
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNi-catalyzed enantioselective three-component reductive alkylacylation of alkenes: modular access to structurally complex α-amino ketones†
Jichao
Xiao
 a,
Tingting
Jia
a,
Shuang
Chen
a,
Mengxiao
Pan
a and
Xingwei
Li
*ab
a,
Tingting
Jia
a,
Shuang
Chen
a,
Mengxiao
Pan
a and
Xingwei
Li
*ab
aSchool of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an, Shanxi 710062, China. E-mail: lixw@snnu.edu.cn
bInstitute of Molecular Science and Engineering, Institute of Frontier and Interdisciplinary Sciences, Shandong University, Qingdao, Shandong 266237, China
First published on 27th August 2024
Abstract
Chiral alpha-amino ketones have found extensive applications as functional molecules. A nickel-catalyzed, enantioselective, and fully intermolecular three-component 1,2-alkylacylation of N-acyl enamides has been realized with tertiary alkyl bromides and carboxylic acid-derived electrophiles as the coupling reagents. This reductive coupling strategy is operationally simple, exhibiting broad substrate scope and excellent functional group tolerance using readily available starting materials and allowing rapid access to structurally complex α-amino ketone derivatives in high enantioselectivity. A suitable chiral biimidazoline ligand together with additional chelation of the amide carbonyl group in a Ni alkyl intermediate facilitates the enantioselective control by suppressing the background reaction, accounting for the excellent enantioselectivity. Mechanistic studies indicated intermediacy of radical species.
Introduction
Chiral α-amino ketones are a key structural motif in various bioactive molecules and pharmaceuticals, and they are also valuable building blocks in synthetic chemistry (Scheme 1A).1–4 Accordingly, the development of efficient asymmetric approaches for synthesis of chiral α-amino ketones has been a long-lasting pursuit in organic chemistry.5–8 Reported classic synthetic routes typically involve nucleophilic amination of α-halogenated ketones, electrophilic amination of suitable enolates, and homologation of chiral α-amino acids.9–19 Nevertheless, these two-electron-based strategies often suffer from limitations, such as poor availability of the nitrogen source, racemic synthesis or racemization of product, requirement of multiple steps, and/or harsh reaction conditions. Owing to the unique properties of nickel catalysts, recently, the groups of Melchiorre,20 Huo,21 and Baran22 elegantly developed nickel-catalyzed asymmetric synthesis of enantio-enriched α-amino ketones via the coupling of an amine-containing reagent and an electrophilic acyl source (Scheme 1B). Meanwhile, the Zhu group reported synthesis of α-amino ketones via reductive coupling of olefins with a carbonyl source and a redox-active ester.23 Despite the advances, there remains an increasing demand for the development of efficient and concise protocols that permit a rapid assembly of α-amino ketones with high levels of complexity, modularity, and diversity from readily available commodities.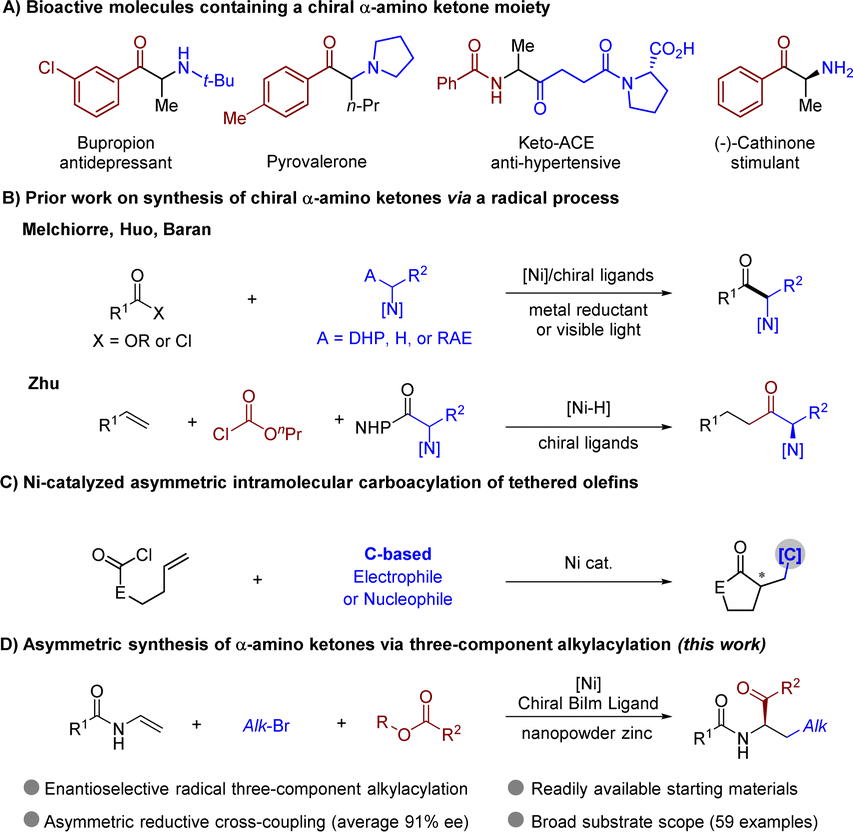 | ||
| Scheme 1 Chiral α-amino ketones and enantioselective carboacylation of olefins. | ||
On the other hand, nickel-catalyzed asymmetric functionalization of olefins stands out as a prominent strategy toward facile construction of chiral complex molecules owing to the unique roles of Ni catalysts that readily participate in single electron transfer (SET) and reductive elimination.24–26 Consequently, catalytic three-component radical difunctionalizations of alkenes represent a powerful strategy for rapid enhancement of molecular complexity by simultaneously forging two functionalities in one single operation, thus enriching the synthetic toolbox to access synthetic building blocks, medicines, and bioactive molecules.27–41 However, enantioselective control of the target stereogenic center remains a formidable challenge. Impressive progress has been made in nickel-catalyzed asymmetric radical three-component difunctionalizations of alkenes. These coupling systems include diarylation,42 alkyl–arylation,43–49 alkyl–alkenylation,50,51 aryl–alkylation,52 and sulfonyl–carbolation53,54 under redox-neutral or reductive conditions, where organohalides serve as the terminal (the 3rd) component.55,56 However, as an important class of dicarbofunctionalization, asymmetric carboacylation reactions have received less attention and are restricted to two categories of intramolecular couplings. A carbamoyl halide-tethered olefin may react with a carbon terminating reagent via cyclization and C–C coupling under redox-neutral or reductive conditions (Scheme 1C).57–66 Alternatively, an aryl iodide-tethered olefin may react with an acylating reagent to fulfil the similar type of reaction.67,68 More recently, the groups of Chu,69 Stanley,70 Wang,71 Yuan72 and other73–77 have developed various methods for fully three-component racemic carboacylation of olefins. In most cases, a nickel acyl species captured a secondary radical species to deliver the final product through reductive elimination. However, the exceptionally high instability and reactivity of open-shell radical intermediates poses significant challenges in enantioselective control in these three-component reactions in general.78–82 In addition, the carbon radical intermediate may also undergo uncatalyzed C–C coupling with a reactive acylating regent, leading to a background reaction. To the best of our knowledge, enantioselective fully three-component 1,2-alkylacylation of alkenes remains unknown. Therefore, the development of 1,2-alkylacylation reactions toward asymmetric synthesis of α-amino ketones will be an intriguing but challenging task.
Herein, we now describe modular synthesis of enantio-enriched α-amino ketones via an efficient nickel-catalyzed enantioselective 1,2-alkylacylation of enamides with an alkyl bromide and an acylating regent, which demonstrates an example of the integration of fully three-component carboacylation of alkenes with a high level of enantioselectivity for the first time (Scheme 1D).
Results and discussion
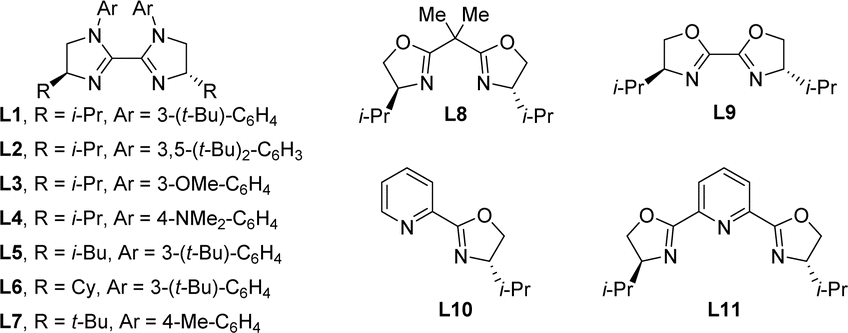
By taking advantage of the chelating effect during the formation of an organonickel intermediate, our initial investigation of asymmetric carboacylation was conducted using N-vinylbenzamide (1a) as the olefin and 2-bromo-2-methylpropane (2a) and 4-chlorobenzoic anhydride (3a) as the coupling components (Table 1). Through systematic investigation of the reaction parameters, the final optimal conditions were found to comprise a chiral biimidazoline (BiIm) ligand L1, NiBr2·diglyme as a precatalyst, nanopowder zinc (40–60 nm) as a reductant, and MgCl2 and 2-picoline as additives in THF/DMAc (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0.1 M) at room temperature for 48 h, from which conditions the α-amino ketone 4 was isolated in 81% yield and 92% ee (entry 1). A ligand screen revealed that chiral BiIm ligands (L1–L7, entries 1–7) were consistently superior to other oxazoline-based ligands (L8–L11, entries 8–11). BiIm skeletons with other alkyl chains or other aryl protecting groups (L2–L7) worked with comparable efficiency but with slightly decreased enantioselectivity, while Box ligand (L8), BiOx (L9), Pyrox (L10), and Pybox (L11) all gave sluggish reactions. Control experiments indicated that the nickel catalyst, ligand, nanopowder zinc, magnesium chloride, 2-picoline and the solvent (entries 13–19) each played a pivotal role in this transformation. The racemic α-amino ketone product can still be obtained in low yield in the absence of the ligand L1 or nickel catalyst (entries 13 and 14), indicating a background reaction. Of note, the particle size of the Zn reductant is vital; nanopowder zinc can dramatically improve the efficiency when compared with other reductants (entry 12, see the ESI† for more information). Furthermore, omitting the MgCl2 and 2-picoline additives led to a substantial decrease of the reaction efficiency, albeit with no erosion of the enantioselectivity (entries 16 and 17). Screening of the solvent indicated that comparable results can be obtained in a single DMAc solvent, but introduction of THF further improved the enantioselectivity (entries 18 and 19, see the ESI† for more information).
:1, 0.1 M) at room temperature for 48 h, from which conditions the α-amino ketone 4 was isolated in 81% yield and 92% ee (entry 1). A ligand screen revealed that chiral BiIm ligands (L1–L7, entries 1–7) were consistently superior to other oxazoline-based ligands (L8–L11, entries 8–11). BiIm skeletons with other alkyl chains or other aryl protecting groups (L2–L7) worked with comparable efficiency but with slightly decreased enantioselectivity, while Box ligand (L8), BiOx (L9), Pyrox (L10), and Pybox (L11) all gave sluggish reactions. Control experiments indicated that the nickel catalyst, ligand, nanopowder zinc, magnesium chloride, 2-picoline and the solvent (entries 13–19) each played a pivotal role in this transformation. The racemic α-amino ketone product can still be obtained in low yield in the absence of the ligand L1 or nickel catalyst (entries 13 and 14), indicating a background reaction. Of note, the particle size of the Zn reductant is vital; nanopowder zinc can dramatically improve the efficiency when compared with other reductants (entry 12, see the ESI† for more information). Furthermore, omitting the MgCl2 and 2-picoline additives led to a substantial decrease of the reaction efficiency, albeit with no erosion of the enantioselectivity (entries 16 and 17). Screening of the solvent indicated that comparable results can be obtained in a single DMAc solvent, but introduction of THF further improved the enantioselectivity (entries 18 and 19, see the ESI† for more information).
|
|
|||
|---|---|---|---|
| Entry | Deviation from standard conditions | Yieldb (%) | eec (%) |
| a Optimal reaction conditions: enamide (1a, 0.1 mmol), alkyl bromide (2a, 0.2 mmol), acid anhydride (3a, 0.15 mmol), NiBr2·diglyme (10 mol%), L1 (15 mol%), picoline, MgCl2, nanopowder Zn (4.0 equiv.), and DMAc/THF was stirred at 25 °C for 48 h. b Isolated yield. c The ee was determined using HPLC with a chiral stationary phase. | |||
| 1 | None | 82 | 92 |
| 2 | L2 instead of L1 | 81 | 91 |
| 3 | L3 instead of L1 | 75 | 87 |
| 4 | L4 instead of L1 | 77 | 87 |
| 5 | L5 instead of L1 | 65 | 77 |
| 6 | L6 instead of L1 | 70 | 79 |
| 7 | L7 instead of L1 | 23 | 21 |
| 8 | L8 instead of L1 | 43 | 12 |
| 9 | L9 instead of L1 | 67 | 31 |
| 10 | L10 instead of L1 | 32 | 15 |
| 11 | L11 instead of L1 | 13 | 45 |
| 12 | Zinc instead of nanopowder zinc | 53 | 91 |
| 13 | No L1 | 26 | 0 |
| 14 | No NiBr2·diglyme | 21 | 0 |
| 15 | No nanopowder zinc | 0 | — |
| 16 | No MgCl2 | 18 | 93 |
| 17 | No 2-picoline | 44 | 92 |
| 18 | THF only | 38 | 72 |
| 19 | DMAC only | 85 | 89 |
|
|||
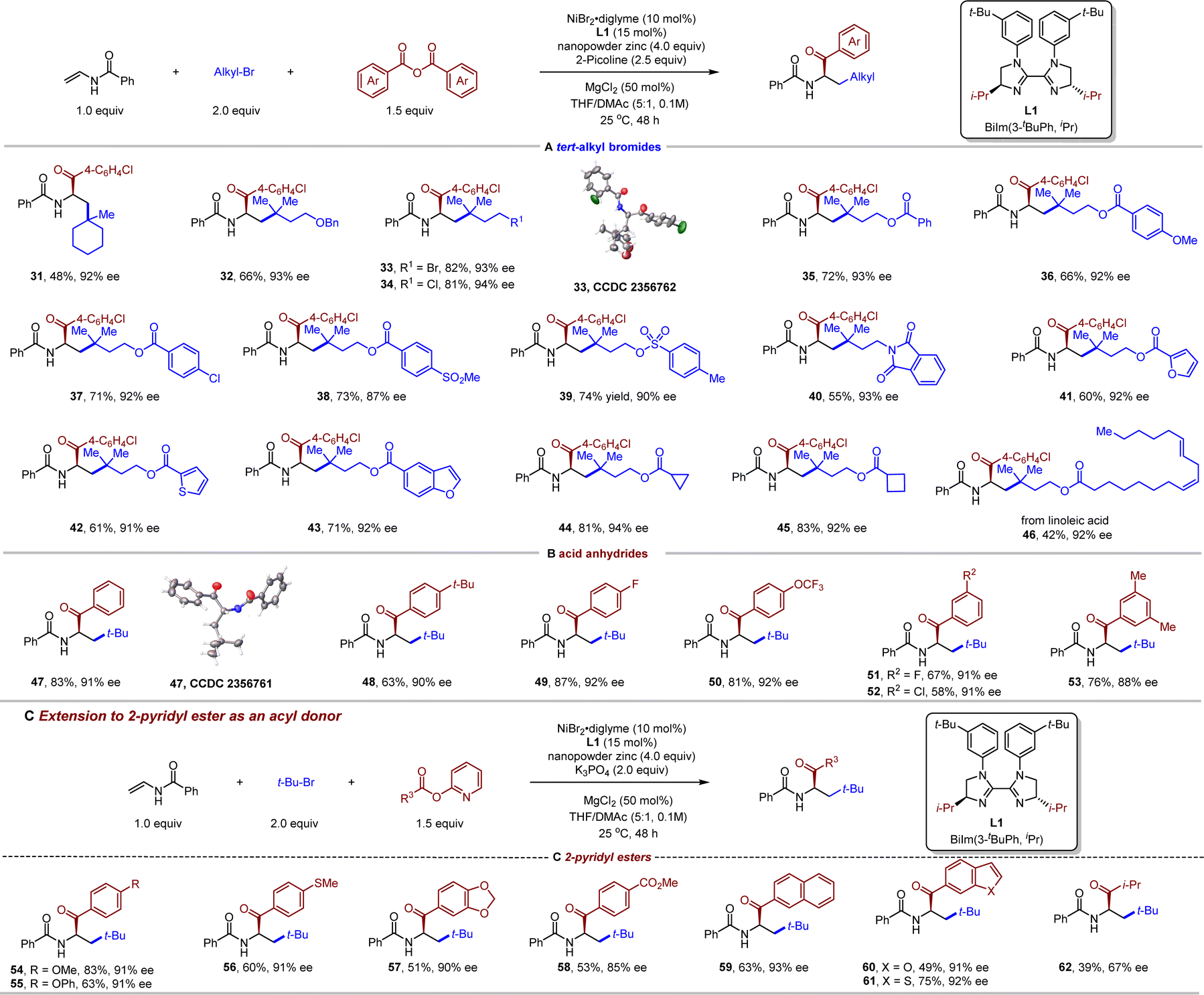
With the optimized conditions in hand, we next investigated the generality of this three-component carboacylation. As illustrated in Table 2, vinyl amides bearing a wide variety of substituents delivered the desired products in good to excellent yields with good to excellent enantioselectivities. Enamides with an electron-donating (5–9 and 21) or -withdrawing (10–20 and 30) substituent at different positions of the benzene ring coupled efficiently, affording the α-amino ketones in 55–90% yields and 87–94% ee. Such mild reaction conditions allowed the use of a diverse spectrum of functional groups, including ether (7 and 12), thiolether (8), amine (9), ketone (13), ester (14), nitrile (15), sulfone (16), and aminosulfonyl (30). Notably, some potentially reactive functionality such as aryl bromide (17) and chlorides (18, 20 and 25) were left intact, offering opportunities for further useful transformations. A series of heterocycles including furan (23), thiophene (24), pyridine (25), benzofuran (26), and pyrimidine (27), which are frequently found in pharmaceutically active molecules, were also compatible. To our delight, cycloalkyl-substituted amides were also viable (28 and 29), albeit with attenuated enantioselectivity (82–90% ee). In contrast, 1,1- and 1,2-disubstituted internal olefins turned out to inapplicable under the standard conditions (see the ESI† for unsuccessful olefins).
| a Reaction conditions: enamide (0.1 mmol), alkyl bromide (0.2 mmol), acid anhydride (0.15 mmol), NiBr2·diglyme (10 mol%), ligand L1 (15 mol%), picoline, MgCl2, and nanopowder Zn (4.0 equiv.) in DMAc/THF was stirred at 25 °C for 48 h. Isolated yield. The ee was determined using HPLC with a chiral stationary phase. |
|---|

|
After defining the scope of vinyl amides, attention was then turned to the scope of the alkyl bromide component. Both cyclic tertiary bromide (31) and open-chained tertiary bromides (32–46) proved to be amenable to the coupling conditions (Table 3 (A)). We were pleased to find excellent functional group compatibility for the tertiary alkyl bromides component. Functional groups such as ether (32), primary alkyl bromide/chloride (33 and 34), aryl chloride (37), sulfone (38), tosylate (39), phthalimide (40), strained ring (44 and 45), and an internal alkene (46) were compatible, affording the corresponding products in moderate to good yields (58–82%) and excellent enantioselectivities (up to 94% ee). The absolute configuration for products 33 and 47 has been confirmed by X-ray crystallographic analyses. Interestingly, when the substrate contains both tertiary and primary alkyl bromide, the reaction is completely selective for the more sterically congested C–Br bond to give product 33 in 82% yield and 93% ee, leaving primary alkyl bromide unchanged, indicative of a stable radical species. Notably, heterocyclic substrates such as furan (41), thiophene (42), and benzofuran (43) were equally suitable for this chemistry. Unfortunately, primary and secondary alkyl bromides were found to be unreactive in our protocol, giving rise to only two-component coupling reaction with the benzoic acid anhydride.
| a Reaction conditions: enamide (0.1 mmol), alkyl bromide (0.2 mmol), acid anhydride or 2-pyridyl ester (0.15 mmol), NiBr2·diglyme (10 mol%), ligand L1 (15 mol%), picoline (or K3PO4), MgCl2, and nanopowder Zn (4.0 equiv.) in DMAc/THF was stirred at 25 °C for 48 h. Isolated yield. The ee was determined using HPLC with a chiral stationary phase. |
|---|
|
We next demonstrated the substrate spectrum with respect to the acyl donors. Both electron-rich and -deficient groups on the different positions of the aromatic ring of the benzoic anhydrides were tolerated, furnishing the desired α-amino ketones 47–53 in moderate to good efficiency (Table 3 (B)). Additionally, we wondered whether other acyl precursors are suitable for this asymmetric three-component reductive alkylacylation. Gratifyingly, 2-pyridyl esters66 were identified to be compatible with our protocol after subtle changes of the reaction conditions (K3PO4 instead of 2-picoline as a base, Table 3 (C)). Accordingly, a variety of 2-pyridyl esters were examined. A series of functional groups, including ether (54 and 55), thioether (56), ester (58), 2-naphthyl (59), benzofuran (60), and benzothiophene (61), were nicely accommodated. Besides aryl 2-pyridyl esters, an aliphatic acid-derived 2-pyridyl ester (62) also exhibited moderate reactivity.
To further demonstrate the synthetic utility of our protocol, a gram-scale (5 mmol scale) reaction was carried out. Product 4 was isolated in a synthetically useful yield (1.11 g) with excellent enantioselectivity (92% ee, Scheme 2A). Chemoselective reduction of the ketone carbonyl group afforded product 63 in good yield and high diastereoselectivity (Scheme 2B). The reduction-Mitsunobu reaction of 4 afforded a chiral oxazoline 64 bearing two chiral centers in high dr, which could be a potential chiral ligand. Reduction of the carbonyls in both the ketone and the amide moieties afforded the amino alcohol 65 in 7.5:1 dr. Condensation between 4 and hydroxyamine followed by a Beckmann rearrangement afforded amide 66 in good yield. In all cases, only slight, if any, erosion of the enantiopurity was observed.
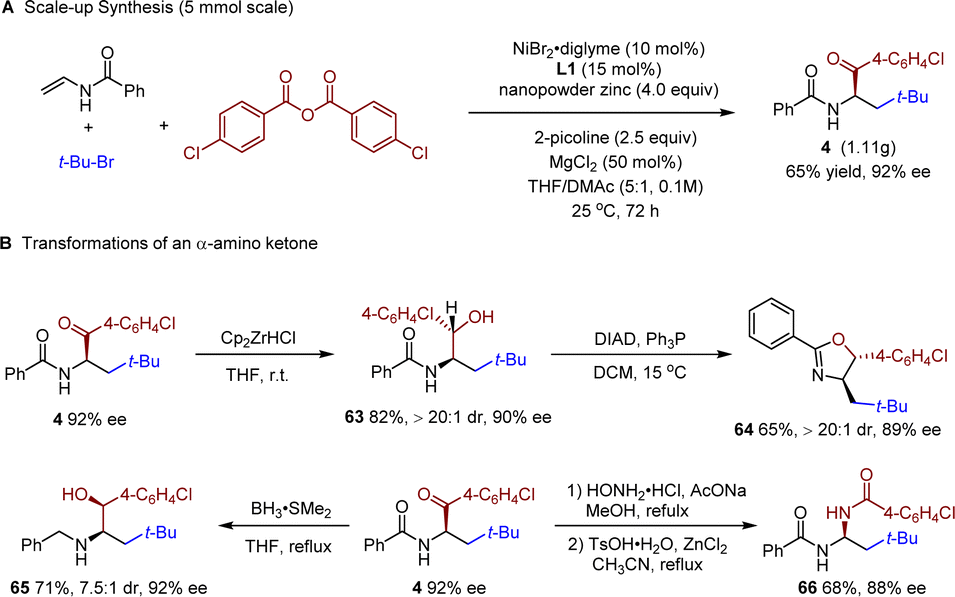 | ||
| Scheme 2 Gram-scale experiment and synthetic transformations. | ||
To shed light on the mechanism of this novel nickel-catalyzed asymmetric alkylacylation system, a series of experiments were conducted. The desired alkylacylation reaction was completely inhibited when a radical scavenger was added (Scheme 3A). Next, a radical-clock experiment using a α-cyclopropyl styrene 67 was conducted (Scheme 3B), and only the ring-opened product 68 was obtained through a sequential radical addition, ring opening, and acylation process, whereas the formation of the direct cross-coupled product 69 was not observed. This result indicated the formation of a tert-butyl radical which, upon addition to the alkene, delivers a new carbon radical that can be intercepted by the acyl-Ni species. The formation of radical species was further confirmed by an electron paramagnetic resonance (EPR) study using a spin-trapping agent phenyl tert-butyl nitrone (PBN, Scheme 3C). Additionally, a nickel(II) acyl complex 72 (ref. 83) was prepared to elucidate the nature of the active nickel species in the catalytic cycle. The stoichiometric reaction of Ni(II)-complex 72, N-vinylbenzamide 1a, and 2-bromo-2-methylpropane 2a in the absence of any zinc reductant gave the desired cross-coupled product 62 in 18% yield, whereas only traces of 62 were detected when a stoichiometric amount of nanopowder zinc was introduced (Scheme 3D). Taken together, these results suggest that the putative acyl-Ni(II) complex could be a productive intermediate in this catalytic cycle. No corresponding product 4 could be observed when employing stoichiometric Ni(cod)2 but lacking the zinc reductant, whereas catalytic amount Ni(cod)2 with stoichiometric zinc resulted in the formation of product 4 in 63% yield (Scheme 3E). These outcome suggests that the reduction of Ni(III) acyl species III to Ni(I) acyl intermediate IV is likely required in the current protocol.
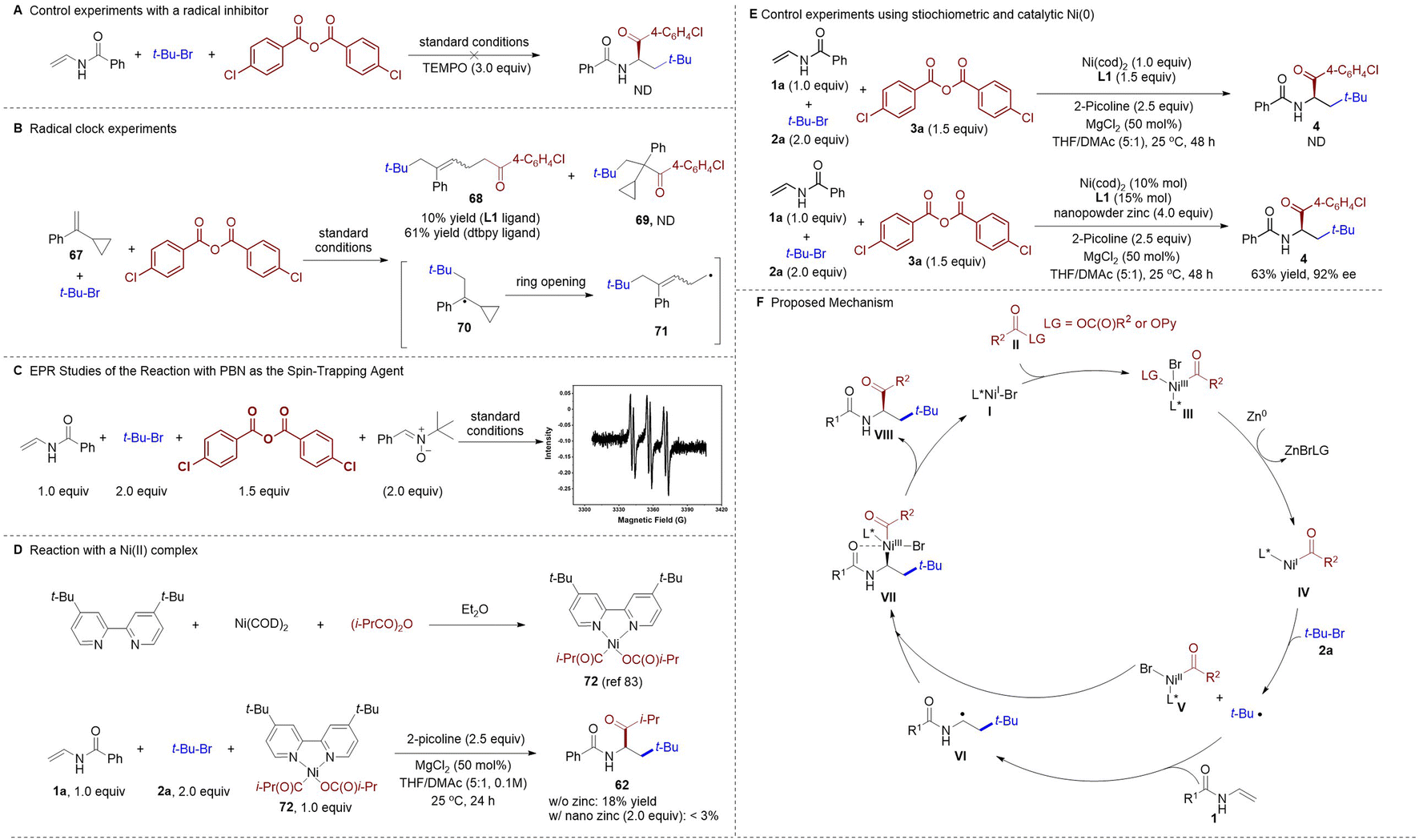 | ||
| Scheme 3 Mechanistic studies. | ||
On the basis of our mechanistic investigations and insight from prior studies,71,72,83,84 a plausible mechanism of this asymmetric three-component alkylacylation reaction is proposed (Scheme 3F). Initially, Ni(I) species I may be formed via reduction of the Ni(II) precatalyst. Oxidative addition of the Ni(I) species I to the anhydrides or 2-pyridyl esters II generates a Ni(III) intermediate III, which reduced by nanopowder zinc as facilitated by MgCl2 (ref. 83 and 84) to give Ni(I) intermediate IV. Then, the tertiary alkyl bromide 2a is reduced by a Ni(I) complex IV to give a tert-butyl radical together with a Ni(II) species V (reduction of 2a by nanopowder zinc to give the tertiary alkyl radical cannot be rule out now). Subsequent addition of the tert-butyl radical to the vinyl amides 1 affords a secondary alkyl radical VI. At this juncture, oxidative addition of the radical VI to Ni(II) V delivers Ni(III) species VII, which then undergoes reductive elimination to release the chiral α-amino ketone product VIII and regenerates the Ni(I) species I to complete the catalytic cycle. We anticipate that the chelation of the carbonyl group to the BiIM-ligated nickel center would be beneficial to control the stereoselectivity in the radical capture step (V + VI → VII).43–45,50,85
Conclusions
In conclusion, we present herein the first enantioselective, fully intermolecular three-component 1,2-alkylacylation of alkenes using tertiary alkyl bromides and an anhydride or a 2-pyridyl esters under reductive conditions. This mild and efficient protocol allows for the straightforward construction of structurally diverse enantioenriched α-amino ketones with good efficiency and excellent enantioselectivity from readily available electrophiles, avoiding the using of preformed air- or moisture-sensitive organometallic reagents. The amide group in the olefin substrate displays a vital role in enhancing regio- and enantioselectivity as well as the reactivity. Studies to further uncover the detailed mechanism of this transformation and expansion to other olefins and electrophiles are ongoing in our laboratory.Data availability
The authors declare that all data supporting the findings of this study, including experimental procedures and compound characterization, NMR, HPLC, and X-ray analyses, are available within the article and its ESI† or from the corresponding author. The crystallographic data used in this study are available in the Cambridge Crystallographic Database under accession codes CCDC 2356762 (33) and 2356761 (47).Author contributions
X. L. and J. X. conceived the concept and directed the project. J. X. and X. L. co-wrote the manuscript, analyzed the data, discussed the results, and commented on the manuscript. J. X., T. J., S. C., and M. P. conducted the experiments. All authors contributed to discussions.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22301171 and 22371175), and research fund from the SNNU.Notes and references
- R. G. Almquist, W.-R. Chao, M. E. Ellis and H. L. Johnson, J. Med. Chem., 1980, 23, 1392–1398 CrossRef CAS PubMed.
- D. M. Perrine, J. T. Ross, S. J. Nervi and R. H. Zimmerman, J. Chem. Educ., 2000, 77, 1479–1480 CrossRef CAS.
- A. T. Nchinda, K. Chibale, P. Redelinghuys and E. D. Sturrock, Bioorg. Med. Chem. Lett., 2006, 16, 4612–4615 CrossRef CAS PubMed.
- F. I. Carroll, et al. , J. Med. Chem., 2009, 52, 6768–6781 CrossRef CAS PubMed.
- A. M. R. Smith and K. K. Hii, Chem. Rev., 2010, 111, 1637–1656 CrossRef PubMed.
- F. Zhou, F.-M. Liao, J.-S. Yu and J. Zhou, Synthesis, 2014, 46, 2983–3003 CrossRef CAS.
- A. d. l. Torre, V. Tona and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 12416–12423 CrossRef PubMed.
- L. A. T. Allen, R.-C. Raclea, P. Natho and P. J. Parsons, Org. Biomol. Chem., 2021, 19, 498–513 RSC.
- F. J. Urban and V. J. Jasys, Org. Process Res. Dev., 2004, 8, 169–175 CrossRef CAS.
- T. L. Suyama and W. H. Gerwick, Org. Lett., 2006, 8, 4541–4543 CrossRef CAS PubMed.
- W. Li, et al. , J. Am. Chem. Soc., 2011, 133, 15268–15271 CrossRef CAS PubMed.
- T. Takeda and M. Terada, J. Am. Chem. Soc., 2013, 135, 15306–15309 CrossRef CAS PubMed.
- B. Xu, S. F. Zhu, X. D. Zuo, Z. C. Zhang and Q. L. Zhou, Angew. Chem., Int. Ed., 2014, 53, 3913–3916 CrossRef CAS PubMed.
- D. H. Miles, J. Guasch and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 7632–7635 CrossRef CAS PubMed.
- X. Yang and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 3205–3208 CrossRef CAS PubMed.
- R. A. Lamb, N. S. Aberle, N. T. Lucas, G. Lessene and B. C. Hawkins, Angew. Chem., Int. Ed., 2017, 56, 14663–14666 CrossRef CAS PubMed.
- E. M. Saied, T. L.-S. Le, T. Hornemann and C. Arenz, Bioorg. Med. Chem. Lett., 2018, 26, 4047–4057 CrossRef CAS PubMed.
- R. d. S. Gomes and E. J. Corey, J. Am. Chem. Soc., 2019, 141, 20058–20061 CrossRef PubMed.
- W. Guo, et al. , J. Am. Chem. Soc., 2020, 142, 14384–14390 CrossRef CAS PubMed.
- E. Gandolfo, X. Tang, S. R. Roy and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 16854–16858 CrossRef CAS PubMed.
- X. Shu, L. Huan, Q. Huang and H. Huo, J. Am. Chem. Soc., 2020, 142, 19058–19064 CrossRef CAS PubMed.
- Y. Gao and P. S. Baran, Angew. Chem., Int. Ed., 2023, 62, e202315203 CrossRef CAS PubMed.
- J. Chen and S. Zhu, J. Am. Chem. Soc., 2021, 143, 14089–14096 CrossRef CAS PubMed.
- A. H. Cherney, N. T. Kadunce and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 7442–7445 CrossRef CAS PubMed.
- K. E. Poremba, S. E. Dibrell and S. E. Reisman, ACS Catal., 2020, 10, 8237–8246 CrossRef CAS PubMed.
- L.-M. Chen and S. E. Reisman, Acc. Chem. Res., 2024, 57, 751–762 CrossRef CAS PubMed.
- E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598–6608 RSC.
- R. K. Dhungana, S. KC, P. Basnet and R. Giri, Chem. Rec., 2018, 18, 1314–1340 CrossRef CAS PubMed.
- R. Giri and S. KC, J. Org. Chem., 2018, 83, 3013–3022 CrossRef CAS PubMed.
- J. Lin, R. J. Song, M. Hu and J. H. Li, Chem. Rec., 2018, 19, 440–451 CrossRef PubMed.
- J.-S. Zhang, L. Liu, T. Chen and L.-B. Han, Chem.–Asian J., 2018, 13, 2277–22291 CrossRef CAS PubMed.
- S. O. Badir and G. A. Molander, Chem, 2020, 6, 1327–1339 CAS.
- J. Derosa, O. Apolinar, T. Kang, V. T. Tran and K. M. Engle, Chem. Sci., 2020, 11, 4287–4296 RSC.
- X. Qi and T. Diao, ACS Catal., 2020, 10, 8542–8556 CrossRef CAS PubMed.
- L. M. Wickham and R. Giri, Acc. Chem. Res., 2021, 54, 3415–3437 CrossRef CAS PubMed.
- S. Zhu, X. Zhao, H. Li and L. Chu, Chem. Soc. Rev., 2021, 50, 10836–10856 RSC.
- S. Duan, et al. , Chem. Sci., 2022, 13, 3740–3747 RSC.
- D. Wang and L. Ackermann, Chem. Sci., 2022, 13, 7256–7263 RSC.
- A. Maity and A. Studer, Chem. Sci., 2023, 14, 7675–7680 RSC.
- G. Tan, et al. , Chem. Sci., 2023, 14, 2447–2454 RSC.
- X.-X. Zhang, et al. , Chem. Sci., 2023, 14, 11170–11179 RSC.
- D. Anthony, Q. Lin, J. Baudet and T. Diao, Angew. Chem., Int. Ed., 2019, 58, 3198–3202 CrossRef CAS PubMed.
- L. Guo, et al. , J. Am. Chem. Soc., 2020, 20390–20399 CrossRef CAS PubMed.
- H. Y. Tu, et al. , J. Am. Chem. Soc., 2020, 142, 9604–9611 CrossRef CAS PubMed.
- X. Wei, W. Shu, A. García-Domínguez, E. Merino and C. Nevado, J. Am. Chem. Soc., 2020, 142, 13515–13522 CrossRef CAS PubMed.
- P. Qian, et al. , Nat. Commun., 2021, 12, 6613 CrossRef CAS PubMed.
- Y.-Z. Wang, et al. , J. Am. Chem. Soc., 2023, 145, 23910–23917 CrossRef CAS PubMed.
- X. Hu, I. Cheng-Sánchez, W. Kong, G. A. Molander and C. Nevado, Nat. Catal., 2024, 655–665 CrossRef PubMed.
- Y.-Z. Li, et al. , Nat. Commun., 2022, 13, 5539 CrossRef CAS PubMed.
- F. Wang, S. Pan, S. Zhu and L. Chu, ACS Catal., 2022, 12, 9779–9789 CrossRef CAS.
- X. Li, et al. , Chem, 2023, 9, 154–169 CAS.
- Z. Dong, et al. , ACS Catal., 2024, 14, 4395–4406 CrossRef CAS.
- X. Y. Du, I. Cheng-Sánchez and C. Nevado, J. Am. Chem. Soc., 2023, 145, 12532–12540 CrossRef CAS PubMed.
- M.-S. Liu and W. Shu, JACS Au, 2023, 3, 1321–1327 CrossRef CAS PubMed.
- H. Huang, Y. M. Lin and L. Gong, Chem. Rec., 2023, 23, e202300275 CrossRef CAS PubMed.
- Z. Dong, L. Song and L.-A. Chen, ChemCatChem, 2023, 15, 4395–4406 Search PubMed.
- P. Fan, Y. Lan, C. Zhang and C. Wang, J. Am. Chem. Soc., 2020, 142, 2180–2186 CrossRef CAS PubMed.
- Y. Lan and C. Wang, Commun. Chem., 2020, 3, 45 CrossRef CAS PubMed.
- Y. Li, et al. , J. Am. Chem. Soc., 2020, 142, 19844–19849 CrossRef CAS PubMed.
- A. D. Marchese, et al. , ACS Catal., 2020, 10, 4780–4785 CrossRef CAS.
- X. Wu, J. Qu and Y. Chen, J. Am. Chem. Soc., 2020, 142, 15654–15660 CrossRef CAS PubMed.
- J. Wu and C. Wang, Org. Lett., 2021, 23, 6407–6411 CrossRef CAS PubMed.
- X. Wu, et al. , Angew. Chem., Int. Ed., 2022, 61, e202111598 CrossRef CAS PubMed.
- X. Wu, et al. , Angew. Chem., Int. Ed., 2022, 61, e202207536 CrossRef CAS PubMed.
- C. Zhang, X. Wu, T. Xia, J. Qu and Y. Chen, Nat. Commun., 2022, 13, 5964 CrossRef PubMed.
- F. He, et al. , CCS Chem., 2023, 5, 341–349 CrossRef CAS.
- Y. Jin, P. Fan and C. Wang, CCS Chem., 2022, 4, 1510–1518 CrossRef CAS.
- R. Wang and C. Wang, Chem. Sci., 2023, 14, 6449–6456 RSC.
- X. Zhao, et al. , Nat. Commun., 2018, 9, 3488 CrossRef PubMed.
- A. A. Kadam, T. L. Metz, Y. Qian and L. M. Stanley, ACS Catal., 2019, 9, 5651–5656 CrossRef CAS.
- L. Wang and C. Wang, Org. Lett., 2020, 22, 8829–8835 CrossRef CAS PubMed.
- X. Xi, Y. Chen and W. Yuan, Org. Lett., 2022, 24, 3938–3943 CrossRef CAS PubMed.
- K. Oguma, M. Miura, T. Satoh and M. Nomura, J. Organomet. Chem., 2002, 648, 297–301 CrossRef CAS.
- T. Ishii, K. Ota, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 14073–14077 CrossRef CAS PubMed.
- J. L. Li, et al. , Angew. Chem., Int. Ed., 2019, 59, 1863–1870 CrossRef PubMed.
- M. Zhou, H. Y. Zhao, S. Zhang, Y. Zhang and X. Zhang, J. Am. Chem. Soc., 2020, 142, 18191–18199 CrossRef CAS PubMed.
- C. Wang, N. Liu, X. Wu, J. Qu and Y. Chen, Chin. J. Chem., 2023, 42, 599–604 CrossRef.
- M. P. Sibi, S. Manyem and J. Zimmerman, Chem. Rev., 2003, 103, 3263–3295 CrossRef CAS PubMed.
- A. Lipp, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2020, 60, 1714–1726 CrossRef PubMed.
- R. S. J. Proctor, A. C. Colgan and R. J. Phipps, Nat. Chem., 2020, 12, 990–1004 CrossRef PubMed.
- H. Liang, et al. , Chem. Sci., 2022, 13, 1088–1094 RSC.
- D. Sun, X. Tao, G. Ma, J. Wang and Y. Chen, Chem. Sci., 2022, 13, 8365–8370 RSC.
- C. Zhao, X. Jia, X. Wang and H. Gong, J. Am. Chem. Soc., 2014, 136, 17645–17651 CrossRef CAS PubMed.
- S. Ni, et al. , J. Am. Chem. Soc., 2019, 141, 6726–6739 CrossRef CAS PubMed.
- Y. He, H. Song, J. Chen and S. Zhu, Nat. Commun., 2021, 12, 638 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2356761 and 2356762. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04561k |
| This journal is © The Royal Society of Chemistry 2024 |