
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cancer immunoprofiling strategy using mass spectrometry coupled with bioorthogonal cleavage†
Maxime
Ribéraud‡
a,
Estelle
Porret‡
b,
Alain
Pruvost
 a,
Frédéric
Theodoro
a,
Anvi Laëtitia
Nguyen
a,
Simon
Specklin
b,
Dimitri
Kereselidze
b,
Caroline
Denis
b,
Benoit
Jego
b,
Peggy
Barbe
a,
Mathilde
Keck
a,
Timothée
D'Anfray
a,
Bertrand
Kuhnast
b,
Davide
Audisio
a,
Charles
Truillet
*b and
Frédéric
Taran
*a
a,
Frédéric
Theodoro
a,
Anvi Laëtitia
Nguyen
a,
Simon
Specklin
b,
Dimitri
Kereselidze
b,
Caroline
Denis
b,
Benoit
Jego
b,
Peggy
Barbe
a,
Mathilde
Keck
a,
Timothée
D'Anfray
a,
Bertrand
Kuhnast
b,
Davide
Audisio
a,
Charles
Truillet
*b and
Frédéric
Taran
*a
aUniversité Paris Saclay, CEA, INRAE, Département Médicaments et Technologies pour la Santé (DMTS), 91191 Gif-sur-Yvette, France. E-mail: frederic.taran@cea.fr
bUniversité Paris-Saclay, CEA, CNRS, Inserm, BioMaps, France. E-mail: charles.truillet@cea.fr
First published on 17th October 2024
Abstract
The accurate quantification of biomarkers is paramount in modern medicine, particularly in cancer where precise diagnosis is imperative for targeted therapy selection. In this paper we described a multiplexed analysis diagnostic approach based on cleavable MS-tagged antibodies. The technology uses MS-tag isotopologues and the sydnonimine-cyclooctyne click-and-release bioorthogonal reaction. In a proof of concept study, we demonstrated the potential of this approach for cancer cell immunoprofiling in culture cells, tissues and in vivo as well, thereby unveiling promising diagnostic avenues.
Introduction
Extensive efforts have been made in recent years to reduce the side effects of cancer therapies by developing targeted treatment strategies based on cell receptors overexpressed within tumours. Among the possible targeted therapies, those employing monoclonal antibodies (mAbs) are certainly the most important to date.1 Several therapeutic mAbs are routinely used for cancer immunotherapies, a dozen antibody–drug conjugates have been already approved by the FDA and many others are advancing through the pipeline, promising enhanced treatment options across various cancer types.2 It is thus very likely that in the coming years a considerable number of targeted therapies will be available, which is an excellent perspective for patient health care but raises the problem of selecting the best therapy among those available. Furthermore, the levels of overexpressed protein receptors not only differ within the cancer types (104 to 106 receptors per cell) but also vary within the different patient tumour lesions as well as from patient to patient.3 In this context, the molecular analysis of cancer cells involving quantification of a maximum number of protein receptors is crucial in establishing precise diagnoses for choosing and guiding available treatments.4 Among the described methods used to quantify the abundance of protein receptors in cells and in biopsy tissues,5 cyclic immunofluorescence appears to be one of the most promising. This approach involves the use of fluorescent antibodies which, after incubation and staining, need to be de-stained to allow a new cycle of detection with new fluorescent antibodies (Fig. 1A). Several approaches have been explored to inactivate the fluorophores attached to antibodies. Photobleaching6 and the use of harsh chemical agents (NaOH–H2O2 for example)7 are effective approaches but these conditions may induce degradation of biological materials and often result in interference with subsequent cycles of staining.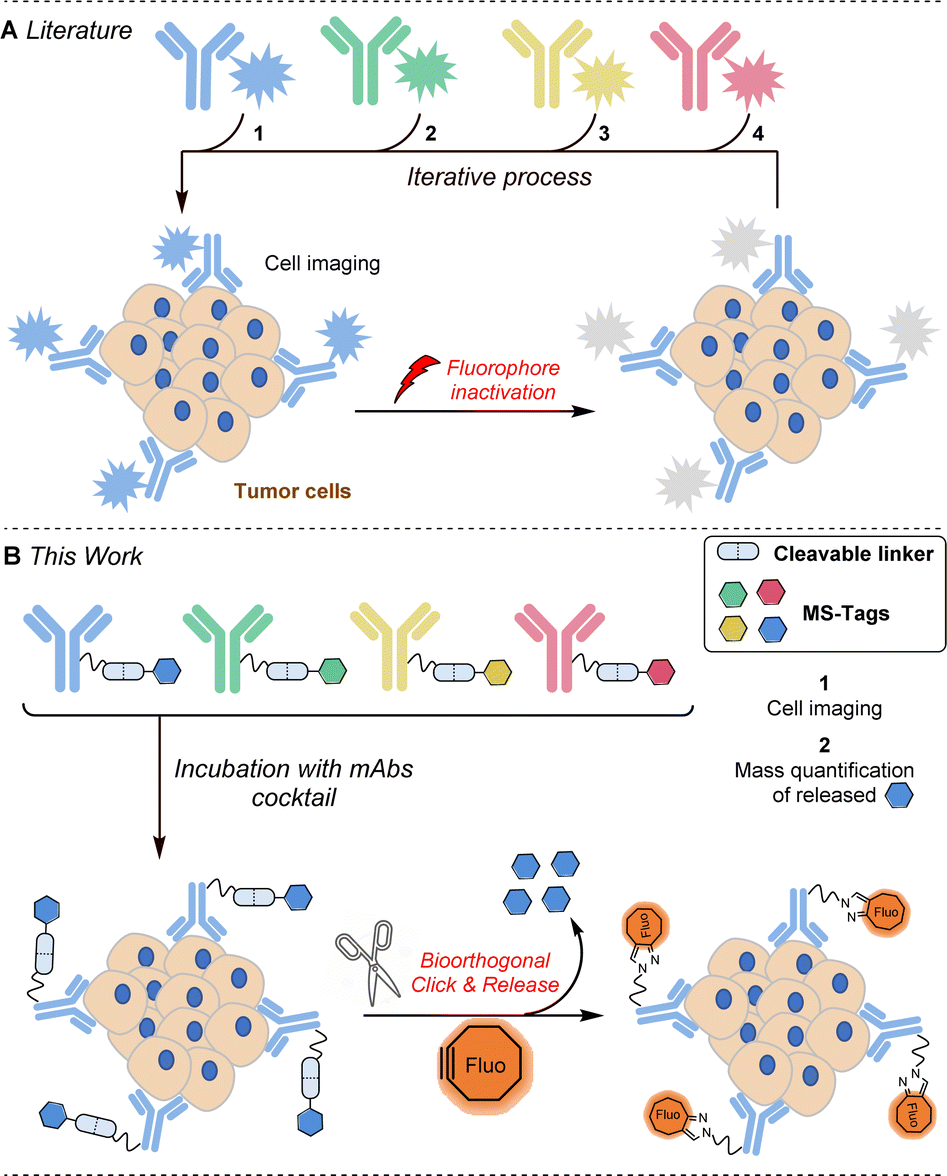 | ||
| Fig. 1 Strategies for multiplexed cancer cell profiling. | ||
A significant improvement of this approach was recently described by Halabi and Weissleder by the use of a photo-immolating triazene linker allowing milder deactivation of fluorophores through a single light pulse at 405 nm.8 DNA displacement has also been successfully investigated to erase fluorescence but nonspecific binding between DNA and endogenous biomolecules may lead to increased background.9 The use of click chemistry has recently emerged as a powerful additional strategy for mild and robust cyclic immunofluorescence. The group of Guo has notably successfully used fluorescent antibodies constructed with an azide-based linker that can be cleaved upon addition of a phosphine to remove the fluorescent label.10 More recently, Carlson, Weissleder et al. nicely exploited the powerful inverse electron demand Diels–Alder reaction to quench fluorescent antibodies derivatized by trans-cyclooctenes with tetrazine quenchers.11 This same group then remarkably expanded the approach by employing tetrazines that not only quench but also completely eliminate fluorescent signals from antibody-labeled cells in very short cycle times.12 These cutting-edge technologies significantly enhance cyclic immunofluorescence, yet the number of biomarkers detectable per cycle remains constrained by the limited number of channels available on microscopes.
Herein, we report a new strategy for multiplexed protein analysis of cancer cells and biopsy tissues using cleavable antibodies labelled with mass spectrometry enhancers, MS-tags (Fig. 1B). With the help of a click-and-release bioorthogonal reaction, the method allows (i) localisation of the antigen receptors set through fluorescent labelling and (ii) specific identification and quantification of the different antigen receptors through the release of MS-tags. Compared to cyclic immunofluorescence, this strategy does not provide spatial information on the different receptors but may offer other key advantages: (i) higher biomarker detection, (ii) (semi) quantification of receptor expression levels and (iii) the potential to use the technology in vivo.
The use of chemical tagging in mass spectrometry (MS) is a well-known approach to increase the sensitivity of the quantification. Moreover, the possibility to label the MS-tag with stable isotopes allows powerful multiplexed MS analysis.13 The choice of the MS-tag is of prime importance as its structure impacts both on signal improvement, by 102 – to 103 – fold depending on the type of tag, and on the possibilities of isotope incorporation, and thus on multiplexed analyse capacities.
In this work, we selected tris(2,4,6,-trimethoxyphenyl)-phosphonium (TMPP) as a tag for LC-MS/MS analysis. Because of its moderate hydrophobicity and the presence of a permanent positive charge, TMPP is known to be highly suitable for proteomic studies using MALDI analysis.14 Importantly, TMPP has 9 methoxy groups, 6 aromatic hydrogens and 27 carbon atoms in its structure which represent as many possibilities for deuterium and carbon 13 labelling. In total there are 60 atoms that can be isotopically labelled, which gives the possibility to theoretically generate 61 TMPP-isotopologues (Fig. 2).
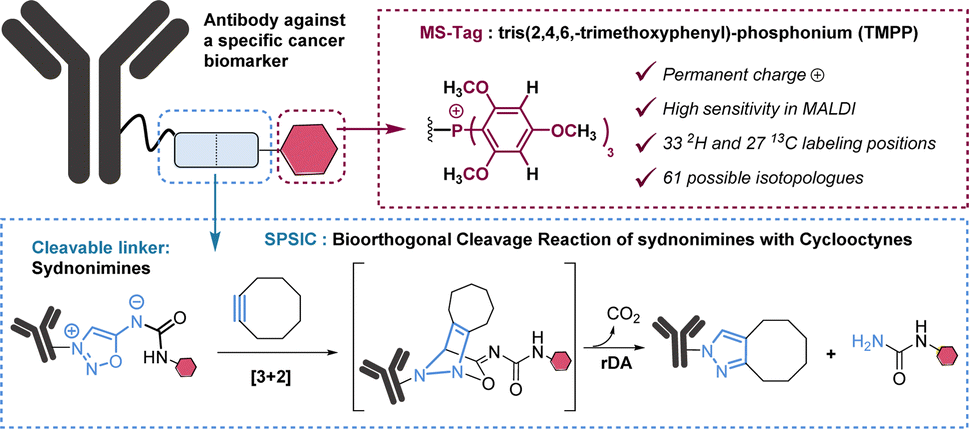 | ||
| Fig. 2 Bioconjugates used in this study. Our strategy is based on the combination of two technologies: the multiplexed MS-detection of TMPP- isotopologues and sydnonimine cleavable linkers. rDA: retro-Diels-Alder. | ||
Results and discussion
Preparation of MS-tagged antibodies
The synthetic approach we followed for the synthesis of MS-tags allows easy access to at least 15 among the 61 possible labelled TMPP (Scheme S1†). As a proof of concept, we performed our study with 4 isotopologues of TMPP which were synthesized in 6 steps from sydnonimine 1, prepared according to previous protocols.15 These four isotopically labelled TMPP tags were attached to antibodies through sydnonimine cleavable linkers we developed in our laboratory.16 Sydnonimines are mesoionic compounds described by our group to undergo bioorthogonal Strained-Promoted SydnonImine-cyclooctyne Cycloaddition (SPSIC) reactions.17 Upon SPSIC reaction, sydnonimines are rapidly and quantitatively cleaved to release a urea-TMPP product and to form a pyrazole product clicked to the antibody (Fig. 2). Short PEG spacers were introduced into the sydnonimine core leading to compound 3 whose alcohol was then tosylated to allow reaction with trimethoxybenzene phosphine and three of its deuterated analogues to obtain four phosphoniums 5 bearing TMPP isotopologues, TMPP-H33, H24, H15 and H6 (Scheme 1). | ||
| Scheme 1 Synthesis of TMPP tags. | ||
Using standard peptide coupling, the TMPP tags were then attached to four FDA-approved therapeutic antibodies (mAbs-TMPP) raised against biomarkers located at the surface of the cell: Trastuzumab (TRZ, anti-Her2), Cetuximab (CTX, anti-EGFR), Durvalumab (DUR, anti-PDL1) and Bevacizumab (BVZ, anti-VEGF). TRZ, CTX, and DUR target extracellular tumour receptors, whereas BVZ targets the growth factor VEGF-A secreted by the cells and bound at their surface. The significant presence of VEGF-A at the surface of tumoral cells has been evidenced by numerous studies.18 The mean number of TMPP linked to the antibodies, TAR for Tag-Antibody-Ratio, was determined (TAR from 2 to 4, Fig. 3 and Table S1†). Control experiments demonstrated excellent stability of tagged antibodies in blood plasma (Fig. S2†).To control the reactivity of the sydnonimine linker, we performed the cleavage of each tagged antibody in solution using the SPSIC reaction. The monitoring of the reaction was carried out by LC-HRMS measurements of the released tags.
 | ||
| Fig. 3 Preparation and characterization of tagged mAbs. TAR = Tag-Antibody-Ratio. TMPP tag release from antibodies upon SPSIC reaction with DBCO. | ||
We observed quantitative detachment of the TMPP tags from the antibodies in a few minutes using excess cyclooctyne DBCO. High rate constant values k were found in all cases: between 85 and 325 M−1 s−1 depending on the antibody (Fig. 3 and S6†). The limits of detection and quantification in solution of each TMPP-tag were then determined by LC-MS/MS analysis and found to be ∼20 pM and ∼70 pM respectively in DMEM cell media, whatever is the tag isotopologue (Table S3†). In cells overexpressing a receptor, we estimate that this detection limit would enable the detection of approximately 105 cells per mL.
Demonstration of the method's feasibility
Having these tools in our hands, we first carried out a series of experiments aimed at assessing the correlation between the quantification of released TMPP tags and antigen expression. To this end, in vitro experiments were performed using CTX-H24 as the model TMPP-mAb and the human epidermoid carcinoma cells A431 that overexpress the epidermal growth factor receptor (EGFR), as confirmed by western blot analysis (Fig. S8A†). CTX-H24 was incubated for 2 h with A431 cells, followed by the addition of DBCO-TAMRA, which was allowed to react for 15 minutes. Epifluorescence microscopy confirmed a specific fluorescent signal at the cell membrane highlighting the efficient click reaction of DBCO-TAMRA to CTX-H24 bound to A431 cells (Fig. 4B). However, a slight non-specific signal coming from the DBCO-TAMRA probe was also observed in the cell cytoplasm. Co-localization experiments conducted with a membrane tracker (CellBrite Fix 640) confirmed the effective TAMRA-labelling of Cetuximab bound to the EGFR embedded in the cell membrane. The specific versus non-specific binding were evaluated by fluorescence measurements of CTX-H24 displacement by a high dose of unlabelled CTX (Fig. 4C). The results showed high and displaceable binding of CTX-H24 to A431 either at 37 °C or 4 °C, and thus confirmed that CTX keeps its ability to specifically interact with the EGFR present on the cell membrane after the introduction of the TMPP-H24 tag. According to microscopy experiments, when cells were incubated with DBCO-TAMRA alone, a significant background signal was observed due to non-specific binding of DBCO to the cell. However, this phenomenon does not alter the selective release of the MS-tag TMPP-H24 from CTX. A noticeable increase of fluorescence was clearly observed when DBCO-TAMRA was added after incubation of CTX-H24 and TMPP-H24 was easily detected and quantified by MS (Fig. 4D). The binding of CTX-H24 to the EGFR present on A431 was significantly displaceable by excess of CTX (p = 0.0024) which confirmed the specificity of the reaction: quantification of the TMPP-H24 tag was ∼43 pM and ∼103 pM with and without excess CTX respectively. A saturation binding assay using released TMPP-H24 detection was performed to determine the apparent dissociation constant (Kd = 25 nM) and the maximum number of binding sites (Bmax = 5.5 nM). Around 6.6 × 106 EGFR receptors per cell in the A431 cell line were quantified (Fig. S10A†), a value consistent with literature data.19 The conjugation of TMPP-H24 to CTX has been shown to have minimal impact on CTX binding when compared to its fluorescent counterpart (Fig. S10C and D†). We then verified the preservation of the binding properties and quantified the released TMPP tags of the three other TMPP-mAbs, namely TRZ-H33, DUR-H15 and BVZ-H6. LC-MS/MS quantification of each TMPP-tag after addition of DBCO on TMPP-mAbs incubated with cells overexpressing the specific receptor: SkBR3, H1975 and U87 cell lines respectively, confirmed the good binding properties of each bioconjugate and the specific detection of each receptor (Fig. S10A†). | ||
| Fig. 4 Proof of concept of the click and release approach combining bioorthogonal click labeling and fluorescence imaging with tag release and LC-MS/MS-analysis. (a) Reagent structures and schematic principle of the technology; (b) fluorescence imaging of A431 cells incubated (2 h) or not with CTX-H24 followed by 15 min reaction with TAMRA-DBCO; (c) fluorescence binding assay to evaluate the specific versus non-specific binding of CTX-H24 and DBCO-TAMRA. A431 cells were incubated with or without excess of CTX at 37 °C or 4 °C; (d) LC-MS/MS quantification of the released TMPP-H24 tag after binding of CTX-H24 on A431 cells and subsequent treatment with TAMRA-DBCO with or without excess of CTX (2 h, 37 °C). | ||
Altogether, these results validated our click-and-release approach and showed the complementarity of LC-MS/MS analysis of released tags allowing the selective quantification of each of the four receptors (EGFR, Her2, PDL1 and VEGF) with fluorescence imaging for their global localization on the cells.
Immuno-profiling on tissue sections
Following this validation study, we carried out a series of experiments with the goal of quantifying several receptors simultaneously on biopsy tissues. This multiplex approach consists of adding the mixture of the four labelled antibodies on tissue slices (14 μm thick) from mice bearing subcutaneous A431 tumours and quantifying the four released TMPP tags after addition of DBCO. The cocktail of the four TMPP-mAbs, TRZ-H33, CTX-H24, DUR-H15 and BVZ-H6 was thus incubated on tissue sections and the abundance of each released TMPP-tag was determined by MS after addition of DBCO (Fig. 5). Three adjacent tumour sections from an A431 subcutaneous tissue were pulled together with the aim of quantifying them by LC-MS/MS analysis. Fluorescence imaging after addition of DBCO-TAMRA confirmed efficient and selective click-and-release reaction on tissue sections (Fig. 5B).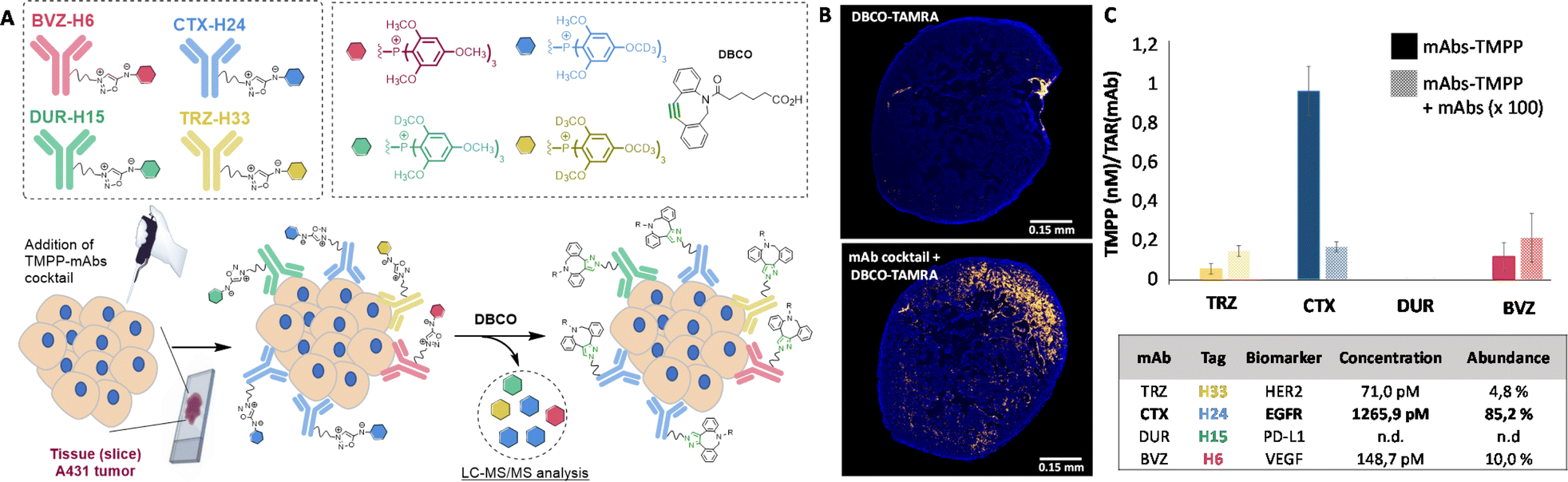 | ||
| Fig. 5 Multiplexed LC-MS/MS detection of cell membrane receptors on tissue slices, using our click-and-release approach. (A) Principle of multiplex detection. (B) Fluorescence imaging of biomarkers after addition of DBCO-TAMRA in the absence or presence of antibodies. (C) Quantification of biomarkers using LC-MS/MS. | ||
As shown in Fig. 5C, LC-MS/MS analysis confirmed that TMPP-H24 is the most abundant representing more than 85% of all released tags. This result confirmed that EGFR was present in a higher proportion on cell membrane compared to the other three receptors. Control experiments conducted with excess CTX confirmed the specificity of EGFR quantification and immunofluorescence imaging of the tissue sections confirmed the overexpression of CTX compared to the other receptors (Fig. S13†). These results showed that our approach can be used to profile tumor biopsies by detecting several biomarkers in one single process.
Immuno-profiling in vivo: proof of concept
Finally, we decided to evaluate if our click-and-release strategy might work in vivo. This is a tremendous challenge as it involves the intravenous (i.v.) injection of the antibody cocktail followed, 3 days later, by the injection of the cyclooctyne. For the strategy to be successful, the cyclooctyne should react in vivo with the tagged antibodies fixed at the tumor site, and not with circulating antibodies, detach the tags which then must be excreted in urine, at least in part, in order to be detected and quantified by LC-MS/MS. The obstacles of this approach are therefore extremely difficult to overcome but the stakes are high. Indeed, biopsy analysis may not fully reflect the state of an entire tumor which may be heterogeneous. To date, there is no method allowing multiplexed in vivo analysis of biomarker expression and we were eager to see if our method could possibly meet this need. To limit the obstacles to overcome, we decided to use intratumoral injection of the cyclooctyne (Fig. 6A). This type of injection allowed the cyclooctyne to be concentrated at the tumor site and limited its diffusion as showed by PET imaging of mice after intratumoral injection of radiolabelled DBCO [18F]6 (Fig. 6B and S14, 15†). Most of the [18F]6 uptake remained on the tumor even 4 hours post-injection. The pharmacokinetics of TMPP-tags was also examined by blood sampling and showed fast blood clearance of the tags and sufficient excretion in urine to be detected and quantified by LC-MS/MS after 24 h. Importantly, the four tags displayed the same pharmacokinetics and excretion profiles (Fig. S16†). Reinforced by this observation, mice bearing left and right A431 tumors were injected intravenously with a cocktail of the four TMPP-mAbs (TRZ-H33, CTX-H24, DUR-H15 and BVZ-H6, 100 μg each). Three days post-injection, which corresponds to the maximum antibody accumulation in the tumor, an excess of non-radiolabeled DBCO [19F]6 was administrated via intratumoral injection in the left tumor. 24 hours later, the urine sampling revealed an important release of the TMPP-H24 compared to the TMPP-H33 and TMPP-H6 tags that can be correlated to the higher expression of EGFR in the A431 model. | ||
| Fig. 6 In vivo immunoprofiling using our click-and-release approach. (A) Principle of the experimental in vivo procedure. (B) Biodistribution of [18F]-DBCO 6 determined by PET imaging (p.i.: post-intratumoral injection). (C) Ex vivo analysis of TMPP-tags released in urine before and after injection of [19F]6. Concentration values were normalized as a function of the TAR. H24 and H15 in urine were significantly higher than H33 and H6 (p < 0.01, Kruskal–Wallis test). | ||
Surprisingly, the amount of TMPP-H15 was also significantly high. The expression of PD-L1 was much lower than EGFR as determined via immunofluorescence analysis on ex vivo A431 tumor slices with the different antibodies of interest. The high in vivo tag release can be explained by the fact that the in vivo uptake is not only related to the expression level but also to the avidity of the antibody to its target20 and/or non-specific bioaccumulation of mAbs through an enhanced permeability and retention (EPR) effect.21 To confirm these in vivo results, we collected the tumors and measured the concentration of TMPP-tags that were still inside the tumor tissue and found a similar profile to the one obtained in urine excretion (Fig. S18A†). The concentration of the tags in the right tumor (which did not receive DBCO injection) was markedly lower, reaching the limit of MS detection. This observation highlights a limited diffusion of the DBCO following intratumoral injection (Fig. S18A†). Urine does not contain any traces of tags before [19F]6 injection, indicating good in vivo stability of the antibody-tag conjugates (Fig. S19†). The urine concentration of the tags, obtained through metabolism cages, may offer a more representative measure of tag release compared to blood samples. This is because the rapid elimination kinetics of the tag from the blood compartment make it challenging to accurately assess the total tag release (Fig. S18B†).
Further in vivo investigations would be necessary to confirm these preliminary results but we think this immuno-profiling approach might be more efficient than classic tumor biopsy immune-histochemical scoring as it relates the full complexity of the in vivo biology mechanisms.
Conclusions
In conclusion, we have developed a technique based on a bioorthogonal click-and-release reaction allowing both localisation by fluorescence imaging, identification and quantification by LC-MS/MS analysis of cell receptors. The main advantages of this technique rely on the very mild conditions used and its high potential for multiplexing. In a proof of concept study, we demonstrated the feasibility of this approach by quantifying four different receptors in a single analysis on culture cells as well as on tissues. The potential of this technique is in principle much higher: as more than 60 TMPP isotopologues are chemically accessible, the technique allows theoretically the multiplexed detection of 61 biomarkers in one single LC-MS/MS analysis. In addition, as the chemistry behind this technology allows the bioorthogonal, mild detachment of tags from antibodies (no apparent cell toxicity of DBCO was found at concentrations up to 400 μM, Fig. S12†), cyclic MS detection is theoretically possible. For these reasons, we think this approach might be a valuable new tool to access at the expression of a high number of different cell receptors. Finally, the technique was tested in vivo with the hope of characterizing tumours in their native environment and without the need for tissue removal. Although the experiments carried out are preliminary and need to be confirmed by further investigations, the results are encouraging. We think this approach may be an interesting alternative to tumour characterization in the future and might help in selecting and monitoring the right immunotherapy for patients.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
M. R. performed the synthesis of tags and the preparation of antibody conjugates. T. D’A. performed the kinetic studies. E. P. and B. J. performed the experiments with cells and with tissues. S. S. performed the synthesis of radiolabelled DBCO. A. P., F. T. and A. L. N. performed the LC/MS/MS analysis. P. B., D. K., C. D. and M. K. performed the in vivo experiments. D. A. supervised the synthesis of tags. C. T. designed and supervised the biological part of the work and participated in the writing of the manuscript. F. T. conceived the idea, acquired funding, guided the experimental work and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Fondation pour la Recherche Médicale (DCM20181039570). The authors thank David-Alexandre Buisson and Sabrina Lebrequier for excellent analytical support.Notes and references
- D. Zahavi and L. Weiner, Antibodies, 2020, 9, 34 CrossRef CAS PubMed.
- See for example: (a) A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Nat. Rev. Drug Discovery, 2017, 16, 315 CrossRef CAS PubMed; (b) C. H. Chau, P. S. Steeg and W. D Figg, Lancet, 2019, 10200, 793 CrossRef PubMed.
- (a) Y. Imai, C. K. Leung, H. G. Friesen and R. P. Shiu, Cancer Res., 1982, 42, 4394 CAS; (b) H. Haigler, J. F. Ash, S. J. Singer and S. Cohen, Proc. Natl. Acad. Sci., 1978, 75, 3317–3321 CrossRef CAS PubMed; (c) G. Carpenter, K. J. Lembach, M. Morrison and S. Cohen, J. Biol. Chem., 1975, 250, 4297 CrossRef CAS PubMed; (d) B. M. Fendly, M. Winget, R. M. Hadziak, M. T. Lipari, M. A. Napier and A. Ullrich, Cancer Res., 1990, 50, 1550–1558 CAS.
- R. Weissleder, M. C. Schwaiger, S. S. Gambhir and H. Hricak, Sci. Transl. Med., 2016, 8, 355ps16 Search PubMed.
- See for example: (a) S. C. Bendall, E. F. Simonds, P. Qiu, El-ad D. Amir, P. O. Krutzik, R. Finck, R. V. Bruggner, R. Melamed, A. Trejo, O. I. Ornatsky, R. S. Balderas, S. K. Plevritis, K. Sachs, D. Pe'er, S. D. Tanner and G. P. Nolan, Science, 2011, 332, 687 CrossRef CAS PubMed; (b) R. Fan, O. Vermesh, A. Srivastava, B. K. H. Yen, L. Qin, H. Ahmad, G. A. Kwong, C.-C. Liu, J. Gould, L. Hood and J. R. Heath, Nat. Biotechnol., 2008, 26, 1373 CrossRef CAS PubMed; (c) Y. Lu, Q. Xue, M. R. Eisele, E. S. Sulistijo, K. Brower, L. Han, E. D. Amir, D. PeQer, K. Miller-Jensen and R. Fan, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 607 CrossRef PubMed; (d) C. Giesen, H. A. O. Wang, D. Schapiro, N. Zivanovic, A. Jacobs, B. Hattendorf, P. J. Schüffler, D. Grolimund, J. M. Buhmann, S. Brandt, Z. Varga, P. J. Wild, D. Günther and B. Bodenmiller, Nat. Methods, 2014, 11, 417 CrossRef CAS PubMed; (e) M. Angelo, S. C. Bendall, R. Finck, M. B. Hale, C. Hitzman, A. D. Borowsky, R. M. Levenson, J. B. Lowe, S. D. Liu, S. Zhao, Y. Natkunam and G. P. Nolan, Nat. Med., 2014, 20, 436 CrossRef CAS PubMed.
- W. Schubert, B. Bonnekoh, A. J. Pommer, L. Philipsen, R. Bçckelmann, Y. Malykh, H. Gollnick, M. Friedenberger, M. Bode and A. W. M. Dress, Nat. Biotechnol., 2006, 24, 1270 CrossRef CAS PubMed.
- (a) M. J. Gerdes, C. J. Sevinsky, A. Sood, S. Adak, M. O. Bello, A. Bordwell, A. Can, A. Corwin, S. Dinn, R. J. Filkins, D. Hollman, V. Kamath, S. Kaanumalle, K. Kenny, M. Larsen, M. Lazare, Q. Li, C. Lowes, C. C. McCulloch, E. McDonough, M. C. Montalto, Z. Pang, J. Rittscher, A. Santamaria-Pang, B. D. Sarachan, M. L. Seel, A. Seppo, K. Shaikh, Y. Sui, J. Zhang and F. Ginty, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11982 CrossRef CAS PubMed; (b) J. R. Lin, M. Fallahi-Sichani and P. K. Sorger, Nat. Commun., 2015, 6, 8390 CrossRef CAS PubMed.
- E. A. Halabi and R. Weissleder, J. Am. Chem. Soc., 2023, 145, 8455 CrossRef CAS PubMed.
- (a) R. M. Schweller, J. Zimak, D. Y. Duose, A. A. Qutub, W. N. Hittelman and M. R. Diehl, Angew. Chem., Int. Ed., 2012, 51, 9292 CrossRef CAS PubMed; (b) D. Y. Duose, R. M. Schweller, J. Zimak, A. R. Rogers, W. N. Hittelman and M. R. Diehl, Nucleic Acids Res., 2012, 40, 3289 CrossRef CAS PubMed.
- M. Mondal, R. Liao, L. Xiao, T. Eno and J. Guo, Angew. Chem., Int. Ed., 2017, 56, 2636 CrossRef CAS PubMed.
- J. Ko, J. Oh, M. S. Ahmed, J. C. T. Carlson and R. Weissleder, Angew. Chem., Int. Ed., 2020, 59, 6839 CrossRef CAS PubMed.
- J. Ko, M. Wilkovitsch, J. Oh, R. H. Kohler, E. Bolli, M. J. Pittet, C. Vinegoni, D. B. Sykes, H. Mikula, R. Weissleder and J. C. T. Carlson, Nat. Biotechnol., 2022, 40, 1654 CrossRef CAS PubMed.
- T. Huang, M. R. Armbruster, J. B. Coulton and J. L. Edwards, Anal. Chem., 2019, 91(1), 109 CrossRef CAS PubMed.
- (a) W. J. Leavens, S. J. Lane, R. M. Carr, A. M. Lockie and I. Waterhouse, Rapid Commun. Mass Spectrom., 2002, 16, 433 CrossRef CAS PubMed; (b) A. Karnezis, C. K. Barlow, R. A. J. O'Hair and W. D. McFadyen, Rapid Commun. Mass Spectrom., 2006, 20, 2865 CrossRef CAS PubMed; (c) P. J. Lee, W. Chen and J. C. Gebler, Anal. Chem., 2004, 76, 4888 CrossRef CAS PubMed; (d) C. Nakajima, H. Kuyama, T. Nakazawa, O. Nishimura and S. Tsunasawa, Analyst, 2011, 136, 113 RSC; (e) O. Koniev, G. Leriche, M. Nothisen, J.-S. Remy, J.-M. Strub, C. Schaeffer-Reiss, A. Van Dorsselaer, R. Baati and A. Wagner, Bioconjugate Chem., 2014, 25, 202 CrossRef CAS PubMed.
- M. Riomet, K. Porte, L. Madegard, P. Thuéry, D. Audisio and F. Taran, Org. Lett., 2020, 22(6), 2403 CrossRef CAS PubMed.
- M. Riomet, E. Decuypere, K. Porte, S. Bernard, L. Plougastel, S. Kolodych, D. Audisio and F. Taran, Chem.–Eur. J., 2018, 34, 8535 CrossRef PubMed.
- S. Bernard, D. Audisio, M. Riomet, S. Bregant, A. Sallustrau, L. Plougastel, E. Decuypere, S. Gabillet, R. A. Kumar, J. Elyian, M. N. Trinh, O. Koniev, A. Wagner, S. Kolodych and F. Taran, Angew. Chem., Int. Ed., 2017, 56, 15612 CrossRef CAS PubMed.
- W. B. Nagengast, M. N. Lub-de Hooge, S. F. Oosting, W. F. A. den Dunnen, F.-J. Warnders, A. H. Brouwers, J. R. de Jong, P. M. Price, H. Hollema, G. A. P. Hospers, P. H. Elsinga, J. W. Hesselink, J. A. Gietema and E. G. E. de Vries, Cancer Res., 2011, 71, 143 CrossRef CAS PubMed.
- D. Patel, A. Lahiji, S. Patel, M. Franklin, X. Jimenez, D. J. Hicklin and X. Kang, Anticancer Res., 2007, 27(5A), 3355 CAS.
- S. I. Rudnick and G. P. Adams, Cancer Biother.Radiopharm., 2009, 24, 155 CrossRef CAS PubMed.
- J. Wu, J. Pers. Med., 2021, 11, 771 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Procedure and analytical data for compounds, kinetic studies, in vitro, ex vivo and in vivo experiments. See DOI: https://doi.org/10.1039/d4sc04471a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |