
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermo-responsive emission induced by different delocalized excited-states in isomorphous Pd(II) and Pt(II) one-dimensional chains†
Tomoya
Saito
a,
Masaki
Yoshida
 *b,
Kaito
Segawa
b,
Daisuke
Saito
ab,
Junichi
Takayama
c,
Satoshi
Hiura
c,
Akihiro
Murayama
c,
Nishshanka M.
Lakshan
d,
W. M. C.
Sameera
de,
Atsushi
Kobayashi
a and
Masako
Kato
*b
*b,
Kaito
Segawa
b,
Daisuke
Saito
ab,
Junichi
Takayama
c,
Satoshi
Hiura
c,
Akihiro
Murayama
c,
Nishshanka M.
Lakshan
d,
W. M. C.
Sameera
de,
Atsushi
Kobayashi
a and
Masako
Kato
*b
aDepartment of Chemistry, Faculty of Science, Hokkaido University, North-10 West-8, Kita-ku, Sapporo, Hokkaido 060-0810, Japan
bDepartment of Applied Chemistry for Environment, School of Biological and Environmental Sciences, Kwansei Gakuin University, 1 Gakuen-Uegahara, Sanda, Hyogo 669-1330, Japan. E-mail: masaki.yoshida@kwansei.ac.jp; katom@kwansei.ac.jp
cFaculty of Information Science and Technology, Hokkaido University, North-14 West-9, Kita-ku, Sapporo, Hokkaido 060-0814, Japan
dDepartment of Chemistry, University of Colombo, Kumaratunga Munidasa Mawatha, Colombo 00700, Sri Lanka
eDepartment of Chemistry and Molecular Biology, University of Gothenburg, SE-41390 Gothenburg, Sweden
First published on 13th August 2024
Abstract
The self-assembly of d8 transition metal complexes is essential for the development of optoelectronic and sensing materials with superior photofunctional properties. However, detailed insight into the electronic delocalization of excited states across multiple molecules, particularly in comparing 5d8 (Pt(II)) and 4d8 (Pd(II)) systems, remains ambiguous but important. In this study, we have successfully evaluated the differences in the excited-state delocalization and thermal responses of self-assembled Pt(II) and Pd(II) complexes. Although the complexes presented herein, K[M(CN)2(dFppy)]·H2O (M = Pt or Pd, dFppy = 2-(4,6-difluorophenyl)pyridinate), are crystallographically isomorphous with similarly short metal⋯metal contacts, only the Pt(II) complex exhibited thermal equilibria between delocalized excited states, resulting in a drastic thermochromic luminescence with a red-shift of greater than 100 nm. In contrast, the dimeric localized emission from the Pd(II) complex showed a significant increase in the quantum yield upon cooling, approaching almost unity.
Introduction
Non-covalent interactions between d8 or d10 metal ions, referred to as “metallophilic interactions,” play an essential role in a wide range of fields, including supramolecular chemistry, materials chemistry, and photochemistry.1 In particular, self-assembled Pt(II) complexes with Pt⋯Pt interactions have garnered significant interest for a long time2,3 following the discovery of a paradigmatic one-dimensional Pt⋯Pt chain, K2[Pt(CN)4].4 Their most notable and unique feature is their coloration and luminescence depending on the strength of the Pt⋯Pt interactions, enabling their application in stimuli-sensing systems.2 Such features are attributed to the delocalization of molecular orbitals and excited states over multiple molecules within the Pt⋯Pt chains.5 Furthermore, exciton delocalization across multiple Pt(II) complexes within the self-assembled chains has recently been highlighted as the key to achieving highly efficient near-infrared (NIR) luminescence by suppressing structural displacement during excitation.3 Thus, a better understanding of the electronically delocalized excited states upon assembly of Pt(II) complexes with metallophilic interactions is central to designing highly photofunctional materials for optoelectronic and sensing applications.To gain a precise understanding of the intermolecularly delocalized excited states within a one-dimensional metal chain, exploring the relationship between the characters of metal ions and their excited states based on metallophilic interactions is crucial. For square-planar d8 metal ions (i.e., Pt(II) and Pd(II)), close contacts between metal ions typically results in the formation of bonding dσ(M⋯M) and antibonding dσ*(M⋯M) orbitals via the intermolecular overlap of occupied dz2 orbitals (Scheme S1;† M = Pt, Pd). If the metal ion is coordinated by ligands with low-energy π* orbitals, charge transfer transitions occur between dσ*(M⋯M) and π* orbitals, which is named as the metal–metal-to-ligand charge transfer (MMLCT) transitions. As mentioned above, the 1MMLCT absorption and 3MMLCT emission in Pt(II) complexes has been widely studied due to facile overlap between the large 5dz2 orbitals of Pt (Fig. 1(a)).2,3 In contrast, despite having the same d8 electronic configuration, surprisingly few examples of 3MMLCT emissions from self-assembled Pd(II) complexes exist,6–9 and thus, their 3MMLCT excited-state photophysics remain largely unexplored. This is attributed to the smaller and less diffuse 4dz2 orbital of Pd relative to the 5dz2 orbital of Pt, rendering intermolecular interactions between Pd centers unfavorable (Fig. 1(a)), which is consistent with the smaller van der Waals radius of Pd (rPd = 1.63 Å) relative to Pt (rPt = 1.75 Å).10 Here, one fundamental point of interest arises regarding the differences in excited state electronic delocalization between the smaller 4dz2 orbitals and the larger 5dz2 orbitals. Such comparisons between Pd(II) and Pt(II) should provide crucial, fundamental insight into the metallophilic interactions in coordination complexes and their intermolecularly delocalized 3MMLCT excited states. However, to the best of our knowledge, detailed investigations into the differences in the extension of delocalized excited states between Pd(II) and Pt(II) complexes have yet to be reported.
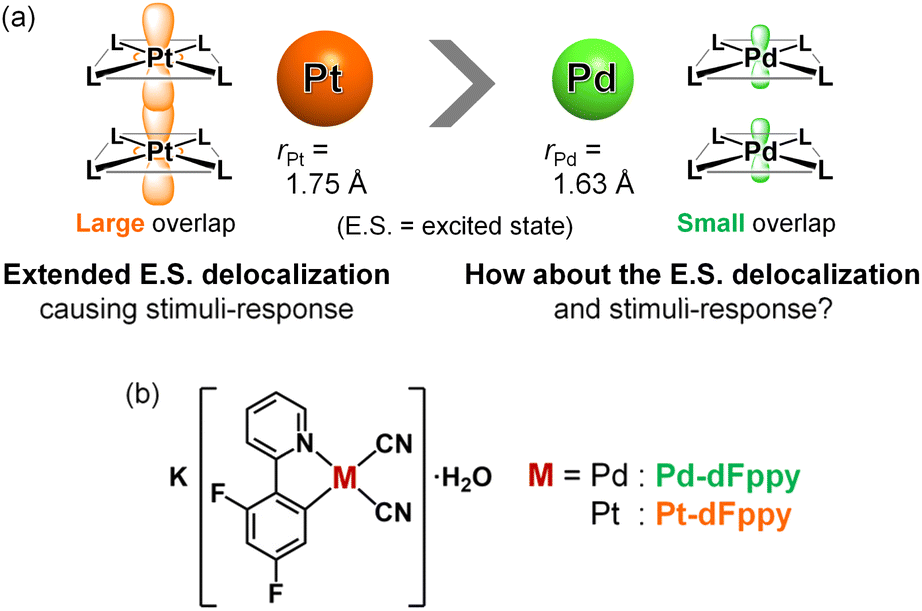 | ||
| Fig. 1 (a) Schematic illustration depicting metallophilic interactions in Pt(II) and Pd(II) complexes. (b) Molecular structures of the complexes investigated in this study. | ||
Here we have succeeded in revealing the differences in electronic delocalization of the 3MMLCT excited state in Pd⋯Pd and Pt⋯Pt interactions in self-assembled Pd(II) and Pt(II) complexes, resulting in differences in their thermal responses. To compare the MMLCT excited states between Pd(II) and Pt(II), we have investigated the photophysical properties of isomorphous Pd(II)/Pt(II) complexes, K[M(CN)2(dFppy)]·H2O (M = Pd (Pd-dFppy) or Pt (Pt-dFppy) in Fig. 1(b); dFppy = 2-(4,6-difluorophenyl)pyridinate) which exhibit one-dimensional metallophilic interactions. Although both Pd-dFppy and Pt-dFppy display 3MMLCT emissions based on metallophilic interactions, only Pt-dFppy exhibits a drastic thermochromic shift in the emission spectrum. In contrast, the 3MMLCT emission of Pd-dFppy shows a significant increase in the quantum yield upon cooling, approaching almost unity, at nearly the same emission wavelength. Detailed variable temperature studies revealed that these differences in emission properties stem from disparities in the delocalization of electron density in 3MMLCT excited states through metallophilic interactions.
Results and discussion
Crystal structures
Single crystals of Pd-dFppy and Pt-dFppy suitable for X-ray crystallography were obtained through recrystallization from non-dehydrated MeCN/tBuOMe. Pd-dFppy was isolated as yellow needle-like crystals, whereas Pt-dFppy was obtained as dark green needle-like crystals. The phase purity of bulk polycrystalline samples after recrystallization was confirmed by elemental analysis, powder X-ray diffraction, and thermogravimetric analysis (Fig. S1 and S2 and Experimental details in the ESI†).The crystal structures of Pd-dFppy and Pt-dFppy were investigated by single crystal X-ray diffraction at 240 K, as shown in Fig. 2 and S3.†Pd-dFppy and Pt-dFppy are isomorphous and comprise a crystallographically independent [M(CN)2(dFppy)]− (M = Pd or Pt) ion, one K+ ion, and one water molecule coordinated to K+. In both complexes, the Pd and Pt centers adopt square-planar geometry coordinated by a dFppy ligand and two CN− ligands. The [M(CN)2(dFppy)]− anions are stacked one-dimensionally along the c-axis in a parallel manner in both crystals (Fig. S3(ii)†), where the electrostatic repulsion between [M(CN)2(dFppy)]− anions is neutralized by K+. This packing arrangement resembles that of K[Pt(CN)2(ppy)]·H2O (ppy = 2-phenylpyridinate).11 Importantly, both Pd-dFppy and Pt-dFppy exhibit short intermolecular metal···metal contacts between the stacked molecules. For Pd-dFppy, the Pd⋯Pd distance (dPd⋯Pd = 3.363(1) Å) was found to be slightly longer than twice the van der Waals radius of Pd (2rPd = 3.26 Å). However, according to previous reports, this Pd⋯Pd distance is short enough to form weak Pd⋯Pd interactions.7,9 On the other hand, the intermolecular Pt⋯Pt distance in Pt-dFppy (dPt⋯Pt = 3.3341(9) Å) is significantly shorter than 2rPt (3.50 Å), revealing the presence of strong intermolecular Pt⋯Pt interactions. Given the proximity of M⋯M distances in the solid-state structures of these complexes, assembly-induced MMLCT absorptions and emissions based on metallophilic interactions were expected.
 | ||
| Fig. 2 Photographs of crystals (top) and ORTEP drawings of molecular (middle) and stacking structures (bottom) of (a) Pd-dFppy and (b) Pt-dFppy at 240 K. Thermal ellipsoids are displayed at the 50% probability level. | ||
Absorption and emission properties
The UV-vis absorption spectra of Pd-dFppy and Pt-dFppy were measured both in the solid state and in solution to evaluate the influence of M⋯M interactions on their electronic structures. Consistent with the colors of the crystals, Pd-dFppy and Pt-dFppy exhibit broad absorption bands at approximately 430 and 610 nm in the solid state, respectively (dashed lines in Fig. 3(a)). These absorption bands are assigned to the 1MMLCT transition arising from metallophilic interactions, as neither complex shows any intense absorption bands in the visible region in the discrete state (i.e., in solution; dashed lines in Fig. 3(b)). The absorption bands observed below 400 nm in the solution state can be assigned to the singlet metal-to-ligand charge transfer (1MLCT) transition without metallophilic interactions for both complexes (Fig. S4–S6†).12 In addition, no new absorption bands are observed for either complex at concentrations up to 100 μM (Fig. S4(b)†), indicating that self-assembly of the complexes in solution is negligible in this concentration range. On the other hand, the addition of a poor solvent to the solutions of complexes resulted in the appearance of characteristic 1MMLCT absorption bands (Fig. S7†), further confirming the importance of the assembly of complexes to exhibit MMLCT transitions.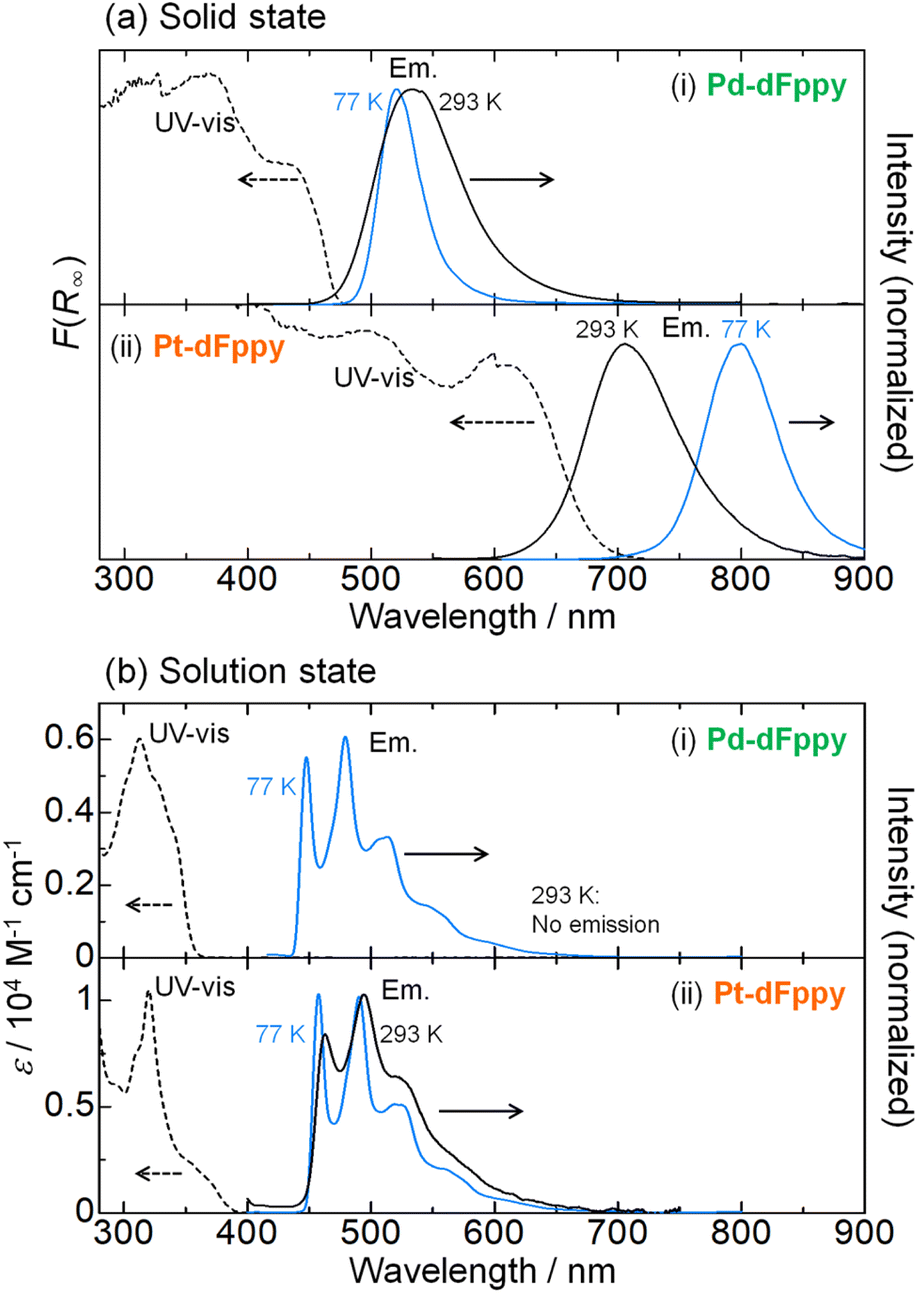 | ||
| Fig. 3 UV-vis absorption (dashed lines) and emission (solid lines) spectra of (i) Pd-dFppy and (ii) Pt-dFppy in (a) the solid state and (b) the solution state (MeOH/EtOH (v/v = 1/1); 5.0 × 10−5 M) at 293 K (black) and 77 K (blue) (λex = 350 nm for solution, 400 nm for Pd-dFppy in the solid state, and 500 nm for Pt-dFppy in the solid state). | ||
Both Pd-dFppy and Pt-dFppy exhibit characteristic 3MMLCT emissions in the solid state (black solid lines in Fig. 3(a)) corresponding to the observed absorptions. As expected, Pd-dFppy exhibits weak green emission and displays a broad structureless emission band (λmax = 534 nm) at 293 K. Conversely, Pt-dFppy displays a similarly broad structureless emission band, but its emission maximum (λmax = 705 nm) is observed at a significantly longer wavelength than that of Pd-dFppy, resulting in a deep red emission color at 293 K. An almost identical 3MMLCT emission band was similarly observed for the aggregate prepared by adding a poor solvent to the solution (Fig. S7(b)†). The red-shifted 3MMLCT emission of Pt-dFppy compared to that of Pd-dFppy suggests the energy of the dσ*(Pt⋯Pt) orbital in Pt-dFppy is much higher than that of the dσ*(Pd⋯Pd) orbital in Pd-dFppy. In addition, the large radiative rate constant (kr) of Pt-dFppy (6.1 × 105 s−1; Table 1) is typical of the 3MMLCT emission of Pt(II) complexes.13,14 In contrast to the solid state data, both Pd-dFppy and Pt-dFppy exhibit approximately the same emission spectra in dilute solutions due to the negligible metallophilic interactions (Fig. 3(b)). The vibronically structured emission bands in the solution state indicate that the emissions originate from a ligand-centered 3ππ* excited state of discrete monomer complexes, consistent with the previous report on (Bu4N)[Pt(CN)2(dFppy)].12 Computational studies further support this assignment (Fig. S8†). Overall, the significant differences in electronic transitions between the solid and solution states undoubtedly support the presence of assembly-induced 3MMLCT excited states arising from metallophilic interactions in both Pt-dFppy and Pd-dFppy. The differences in electronic transitions between the solid and solution states have also been confirmed by the corresponding excitation spectra (Fig. S9†).
| Complex | (a) Solid state | (b) Solution state | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| T/K | λ max /nm | Φ | τ /μs (Ad) | τ av /μs | k r /s−1 | k nr /s−1 | T/K | λ max /nm | Φ | |
| a Emission maxima. b Emission quantum yields. c Emission lifetimes. d Pre-exponential factors. e Averaged emission lifetimes. f Radiative rate constants, kr = Φ/τav. g Nonradiative rate constants, knr = kr(1 − Φ)/Φ. h Non-emissive. i Consistent with literature values for (Bu4N)[Pt(CN)2(dFppy)].12 | ||||||||||
| Pd-dFppy | 293 | 534 | <0.01 | 0.0300 (0.668), 0.168 (0.332) | 0.132 | — | — | 293 | —h | —h |
| 77 | 520 | 0.92 | 8.61 (0.476), 16.5 (0.524) | 14.0 | 6.6 × 104 | 5.7 × 103 | 77 | 448, 479, 513 | 0.46 | |
| Pt-dFppy | 293 | 705 | 0.01 | 0.00819 (0.658), 0.0222 (0.342) | 0.0163 | 6.1 × 105 | 6.1 × 107 | 293 | 463, 494, 521i | <0.01i |
| 77 | 800 | 0.16 | 0.166 (0.473), 1.03 (0.526) | 0.916 | 1.8 × 105 | 9.2 × 105 | 77 | 458, 490, 519i | 0.72 | |
The most notable difference between Pd-dFppy and Pt-dFppy is their temperature-dependent behavior (blue lines in Fig. 3(a)). The 3MMLCT emission energies of typical one-dimensional Pt(II) complexes have been reported to exhibit a red-shift with decreasing temperature,15 and indeed, the emission band of Pt-dFppy shifted into the NIR region (λmax = 800 nm) upon cooling to 77 K. Conversely, the shift of the emission band of Pd-dFppy at 77 K was not significant (λmax = 520 nm), indicating the non-thermochromic nature of Pd-dFppy. Instead, the emission intensity of Pd-dFppy was drastically enhanced to approximate unity upon cooling (Φ < 0.01 at 293 K to Φ = 0.92 at 77 K; Table 1).
Computational studies
For an in-depth investigation into the nature of the intermolecular interactions and MMLCT transitions, computational studies were performed for cluster model systems M2, M4, and M6 (Mn = stacked clusters of the Pd or Pt complexes with n layers) based on the structures of Pd-dFppy and Pt-dFppy. The ground-state structures of the cluster models (Fig. S10†) were fully optimized using the two-layer ONIOM method.16We used the optimized Pd2 and Pt2 systems for energy decomposition analyses (EDA).17 Numerical data from the EDA are summarized in Table 2. The computed interaction energies (ΔEint) in the Pd2 and Pt2 systems are −102.0 and −107.6 kcal mol−1, respectively. Hence, the Pt2 system exhibits relatively strong interactions. The ΔEint term was then decomposed into Pauli repulsion (ΔEPauli), electrostatic attraction (ΔEelstat), orbital interactions (ΔEorb), and dispersion interactions (ΔEdisp). Notably, the computed ΔEPauli for the Pd2 system (47.8 kcal mol−1) is relatively weak compared to the Pt2 system (53.7 kcal mol−1), reflecting a less overlap between the 4dz2 orbitals in Pd2 than the 5dz2 orbitals in Pt2. The computed ΔEorb for the Pd2 system (−83.6 kcal mol−1) is similar to that of the Pt2 system (−82.7 kcal mol−1). In addition, ΔEelstat for the Pt2 system (−40.1 kcal mol−1) is significantly stronger than that of the Pd2 system (−29.5 kcal mol−1), which could be a crucial factor in describing the attractive intermolecular interaction between Pt(II) centers leading to slightly short M⋯M distances compared to the Pd systems.
| Pd2 | Pt2 | |
|---|---|---|
| ΔEint | −102.0 | −107.6 |
| ΔEPauli | 47.8 | 53.7 |
| ΔEelstat | −29.5 | −40.1 |
| ΔEorb | −83.6 | −82.7 |
| ΔEdisp | −36.8 | −38.5 |
The vertical excitations of the M2, M4, and M6 systems were calculated to evaluate the excited states. The computed excitation wavelengths and the corresponding natural transition orbitals are summarized in Table S1 and Fig. S11.† The highest occupied natural transition orbitals for all systems are located on the M⋯M chain, whereas the lowest unoccupied natural transition orbitals are localized on the dFppy ligand (Fig. S11†), clearly indicating the 1MMLCT origin of the absorption bands for both Pd-dFppy and Pt-dFppy. The 1MMLCT excitations in the Pd systems followed the order: Pd2 (389 nm) < Pd4 (419 nm) < Pd6 (458 nm), and those of the Pt systems followed the order: Pt2 (426 nm) < Pt4 (468 nm) < Pt6 (568 nm). Thus, compared with the Pd clusters, the computed 1MMLCT vertical excitations of the Pt clusters occurred at relatively long wavelengths. Even in the Pd6 system (according to previous reports, this would not be favorable),5 the excitation energy (458 nm) was considerably higher than that of Pt6 (568 nm). This tendency is consistent with the experimental observations.
Thermochromic response
As mentioned earlier, the most significant distinction between Pd-dFppy and Pt-dFppy is the temperature-dependent emission shift (i.e., thermochromic luminescence). To gain further insight, variable-temperature X-ray structural analyses and emission measurements were performed (Fig. 4 and S12†). No phase transition was observed in the temperature range of 100–240 K for either Pt-dFppy or Pd-dFppy, however, when the temperature was decreased, anisotropic contraction of the crystals in the direction of the c-axis was observed (Fig. S12(a)†). Since the [M(CN)2(dFppy)]− anions are stacked in the direction of the c-axis, both Pt-dFppy and Pd-dFppy showed similar anisotropic contraction in dM⋯M lengths (∼0.05 Å; Fig. S12(b)†) when the temperature was decreased from 240 K to 100 K. Such shortening in Pt⋯Pt distance upon cooling has been widely considered to be the cause of the thermochromic shift of the 3MMLCT emissions,5a,15 as observed in Pt-dFppy (Fig. 4(b)). In contrast, despite the similar degree of shortening in the Pd⋯Pd distance, only a small shift in the emission maxima was observed for Pd-dFppy upon cooling (Fig. 4(a)), except for the decrease of the spectral width. Although the 3MMLCT emission of Pd-dFppy originates from intermolecular Pd⋯Pd interactions, these results indicate the 3MMLCT emission energy in Pd-dFppy is not sensitive to changes in Pd⋯Pd distance (dPd⋯Pd), unlike Pt-dFppy (Fig. 4(c)). Similar behavior was observed in the excitation spectra (Fig. S13†), indicating that not only the emission from the 3MMLCT state, but also the vertical excitation to the 1MMLCT state reflect differences in metallophilic interactions involving Pd and Pt.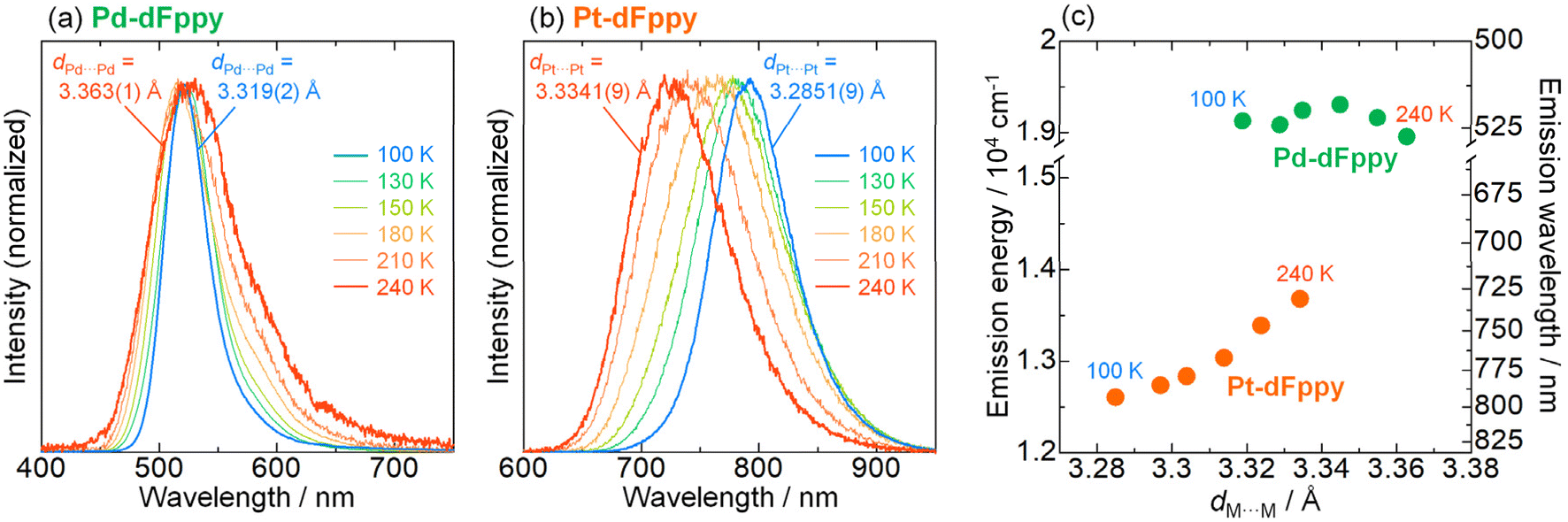 | ||
| Fig. 4 Temperature dependence of the emission spectra of (a) Pd-dFppy (λex = 400 nm) and (b) Pt-dFppy (λex = 500 nm) in the solid state, displayed alongside dM⋯M lengths at 240 and 100 K. (c) Correlation between the emission maxima and the M⋯M distances (dM⋯M) of Pd-dFppy (green) and Pt-dFppy (orange). | ||
To discuss the relationship between metallophilic interactions and thermochromism, the results reported herein are compared with those of previous reports. Lu et al. reported that [Pd(pbpy)(CCim)](PF6) (pbpyH = 6-phenyl-2,2′-bipyridine, CCimH+ = 2-ethynyl-1,3-dimethyl-1H-imidazolium), a complex that exhibits short Pd⋯Pd distances (dPd⋯Pd = 3.295(2) and 3.298(2) Å) comparable to 2rPd (3.26 Å), demonstrates obvious thermochromic 3MMLCT emission (Fig. 5(a)).8a On the other hand, we recently reported a unique case in which [Pt(CN)2(tBu-impy)] (tBu-impyH+ = 1-tert-butyl-3-(2-pyridyl)-1H-imidazolium; see Fig. 5(a)),13a which is a one-dimensional Pt(II) complex with slightly longer dPt⋯Pt than 2rPt, exhibits non-thermochromic 3MMLCT emission.13 Moreno et al. also recently reported that Pt(II) complex [Pt(dFppy)Cl(CNtBu)] with longer Pt⋯Pt distances (3.6876(5) and 3.8176(5) Å) exhibits a smaller thermochromic shift of the 3MMLCT emission.18 These suggest that a drastic thermochromic shift of the 3MMLCT emission occurs if dM⋯M is shorter than 2rM by a certain margin.
 | ||
| Fig. 5 (a) Examples of thermochromic and non-thermochromic Pt(II) and Pd(II) complexes with effective metallophilic interactions. (b) Suggested mechanisms of (i) thermochromic and (ii) non-thermochromic 3MMCLT emission. [a] Ref. 8a. [b] Ref. 13a. [c] Ref. 5a. | ||
Sakaki et al. discussed the thermochromic mechanism for [Pt(CN)2(bpy)] (bpy = 2,2′-bipyridine), which forms a typical one-dimensional stacked arrangement with short dPt⋯Pt (3.35 Å at 293 K).5a According to their explanation, thermochromic luminescence is mainly caused by thermal equilibrium between the trimerically and tetramerically delocalized 3MMLCT excited states, whereby the equilibrium shifts due to changes in dPt⋯Pt caused by changes in temperature (as illustrated in (i) in Fig. 5(b)). Indeed, the full width at half maximum (FWHM) of the emission band of Pt-dFppy showed a two-step temperature dependence: it increased until a temperature of approximately 210 K was reached, then decreased with temperature (Fig. S14(a)†), consistent with the presence of multiple competitive emission components. Based on this explanation, the absence of thermochromism in Pd-dFppy is indicative of a dimeric 3MMLCT excited state without thermal equilibrium with further extended oligomeric excited states (as illustrated in (ii) in Fig. 5(b)), as suggested for [Pt(CN)2(tBu-impy)].13a Certainly, the emission band of Pd-dFppy displayed typical thermal broadening without any decrease in FWHM (Fig. S14(b)†), indicating an absence of any thermal change of the emission components. Thus, the above facts suggest that thermal equilibria between delocalized excited states are achieved when dM⋯M is sufficiently shorter than 2rM. Notably, the van der Waals radii of Pd and Pt (rPd and rPt) were originally proposed based on the change in absorption of crystals of [M(Hdmg)2] (H2dmg = dimethylglyoxime) and related derivatives,10 but in fact, this absorption band can be assigned to the one-dimensionally delocalized dσ*(M⋯M) → pσ(M⋯M)-type transitions of the M⋯M chain.19 In other words, traditional 2rPd and 2rPt distances mean the M⋯M distances sufficient to facilitate the extension of the electronically delocalized 1MMLCT excited state (as well as the 3MMLCT state) across multiple molecules in a one-dimensional chain.20
The difference in the electronic delocalization of the 3MMLCT excited state between the Pd(II) and Pt(II) complexes has been further supported by the energy difference in their emissions. Recently, Strassert et al. compared the photophysical properties of crystallographically isomorphous Pd(II) and Pt(II) complexes PdL and PtL (H2L = N,N-di-[6-(2,6-difluoropyridin-3-yl)-4-methoxypyridin-2-yl]-4-hexylaniline) exhibiting dimeric structures with similar M⋯M distance (3.5025(8) Å for Pd, 3.4748(7) Å for Pt).9a The shift in the dimeric 3MMLCT emission of PtL relative to that of PdL was found to be approximately 2.0 × 103 cm−1 (Scheme S2†). For the present complexes, the calculated vertical excitation energy of the dimeric model of Pt-dFppy (i.e., Pt2) is similarly red-shifted by 2.2 × 103 cm−1 compared to that of the dimeric model of Pd-dFppy (i.e., Pd2) (see “Computational studies”; Scheme S2†), if both Pd-dFppy and Pt-dFppy form a dimeric excited state. However, the experimental 3MMLCT emission of Pt-dFppy was red-shifted by 4.5 × 103 cm−1 compared to Pd-dFppy (Fig. 3(a)). These results reveal that Pt-dFppy likely forms excited oligomers with more delocalized electron density than Pd-dFppy due shorter dPt⋯Pt than 2rPt, resulting in thermochromism via a thermal equilibrium between excited oligomers ((i) in Fig. 5(b)).5 In contrast, when dM⋯M is slightly longer than 2rM, such an extension in electron density delocalization is negligible. Therefore, the observed 3MMCLT emission for Pd-dFppy is attributed to a dimeric excited state, leading to a high-energy emission with non-thermochromism ((ii) in Fig. 5(b)).13a
Thermal quenching behavior
Instead of the non-thermochromism, Pd-dFppy shows a drastic increase in emission intensity to almost unity (Φ = 0.92 at 77 K) upon cooling (Fig. 6(a)). As shown in Fig. 6(b), the emission quantum yield for Pd-dFppy increased rapidly from 210 to 100 K, and the emission lifetime increased similarly in this temperature range. Therefore, the increase in emission lifetime is caused by the suppression of thermally activated nonradiative decay. This behavior could be characteristic of the 3MMLCT emission of Pd-dFppy, since such drastic and rapid thermal quenching has not been observed in Pt-dFppy (Fig. S15†). To validate this thermal quenching behavior, the emission lifetime data in the range of 20–300 K was analyzed using the following Arrhenius-type eqn (1) (Fig. 6(c)):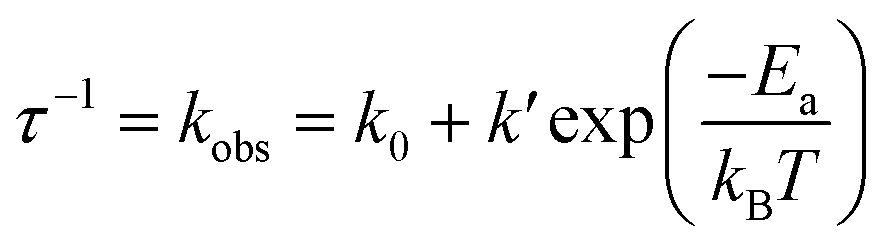 | (1) |
 | ||
| Fig. 6 (a) Temperature dependence of the non-normalized emission spectrum; (b) (i) emission quantum yield, and (ii) average emission lifetime of Pd-dFppy (λex = 400 nm) in the solid state. The emission quantum yield values at temperatures other than 77 K were determined by measuring the relative peak areas of the emission bands and were set in relation to the value for absolute emission quantum yield at 77 K (Φ = 0.92). (c) Plot of the decay rate constant (τ−1) as a function of temperature (20–300 K). The red line indicates the fitted curves based on eqn (1), and the resulting fitting parameters are shown in the inset. | ||
In contrast, the change in emission lifetime below 20 K was not accompanied by a change in the emission quantum yield for Pd-dFppy (Fig. 6(b)). Therefore, the temperature dependence observed below 20 K was not due to thermal deactivation, but mainly due to the zero-field splitting (ZFS) of the T1 (i.e., 3MMLCT) state, as described below.
Temperature dependence at very low temperatures
Finally, we evaluated the ZFS values of the T1 states of Pd-dFppy and Pt-dFppy, which reflect the degree of spin–orbit coupling.24 For this purpose, the emission lifetime data at very low temperatures (<50 K) were analyzed using eqn (2) (ref. 25) (Fig. 7).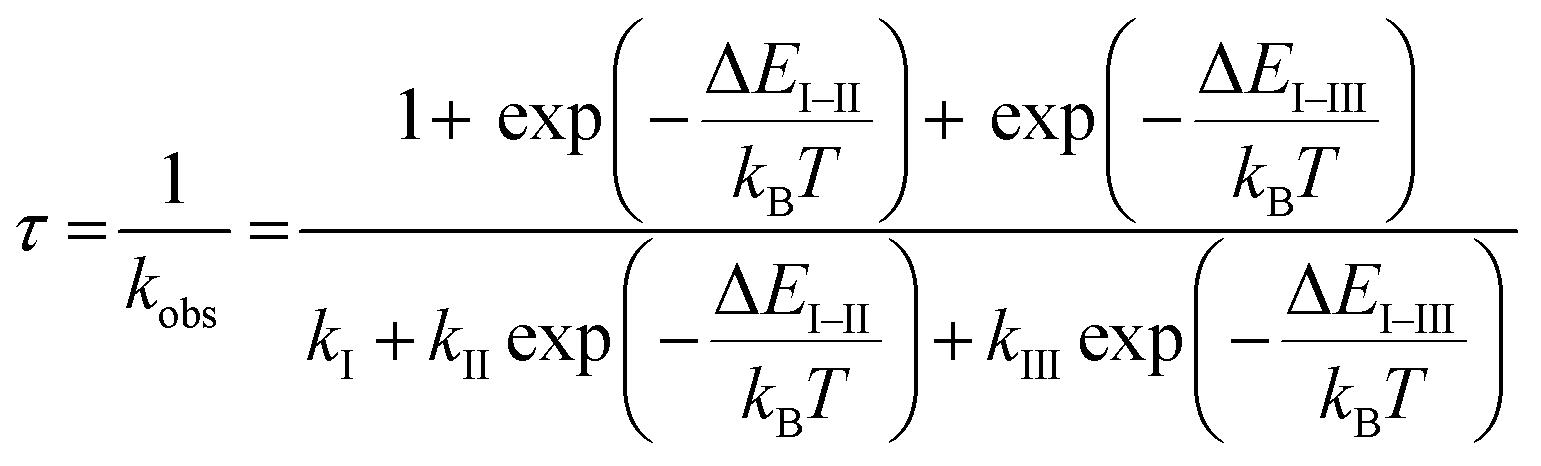 | (2) |
 | ||
| Fig. 7 Temperature dependence of emission lifetimes of (a) Pd-dFppy and (b) Pt-dFppy. Red lines represent the fitted curves based on eqn (2). Schematic energy diagrams showing the optimized fitting parameters for (c) Pd-dFppy and (d) Pt-dFppy (see also Table S2†). | ||
The analysis for Pd-dFppy was slightly more complicated compared to Pt-dFppy. Below 20 K, Pd-dFppy exhibited competitive emission between 3MMLCT and slightly higher-lying 3ππ* states, as indicated by the global fitting analysis of emission decays (Fig. S16†). Therefore, at temperatures below 20 K, only the 3MMLCT-derived decay component was used for fitting the data (Fig. 7(a)). During the fitting, the ΔEI–II value was found to be very small (<1 cm−1); thus, this value was approximated as 0 to optimize the other fitting parameters as shown in Fig. 7(c). The optimized ZFS value (ΔEI–III) of the T1 state of Pd-dFppy was found to be only ∼4 cm−1, indicating weaker spin–orbit coupling for Pd compared to Pt. This ZFS value is comparable to those of the triplet metal-to-ligand charge-transfer (3MLCT) states of Cu(I) complexes, which are known to exhibit weaker spin–orbit coupling.27 Such a small ZFS for Pd-dFppy is reasonable considering a previous report indicating the ZFS of a 3ππ*/3MLCT state of [Pd(thpy)2] (0.0962 cm−1; thpy = 2-thyenylpyridinate), a discrete Pd(II) complex, is much smaller than that of [Pt(thpy)2] (16 cm−1).28 However, this is the first example of ZFS of the 3MMLCT state of a self-assembled Pd(II) complex.
Conclusions
In conclusion, we have successfully evaluated the differences in the electronic delocalization of the 3MMLCT excited state, which drastically governs thermal responsiveness (Fig. 8). Isomorphous Pd(II) and Pt(II) complexes that exhibit a one-dimensional stacking arrangement with similar M⋯M distances, Pd-dFppy and Pt-dFppy, show green and deep red 3MMLCT emissions, respectively, which depend on the strength of metallophilic interactions. Despite their structural similarity, only Pt-dFppy displayed obvious thermochromic luminescence, whereas Pd-dFppy demonstrated drastic thermal quenching behavior. A comprehensive comparison with present and previous studies indicated that the 3MMLCT excited state should be oligomerically delocalized through the metallophilic interactions (Fig. 8(a)) if the M⋯M distance (dM⋯M) is sufficiently shorter than twice the van der Waals radii (2rM). Thus, Pt-dFppy, which has a sufficiently short dPt⋯Pt relative to 2rPt, showed a large thermochromic shift due to thermal equilibrium between excited oligomers. Although dPd⋯Pd is slightly longer than 2rPd, Pd-dFppy still showed 3MMLCT emission, given the van der Waals radii of Pt and Pd are still ambiguous.10,29 However, the electron density of the 3MMLCT excited state of Pd-dFppy is localized between two neighboring molecules without thermal equilibrium (Fig. 8(b)), resulting in an emission that shows drastic thermal quenching owing to structural deformation during excitation. Despite the recent revelation of the solution-state equilibria of excited oligomers of Pt(II) complexes,30 this work provides a crucial experimental evidence for the thermal equilibria of intermolecularly delocalized 3MMLCT excited states within one-dimensional metal chains in the solid state, resulting in a thermochromic response. While 3MMLCT-emissive materials have been widely studied, the results presented herein shed light on the limiting factor for extending the degree of delocalization of 3MMLCT excited states over multiple molecules through metallophilic interactions for the first time, providing insight for controlling the functionalities of molecular-based materials. | ||
| Fig. 8 Schematic illustrations of the differences in electronic delocalization in the excited state, and the resulting thermal response for (a) Pt-dFppy and (b) Pd-dFppy. | ||
Data availability
All the data supporting this study are included in the main text and the ESI.†Author contributions
M. Y. and M. K. conceived and directed the project. T. S. and K. S. performed the synthesis, characterizations, and all measurements. D. S. and A. K. supported experiments, and evaluated and validated the research. J. T., S. H., and A. M. contributed to the variable-temperature emission measurements. N. M. L. and W. M. C. S. conducted the computational studies. M. Y., W. M. C. S., and M. K. co-wrote the manuscript. All authors discussed the results, and reviewed and approved the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by JSPS KAKENHI grant numbers JP17H06367, JP20H05082, JP21K05094, JP22H02098, JP23K23366, and JP24K08455, and also the Hyogo Science and Technology Association. Supercomputing resources at the Research Center for Computational Science, Okazaki, Japan are also acknowledged (Project: 23-IMS-C031).Notes and references
- (a) J. J. Novoa, G. Aullh, P. Alemany and S. Alvarez, J. Am. Chem. Soc., 1995, 117, 7169–7171 CrossRef CAS; (b) P. Pyykkö, Chem. Rev., 1997, 97, 597–636 CrossRef PubMed; (c) A. Poater, S. Moradell, E. Pinilla, J. Poater, M. Solà, M. Á. Martíne and A. Llobet, Dalton Trans., 2006, 1188–1196 RSC; (d) Q. Zheng, S. Borsley, G. S. Nichol, F. Duarte and S. L. Cockroft, Angew. Chem., Int. Ed., 2019, 58, 12617–12623 CrossRef CAS PubMed; (e) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931–1951 RSC.
- (a) M. Kato, Bull. Chem. Soc. Jpn., 2007, 80, 287–294 CrossRef CAS; (b) O. S. Wenger, Chem. Rev., 2013, 113, 3686–3733 CrossRef CAS PubMed; (c) Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC; (d) K. M.-C. Wong and V. W.-W. Yam, Acc. Chem. Res., 2011, 44, 424–434 CrossRef CAS PubMed; (e) A. Kobayashi and M. Kato, Eur. J. Inorg. Chem., 2014, 4469–4483 CrossRef CAS; (f) V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed; (g) A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed; (h) A. Aliprandi, D. Genovese, M. Mauro and L. De Cola, Chem. Lett., 2015, 44, 1152–1169 CrossRef CAS; (i) M. Kato, H. Ito, M. Hasegawa and K. Ishii, Chem.–Eur. J., 2019, 25, 5105–5112 CrossRef CAS PubMed; (j) M. Yoshida and M. Kato, Coord. Chem. Rev., 2020, 408, 213194 CrossRef CAS; (k) M. A. Soto, R. Kandel and M. J. MacLachlan, Eur. J. Inorg. Chem., 2021, 894–906 CrossRef CAS; (l) M. Sadeghian, M. G. Haghighi, E. Lalinde and M. T. Moreno, Coord. Chem. Rev., 2022, 452, 214310 CrossRef CAS; (m) Z.-L. Gong, Z.-Q. Li and Y.-W. Zhong, Aggregate, 2022, 3, e177 CrossRef CAS; (n) S.-Y. Yang, Y. Chen, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2024, 53, 5366–5393 RSC; (o) M. Kato, IUCrJ, 2024, 11, 442–452 CrossRef CAS PubMed.
- (a) Y.-C. Wei, K.-H. Kuo, Y. Chi and P.-T. Chou, Acc. Chem. Res., 2023, 56, 689–699 CrossRef CAS PubMed; (b) P. L. dos Santos, P. Stachelek, Y. Takeda and P. Pander, Mater. Chem. Front., 2024, 8, 1731–1766 RSC; (c) M. Wałęsa-Chorab, J. Photochem. Photobiol., C, 2024, 59, 100664 CrossRef; (d) Y.-C. Wei, S. F. Wang, Y. Hu, L.-S. Liao, D.-G. Chen, K.-H. Chang, C.-W. Wang, S.-H. Liu, W.-H. Chan, J.-L. Liao, W.-Y. Hung, T.-H. Wang, P.-T. Chen, H.-F. Hsu, Y. Chi and P.-T. Chou, Nat. Photonics, 2020, 14, 570–577 CrossRef CAS; (e) S.-F. Wang, B.-K. Su, X.-Q. Wang, Y.-C. Wei, K.-H. Kuo, C.-H. Wang, S.-H. Liu, L.-S. Liao, W.-Y. Hung, L.-W. Fu, W.-T. Chuang, M. Qin, X. Lu, C. You, Y. Chi and P.-T. Chou, Nat. Photonics, 2022, 16, 843–850 CrossRef CAS.
- G. Gliemann and H. Yersin, Struct. Bonding, 1985, 62, 87–153 CrossRef CAS.
- (a) M. Nakagaki, S. Aono, M. Kato and S. Sakaki, J. Phys. Chem. C, 2020, 124, 10453–10461 CrossRef CAS; (b) J.-J. Zheng and S. Sakaki, J. Photochem. Photobiol., C, 2022, 51, 100482 CrossRef CAS.
- D. Dalmau and E. P. Urriolabeitia, Molecules, 2023, 28, 2663 CrossRef CAS PubMed.
- (a) Q. Wan, W.-P. To, C. Yang and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 3089–3093 CrossRef CAS PubMed; (b) Q. Wan, W.-P. To, X. Chang and C.-M. Che, Chem, 2020, 6, 945–967 CrossRef CAS; (c) Q. Wan, D. Li, J. Zou, T. Yan, R. Zhu, K. Xiao, S. Yue, X. Cui, Y. Weng and C.-M. Che, Angew. Chem., Int. Ed., 2022, 61, e202114323 CrossRef CAS PubMed.
- (a) C. Zou, J. Lin, S. Suo, M. Xie, X. Chang and W. Lu, Chem. Commun., 2018, 54, 5319–5322 RSC; (b) N. Zhou, C. Zou, S. Suo, Y. Liu, J. Lin, X. Zhang, M. Shi, X. Chang and W. Lu, Dalton Trans., 2023, 52, 5503–5513 RSC; (c) N. Zhou, M. Xie, C. Zheng, J. An, C. Zou and W. Lu, Adv. Opt. Mater., 2024, 12, 2400295 CrossRef CAS.
- (a) T. Theiss, S. Buss, I. Maisuls, R. López-Arteaga, D. Brünink, J. Kösters, A. Hepp, N. L. Doltsinis, E. A. Weiss and C. A. Strassert, J. Am. Chem. Soc., 2023, 145, 3937–3951 CrossRef CAS PubMed; (b) L. Cao, K. Klimes, Y. Ji, T. Fleetham and J. Li, Nat. Photonics, 2021, 15, 230–237 CrossRef CAS; (c) M. V. Kashina, K. V. Luzyanin, D. V. Dar'in, S. I. Bezzubov and M. A. Kinzhalov, Inorg. Chem., 2024, 63, 5315–5319 CrossRef CAS PubMed; (d) D. Saito, K. Segawa, M. Kikkawa, M. Yoshida, A. Kobayashi and M. Kato, Cryst. Growth Des., 2024 DOI:10.1021/acs.cgd.4c00817.
- (a) A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS; (b) C. V. Banks and D. W. Barnum, J. Am. Chem. Soc., 1958, 80, 3579–3582 CrossRef CAS.
- B. D. Belviso, F. Marin, S. Fuertes, V. Sicilia, R. Rizzi, F. Ciriaco, C. Cappuccino, E. Dooryhee, A. Falcicchio, L. Maini, A. Altomare and R. Caliandro, Inorg. Chem., 2021, 60, 6349–6366 CrossRef CAS PubMed.
- A. F. Rausch, U. V. Monkowius, M. Zabel and H. Yersin, Inorg. Chem., 2010, 49, 7818–7825 CrossRef CAS PubMed.
- (a) D. Saito, T. Ogawa, M. Yoshida, J. Takayama, S. Hiura, A. Murayama, A. Kobayashi and M. Kato, Angew. Chem., Int. Ed., 2020, 59, 18723–18730 CrossRef CAS PubMed; (b) K. Sasaki, D. Saito, M. Yoshida, F. Tanaka, A. Kobayashi, K. Sada and M. Kato, Chem. Commun., 2023, 59, 6745–6748 RSC.
- (a) C.-W. Hsu, K. T. Ly, W.-K. Lee, C.-C. Wu, L.-C. Wu, J.-J. Lee, T.-C. Lin, S.-H. Liu, P.-T. Chou, G.-H. Lee and Y. Chi, ACS Appl. Mater. Interfaces, 2016, 8, 33888–33898 CrossRef CAS PubMed; (b) K. T. Ly, R.-W. Chen-Cheng, H.-W. Lin, Y.-J. Shiau, S.-H. Liu, P.-T. Chou, C.-S. Tsao, Y.-C. Huang and Y. Chi, Nat. Photonics, 2017, 11, 63–68 CrossRef.
- (a) W. B. Connick, L. M. Henling, R. E. Marsh and H. B. Gray, Inorg. Chem., 1996, 35, 6261–6265 CrossRef CAS; (b) M. Kato, C. Kosuge, K. Morii, J. S. Ahn, H. Kitagawa, T. Mitani, M. Matsushita, T. Kato, S. Yano and M. Kimura, Inorg. Chem., 1999, 38, 1638–1641 CrossRef CAS.
- (a) L. W. Chung, W. M. C. Sameera, R. Ramozzi, A. J. Page, M. Hatanaka, G. P. Petrova, T. V. Harris, X. Li, Z. Ke, F. Liu, H.-B. Li, L. Ding and K. Morokuma, Chem. Rev., 2015, 115, 5678–5796 CrossRef CAS PubMed; (b) W. M. C. Sameera and F. Maseras, J. Chem. Inf. Model., 2018, 58, 1828–1835 CrossRef CAS PubMed.
- (a) T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef CAS; (b) Q. Wan, J. Yang, W.-P. To and C.-M. Che, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e201926511 Search PubMed.
- M. Martínez-Junquera, E. Lalinde and M. T. Moreno, Inorg. Chem., 2022, 61, 10898–10914 CrossRef PubMed.
- (a) Y. Ohashi, I. Hanazaki and S. Nagakura, Inorg. Chem., 1970, 9, 2551–2556 CrossRef; (b) K. Takeda, T. Sasaki, J. Hayashi, S. Kagami, I. Shirotani and K. Yakushi, J. Phys.: Conf. Ser., 2010, 215, 012065 CrossRef.
- Although a few self-assembled Pt(II) complexes with slightly long Pt⋯Pt distances (dPt···Pt = ca. 3.6–3.7 Å) have also been reported to exhibit larger thermochromic shift of 3MMLCT emission, in these cases the delocalization of the excited state could be assisted by significant π–π interactions between the aromatic ligands. For example: (a) H. Matsukawa, M. Yoshida, T. Tsunenari, S. Nozawa, A. Sato-Tomita, Y. Maegawa, S. Inagaki, A. Kobayashi and M. Kato, Sci. Rep., 2019, 9, 15151 CrossRef PubMed; (b) B.-S. Su, Y.-C. Wei, W.-T. Chuang, S.-C. Weng, S.-F. Wang, D.-G. Chen, Z.-X. Huang, Y. Chi and P.-T. Chou, J. Phys. Chem. Lett., 2021, 12, 7482–7489 CrossRef CAS PubMed.
- (a) T. Sajoto, P. I. Djurovich, A. B. Tamayo, J. Oxgaard, W. A. Goddard III and M. E. Thompson, J. Am. Chem. Soc., 2009, 131, 9813–9822 CrossRef CAS PubMed; (b) L. S. Forster, Coord. Chem. Rev., 2002, 227, 59–92 CrossRef CAS; (c) C. B. Blanton, Z. Murtaza, R. J. Shaver and D. P. Rillema, Inorg. Chem., 1992, 31, 3230–3235 CrossRef CAS; (d) T. Ogawa, W. M. C. Sameera, D. Saito, M. Yoshida, A. Kobayashi and M. Kato, Inorg. Chem., 2018, 57, 14086–14096 CrossRef CAS PubMed.
- (a) A. Islam, N. Ikeda, K. Nozaki, Y. Okamoto, B. Gholamkhass, A. Yoshimura and T. Ohno, Coord. Chem. Rev., 1998, 171, 355–363 CrossRef CAS; (b) T. Ogawa, W. M. C. Sameera, M. Yoshida, A. Kobayashi and M. Kato, Chem. Phys. Lett., 2020, 739, 137024 CrossRef CAS.
- D. Escudero and W. Thiel, Inorg. Chem., 2014, 53, 11015–11019 CrossRef CAS PubMed.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS.
- G. D. Hager and G. A. Crosby, J. Am. Chem. Soc., 1975, 97, 7031–7037 CrossRef CAS.
- J. C. Deaton, A. Chakraborty, R. Czerwieniec, H. Yersin and F. N. Castellano, Phys. Chem. Chem. Phys., 2018, 20, 25096–25104 RSC.
- (a) T. Hofbeck, U. Monkowius and H. Yersin, J. Am. Chem. Soc., 2015, 137, 399–404 CrossRef CAS PubMed; (b) R. Czerwieniec and H. Yersin, Inorg. Chem., 2015, 54, 4322–4327 CrossRef CAS PubMed; (c) A. Schinabeck, M. J. Leitl and H. Yersin, J. Phys. Chem. Lett., 2018, 9, 2848–2856 CrossRef CAS PubMed.
- (a) H. Yersin, S. Schüetzenmeier, H. Wiedenhofer and A. von Zelewsky, J. Phys. Chem., 1993, 97, 13496–13499 CrossRef CAS; (b) H. Yersin and D. Donges, Top. Curr. Chem., 2001, 214, 81–186 CrossRef CAS.
- (a) S. S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS; (b) S. Alvarez, Dalton Trans., 2013, 42, 8617–8636 RSC; (c) N. Harris, A. K. Sakr, H. V. Snelling and N. A. Young, J. Mol. Struct., 2018, 1172, 80–88 CrossRef CAS.
- (a) M. Iwamura, A. Fukui, K. Nozaki, H. Kuramochi, S. Takeuchi and T. Tahara, Angew. Chem., Int. Ed., 2020, 59, 23154–23161 CrossRef CAS PubMed; (b) S. Hattori, T. Nakano, N. Kobayashi, Y. Konno, E. Nishibori, T. Galica and K. Shinozaki, Dalton Trans., 2022, 51, 15830–15841 RSC; (c) H. Watanabe, M. Iwamura and K. Nozaki, Inorg. Chem., 2024, 63, 5580–5585 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental detail, supplementary X-ray crystallographic, spectral, and theoretical data. CCDC 2357833–2357844. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04497e |
| This journal is © The Royal Society of Chemistry 2024 |