
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic dehydrogenative synthesis of α,β-unsaturated secondary amides without external oxidants†
Shaokang
An
a,
Guoyin
Lai
b and
Wenbo H.
Liu
 *a
*a
aSchool of Chemistry, Sun Yat-sen University, Guangzhou 510006, China. E-mail: liuwb29@mail.sysu.edu.cn
bGuangzhou Flower Flavours & Fragrances Co., Ltd, Guangzhou 510442, China
First published on 23rd August 2024
Abstract
Direct dehydrogenative synthesis of α,β-unsaturated secondary amides still represents an elusive transformation. Herein we describe a palladium-catalyzed redox-neutral desaturation to prepare α,β-conjugated secondary amides. Without external oxidants, this approach relies on the N–O bond cleavage as the driving force to achieve formal dehydrogenation. Complementary to known protocols, this transformation is enabled by the unique reactivity of hydroxamate, thereby representing a novel strategy to accomplish carbonyl desaturation. Desired conjugated secondary amides can be efficiently synthesized in the presence of more reactive esters and even ketones, thus providing a solution to the long-standing issue of α,β-unsaturated secondary amides via C–C desaturation.
α,β-Unsaturated carbonyls are important building blocks in organic chemistry, and are useful in a wide range of transformations such as the Michael addition, radical addition, cycloaddition and reduction/oxidation. Consequently, desaturation of carbonyl compounds to α,β-unsaturated carbonyls is a fundamental process in organic synthesis.1,2 Although robust protocols for ketone and aldehyde desaturation have been established, efficient methods for amides are scarce due to the significantly reduced α-acidity.3 Nonetheless, notable advancements have been reported recently. For example, by employing strong bases to convert amides to enolates, Newhouse et al. developed elegant Pd/Ni-catalyzed methods to afford unsaturated amides.4,5 Besides hard enolization, Dong et al. leveraged soft enolization to enable the α,β-desaturation of protected-lactams.6–8 In addition to metal-catalyzed protocols, the transition-metal-free approach for amide dehydrogenation is also known.9Via electrophilic amide activation, Maulide et al. achieved the α,β-desaturation of tertiary amides9a (Fig. 1A). These examples represent the state of the art in the field of amide α,β-desaturation. However, only tertiary amides and protected lactams are feasible substrates and external oxidants are all required in these protocols. A general and efficient method to access α,β-unsaturated secondary amides via desaturation without external oxidants remains elusive.10
 | ||
| Fig. 1 Background and this work. | ||
In terms of the overall redox change, dehydrogenation to generate a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond from a C–C bond is analogous to the transformation of forging a C–C bond from two C–H bonds, which is known as cross-dehydrogenative coupling (CDC).11–13 Despite being termed dehydrogenative coupling, hydrogen gas is usually not liberated because of the less favorable thermodynamics. Thus, most CDC processes require external sacrificial oxidants as the driving force. Regarding the oxidants in CDC reactions, when N-methoxy amide and hydroxamates serve as the directing groups,14,15 the N–O bond promotes the catalyst turnover as the internal oxidant. Without external oxidants, undesired side reactions can be suppressed when oxidizable functionalities are present, therefore representing a notable advantage compared to the presence of external oxidants.14 Although this internal-oxidant concept has been widely applied in CDC for heterocycle synthesis,16–19 it is rarely employed for the desaturation of the C–C bond to afford a CC bond. Recently, Yu et al. developed a redox-neutral γ,δ-desaturation of a primary amide by using N–OMe as the internal oxidant20 (Fig. 1B). Besides the N–O bond as the internal oxidant, aryl iodide as the aryl radical precursor can also be employed for the desaturation via a similar 1,5-hydrogen atom transfer (HAT) process, which is followed by radical capture and elimination.21 Herein, we describe an α,β-desaturation of a secondary amide without the external oxidant, which is complementary to Yu's γ,δ-desaturation (Fig. 1C).
C bond from a C–C bond is analogous to the transformation of forging a C–C bond from two C–H bonds, which is known as cross-dehydrogenative coupling (CDC).11–13 Despite being termed dehydrogenative coupling, hydrogen gas is usually not liberated because of the less favorable thermodynamics. Thus, most CDC processes require external sacrificial oxidants as the driving force. Regarding the oxidants in CDC reactions, when N-methoxy amide and hydroxamates serve as the directing groups,14,15 the N–O bond promotes the catalyst turnover as the internal oxidant. Without external oxidants, undesired side reactions can be suppressed when oxidizable functionalities are present, therefore representing a notable advantage compared to the presence of external oxidants.14 Although this internal-oxidant concept has been widely applied in CDC for heterocycle synthesis,16–19 it is rarely employed for the desaturation of the C–C bond to afford a CC bond. Recently, Yu et al. developed a redox-neutral γ,δ-desaturation of a primary amide by using N–OMe as the internal oxidant20 (Fig. 1B). Besides the N–O bond as the internal oxidant, aryl iodide as the aryl radical precursor can also be employed for the desaturation via a similar 1,5-hydrogen atom transfer (HAT) process, which is followed by radical capture and elimination.21 Herein, we describe an α,β-desaturation of a secondary amide without the external oxidant, which is complementary to Yu's γ,δ-desaturation (Fig. 1C).
To begin the study, we outlined that the key challenge for such a transformation (Fig. 1C) is how to correlate the α,β-positions of carbonyls with the N–O bond. In Yu's remote dehydrogenation,20 the connection is via [1,5]-HAT, which is a robust process in radical chemistry.21 Once the N–O bond is reduced to the amidyl radical for the subsequent HAT, the oxidation state of the N–O bond is transferred to the remote positions. To correlate the N–O bond of the hydroxamates with α,β positions, we proposed that the α-lactam intermediate I may be feasible as the bridge22,23 (Fig. 2). From starting compound 1 to α-lactam I, the N–O bond is reduced but the α-C–H bond is oxidized by forming the C–N bond, thereby implying that the redox transfer has been realized. However, due to the ring-strain, only specific α-lactams with bulky substituents at both α-C and N have been synthesized so far and α-lactams like I are unknown.24–26 Moreover, α-lactam has been proposed from hydroxamate by Hoffman, which is unfortunately only suitable for α-aryl substituted substrates.22b Therefore, employing the regular α-lactam without bulky and aryl substituents as the isolable intermediate seems problematic. To solve this issue, we hypothesized that adding an appropriate reagent to trap the transient α-lactam in situ may outweigh the unproductive decomposition pathways. Via the trapped intermediate II, the oxidation state of the N–O bond can still be transferred to the α-position of the carbonyl for the following dehydrogenation. A simple and qualified candidate for this purpose is the halide ion considering that the generated halo compound can be transformed into the olefin27 either through transition metal-catalyzed27a or base-promoted elimination27b (Fig. 2).
 | ||
| Fig. 2 The internal oxidant concept for amide α,β-dehydrogenation. | ||
To test our hypothesis, we selected the transformation of 1a to 2a as the target. After a comprehensive investigation, the optimized conditions were identified. Upon the synergistic activation of MgBr2·Et2O and iPr2NEt, the desired 2a was isolated in 75% yield (E/Z > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with Pd(OAc)2/Xantphos as the catalyst and K2CO3 as the base. To understand how the components under the optimized conditions impact this reaction, further control experiments were carried out. A Lewis acid, Lewis base and palladium salt are essential since these reactions cannot proceed without them (entries 2–4). Removing the ligand and the base from the standard conditions decreased the yield to 15% and 11% respectively (entries 5 and 6). Switching Pd(OAc)2 with other palladium sources furnished lower yields (entries 7 and 8). Replacing the optimal ligand Xantphos with other phosphorus ligands resulted in poor delivery of the products in all instances (entries 9–20). Other bases including Na2CO3 and Cs2CO3 are less efficient than K2CO3 (entries 21 and 22). These control experiments suggest that the Lewis acid, Lewis base, palladium complex and inorganic base are all necessary to afford the α,β-unsaturated amide in good yields (Table 1). It is important to note that without adding the palladium complex, 1a is still completely consumed, and is converted to alpha-bromo amide in high yield as proposed in Fig. 2.
:1) with Pd(OAc)2/Xantphos as the catalyst and K2CO3 as the base. To understand how the components under the optimized conditions impact this reaction, further control experiments were carried out. A Lewis acid, Lewis base and palladium salt are essential since these reactions cannot proceed without them (entries 2–4). Removing the ligand and the base from the standard conditions decreased the yield to 15% and 11% respectively (entries 5 and 6). Switching Pd(OAc)2 with other palladium sources furnished lower yields (entries 7 and 8). Replacing the optimal ligand Xantphos with other phosphorus ligands resulted in poor delivery of the products in all instances (entries 9–20). Other bases including Na2CO3 and Cs2CO3 are less efficient than K2CO3 (entries 21 and 22). These control experiments suggest that the Lewis acid, Lewis base, palladium complex and inorganic base are all necessary to afford the α,β-unsaturated amide in good yields (Table 1). It is important to note that without adding the palladium complex, 1a is still completely consumed, and is converted to alpha-bromo amide in high yield as proposed in Fig. 2.
|
|
||
|---|---|---|
| Entry | Change from “standard conditions” | Yielda |
| a The yields were determined by 1H NMR. b This refers to the isolated pure compound and the E/Z ratio was determined by 1H NMR. | ||
| 1 | No change | 82% (75%) (E/Z > 20:1)b |
| 2 | No MgBr2·Et2O | 0% |
| 3 | No iPr2NEt | 0% |
| 4 | No Pd(OAc)2 | 0% |
| 5 | No Xantphos | 15% |
| 6 | No K2CO3 | 11% |
| 7 | Pd(PPh3)4 instead of Pd(OAc)2 | 26% |
| 8 | Pd2(dba)3 instead of Pd(OAc)2 | 67% |
| 9 | X-phos instead of Xantphos | 15% |
| 10 | S-Phos instead of Xantphos | 13% |
| 11 | dppb instead of Xantphos | 12% |
| 12 | dppp instead of Xantphos | 9% |
| 13 | P(tBu)3·HBF4 instead of Xantphos | 41% |
| 14 | P(Cy)3·HBF4 instead of Xantphos | 7% |
| 15 | BINAP instead of Xantphos | 29% |
| 16 | PPh3 instead of Xantphos | 21% |
| 17 | DPEPhos instead of Xantphos | 63% |
| 18 | dppf instead of Xantphos | 45% |
| 19 | P(o-Me-Ph)3 instead of Xantphos | 36% |
| 20 | P(p-Cl-Ph)3 instead of Xantphos | 21% |
| 21 | Na2CO3 instead of K2CO3 | 21% |
| 22 | Cs2CO3 instead of K2CO3 | 7% |

With the optimized conditions established, we next evaluated the generality of this redox-neutral dehydrogenation. Various structurally diversified α,β-unsaturated secondary amides could be efficiently prepared through this approach (Fig. 3). For example, numerous cinnamic acid-derived amides with different substituents are obtained in good to high yields (2b–2e, 42–71%). Besides the regular aryl substituents, a heteroaryl substituted-unsaturated amide was synthesized in high yield although the E/Z selectivity was poor (2f, 88%, E/Z = 1:1). The low E/Z selectivity for this particular case is probably due to the coordination effect of the nitrogen heteroatom of the oxazole. In addition to the cinnamic type amides, regular aliphatic unsaturated amides were also produced smoothly (2g–2m, 41–90%). Both the methylene (2h–2k) and methine-type (2l, 2m) substituents at the β-positions are tolerated. Various substituents on the amide nitrogen are also compatible (2n–2p). Finally, several unsaturated amides derived from complicated molecules were successfully accessed in synthetically useful yields (2q–2t, 41–49%). It is important to highlight that in most cases, the E/Z selectivity is higher than 20:1. Unfortunately, substrates with substituents at the alpha and beta positions are not feasible under the current conditions (2u, 2v).
 | ||
| Fig. 3 Scope of the substrates. [a]The yields refer to the isolated pure compounds and the E/Z ratio was calculated based on 1H NMR. [b]Pd(OAc)2 (10 mol%), Xantphos (20 mol%) and K2CO3 (2.0 equiv.), 80 °C, 16 h instead of the standard Pd conditions. [c]DBU (3.0 equiv.), DMSO (1 mL), 80 °C, 16 h instead of the Pd conditions. | ||
Moreover, as proposed in Fig. 2, the chemoselectivity of the dehydrogenation is determined by the hydroxamate functionality instead of the acidity of the α-C–H of carbonyls. Therefore, to illustrate the synthetic advantages of this redox-neutral dehydrogenation, substrates with multiple carbonyls were subjected to the standard conditions, which all generated the corresponding conjugated secondary amides in good yields. For instance, in the presence of more reactive esters (1w, 1x, 1z) and even a ketone (1y), the desaturation of the hydroxamates to furnish the amides proceeded smoothly. These structures are challenging to access through other known dehydrogenative approaches1 (Fig. 4).
 | ||
| Fig. 4 Chemoselective synthesis of unsaturated secondary amides. | ||
To further demonstrate the synthetical practicality of this dehydrogenation, the model reaction was performed on a gram scale (2a, 0.77 g, 68%, Fig. 5A). Since the α,β-unsaturated amide is a versatile functionality, additional derivatizations were performed. Under basic conditions, the secondary amide was alkylated into a tertiary amide (3, 75%). This represents a particular advantage compared to the desaturation of tertiary amides considering that a secondary amide can be converted into a tertiary amide but the reverse process is unlikely. Under copper catalysis conditions, the β-borylated amide was prepared in high yield (4, 92%). Moreover, the unsaturated amide was efficiently converted into other functionalities such as an unsaturated ester (6, 87%) via a Boc-protected intermediate (5, 81%). These examples only represent a small portion of transformations that α,β-unsaturated amides can participate in (Fig. 5B). As another application in the real synthetic context, this dehydrogenation was leveraged as the key step to prepare the drug llepcimide for diarrhea treatment.28 Starting from commercially available acid 7, hydroxamate 8 was prepared in excellent yield (96%), and was further converted to the desaturated amide 9via this dehydrogenation strategy (80%). Through a one-pot two-step protocol, the transamination allows for the synthesis of llepcimide in good yield (62%) (Fig. 5C).
 | ||
| Fig. 5 Synthetic applications. | ||
To obtain more insights on this redox-neutral desaturation, additional control experiments were designed. When the –OTs group was changed to other common leaving groups including –Cl, –OAc and –OMe, no desired products were detected (Fig. 6A). The reactivity difference of these groups may be attributed to their different nucleofugality as the leaving groups. The proposed intermediate (10) was independently synthesized, and was subjected to the palladium-catalysis conditions. The desired unsaturated amide 2a was isolated in 79% yield, which suggests that α-bromo amide is a potential intermediate in this process (Fig. 6B). Since the α-bromo amide is formed prior to the elimination, to understand the kinetics of this transformation in detail, the kinetic isotope effect was investigated through two pairs of parallel reactions with two deuterium atoms at the α,β positions respectively (Fig. 6C). The control experiments imply that the Pd-catalyzed elimination is not the rate determining step.
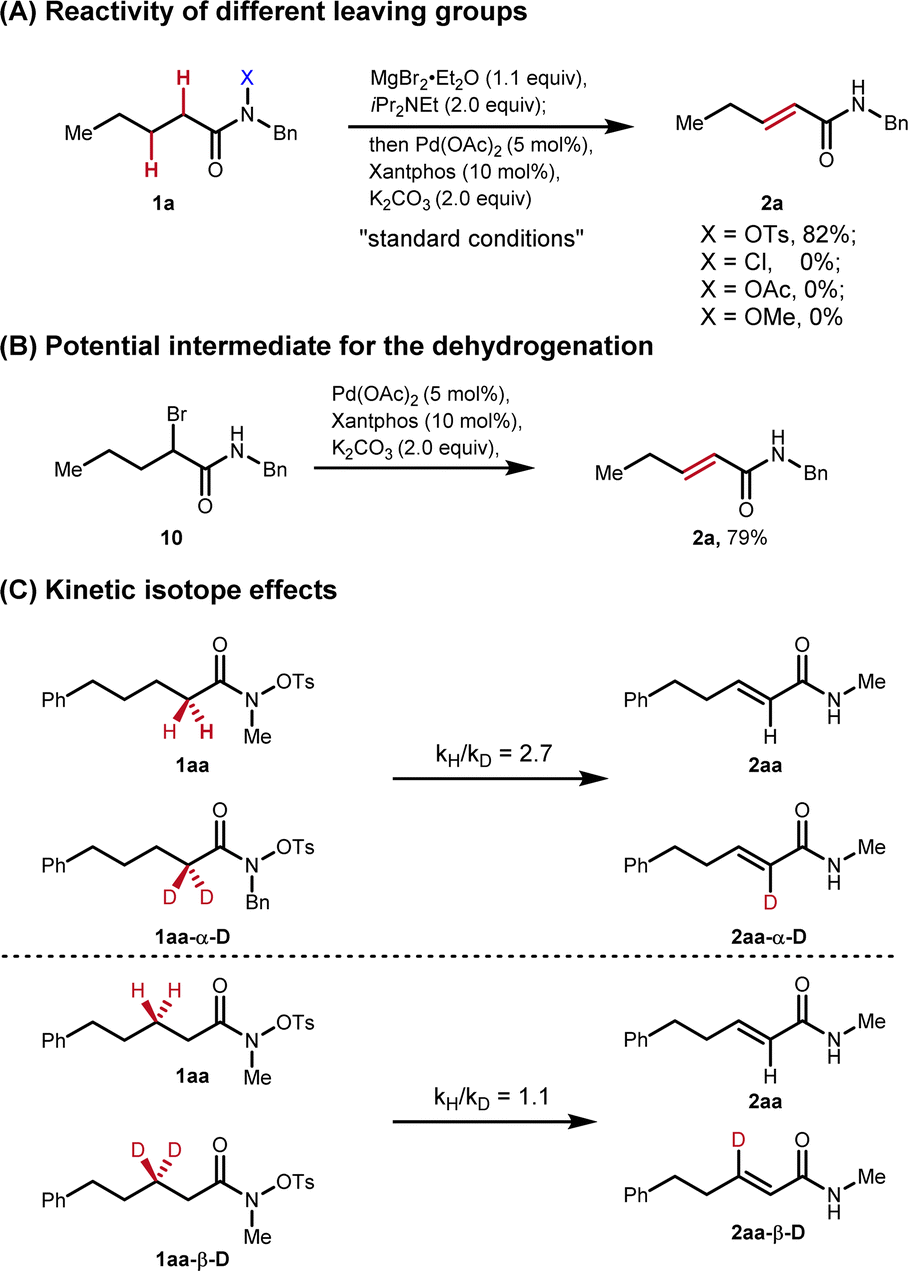 | ||
| Fig. 6 Control experiments for the mechanistic studies. Please see the ESI† for the experimental details. | ||
Based on the control experiments, the following mechanism was proposed to rationalize this dehydrogenation (Fig. 7). Upon coordination with Lewis acid Mg2+, 1 was converted to Complex-I. This coordination with the Lewis acid acidifies the α-C–H bond of carbonyl, which can be deprotonated by a mild base (iPr2NEt) to furnish Int-I. Because of the potent leaving ability of -OTs, Int-I rearranges into α-lactam intermediate I. Due to the high ring strain, bromide rapidly reacts with I to deliver the α-Br secondary amide II. Subsequently, the in situ formed Pd(0) undergoes oxidative addition to produce III. Regarding the following β-H elimination, the H should be cis with Pd. Among two possible conformations, III-A is more favorable than III-B due to the less steric repulsion. Therefore, trans-olefin 2 is produced and Pd(0) is released for the next catalytic cycle. All the experimental results are consistent with this proposal.
 | ||
| Fig. 7 Proposed mechanistic cycle. | ||
Conclusions
In conclusion, a novel and redox-neutral desaturation strategy to synthesize an α,β-conjugated secondary amide was developed. This approach does not rely on the α-acidity of carbonyls, thereby showing complementary chemoselectivity to established dehydrogenation methods. Without strong bases/acids, this protocol tolerates various functional groups. In the presence of more reactive esters and ketones, desaturation still occurs precisely. In contrast to previous dehydrogenation logic, this strategy does not involve any external oxidant and sulfur/selenium-type reagents. Considering the ready accessibility of starting materials, the simple manipulation of the transformation and versatility of the product, it is expected that this transformation will be useful for synthetic chemists.Data availability
Experimental details and characterization data of compounds can be found in the ESI.†Author contributions
S. A. performed the experiments. W. H. L. designed the experiments and prepared the manuscript. All the authors contributed to the discussion of the results.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22201311), Guangdong Basic and Applied Basic Research Foundation (2023A0505050100) and Sun Yat-sen University. We are grateful to Mr Rui Wang (Sun Yat-sen University) for the initial results in this project.Notes and references
- S. Gnaim, J. C. Vantourout, F. Serpier, P.-G. Echeverria and P. S. Baran, ACS Catal., 2021, 11, 883–892 CrossRef CAS.
- D. Huang and T. R. Newhouse, Acc. Chem. Res., 2021, 54, 1118–1130 CrossRef CAS PubMed.
- Y. Chen, J. P. Romaire and T. R. Newhouse, J. Am. Chem. Soc., 2015, 137, 5875–5878 CrossRef CAS.
- Y. Chen, A. Turlik and T. R. Newhouse, J. Am. Chem. Soc., 2016, 138, 1166–1169 CrossRef CAS.
- D. Huang, S. M. Szewczyk, P. Zhang and T. R. Newhouse, J. Am. Chem. Soc., 2019, 141, 5669–5674 CrossRef CAS PubMed.
- M. Chen and G. Dong, J. Am. Chem. Soc., 2017, 139, 7757–7760 CrossRef CAS PubMed.
- M. Chen, A. J. Rago and G. Dong, Angew. Chem., Int. Ed., 2018, 57, 16205–16209 CrossRef CAS PubMed.
- M. Chen and G. Dong, J. Am. Chem. Soc., 2019, 141, 14889–14897 CrossRef CAS.
- (a) C. J. Teskey, P. Adler, C. R. Gonçalves and N. Maulide, Angew. Chem., Int. Ed., 2019, 58, 447–451 CrossRef CAS PubMed; (b) A. Bauer and N. Maulide, Chem. Sci., 2019, 10, 9836–9840 RSC; (c) M.-M. Wang, G.-H. Sui, X.-C. Cui, H. Wang, J.-P. Qu and Y.-B. Kang, J. Org. Chem., 2019, 84, 8267–8274 CrossRef CAS PubMed.
- (a) Z. Wang, Z. He, L. Zhang and Y. Huang, J. Am. Chem. Soc., 2018, 140, 735–740 CrossRef CAS; (b) R. Giri, N. Maugel, B. M. Foxman and J.-Q. Yu, Organometallics, 2008, 27, 1667–1670 CrossRef CAS.
- C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS.
- C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed.
- S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS PubMed.
- N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449–6457 CrossRef CAS.
- N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908–6909 CrossRef CAS.
- Y. Fukui, P. Liu, Q. Liu, Z.-T. He, N.-Y. Wu, P. Tian and G.-Q. Lin, J. Am. Chem. Soc., 2014, 136, 15607–15614 CrossRef CAS PubMed.
- Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 3676–3677 CrossRef CAS PubMed.
- H. Wang, C. Grohmann, C. Nimphius and F. Glorius, J. Am. Chem. Soc., 2012, 134, 19592–19595 CrossRef CAS PubMed.
- J. Wu, X. Cui, L. Chen, G. Jiang and Y. Wu, J. Am. Chem. Soc., 2009, 131, 13888–13889 CrossRef CAS PubMed.
- S. Zhou, Z.-J. Zhang and J.-Q. Yu, Nature, 2024, 629, 363–369 CrossRef CAS PubMed.
- (a) S. Yang, H. Fan, L. Xie, G. Dong and M. Chen, Org. Lett., 2022, 24, 6460–6465 CrossRef CAS; (b) W.-L. Yu, Z.-G. Ren, K.-X. Ma, H.-Q. Yang, J.-J. Yang, H. Zheng, W. Wu and P.-F. Xu, Chem. Sci., 2022, 13, 7947–7954 RSC; (c) S. Z. Zard, Chem. Soc. Rev., 2008, 37, 1603–1618 RSC.
- (a) I. Lengyel and J. C. Sheehan, Angew. Chem., Int. Ed., 1968, 7, 25–36 CrossRef CAS; (b) R. V. Hoffman, N. K. Nayyar and W. Chen, J. Am. Chem. Soc., 1993, 115, 5031–5034 CrossRef CAS.
- G. L'Abbé, Angew. Chem., Int. Ed., 1980, 19, 276–289 CrossRef.
- H. E. Baumgarten, N. C. R. Chiang, V. J. Elia and P. V. Beum, J. Org. Chem., 1985, 50, 5507–5512 CrossRef CAS.
- H. E. Baumgarten, J. Am. Chem. Soc., 1962, 84, 4975–4976 CrossRef CAS.
- J. C. Sheehan and I. Lengyel, J. Am. Chem. Soc., 1964, 86, 1356–1359 CrossRef CAS.
- (a) A. C. Bissember, A. Levina and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 14232–14237 CrossRef CAS; (b) For the substrates with more acidic C–H bonds at the β position, the direct base-promoted elimination may be more favorable. Regarding the less acidic substrates, the simple base-promoted elimination is challenging considering that more acidic α-C–H and N–H adjacent to carbonyl are present..
- For the details about this drug, please see https://www.drugs.com/loperamide.html.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04419c |
| This journal is © The Royal Society of Chemistry 2024 |