
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-emissive luminescence in OIHMH single crystals: tunable red-green emissions via Mn2+ doping and theoretical insights†
Qianrong
Jin
 a,
Rui
Wu
a,
Yuexiao
Pan
*a,
Yihong
Ding
a,
Hongzhou
Lian
b,
Jun
Lin
*b and
Liyi
Li
*c
a,
Rui
Wu
a,
Yuexiao
Pan
*a,
Yihong
Ding
a,
Hongzhou
Lian
b,
Jun
Lin
*b and
Liyi
Li
*c
aKey Laboratory of Carbon Materials of Zhejiang Province, College of Chemistry and Materials Engineering, Wenzhou University, Wenzhou 325035, P.R. China. E-mail: yxpan@wzu.com
bState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: jlin@ciac.ac.cn
cResearch Center for Eco-environmental Engineering, Dongguan University of Technology, Dongguan, 523808, P. R. China. E-mail: liliyi@dgut.edu.cn
First published on 18th September 2024
Abstract
The burgeoning demand for materials with tunable photoluminescence properties for applications has necessitated the exploration of novel luminescent materials. This study presents the synthesis and characterization of a novel 0D organic–inorganic hybrid metal halide (OIHMH) single crystal, C6H14N2CdBr4, which exhibits intriguing luminescent properties upon Mn2+ doping. The introduction of single Mn2+ ions results in a dual-emission, with the unexpected emergence of a red emission peak at 627 nm, in addition to the conventional tetrahedral green emission at 525 nm. This dual-emission is attributed to the distinct inter-tetrahedral distances within the crystal structure of C6H14N2CdBr4 with varying distances between [CdBr4]2− tetrahedra, influencing the spatial distribution and interaction of the [MnBr4]2− tetrahedra. Electron paramagnetic resonance (EPR) spectroscopy and theoretical calculations reveal the 627 nm and 525 nm emissions are attributed to the d–d transition of magnetic coupled Mn2+–Mn2+ pairs and isolated Mn2+ ions, respectively. This study not only advances the understanding of the luminescence mechanisms of Mn2+ but also paves the way for the development of tunable luminescent materials.
1 Introduction
Organic–inorganic hybrid metal halides (OIHMHs) are renowned for their ability to integrate the superior charge transport properties of organic materials with the robust structural characteristics of their inorganic counterparts, giving rise to a class of materials with tunable bandgaps and high photoluminescence quantum yields (PLQYs).1–3 A common strategy in the synthesis of OIHMHs involves the substitution of A-site cations in the perovskite structure with monovalent organic ammonium ions, such as CH3NH3+ and CH(NH2)2+. This substitution often leads to the formation of 0D structures where larger organic molecules effectively isolate central ions, thereby enhancing the electron–phonon coupling effect feature which is highly desirable for luminescent materials.4–7Dual-emission materials are particularly intriguing because they can emit light in two distinct color bands, which can be tuned independently for various applications. Researchers have explored the incorporation of different metal ions, the use of co-doping strategies, to fine-tune the emission spectra.8–11 A dual-band tunable multicolor luminescence system has been developed by co-doping Bi3+ and Te4+ into a 0D hybrid perovskite, (C4H12N)2HfCl6.8 The integration of Mn2+ and Er3+ ions through codoping within Cs2AgInCl6 endows the material with dual-band emission and innovative anti-counterfeiting features.10 However, the identification of materials with dual-emission properties stemming from single-ion doping is relatively rare.
With an electron configuration of [Ar] 3d5, the Mn2+ ions possess five 3d electrons that occupy the d orbitals in various spin and orbital momentum states. The interplay between electron spin and orbital angular momentum results in a multitude of electronic states, which, under the influence of a crystal field, give rise to energy level splitting. This splitting is pivotal in determining the absorption and emission spectral characteristics of Mn2+ ions.12–20 As a luminescent center, Mn2+ ions exhibit emission properties that are highly dependent on the crystal field environment in which they reside. Traditionally, Mn2+ ions in a tetrahedral coordination emit green light, while in an octahedral coordination, they emit red light.13–15 For example, the Mn2+ doped {TETA[Pb2Br6]}n 1 and {TETA[Pb2Cl6]}n 2 show that the emissions vary from 551 to 615 nm and from 447 to 660 nm, respectively.13 A novel pseudo-2D layered (MTP)2MnBr4 was reported, which exhibits intense green emission under UV/blue light or X-ray excitation with a near-unity PLQY.15 It can be seen that the doping strategies of divalent Mn2+ ions and synthesis of the manganese halides have expanded the luminescence performance of OIHMHs to different degrees.
In the context of tetrahedral structures, the close proximity of Mn2+ ions to their neighbors can lead to the formation of exchange-coupled Mn2+–Mn2+ dimers or pairs, which is a phenomenon that can result in red light emission.16–20 This occurrence challenges the conventional understanding of Mn2+ luminescence, which typically associates red emission with octahedral coordination. For instance, the innovative work by Song et al. achieves a dual-wavelength (530/650 nm) in a tetrahedral environment via tuning the Mn2+ concentration, which is beneficial from the Mn2+–Mn2+ super-exchange interactions.17 Red lights have also been observed in tetrahedral structures due to the magnetic coupling effect in Mn2+–Mn2+ pairs in C4H14N2MnBr4 metal halide single crystals with only tetrahedra, which is attributed to magnetic interactions.18
Our approach to synthesizing a novel OIHMH C6H14N2CdBr4 crystal involved a divalent cation occupying the A-site. This synthetic strategy yielded a material with an intriguing dual-emission characteristic, which is attributed to the distinct inter-tetrahedral distances within the crystal structure of C6H14N2CdBr4. These varying distances at 9.87 Å, 7.78 Å, and 6.90 Å influence the spatial distribution and interaction of the [MnBr4]2− tetrahedra, thereby providing a unique platform for Mn2+ doping to produce a diverse array of luminescent properties.
2 Experimental
2.1. Chemicals
The reagents employed in this study encompassed high-purity cadmium oxide (CdO, 99.99%), manganous bromide (MnBr2, 99.99%), 1,4-diazabicyclo[2.2.2]octane (C6H12N2, 98%), hydrobromic acid (HBr, 37 wt%), and ethanol (96%). All these materials were procured from Aladdin Chemistry Co. Ltd (China) and were utilized in their received state, precluding the necessity for additional purification processes.2.2. Fabrication of C6H14N2CdBr4 and C6H14N2Cd1−xMnxBr4 crystalline structures
A series of C6H14N2CdBr4 and C6H14N2Cd1−xMnxBr4 crystalline entities were synthesized via a conventional hydrothermal synthesis approach. For the synthesis of C6H14N2CdBr4, 1 mmol of CdO and 2 mmol of C6H12N2 were mixed in a 4 mL HBr solution in a 25 mL stainless steel autoclave. Subsequently, the mixture was subjected to thermal treatment at 180 °C for a duration of 12 hours, followed by a controlled cooling to ambient temperature. Then the crystals were washed three times with ethanol and dried at 60 °C for 12 hours. For the synthesis of the C6H14N2Cd1−xMnxBr4 variants, the following steps were involved: 2 mmol of C6H12N2, 1 − x mmol of CdO, and x mmol of MnBr2 (x varied between 0% and 85%) were mixed with 4 mL HBr in a 25 mL Teflon liner. The resulting solution was subjected to hydrothermal treatment at 180 °C for 12 hours. After completion, the autoclave was slowly cooled to room temperature at a decremental rate of 10 °C per hour. Subsequently, the crystalline products were isolated subsequent to the cooling process, with purification achieved through repetitive washing with ethanol, followed by desiccation in an oven maintained at 60 °C.2.3. Characterization techniques
The crystallographic data for single crystals were obtained using a state-of-the-art SMART APE II DUO X-ray four-circle single-crystal diffractometer, equipped with a CCD detector, a graphite monochromator, and a Cu Kα radiation source. Powder X-ray diffraction (XRD) patterns were captured on a Bruker D8 Advance XRD apparatus, utilizing Cu Kα radiation (λ = 0.15418 nm) and scanning at a rate of 10° per minute over a 2θ range spanning from 10 to 60°. The chemical compositional analysis of the particle surfaces was conducted using X-ray photoelectron spectroscopy (XPS) on an ESCALAB 250Xi VersaProbe spectrometer, employing Al Kα radiation (hν = 486.6 eV) as the monochromatic X-ray source. Photoluminescence (PL) spectra, PL quantum yield (PLQY), and temperature-dependent PL spectra were recorded using a HORIBA Jobin Yvon FluoroMax-4 fluorescence spectrometer, which was equipped with a 150 W xenon lamp serving as the excitation source. Thermogravimetric analysis (TGA/SDTA851) of the samples was performed from room temperature to 900 °C at a heating rate of 10 °C min−1. Electron paramagnetic resonance (EPR) spectra were acquired on a Bruker A300 spectrometer operating at X-band (9.44 GHz) at room temperature.2.4. Computational methodology
A depiction of the computational framework was utilized in this study, illustrating the application of ab initio simulations grounded in spin-polarized Density Functional Theory (DFT), as implemented in the Vienna Ab initio Simulation Package (VASP).21 The electronic wave functions were delineated using a plane-wave basis set, with a defined energy cutoff of 350 eV. The Brillouin zone was meticulously sampled with a k-mesh, focusing exclusively on the Γ point. The unit cell configurations for C6H14N2CdBr4 and its Mn2+-doped variants were established with a 3 × 2 × 2 k-point grid, whereas the supercell for Mn2+-doped C6H14N2CdBr4 was delineated with a 1 × 1 × 2 k-point grid, accommodating a total of 144 atoms for the comprehensive analysis. The C6H14N2Cd1−xMnxBr4 system, with varying Mn2+ concentrations (x = 0.50, 0.25, and 0.125), was crafted through the simulation of Cd2+ atom substitution with Mn2+ atoms. The exchange–correlation potential was approximated using the Generalized Gradient Approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional, and electron–ion interactions were delineated through projector-augmented wave (PAW) pseudopotentials.22 The PBE+U methodology was employed to refine the DFT calculations.23,24 The magnetic moments were initialized in a high-spin configuration, guided by the MAGMOM directive.25 A stringent convergence criterion for the maximal interatomic force was set at 0.03 eV Å−1. The refined structures were subjected to Crystal Orbital Hamiltonian Population (COHP) analysis, facilitated by VASP in tandem with LOBSTER 4.1.0, with basis functions specified according to the KOGA basis set. The computational study comprehensively included pure C6H14N2CdBr4 and its Mn2+ doped derivatives, ensuring absolute charge spilling was maintained below 1%, thereby substantiating the precision of the computational methodology employed.3 Results and discussion
3.1. The structure and composition confirmation of C6H14N2CdBr4 based crystals
The crystal structure, elucidated by single-crystal XRD of the 0D single crystal C6H14N2CdBr4, revealed a divalent cation at the A-site, which is relatively rare and possesses a high polarity, making it a valuable addition to the perovskite family. Fig. 1a presents the structural diagram of the 0D single crystal, where each Cd2+ ion is coordinated with four Br− ions to form [CdBr4]2− tetrahedral units. These units are surrounded by [C6H14N2]2+ organic cations, creating the characteristic 0D structure. Analysis of the single-crystal results indicates that the C6H14N2CdBr4 crystal belongs to the Pnma space group, with unit cell parameters of a = 13.8382(7) Å, b = 8.5330(9) Å, and c = 11.8654(8) Å. Fig. 1b provides detailed distances between inter-tetrahedral [CdBr4]2− within the crystal structure, highlighting three distinct distances: d1 = 9.87 Å, d2 = 7.78 Å, and d3 = 6.90 Å. The observed inter-tetrahedral distances are of particular interest, as the shortest distance is notably close to the critical distance of 6.6 Å reported for ferromagnetic Mn2+–Mn2+ pairs, suggesting a potential magnetic interaction that may influence the PL properties of Mn2+.18,20,26 Additionally, the ball-and-stick model of the [CdBr4]2− unit illustrates Cd–Br bond lengths ranging from 2.612 to 2.643 Å, which is consistent with expected values for such bonds. The XRD pattern corroborates the single-crystal results, confirming the purity of the synthesized sample (Fig. 1c). | ||
| Fig. 1 (a) Structure diagram of the 0D single crystal C6H14N2CdBr4. (b) The detailed diagram of the different distances between the [CdBr4]2− tetrahedra and ball-and-stick diagram of an individual [CdBr4]2− unit and organic cation. (c) The experimental and simulated XRD patterns of C6H14N2CdBr4. (d) XPS full spectrum of C6H14N2CdBr4:Mn2+. | ||
The XPS survey spectrum of C6H14N2CdBr4:Mn2+ revealed the presence of C, N, Cd, Mn, and Br elements (Fig. 1d). The absence of hydrogen in the XPS spectrum is attributed to its low atomic weight, which typically precludes detection by this technique. Fig. S1† presents the high-resolution XPS spectra of the Mn 2p, Cd 3d, and Br 3d levels. Notably, the Mn 2p spectrum displays two distinct peaks corresponding to the Mn 2P1/2 and Mn 2P3/2 spin–orbit components at binding energies of 653 eV and 640.9 eV, respectively. These values are characteristic of Mn2+ ions, thereby substantiating the successful incorporation of Mn2+ into the host lattice without altering its oxidation state.
The XRD patterns of the samples doped with varying concentrations of Mn2+ were found to be in good agreement with the reference pattern of the pristine C6H14N2CdBr4, indicating the formation of single-phase materials (Fig. S2†). The absence of impurity peaks in the XRD spectra, even at the highest doping concentration of 85%, underscores the remarkable tolerance of the C6H14N2CdBr4 crystal lattice to accommodate Mn2+ ions without introducing structural impurities. Moreover, the observation of a shift in the diffraction peaks towards higher angles with increasing Mn2+ concentration is indicative of a lattice contraction. This phenomenon can be attributed to the smaller ionic radius of Mn2+ (r = 0.80 Å) compared to that of Cd2+ (r = 0.97 Å).27,28 The successful integration of Mn2+ ions into the crystal lattice without disrupting the crystallographic symmetry suggests that the material possesses a high degree of structural stability and tolerance, which is critical for its potential applications in optoelectronics and other advanced technologies. The high tolerance for Mn2+ substitution observed in the C6H14N2CdBr4 perovskite is a significant finding, as it suggests that this material system can be finely tuned by doping to achieve desired properties.
3.2. The dual-emissions of single Mn2+ doped C6H14N2CdBr4 and its mechanism
The PLE spectra monitored at 525 nm (Fig. 2a) and 627 nm (Fig. 2b) indicate that both the green and red emissions originate from the d–d transitions of Mn2+.16,17 The presence of multiple excitation peaks at 277, 363, and 455 nm is attributed to the electronic transitions of Mn2+ from 6A1(S) to 4A2(F), 4E(D), and 4T2(G) states, respectively.13,15 As the Mn2+ doping concentration increases, the intensities of these excitation peaks augment, reaching a maximum at a doping level of 20% (x = 0.20) for the green emission and 65% (x = 0.65) for the red emission, beyond which a decline is observed. This suggests an optimal doping concentration for achieving peak luminescence, above which concentration quenching effects may come into play. | ||
| Fig. 2 (a and b) PLE, (c) PL spectra, (d and e) decay curves, and (f) CIE color coordinates of C6H14N2Cd1−xMnxBr4 crystals changing with the x value, respectively (the insets are the corresponding photographs of the Mn2+-doped samples under a 365 nm UV lamp). | ||
The PL spectra further elucidate the emission behavior of the C6H14N2Cd1−xMnxBr4 crystals under 363 nm excitation (Fig. 2c). At low Mn2+ doping concentration (x = 0.03), the samples predominantly exhibit green emission at 525 nm, with a negligible red emission at 627 nm. As the doping concentration increases, both the green and red emission intensities rise, with the green emission peaking at a doping concentration of 20% (x = 0.20). Beyond this concentration, while the green emission intensity diminishes, the red emission continues to intensify, reaching its maximum at a doping concentration of 65% (x = 0.65), after which it declines at 85% (x = 0.85).
In the context of the C6H14N2CdBr4 crystal structure, which only contains [CdBr4]2− tetrahedral units, the introduction of Mn2+ to replace Cd2+ results in the formation of [MnBr4]2− tetrahedra. The green emission at 525 nm is ascribed to the typical d–d transition of Mn2+ under a low crystal field, indicative of tetrahedral coordination. However, the origin of the red emission at 627 nm is less straightforward, given the uniform tetrahedral environment. The crystal structure analysis reveals three distinct inter-tetrahedral distances, with the shortest being 6.90 Å, which approaches the critical distance for magnetic coupling in prior studies.18,26 At high doping concentrations, such as 65%, where the [MnBr4]2− tetrahedra are in close proximity, magnetic coupling between adjacent Mn2+ ions could result in a strong crystal field, leading to the observed strong red emission.19,20
The PL spectra of the C6H14N2Cd1−xMnxBr4 samples, as depicted in Fig. S3 and S4,† under 277 and 455 nm excitation, respectively, exhibit identical spectral shapes and positions aside from variations in intensity. This uniformity in spectral characteristics, excluding intensity, suggests that the two emission peaks originate from the same emission center. Notably, the maximum emission at 525 nm for green light and at 627 nm for red light is observed at x = 0.20 and x = 0.65, respectively, under the two different excitation wavelengths. This reveals an increase in PLQY with the rise in doping concentration, reaching a maximum value of 13% (Fig. S5†).
Specifically, the lifetime at 525 nm decreases from 9.24 ms to 4.90 ms, while at 627 nm, it diminishes from 4.28 ms to 1.61 ms with increasing Mn2+ doping concentration from 3% to 65% in Fig. 2d and e. This reduction in lifetime is attributed to an increase in non-radiative recombination processes as the doping concentration escalates. Furthermore, the faster decay observed at 627 nm compared to 525 nm is indicative of the magnetic coupling effect between Mn2+ ions, which results in a shorter luminescence lifetime for Mn2+–Mn2+ pairs than for isolated Mn2+ ions.20,29 The CIE color coordinates in Fig. 2f illustrate the tunability of the emission color from yellow-green to orange-red as x increases. The coordinates shift from (0.36, 0.58) to (0.49, 0.47), confirming the successful manipulation of the luminescence color through doping concentration variation. The insets in Fig. 2f provide visual representations of the Mn2+-doped samples under 365 nm UV illumination, further corroborating the color tunability. Thus, the systematic study of the photoluminescence properties of C6H14N2Cd1−xMnxBr4 crystals as a function of Mn2+ doping has unveiled a complex interplay between doping concentration, crystal field strength, and magnetic coupling effects.
EPR measurements were conducted on crystals with 20% (x = 0.20) and 65% (x = 0.65) Mn2+ doping concentrations to further substantiate the magnetic coupling effect responsible for the red emission at 627 nm (Fig. 3a). At a doping level of 20% (x = 0.20), six clear peaks are observed, indicative of the high-spin Mn2+ ions with five unpaired electrons, which confers EPR activity. The hyperfine splitting into six lines is attributed to the nuclear spin I = 5/2 of the Mn2+ ions. The g-value of approximately 2.01 is noted, which is close to that of a free electron.30 In contrast, at a doping concentration of 65% (x = 0.65), a continuous broad dipole peak replaces the six hyperfine splitting patterns of the individual Mn2+ ions, suggesting the presence of weak ferromagnetic interactions between adjacent Mn2+ ions, indicative of Mn2+–Mn2+ pair interactions. Based on the above findings, a luminescence mechanism has been proposed, as depicted in Fig. 3b. The green emission at 525 nm is attributed to the 4T1-6A1 transition of isolated Mn2+ ions, while the red emission at 627 nm is ascribed to the magnetic coupling effect of Mn2+–Mn2+ pairs. When the concentration of dopants is excess 50% (x = 0.50) within a crystal, adjacent Mn2+ ions may couple magnetically. This results in an antiferromagnetic arrangement of the Mn2+ ion ground states, splitting the 6A1 state into six states (S = 0 to 5) and the 4T1 excited state into four states (S = 1 to 4). Transitions between split energy levels with the same S, one from 4T1 and the other from 6A1, are partially allowed.19,20,29
 | ||
| Fig. 3 (a) EPR spectra of Mn2+-doped C6H14N2CdBr4 with different dopant concentrations. (b) Schematic illustration of the energy levels and optical transitions in the tetrahedrally coordinated isolated Mn2+ ions and the Mn2+–Mn2+ pairs. | ||
3.3. Insights into the electronic properties of Mn2+-doped C6H14N2CdBr4
The electronic properties of Mn2+-doped C6H14N2CdBr4 single crystals have been investigated through DFT calculations to gain a deeper understanding of their characteristics. Fig. S6† presents the charge density distribution, electronic band structure, and DOS of the undoped matrix C6H14N2CdBr4. The calculated electronic band structure reveals a bandgap of 3.70 eV for the matrix. The charge density distribution and DOS indicate that the VBM (valence band maximum) is primarily contributed by Cd d, Cd p, and Br p orbitals, while the CBM (conduction band maximum) is mainly contributed by Cd s and Br p orbitals. For the Mn2+ ion doped crystals, the electronic structures of the crystals at different Mn2+ ion concentrations, x = 0.50, 0.125, and 0.25, have been calculated and analyzed. At a concentration of x = 0.50, there are three distances between adjacent [MnBr4]2− tetrahedral units, and the focus is on the farthest (9.87 Å) and closest (6.90 Å) conditions, examining the charge distribution of Mn d electrons in the spin-up and spin-down states.Fig. 4 illustrates the charge density distribution, electronic band structure, and DOS calculated by DFT for the crystal at a theoretical doping concentration of x = 0.50, where Mn2+–Mn2+ distances of 6.90 Å and 9.87 Å are considered. The use of spin-polarized magnetism provides a better description of the interactions between Mn 3d electrons. The presence of magnetism in Mn results in different charge distributions and DOS for spin-down and spin-up states. The CBM in both cases exhibits a moderate curvature, while the VBM shows an almost flat band, suggesting weak inter-motif coupling in the 0D metal halide C6H14N2Cd0.50Mn0.50Br4 single crystals. Fig. 4a and b correspond to the spin-up state with an Mn2+–Mn2+ distance of 6.90 Å, showing the charge density distribution, electronic band structure, and DOS. Fig. 4a indicates that the VBM charge is distributed on Mn2+ and nearby Br−, with no charge distribution on [CdBr4]2−. Conversely, the CBM charge has some distribution on [CdBr4]2−, but it is primarily concentrated on Mn2+, with Br− having negligible charge distribution. As shown in Fig. 4b, the calculated bandgap value is 3.69 eV, which is slightly reduced compared to the matrix bandgap. This decrease is attributed to lattice distortion caused by Mn2+ doping, leading to the formation of local energy levels and enhanced electron correlation effects that influence the electronic band structure and, consequently, reduce the bandgap. The DOS distribution shows that the VBM is mainly contributed by Cd p, Mn d, and Br p orbitals, while the CBM is contributed by Cd s and Mn d orbitals.
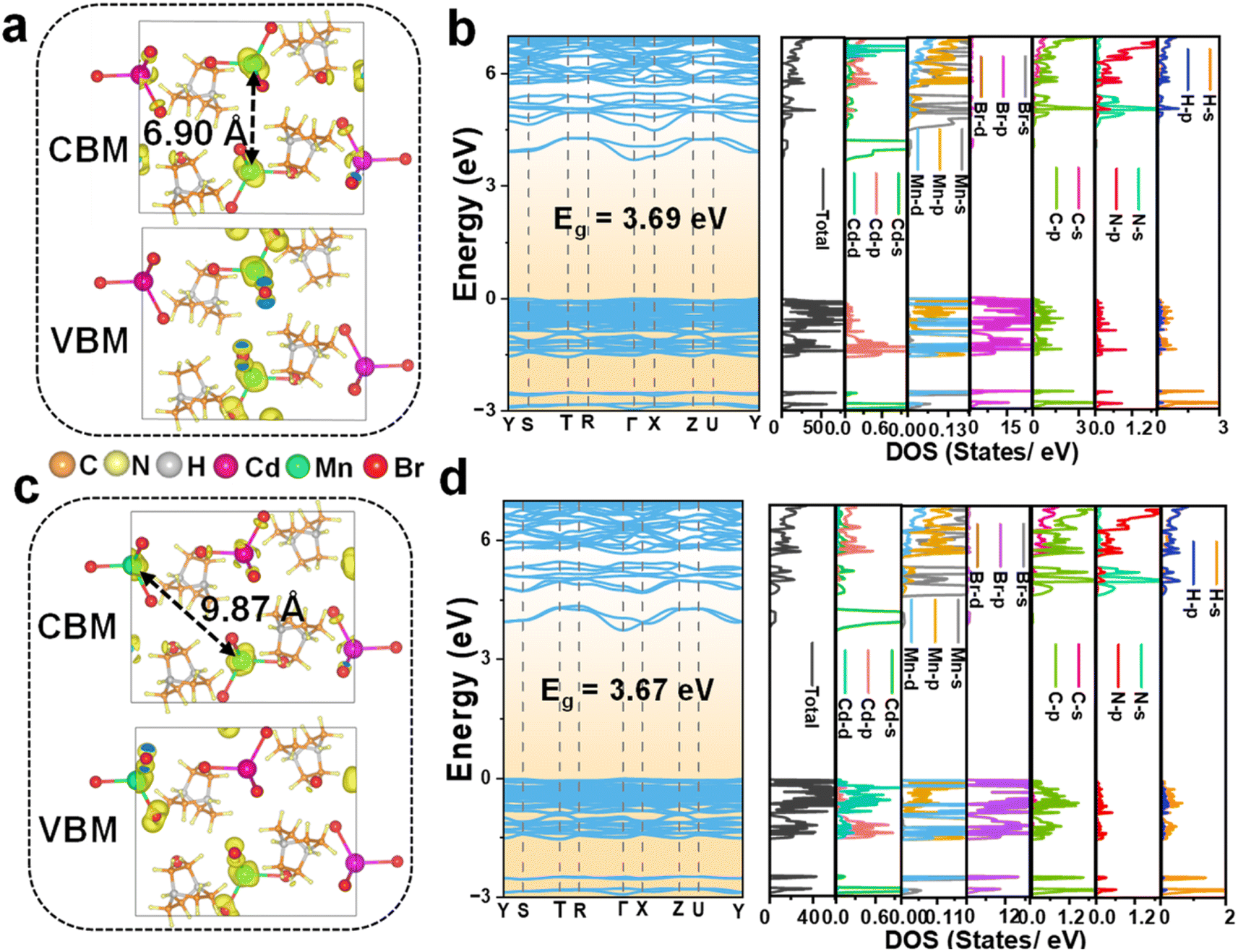 | ||
| Fig. 4 Charge density distribution of the CBM and VBM, and calculated electronic band structure and partial density of states of C6H14N2Cd0.50Mn0.50Br4 with different Mn2+–Mn2+ distances at (a and b) 6.90 Å and (c and d) 9.87 Å. | ||
The charge distribution and DOS for the spin-down configuration at a Mn2+–Mn2+ distance of 6.90 Å, as depicted in Fig. S7,† reveal distinct characteristics compared to the spin-up counterpart. Notably, the VBM charge is predominantly localized on the Br− ions, with no significant distribution observed on Mn2+ and Cd2+ ions. Conversely, the CBM charge is concentrated on both Br− and Cd2+ ions. This distribution differs markedly from that observed in the spin-up state, where the VBM and CBM charges are localized on Mn2+ and nearby Br− ions and Cd2+ ions, respectively. The VBM in the spin-down state is primarily composed of contributions from Cd d, Mn d, and Br p orbitals, while the CBM is characterized by contributions from Br p, Cd s, and Mn d orbitals.
Further insights are provided by the analysis of the spin-up configuration at a greater Mn2+–Mn2+ distance of 9.87 Å, as shown in Fig. 4c and d. The charge distribution at this distance is strikingly similar to that at 6.90 Å, with VBM charge localized on Mn2+ and nearby Br− ions, with negligible distribution on [CdBr4]2−. The CBM charge, while present on [CdBr4]2−, is predominantly concentrated on Mn2+ ions with minimal distribution on Br−. The DOS at 9.87 Å exhibits a slight variation from the 6.90 Å one, with the VBM and CBM being contributed by Cd d, Mn d, and Br p orbitals, and Cd s and Mn d orbitals, respectively. The calculated bandgap value of 3.67 eV at this distance is consistent with the trend of reduced bandgap values compared to the pristine matrix, suggesting the influence of doping on the electronic structure. The spin-down charge distribution and DOS at the 9.87 Å distance, presented in Fig. S8,† are similar to those observed at 6.90 Å.
For x = 0.125, the VBM charge is predominantly localized on the [MnBr4]2− units, with negligible distribution on [CdBr4]2− (Fig. 5a and b). Conversely, the CBM charge is focused on [CdBr4]2−, with minimal distribution on [MnBr4]2−. The calculated bandgap for this doping level is 3.64 eV, which is slightly narrower than that of the undoped matrix. The DOS for the spin-up configuration indicates contributions to the VBM from Cd d, Mn d, and Br p orbitals, while the CBM is influenced by Cd s and Br p orbitals. The spin-down configuration reveals a VBM charge concentration on Br−, with a minor distribution on [MnBr4]2− for the CBM (Fig. S9†). The VBM in the spin-down state is mainly composed of Cd d and Br p orbitals, while the CBM is contributed by Br p, Cd s, and Mn d orbitals. The comparison between spin-up and spin-down configurations suggests that the spin-up state significantly contributes to luminescence due to the concentration of charge on the [MnBr4]2− tetrahedra, facilitating d–d transition emission from Mn2+ ions.
 | ||
| Fig. 5 Charge density distribution of the CBM and VBM, and calculated electronic band structure and partial density of states of (a and b) C6H14N2Cd0.875Mn0.125Br4 and (c and d) C6H14N2Cd0.75Mn0.25Br4. | ||
At a 25% (x = 0.25) Mn2+ doping concentration, the VBM charge distribution remains focused on the [MnBr4]2− units, with an interesting observation of charge distribution in the organic component, particularly on nitrogen atoms (Fig. 5c and d). The CBM charge distribution is similar to that of 12.5% (x = 0.125) doping, being concentrated on [CdBr4]2− with less distribution on [MnBr4]2−. The bandgap value of 3.66 eV for this doping level is consistent with the trend of reduced bandgap values observed with increasing Mn2+ doping. The DOS for the spin-up configuration at this doping level is similar to that of the 12.5% (x = 0.125) doping scenario, with the VBM and CBM influenced by the same orbitals. The spin-down configuration (Fig. S10†) exhibits a VBM charge concentration on Br− and a CBM charge concentration on [CdBr4]2−, with a partial distribution on [MnBr4]2−. The orbital contributions to the VBM and CBM in the spin-down state are analogous to those at the 12.5% (x = 0.125) doping level. Thus, the DFT calculations provide insights into the electronic structure and the effects of Mn2+ doping on the bandgap and charge distribution in C6H14N2CdBr4 single crystals. The findings elucidate the role of inter-motif coupling and the impact of Mn2+-doping on the electronic properties, which are crucial for understanding the luminescent behavior of Mn2+.
3.4. Investigation on the thermal stability of Mn2+-doped C6H14N2CdBr4
The investigation into the temperature-dependent luminescent properties of C6H14N2Cd1−xMnxBr4 provides valuable insights into the performance under varying thermal conditions. The emission spectra of the C6H14N2Cd0.50Mn0.50Br4, in particular, exhibit intriguing behavior across the 298–453 K temperature range (Fig. 6a and b). As the temperature increases, the 525 nm (peak 1) emission intensifies, while the 627 nm (peak 2) emission diminishes, with the latter almost vanishing at 453 K, leaving the green emission peak dominant. This observation contradicts the typical luminescence quenching effect observed with increasing temperature. Instead, the crystal continues to emit a bright green light even at elevated temperatures. This phenomenon can be attributed to lattice expansion at higher temperatures, which increases the Mn2+–Mn2+ distance, thereby reducing the probability of concentration quenching.26,31 The unusual increase in intensity of the 525 nm peak (peak 1) at high temperatures suggests that the material possesses unique properties that could be harnessed for information encryption under extreme temperature conditions. The normalized integrated PL intensity of the two emissions demonstrates the reversible thermal effect on the luminescence intensity (Fig. 6c and d). The intensity of both emissions varies with temperature, and after a complete heating and cooling cycle, the luminescence intensity of the sample is largely restored to its original state. This reversibility indicates the robustness of the PL properties of the as-obtained crystal against thermal cycling, which is a desirable trait for applications requiring thermal stability. | ||
| Fig. 6 (a and b) Temperature-dependent emission spectra of C6H14N2Cd0.50Mn0.50Br4 during the heating and cooling processes. (c and d) The corresponding normalized integrated PL intensity of peak 1 and peak 2 at various temperatures during heating and cooling, respectively. (e) PL spectra and the corresponding (f) FWHM of C6H14N2Cd0.50Mn0.50Br4 crystals varied from 77 to 298 K. | ||
The low-temperature spectra of the C6H14N2Cd0.50Mn0.50Br4 sample reveal that as the temperature decreases from 298 K to 77 K, both the 525 nm (peak 1) and 627 nm (peak 2) emission peaks increase in intensity, with the 525 nm (peak 1) emission showing a significantly greater enhancement (Fig. 6e). At 77 K, the green emission is dominant, and this enhancement is attributed to the reduced lattice vibrations which facilitate a more efficient radiative recombination process in isolated [MnBr4]2− tetrahedra.32,33 In summary, the temperature-dependent studies highlight the unique luminescent behavior of C6H14N2Cd0.50Mn0.50Br4, which includes the counterintuitive increase in emission intensity at high temperatures and the reversible luminescence response to thermal cycling.
The Huang–Rhys factor (S), which quantifies the electron–phonon coupling strength, is deduced from the fitting of the emission spectra of C6H14N2Cd0.50Mn0.50Br4 (Fig. 6f). To further analyze this interaction, we fit the relationship between the full width at half maximum (FWHM) and temperature using the following equation:33

The thermogravimetric analysis curves for both undoped and 50% Mn2+-doped crystals are largely congruent, showing no significant weight loss below 331 °C (Fig. S11†). Upon further heating, the initial weight loss of 43% is attributed to the decomposition of the organic component. The second stage of weight loss is 46%, with the decomposition temperatures for the undoped and doped samples being 472 °C and 492 °C, respectively. The improved thermal stability observed in the doped sample suggests that the incorporation of Mn2+ ions enhances the overall stability of the material against thermal degradation.
3.5. The potential application of the Mn2+-doped C6H14N2CdBr4
The investigation into the temperature-responsive luminescent properties of C6H14N2Cd0.50Mn0.50Br4, as presented in Fig. 7a, reveals a remarkable color tunability. The CIE color coordinates and corresponding photographs demonstrate a transition from orange-red to green emission as the temperature increases from room temperature to 453 K. This shift is characterized by a change in CIE coordinates from (0.43, 0.48) to (0.24, 0.63), effectively covering the entire visible spectrum and showcasing the potential for wide-range color tuning applications. Leveraging the high-temperature anti-quenching characteristics of C6H14N2Cd0.50Mn0.50Br4, the study explores its application in temperature-sensitive anti-counterfeiting. Fig. 7b displays a designed “Fu” (meaning fortune or happiness in Chinese) pattern, fabricated by etching on a quartz slide. The pattern exhibits a temperature-induced color change from orange-red to yellow and ultimately to pure green at elevated temperatures, with high luminescence intensity maintained even at high temperatures. | ||
| Fig. 7 (a) CIE color coordinates and the corresponding photographs of C6H14N2Cd0.50Mn0.50Br4 under different temperatures. (b) The designed patterns for temperature-sensitive anti-counterfeiting application. (c) Electroluminescence spectra of WLEDs fabricated with the C6H14N2Cd0.50Mn0.50Br4 on the 365 nm ultraviolet chip (the inset is the photo of the device in working condition) and (d) the current-dependent EL spectra. (e) XRD of C6H14N2Cd0.50Mn0.50Br4 after being exposed to air for different number of months. | ||
The EL spectra of white light-emitting diodes (WLEDs) fabricated with C6H14N2Cd0.50Mn0.50Br4 demonstrate the successful generation of white light by combining the material with commercial blue powder BaMgAl10O17:Eu and encapsulating it on a 365 nm UV chip (Fig. 7c). The inset photo illustrates the device in operation, emitting bright white light with CIE coordinates of (0.32, 0.37), a color rendering index (CRI) of 86, and a correlated color temperature (CCT) of 6007 K, which are characteristic of typical white light. Fig. 7d presents the current-dependent EL spectra of the LED, showing an increase in emission intensity with rising current, indicative of controllable LED brightness based on C6H14N2Cd0.50Mn0.50Br4. Notably, the absence of spectral shift with increasing current suggests that the material exhibits strong stability against current-induced variations, a crucial attribute for practical applications. Compared to multi-component systems such as tri-color encapsulation, the dual-peak emission offers advantages in terms of simplicity, efficiency, stability, and simplified encapsulation processes, positioning it as a promising candidate for future lighting and display technologies. The XRD patterns in Fig. 7e, comparing the freshly prepared C6H14N2Cd0.50Mn0.50Br4 sample with that stored in a room-temperature environment for one month, show no significant changes, indicating that the sample maintains good stability under ambient conditions.
4 Conclusions
This study presents a comprehensive exploration into the luminescent properties of a novel 0D OIHMH single crystal, C6H14N2CdBr4, with a focus on the effects of Mn2+ doping. The introduction of Mn2+ ions into this matrix resulted in a dual-emission phenomenon, with the appearance of a red emission peak at 627 nm in addition to the green emission at 525 nm, a characteristic not typically observed in such tetrahedral structures. The interplay between the inter-tetrahedral distances and the doping concentration of Mn2+ was identified as a critical factor influencing the luminescent properties. Theoretical calculations at a 50% doping concentration indicated the presence of three distinct inter-tetrahedral distances, which facilitated magnetic coupling and the emergence of red emission. Theoretical calculations revealed that the spin-up electrons of Mn2+ were primarily responsible for the luminescence, with the charge concentrated within the [MnBr4]2− tetrahedra enabling d–d transitions. This insight into the electronic configuration of Mn2+ ions within the perovskite lattice has deepened our understanding of the luminescence process in hybrid perovskites. The discovery of the dual-emission of Mn2+ singly doped in an OIHMH matrix, along with the elucidation of the underlying mechanisms, opens up new avenues for the design and synthesis of materials with tailored PL properties.Data availability
The data supporting the findings of this study are openly available upon request from the corresponding author. Data will be shared promptly, subject to no privacy or proprietary restrictions. For access to the data, interested parties should submit a brief request outlining the specific data needed and their research purpose. The authors will review and respond to requests within a reasonable time frame. For any inquiries regarding data availability, please contact the corresponding author.Author contributions
Q. Jin undertaken conceptualization, experiments, data curation, writing – original draft preparation. R. Wu measured XPS, TGA and analyze. Y. Pan undertaken supervision, writing – reviewing and editing. Y. Ding taken charge of formal analysis, visualization, software. H. Lian and J. Lin undertaken supervision, funding acquisition, writing – review and editing. L. Li was responsible for calculation and theory analysis. All authors contributed to the general discussion and analysis of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is financially supported by the National Natural Science Foundation (NSF) of China (52172152, 52172166, 51932009), Seedling Program Project of Zhejiang Province (2024R429A013), and Graduate Student Innovation Fund of Wenzhou University (3162024001001, 3162024003043).References
- J. Jin, Y. Z. Wang, K. Han and Z. G. Xia, Angew. Chem., Int. Ed., 2024, 63, e202408653 CrossRef CAS PubMed.
- D. H. Liang, M. Wang, S. Y. Zhao, Z. Y. Xu, M. H. Qaid and Z. G. Zang, Laser Photonics Rev., 2024 DOI:10.1002/lpor.202400244.
- H. L. Huang, Y. L. Yang, S. Y. Qiao, X. D. Wu, Z. Y. Chen, Y. Chao, K. D. Yang, W. P. Guo, Z. Z. Luo, X. R. Song, Q. S. Chen, C. K. Yang, Y. Yu and Z. G. Zou, Adv. Funct. Mater., 2024, 34, 2309112 CrossRef CAS.
- C. Li, Y. Wei, Y. W. Li, Z. S. Luo, Y. L. Liu, M. Y. He, Y. Zhang, X. He, X. Y. Chang and Z. W. Quan, Small, 2024, 20, 2400338 CrossRef PubMed.
- L. Zhao, G. Y. Yao, H. Chen, T. Zeng and Z. Y. Yang, Chem. Mater., 2024, 36, 3277–3284 CrossRef CAS.
- L. L. Jiang, L. K. Wu, H. Y. Sun, H. Y. Yin, Q. H. Zou, J. W. Deng, R. F. Li, H. Y. Ye and J. R. Li, Chem. Eng. J., 2024, 494, 153060 CrossRef CAS.
- X. W. Kong, L. X. Wu, X. Yang, D. Y. Wang, S. X. Wang, S. Y. Li, C. Y. Yue, F. Yu and X. W. Lei, Adv. Opt. Mater., 2024, 12, 2302710 CrossRef CAS.
- X. Y. Yun, J. H. Nie, H. L. Hu, H. Z. Zhong, D. H. Xu, Y. M. Shi and H. N. Li, ACS Appl. Nano Mater., 2024, 7, 12121–12128 CrossRef CAS.
- Z. X. Wang, J. A. Lai, Q. Huang, D. F. Wu, F. Qi, N. Zhang, Y. Y. Pu, C. Q. Tian, W. W. Chen, Y. F. Liu, P. He, K. An, X. S. Tang, F. Wang, Y. Liu and G. Q. Han, Adv. Opt. Mater., 2023, 11, 2300399 CrossRef CAS.
- Z. X. Zeng, G. D. Zhang, Y. S. Wang, M. Zhang, Z. Y. Cheng, H. Z. Lian and J. Lin, Laser Photonics Rev., 2024, 18, 2300983 CrossRef CAS.
- H. Q. Xu, J. Liu, Q. C. Hu, J. Y. Yu, Q. J. Han and W. Z. Wu, Adv. Opt. Mater., 2024, 12, 2302288 CrossRef CAS.
- Y. H. Yu, S. Y. Liu, J. R. Zhang, W. Zhao, Y. Tang, C. F. Han, X. Y. Chen, L. G. Xu, R. F. Chen, M. G. Li, Y. Tao and W. Z. Lv, Inorg. Chem., 2024, 63, 10296–10303 CrossRef CAS PubMed.
- F. Ahmad, M. S. Lassoued, W. P. Chen, G. Y. Gou and Y. Z. Zheng, ACS Appl. Mater. Interfaces, 2024, 16, 31067–31075 CrossRef CAS PubMed.
- H. D. Tang, Y. Q. Xu, X. B. Hu, H. J. Chen, S. H. Wang, W. H. Jiang, Q. Hu, L. J. Wang and W. Jiang, Laser Photonics Rev., 2023, 18, 2300672 CrossRef.
- W. Zhang, P. Sui, W. Zheng, L. Y. Li, S. H. Wang, P. Huang, W. Zhang, Q. Zhang, Y. Yu and X. Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202309230 CrossRef CAS PubMed.
- T. Hang, H. Peng, C. Y. Peng, Y. Tian, S. F. Yao, X. X. Han and B. S. Zou, Nano Energy, 2022, 93, 106863 CrossRef.
- E. H. Song, M. H. Chen, Z. T. Chen, Y. Y. Zhou, W. J. Zhou, H. T. Sun, X. F. Yang, J. L. Gan, S. Ye and Q. Y. Zhang, Nat. Commun., 2022, 13, 2166 CrossRef CAS PubMed.
- H. Peng, T. Huang, B. S. Zou, Y. Tian, X. X. Wang, Y. C. Guo, T. T. Dong, Z. M. Yu, C. J. Ding, F. Yang and J. P. Wang, Nano Energy, 2021, 87, 106166 CrossRef CAS.
- W. Wang, Y. Wei, L. Qiu, M. S. Molokeev, H. Yang, H. R. Liu, R. Gao, M. Y. Guan, D. Tu and G. G. Li, Chem. Mater., 2024, 36, 5574–5586 CrossRef CAS.
- X. L. Yang, C. D. Pu, H. Y. Qin, S. J. Liu, Z. Xu and X. G. Peng, J. Am. Chem. Soc., 2019, 141, 2288–2298 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- B. Zhou, A. Du, D. Ding, Z. Liu, Y. Wang, H. Zhong, H. Li, H. Hu and Y. Shi, Nano-Micro Lett., 2023, 15, 207 CrossRef CAS PubMed.
- Z. Tang, R. Liu, J. Chen, D. Zheng, P. Zhou, S. Liu, T. Bai, K. Zheng, K. Han and B. Yang, Angew. Chem., Int. Ed., 2022, 61, e202210975 CrossRef CAS PubMed.
- H. P. Xu, X. H. Dong, Z. Z. Zhang, L. Huang, H. M. Zeng, Z. Lin and G. H. Zou, J. Mater. Chem. C, 2022, 10, 13844–13850 RSC.
- M. Gao, Y. X. Pan, C. D. Peng, Y. H. Ding, H. Z. Lian, L. Y. Li and J. Lin, Chem. Mater., 2023, 35, 773–782 CrossRef CAS.
- L. Mao, P. Guo, S. Wang, A. K. Cheetham and R. Seshadri, J. Am. Chem. Soc., 2020, 142, 13582–13589 CrossRef CAS PubMed.
- S. S. He, Q. P. Qiang, T. C. Lang, M. S. Cai, T. Han, H. J. You, L. L. Peng, S. X. Cao, B. T. Liu, X. L. Jing and B. Jia, Angew. Chem., Int. Ed., 2022, 61, e202208937 CrossRef CAS PubMed.
- Y. L. Gao, X. X. Han, Q. L. Wei, T. Chang, Y. J. Chen, B. S. Zou, S. Cao, J. L. Zhao and R. S. Zeng, J. Phys. Chem. Lett., 2022, 13, 7177–7184 CrossRef CAS PubMed.
- L. R. Bradshaw, J. W. May, J. L. Dempsey, X. S. Li and D. R. Gamelin, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 115312 CrossRef.
- S. Banerjee, S. Saikia, M. S. Molokeev and A. Nag, Chem. Mater., 2024, 36, 4750–4757 CrossRef CAS.
- G. Zhou, Z. Liu, J. Huang, M. S. Molokeev, Z. Xiao, C. Ma and Z. Xia, J. Phys. Chem. Lett., 2020, 11, 5956 CrossRef CAS PubMed.
- X. Yuan, J. J. Zheng, R. S. Zeng, P. T. Jing, W. Y. Ji, J. L. Zhao, W. Y. Yang and H. B. Li, Nanoscale, 2014, 6, 300–307 RSC.
- M. Wan, H. R. Chen, Y. N. Wang, K. Shi, J. Y. Liu, Z. M. Li, S. Y. Ye, J. Y. Li and L. Z. Chen, Mater. Chem. Front., 2022, 6, 3094–3101 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04706k |
| This journal is © The Royal Society of Chemistry 2024 |