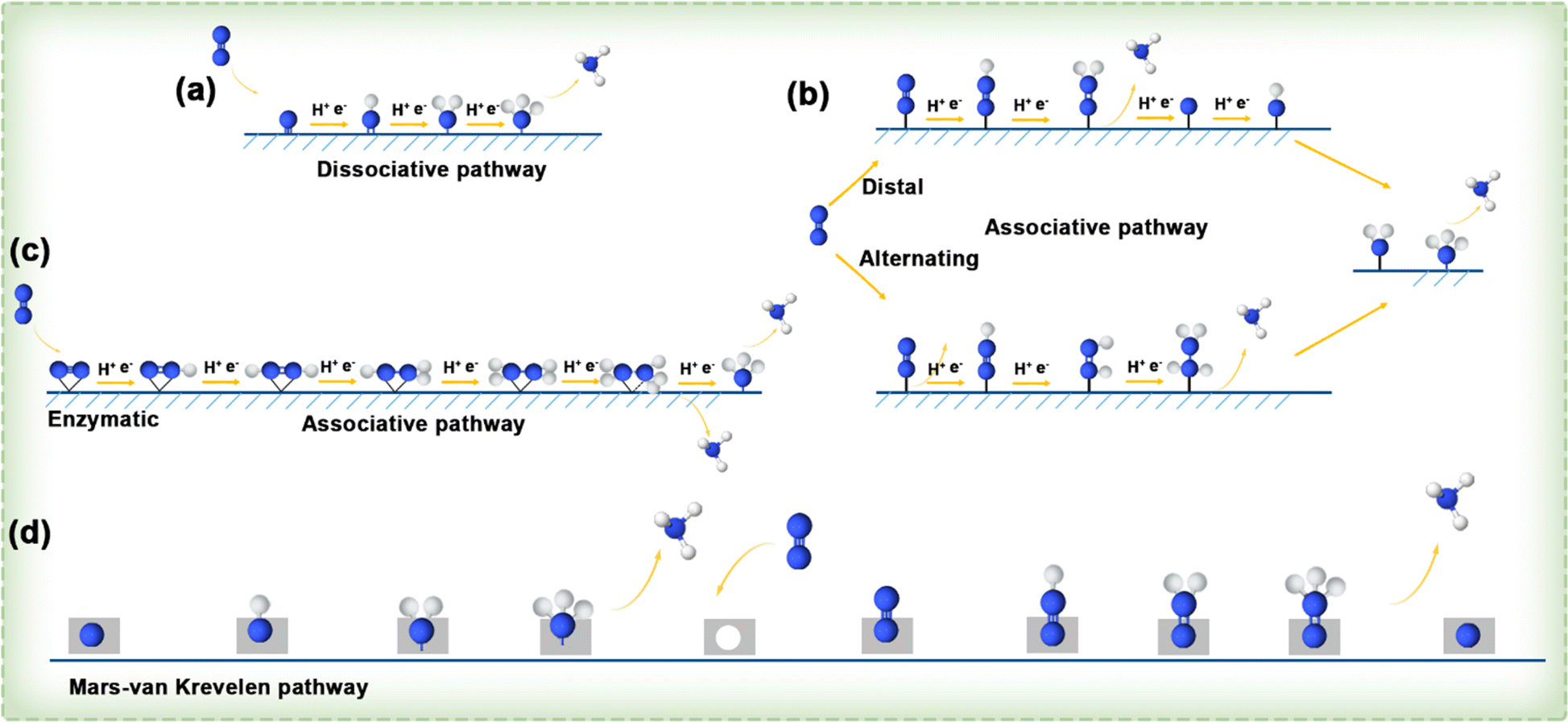 | ||
| Fig. 15 (a–c) Diagram illustrating the dissociative and associative pathways. (d) Illustration depicting MvK reaction pathways.131 | ||
Zheng et al. discovered that Fe SA anchored on the oxide surface layer of Ti foam could improve the NH3 Faradaic efficiency (FE). This enhancement was attributed to oxygen vacancies (OVs) in the titanium oxide layer, which cause spin polarization in both Fe and neighboring Ti atoms.134 They confirmed the electronic structures and spin states of the spin-polarized Fe1–Ti (SP-Fe1–Ti) and spin-depressed Fe1–Ti (SD-Fe1–Ti) through various spectroscopic and magnetic measurements (Fig. 16a–f). DFT calculations were utilized to investigate the NITRR mechanism involving SP-Fe1–Ti pairs. The spin density diagrams (Fig. 16g) show that in SD-Fe1–Ti, only the Fe atom is spin-polarized. In contrast, both the Fe atom and the adjacent Ti atom are spin-polarized in the SP-Fe1–Ti configuration. Additionally, in the SD-Fe1–Ti configuration, the spin-up and spin-down components of the Ti atom are nearly equal. Nevertheless, the introduction of OVs leads to the emergence of unpaired electronic states near the EF (Fig. 16h). The presence of magnetic Ti and Fe atoms plays a crucial role in enhancing the efficiency of the NITRR process. The primary interactions between Fe and NO arise from the dxz/dyz/dz2 orbitals of the Fe atom and the σ/π orbitals of NO (Fig. 16i), consistent with the high-spin-polarization configuration (t2g4 eg2) of Fe. Consequently, the spin-polarized Fe–Ti paired sites could effectively facilitate NO3− deoxygenation to *NO, stabilize *NO and *NHO intermediates, and enhance the subsequent *NO hydrogenation. This also inhibits the HER, resulting in a high activity and selectivity of the SP-Fe1–Ti electrode for the NITRR. As anticipated, SP-Fe1–Ti demonstrated outstanding performance, achieving an impressive NH3 yield rate of 272000 μg per h per mg per Fe with a high NH3 FE of 95.2% at −0.4 V vs. RHE.
 | ||
| Fig. 16 (a) Energy alignment. (b) XPS spectra of Fe 2p; (c) Mössbauer spectra; (d) Ti L-edge soft-XAS spectra; (e) Ti 2p XPS spectra; (f) 1/χmversus temperature plots and (g) optimized structures and spin density diagrams of SD-Fe1–Ti and SP-Fe1–Ti. (h) DOSs of Fe and Ti atoms. (i) Remarkable orbital interactions between the Fe atom and the NO molecule. Reproduced with permission.134 Copyright 2024, Nature. Magnetic susceptibility of (j) FePPc and (k) FeMoPPc. (l) Room-temperature 57Fe Mössbauer spectrum of FeMoPPc. Reproduced with permission.135 Copyright 2024, Science. | ||
Zhang and colleagues designed and created a precisely defined Fe–Mo hetero-single-atom (h-SAs) electrocatalyst for the ENRR, which was obtained by co-anchoring atomically dispersed FeN4 and MoN4 coordination sites within a polyphthalocyanine (FeMoPPc) organic framework using a low-temperature melt polymerization technique.135 ZFC M–T susceptibility measurements were performed to clarify the electronic configuration of the d-orbitals of Fe in FeMoPPc (Fig. 16j and k). The findings showed that the computed μeff values of the Fe sites for adsorption decreased from 2.43 to 1.89 μeff, following the incorporation of Mo. 57Fe Mössbauer spectroscopy was performed and confirmed that the D1 peak could be attributed to FePc-like medium-spin FeIIN4 species, indicating that FeII was predominantly present in a medium-spin state in FeMoPPc through quantitative analysis (Fig. 16l). Notably altering the spin state of the Fe center from high spin to medium spin enhanced the π-backdonation process, aiding the initial hydrogenation of N2. As expected, the energy barrier of the desorption of NH3 on FeN4 sites (0.09 eV) was much smaller than that on MoN4 sites (0.54 eV). Consequently, the synthesized FeMoPPc with medium-spin FeN4 demonstrated exceptional NRR performance. It showed faradaic efficiencies 2.0 and 9.0 times higher, and NH3 yields 2.0 and 17.2 times greater than those of FePPc and MoPPc, respectively.
4. Summary and perspective
Significant attention has been directed toward controlling the spin states at the active centers of DACs. In this review, we delved into the implications of spin state transitions for various electrocatalytic reactions. We described the spin state transition characterizations and descriptors of atomic catalysts, and then proposed strategies for regulating the spin states of DACs' active centers. Finally, we summarized recent research on the spin effect of DACs in the ORR, OER, HER, ECO2RR, and ENRR, providing a foundation for future studies. Despite progress, the research on spin state transitions in DACs is still in its early stage, and some key issues remain that deserve further attention, including the following:(i) The impact of various spin states in DACs on different catalytic reactions remains under-researched. It is unclear whether spin state transitions alter the adsorption configurations of reaction intermediates and reaction pathways. In the future, more advanced characterization techniques need to be integrated to further probe the mechanisms.
(ii) Currently, the primary methods for studying spin state transitions in DACs rely on ex situ characterization techniques. During catalysis, the reaction intermediates adsorbed at active sites are dynamically evolving. Ex situ techniques cannot capture the spin state transitions of the magnetism of metal active centers during reaction processes. In situ characterization methods for detecting the spin states of active centers mainly include in situ Mössbauer spectroscopy and in situ XES. Advancing in situ characterization techniques for the real-time detection of spin states, such as in situ XAS and in situ VSM, is necessary for the on-site monitoring of the spin changes in active sites and reaction intermediates during catalytic reactions.
(iii) There exist limited approaches to modulate the spin states of the active centers of DACs. Additionally, there is a lack of real-time control strategies for spin states during reactions. The real-time regulation of spin states during reactions can help optimize the adsorption and desorption of key reaction intermediates, thereby enhancing the catalyst function. Developing real-time spin state control strategies is a crucial research direction in this field.
(iv) The spin catalytic HER, ENRR, and ECO2RR mechanisms on DACs are rarely reported. To develop more reliable electrocatalysts, it is essential to understand how changes in the spin states of their active centers affect their catalytic performances. Theoretical calculations using computational chemistry can not only assist experimental studies in identifying catalytic active sites, but can also play a crucial role in elucidating the reaction mechanisms, which in turn would help to guide the design of new catalysts. Therefore, future efforts are needed to address this challenge.
(v) Precise designs to take advantage of spin effects need to be explored. Additionally, there is no comprehensive summary on the selection of magnetic catalysts to achieve optimal catalytic performance. In the future, high-throughput theoretical calculations may be employed to analyze catalytic reaction mechanisms from the perspective of the electronic structure. Furthermore, machine learning could be utilized to elucidate the relationship between spin effects and performance, thereby guiding the design of catalysts.
In general, while prominent progress has been made in the spin effect of DACs in the field of electrocatalysis, several issues still need to be addressed. Nevertheless, we anticipate that the advancement of new synthesis methods, sophisticated characterization techniques, regulation strategies, and computational theories will facilitate the rational design of high-performance DACs for energy-storage and -transformation applications.
Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.Author contributions
All of the authors contributed to the literature search, writing and editing of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22075099), and the Natural Science Foundation of Jilin Province (No. 20220101051JC).Notes and references
- W. Zhang, Y. Chao, W. Zhang, J. Zhou, F. Lv, K. Wang, F. Lin, H. Luo, J. Li, M. Tong, E. Wang and S. Guo, Adv. Mater., 2021, 33, 2102576 CrossRef.
- T. He, A. R. P. Santiago, Y. Kong, M. A. Ahsan, R. Luque, A. Du and H. Pan, Small, 2022, 18, 2106091 CrossRef.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2017, 16, 57–69 CrossRef.
- L. Tang, X. Meng, D. Deng and X. Bao, Adv. Mater., 2019, 31, 1901996 CrossRef PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- L. Xiao, Z. Wang and J. Guan, Adv. Funct. Mater., 2024, 34, 2310195 CrossRef.
- L. Xiao, L. Qi, J. Sun, A. Husile, S. Zhang, Z. Wang and J. Guan, Nano Energy, 2024, 120, 109155 CrossRef.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem, 2018, 2, 65–81 CrossRef.
- Y. Liu, L. Zhou, S. Liu, S. Li, J. Zhou, X. Li, X. Chen, K. Sun, B. Li, J. Jiang and H. Pang, Angew. Chem., Int. Ed., 2024, 63, e202319983 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Chem. Rev., 2020, 120, 623–682 CrossRef CAS.
- X. Bai, Y. Wang, J. Han, X. Niu and J. Guan, Appl. Catal., B, 2023, 337, 122966 CrossRef CAS.
- Z. Zhang, C. Feng, D. Wang, S. Zhou, R. Wang, S. Hu, H. Li, M. Zuo, Y. Kong, J. Bao and J. Zeng, Nat. Commun., 2022, 13, 2473 CrossRef CAS.
- R. Li, J. Zhao, B. Liu and D. Wang, Adv. Mater., 2024, 36, 2308653 CrossRef CAS.
- Y. Hu, Z. Li, B. Li and C. Yu, Small, 2022, 18, 2203589 CrossRef.
- J. Zhang, Q.-a. Huang, J. Wang, J. Wang, J. Zhang and Y. Zhao, Chin. J. Catal., 2020, 41, 783–798 CrossRef.
- T. Tang, X. Bai, Z. Wang and J. Guan, Chem. Sci., 2024, 15, 5082–5112 RSC.
- R. R. Burch, E. L. Muetterties, R. G. Teller and J. M. Williams, J. Am. Chem. Soc., 1982, 104, 4257–4258 CrossRef.
- C. Fang, J. Zhou, L. Zhang, W. Wan, Y. Ding and X. Sun, Nat. Commun., 2023, 14, 4449 CrossRef PubMed.
- J. Wang, C.-X. Zhao, J.-N. Liu, Y.-W. Song, J.-Q. Huang and B.-Q. Li, Nano Energy, 2022, 104, 107927 CrossRef.
- T. Tang, Z. Wang and J. Guan, Coord. Chem. Rev., 2023, 492, 215288 CrossRef.
- A. L. Buchachenko, L. V. Ruban, E. N. Step and N. J. Turro, Chem. Phys. Lett., 1995, 233, 315–318 CrossRef CAS.
- W. Zhang, K. Banerjee-Ghosh, F. Tassinari and R. Naaman, ACS Energy Lett., 2018, 3, 2308–2313 CrossRef CAS.
- Z. Fang, W. Zhao, T. Shen, D. Qiu, Y. Lv, X. Hou and Y. Hou, Precis. Chem., 2023, 1, 395–417 CrossRef CAS.
- G. Xu, C. Cai and T. Wang, J. Am. Chem. Soc., 2022, 144, 23089–23095 CrossRef CAS.
- F. A. Garcés-Pineda, M. Blasco-Ahicart, D. Nieto-Castro, N. López and J. R. Galán-Mascarós, Nat. Energy, 2019, 4, 519–525 CrossRef.
- X. Li, Z. Cheng and X. Wang, Electrochem. Energy Rev., 2021, 4, 136–145 CrossRef CAS.
- A. H. Compton, J. Franklin Inst., 1921, 192, 145–155 CrossRef CAS.
- D. Yardimci, P. Serna and B. C. Gates, Chem.–Eur. J., 2013, 19, 1235–1245 CrossRef CAS.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS.
- Y. Sun, J. Wang, Q. Liu, M. Xia, Y. Tang, F. Gao, Y. Hou, J. Tse and Y. Zhao, J. Mater. Chem. A, 2019, 7, 27175–27185 RSC.
- H. Li, J. Wang, R. Qi, Y. Hu, J. Zhang, H. Zhao, J. Zhang and Y. Zhao, Appl. Catal., B, 2021, 285, 119778 CrossRef CAS.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J.-N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- T. He, Y. Chen, Q. Liu, B. Lu, X. Song, H. Liu, M. Liu, Y.-N. Liu, Y. Zhang, X. Ouyang and S. Chen, Angew. Chem., Int. Ed., 2022, 61, e202201007 CrossRef CAS PubMed.
- J. Zhang, Y. Zhao, W. Zhao, J. Wang, Y. Hu, C. Huang, X. Zou, Y. Liu, D. Zhang, X. Lu, H. Fan and Y. Hou, Angew. Chem., Int. Ed., 2023, 62, e202314303 CrossRef CAS PubMed.
- L. Wang, Q. An, X. Sheng, Z. Mei, Q. Jing, X. Zhao, Q. Xu, L. Duan, X. Zou and H. Guo, Appl. Catal., B, 2024, 343, 123509 CrossRef CAS.
- T. S. Metzger, S. Mishra, B. P. Bloom, N. Goren, A. Neubauer, G. Shmul, J. Wei, S. Yochelis, F. Tassinari, C. Fontanesi, D. H. Waldeck, Y. Paltiel and R. Naaman, Angew. Chem., Int. Ed., 2020, 59, 1653–1658 CrossRef CAS.
- K. Song, B. Yang, X. Zou, W. Zhang and W. Zheng, Energy Environ. Sci., 2024, 17, 27–48 RSC.
- R. J. Soulen, J. M. Byers, M. S. Osofsky, B. Nadgorny, T. Ambrose, S. F. Cheng, P. R. Broussard, C. T. Tanaka, J. Nowak, J. S. Moodera, A. Barry and J. M. D. Coey, Science, 1998, 282, 85–88 CrossRef CAS.
- R. G. Lawler, J. Am. Chem. Soc., 1967, 89, 5519–5521 CrossRef CAS.
- E. A. Zhukov, D. R. Yakovlev, A. Schwan, O. A. Yugov, A. Waag, L. W. Molenkamp and M. Bayer, Phys. Status Solidi B, 2014, 251, 1872–1880 CrossRef CAS.
- B. F. Minaev and H. Ågren, J. Quantum Chem., 1996, 57, 519–532 CrossRef CAS.
- R. Chattot, O. Le Bacq, V. Beermann, S. Kühl, J. Herranz, S. Henning, L. Kühn, T. Asset, L. Guétaz, G. Renou, J. Drnec, P. Bordet, A. Pasturel, A. Eychmüller, T. J. Schmidt, P. Strasser, L. Dubau and F. Maillard, Nat. Mater., 2018, 17, 827–833 CrossRef CAS PubMed.
- H. Xue, J. Wang, H. Cheng, H. Zhang, X. Li, J. Sun, X. Wang, L. Lin, Y. Zhang, X. Liao and Y. He, Appl. Catal., B, 2024, 353, 124087 CrossRef CAS.
- Y. Sun, H. Lv, H. Yao, Y. Gao and C. Zhang, Carbon Energy, 2024, e575 CrossRef.
- X. Li and Z. Cheng, Chem Catal., 2022, 2, 2140–2149 CrossRef CAS.
- X. Li, C. Hao, Y. Du, Y. Lu, Y. Fan, M. Wang, N. Wang, R. Meng, X. Wang and Z. J. Xu, Chin. J. Catal., 2023, 55, 191–199 CrossRef CAS.
- S. Sun, Y. Zhang, X. Shi, W. Sun, C. Felser, W. Li and G. Li, Adv. Mater., 2024, 2312524 CrossRef.
- Y. Zhang, D. Wang, G. Wei, B. Li, Z. Mao, S.-M. Xu, S. Tang, J. Jiang, Z. Li, X. Wang and X. Xu, JACS Au, 2024, 4, 1509–1520 CrossRef CAS.
- W. Zhong, Y. Qiu, H. Shen, X. Wang, J. Yuan, C. Jia, S. Bi and J. Jiang, J. Am. Chem. Soc., 2021, 143, 4405–4413 CrossRef CAS PubMed.
- P. Lv, W. Lv, D. Wu, G. Tang, X. Yan, Z. Lu and D. Ma, Phys. Rev. Appl., 2023, 19, 054094 CrossRef CAS.
- K. Liu, J. Fu, Y. Lin, T. Luo, G. Ni, H. Li, Z. Lin and M. Liu, Nat. Commun., 2022, 13, 2075 CrossRef CAS.
- Z. Li, Z. Wang, S. Xi, X. Zhao, T. Sun, J. Li, W. Yu, H. Xu, T. S. Herng, X. Hai, P. Lyu, M. Zhao, S. J. Pennycook, J. Ding, H. Xiao and J. Lu, ACS Nano, 2021, 15, 7105–7113 CrossRef CAS PubMed.
- W. Yang, Z. Jia, B. Zhou, L. Chen, X. Ding, L. Jiao, H. Zheng, Z. Gao, Q. Wang and H. Li, ACS Catal., 2023, 13, 9695–9705 CrossRef CAS.
- H. Bai, J. Feng, D. Liu, P. Zhou, R. Wu, C. T. Kwok, W. F. Ip, W. Feng, X. Sui, H. Liu and H. Pan, Small, 2023, 19, 2205638 CrossRef CAS.
- A. Cao and J. K. Nørskov, ACS Catal., 2023, 13, 3456–3462 CrossRef CAS.
- J. Huang, J. Yu, X. Lu, Y. Wei, H. Song, A. Cao, J. Cai, S.-Q. Zang and S. Lu, Inorg. Chem. Front., 2023, 10, 3955–3962 RSC.
- L. Zhang and Z. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CrossRef CAS.
- M. Tamtaji, Q. Peng, T. Liu, X. Zhao, Z. Xu, P. R. Galligan, M. D. Hossain, Z. Liu, H. Wong, H. Liu, K. Amine, Y. Zhu, W. A. Goddard Iii, W. Wu and Z. Luo, Nano Energy, 2023, 108, 108218 CrossRef CAS.
- Z. Zhang, Z. Xu, J. He and J. Lu, IEEE Trans. Instrum. Meas., 2022, 71, 1–9 Search PubMed.
- X. Li, Y. Sun, F. Ren, Y. Bai and Z. Cheng, Mater. Today Energy, 2021, 19, 100619 CrossRef CAS.
- V. Shukla, Introduction of Vibrating Sample Magnetometer for Magnetic Characterization, Springer, Cham, 2022, pp. 483–505, DOI:10.1007/978-3-030-90948-2_19.
- T. Kahmann, E. L. Rösch, K. Enpuku, T. Yoshida and F. Ludwig, J. Magn. Magn. Mater., 2021, 519, 167402 CrossRef CAS.
- S. Phokha, S. Pinitsoontorn, S. Maensiri and S. Rujirawat, J. Sol-Gel Sci. Technol., 2014, 71, 333–341 CrossRef CAS.
- H. Zhang, H.-C. Chen, S. Feizpoor, L. Li, X. Zhang, X. Xu, Z. Zhuang, Z. Li, W. Hu, R. Snyders, D. Wang and C. Wang, Adv. Mater., 2024, 36, 2400523 CrossRef CAS.
- Y. Liu, X. Liu, Z. Lv, R. Liu, L. Li, J. Wang, W. Yang, X. Jiang, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2022, 61, e202117617 CrossRef CAS.
- Y. Li, Y. Ji, Y. Zhao, J. Chen, S. Zheng, X. Sang, B. Yang, Z. Li, L. Lei, Z. Wen, X. Feng and Y. Hou, Adv. Mater., 2022, 34, 2202240 CrossRef CAS.
- J. Ding, Z. Wei, F. Li, J. Zhang, Q. Zhang, J. Zhou, W. Wang, Y. Liu, Z. Zhang, X. Su, R. Yang, W. Liu, C. Su, H. B. Yang, Y. Huang, Y. Zhai and B. Liu, Nat. Commun., 2023, 14, 6550 CrossRef CAS.
- Z. Chen, H. Niu, J. Ding, H. Liu, P.-H. Chen, Y.-H. Lu, Y.-R. Lu, W. Zuo, L. Han, Y. Guo, S.-F. Hung and Y. Zhai, Angew. Chem., Int. Ed., 2021, 60, 25404–25410 CrossRef CAS.
- V. A. Saveleva, K. Ebner, L. Ni, G. Smolentsev, D. Klose, A. Zitolo, E. Marelli, J. Li, M. Medarde, O. V. Safonova, M. Nachtegaal, F. Jaouen, U. I. Kramm, T. J. Schmidt and J. Herranz, Angew. Chem., Int. Ed., 2021, 60, 11707–11712 CrossRef CAS PubMed.
- A. Brückner, Chem. Soc. Rev., 2010, 39, 4673–4684 RSC.
- T. S. Seifert, S. Kovarik, D. M. Juraschek, N. A. Spaldin, P. Gambardella and S. Stepanow, Sci. Adv., 2020, 6, eabc5511 CrossRef CAS PubMed.
- M. Abbate, J. C. Fuggle, A. Fujimori, L. H. Tjeng, C. T. Chen, R. Potze, G. A. Sawatzky, H. Eisaki and S. Uchida, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 16124–16130 CrossRef CAS PubMed.
- Y. Cao, L. Gao, Z. Lai, C. Wang, Y. Yao, X. Zhu and Z. Zou, Appl. Phys. Lett., 2021, 119, 163902 CrossRef CAS.
- X. Li, H. Liu, Z. Chen, Q. Wu, Z. Yu, M. Yang, X. Wang, Z. Cheng, Z. Fu and Y. Lu, Nat. Commun., 2019, 10, 1409 CrossRef.
- X. Li, Y. Sun, Q. Wu, H. Liu, W. Gu, X. Wang, Z. Cheng, Z. Fu and Y. Lu, J. Am. Chem. Soc., 2019, 141, 3121–3128 CrossRef CAS PubMed.
- U. I. Kramm, M. Lefèvre, N. Larouche, D. Schmeisser and J.-P. Dodelet, J. Am. Chem. Soc., 2014, 136, 978–985 CrossRef CAS.
- J. Li, M. T. Sougrati, A. Zitolo, J. M. Ablett, I. C. Oğuz, T. Mineva, I. Matanovic, P. Atanassov, Y. Huang, I. Zenyuk, A. Di Cicco, K. Kumar, L. Dubau, F. Maillard, G. Dražić and F. Jaouen, Nat. Catal., 2021, 4, 10–19 CrossRef CAS.
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 CrossRef CAS.
- Y. Gao, J. Wang, Y. Yang, J. Wang, C. Zhang, X. Wang and J. Yao, Nano-Micro Lett., 2023, 15, 158 CrossRef CAS PubMed.
- X. Wang, L. Xu, C. Li, C. Zhang, H. Yao, R. Xu, P. Cui, X. Zheng, M. Gu, J. Lee, H. Jiang and M. Huang, Nat. Commun., 2023, 14, 7210 CrossRef CAS PubMed.
- L. Zhao, Q. Cai, B. Mao, J. Mao, H. Dong, Z. Xiang, J. Zhu, R. Paul, D. Wang, Y. Long, L. Qu, R. Yan, L. Dai and C. Hu, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2308828120 CrossRef CAS.
- W. Zou, R. Lu, X. Liu, G. Xiao, X. Liao, Z. Wang and Y. Zhao, J. Mater. Chem. A, 2022, 10, 9150–9160 RSC.
- M. Liu, X. Wang, S. Cao, X. Lu, W. Li, N. Li and X.-H. Bu, Adv. Mater., 2024, 36, 2309231 CrossRef CAS.
- S. Ye, S. Xie, Y. Lei, X. Yang, J. Hu, L. Zheng, Z. Chen, Y. Fu, X. Ren, Y. Li, X. Ouyang, Q. Zhang, J. Liu and X. Sun, Nano Res., 2023, 16, 1869–1877 CrossRef CAS.
- T. Ding, X. Liu, Z. Tao, T. Liu, T. Chen, W. Zhang, X. Shen, D. Liu, S. Wang, B. Pang, D. Wu, L. Cao, L. Wang, T. Liu, Y. Li, H. Sheng, M. Zhu and T. Yao, J. Am. Chem. Soc., 2021, 143, 11317–11324 CrossRef CAS PubMed.
- M. Liu, S. Liu, Q. Xu, Q. Miao, S. Yang, S. Hanson, G. Z. Chen, J. He, Z. Jiang and G. Zeng, Carbon Energy, 2023, 5, e300 CrossRef CAS.
- X. Sun, Y. Song, G. Jiang, X. Lan and C. Xu, Sci. China Mater., 2024, 67, 1–17 CrossRef CAS.
- P. Zhu, Z.-Y. Wu, A. Elgazzar, C. Dong, T.-U. Wi, F.-Y. Chen, Y. Xia, Y. Feng, M. Shakouri, J. Y. Kim, Z. Fang, T. A. Hatton and H. Wang, Nature, 2023, 618, 959–966 CrossRef CAS PubMed.
- C. Cai, K. Liu, L. Zhang, F. Li, Y. Tan, P. Li, Y. Wang, M. Wang, Z. Feng, D. Motta Meira, W. Qu, A. Stefancu, W. Li, H. Li, J. Fu, H. Wang, D. Zhang, E. Cortés and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202300873 CrossRef CAS PubMed.
- J. Pei, L. Yang, J. Lin, Z. Zhang, Z. Sun, D. Wang and W. Chen, Angew. Chem., Int. Ed., 2024, 63, e202316123 CrossRef CAS.
- S. Biswas, J. Zhou, X.-L. Chen, C. Chi, Y.-A. Pan, P. Cui, J. Li, C. Liu and X.-H. Xia, Angew. Chem., Int. Ed., 2024, 63, e202405493 CrossRef CAS.
- L. Li, J. Zhu, F. Kong, Y. Wang, C. Kang, M. Xu, C. Du and G. Yin, Matter, 2024, 7, 1517–1532 CrossRef CAS.
- G. Chen, Y. Sun, R. R. Chen, C. Biz, A. C. Fisher, M. P. Sherburne, J. W. Ager Iii, J. Gracia and Z. J. Xu, J. Phys.: Energy, 2021, 3, 031004 CAS.
- X. Li, Z. Cheng and X. Wang, Electrochem. Energy Rev., 2021, 4, 136–145 CrossRef CAS.
- H. He, S. Liu, Y. Liu, L. Zhou, H. Wen, R. Shen, H. Zhang, X. Guo, J. Jiang and B. Li, Green Chem., 2023, 25, 9501–9542 RSC.
- K. Song, B. Yang, X. Zou, W. Zhang and W. T. Zheng, Energy Environ. Sci., 2023, 17, 27–48 RSC.
- L. Han, H. Cheng, W. Liu, H. Li, P. Ou, R. Lin, H.-T. Wang, C.-W. Pao, A. R. Head, C.-H. Wang, X. Tong, C.-J. Sun, W.-F. Pong, J. Luo, J.-C. Zheng and H. L. Xin, Nat. Mater., 2022, 21, 681–688 CrossRef CAS.
- X. Han, T. Zhang, W. Chen, B. Dong, G. Meng, L. Zheng, C. Yang, X. Sun, Z. Zhuang and D. Wang, Adv. Energy Mater., 2021, 11, 2002753 CrossRef CAS.
- Y. Chen, J. Lin, B. Jia, X. Wang, S. Jiang and T. Ma, Adv. Mater., 2022, 34, 2201796 CrossRef CAS.
- T. Tang, Y. Wang, J. Han, Q. Zhang, X. Bai, X. Niu, Z. Wang and J. Guan, Chin. J. Catal., 2023, 46, 48–55 CrossRef CAS.
- C. Chen, Y. Li, A. Huang, X. Liu, J. Li, Y. Zhang, Z. Chen, Z. Zhuang, Y. Wu and W.-C. Cheong, J. Am. Chem. Soc., 2023, 145, 21273–21283 CrossRef CAS PubMed.
- H. Li, S. Di, P. Niu, S. Wang, J. Wang and L. Li, Energy Environ. Sci., 2022, 15, 1601–1610 RSC.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, 1806296 CrossRef.
- J. Fu, R. Liang, G. Liu, A. Yu, Z. Bai, L. Yang and Z. Chen, Adv. Mater., 2019, 31, 1805230 CrossRef.
- X. Bai, J. Han, S. Chen, X. Niu and J. Guan, Chin. J. Catal., 2023, 54, 212–219 CrossRef CAS.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS PubMed.
- X. Zhu, D. Zhang, C.-J. Chen, Q. Zhang, R.-S. Liu, Z. Xia, L. Dai, R. Amal and X. Lu, Nano Energy, 2020, 71, 104597 CrossRef CAS.
- S. Sarkar, A. Biswas, E. E. Siddharthan, R. Thapa and R. S. Dey, ACS Nano, 2022, 16, 7890–7903 CrossRef CAS.
- L. Bai, C.-S. Hsu, D. T. Alexander, H. M. Chen and X. Hu, Nat. Energy, 2021, 6, 1054–1066 CrossRef CAS.
- T. Tang, J. Han, Z. Wang, X. Niu and J. Guan, Nano Res., 2024, 17, 3794–3800 CrossRef CAS.
- T. Tang, Z. Duan, D. Baimanov, X. Bai, X. Liu, L. Wang, Z. Wang and J. Guan, Nano Res., 2023, 16, 2218–2223 CrossRef CAS.
- W. Wan, Y. Zhao, S. Wei, C. A. Triana, J. Li, A. Arcifa, C. S. Allen, R. Cao and G. R. Patzke, Nat. Commun., 2021, 12, 5589 CrossRef CAS PubMed.
- J. Zhang, Y. Zhao, W. Zhao, J. Wang, Y. Hu, C. Huang, X. Zou, Y. Liu, D. Zhang and X. Lu, Angew. Chem., Int. Ed., 2023, 62, e202314303 CrossRef CAS PubMed.
- K. Yuan, D. Lützenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. Leung and Y. Chen, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS PubMed.
- B. Hu, A. Huang, X. Zhang, Z. Chen, R. Tu, W. Zhu, Z. Zhuang, C. Chen, Q. Peng and Y. Li, Nano Res., 2021, 14, 3482–3488 CrossRef CAS.
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- F. Sun, Q. Tang and D.-e. Jiang, ACS Catal., 2022, 12, 8404–8433 CrossRef CAS.
- L. Wan, Z. Xu, Q. Xu, M. Pang, D. Lin, J. Liu and B. Wang, Energy Environ. Sci., 2023, 16, 1384–1430 RSC.
- Y. Chen, K. Yang, B. Jiang, J. Li, M. Zeng and L. Fu, J. Mater. Chem. A, 2017, 5, 8187–8208 RSC.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Adv. Sci., 2018, 5, 1700275 CrossRef.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- J. Han, X. Bai, X. Xu, A. Husile, S. Zhang, L. Qi and J. Guan, Chem. Sci., 2024, 15, 7870–7907 RSC.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li and Y. Wang, Nature, 2020, 577, 509–513 CrossRef CAS.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang and Y. Lum, Science, 2021, 372, 1074–1078 CrossRef CAS.
- S. Cao, S. Zhou, H. Chen, S. Wei, S. Liu, X. Lin, X. Chen, Z. Wang, W. Guo and X. Lu, Energy Environ. Mater., 2023, 6, e12287 CrossRef CAS.
- Z. Jin, M. Yang, Y. Dong, X. Ma, Y. Wang, J. Wu, J. Fan, D. Wang, R. Xi and X. Zhao, Nano-Micro Lett., 2024, 16, 4 CrossRef CAS PubMed.
- X. Kong, J. Ke, Z. Wang, Y. Liu, Y. Wang, W. Zhou, Z. Yang, W. Yan, Z. Geng and J. Zeng, Appl. Catal., B, 2021, 290, 120067 CrossRef CAS.
- M. He, W. An, Y. Wang, Y. Men and S. Liu, Small, 2021, 17, 2104445 CrossRef CAS.
- X. Zhao, G. Hu, G. F. Chen, H. Zhang, S. Zhang and H. Wang, Adv. Mater., 2021, 33, 2007650 CrossRef CAS.
- Y. Ashida, K. Arashiba, K. Nakajima and Y. Nishibayashi, Nature, 2019, 568, 536–540 CrossRef CAS PubMed.
- X. Lv, W. Wei, B. Huang, Y. Dai and T. Frauenheim, Nano Lett., 2021, 21, 1871–1878 CrossRef CAS PubMed.
- J. Dai, Y. Tong, L. Zhao, Z. Hu, C.-T. Chen, C.-Y. Kuo, G. Zhan, J. Wang, X. Zou and Q. Zheng, Nat. Commun., 2024, 15, 88 CrossRef PubMed.
- Y. Wang, W. Cheng, P. Yuan, G. Yang, S. Mu, J. Liang, H. Xia, K. Guo, M. Liu, S. Zhao, G. Qu, B.-A. Lu, Y. Hu, J. Hu and J.-N. Zhang, Adv. Sci., 2021, 8, 2102915 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |