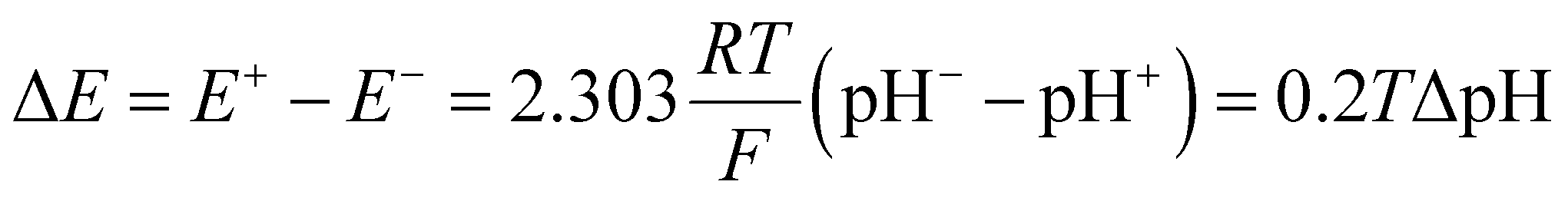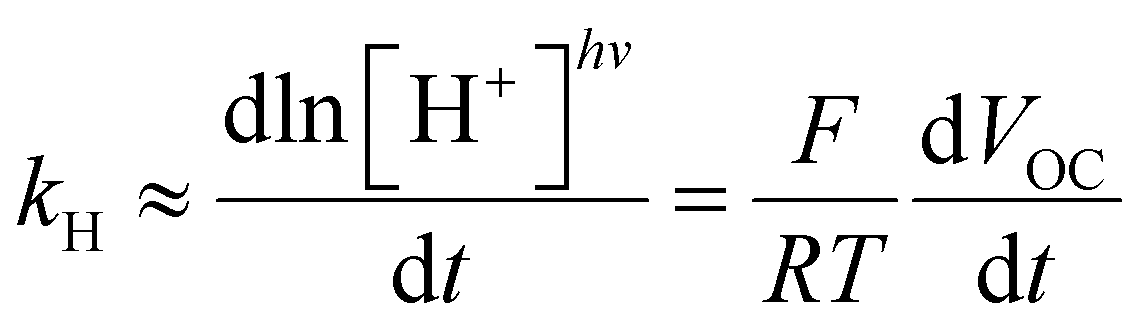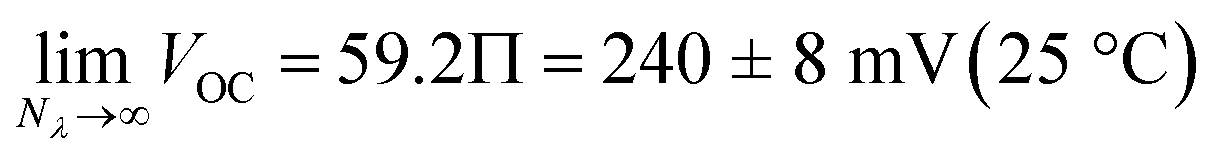DOI:
10.1039/D4SC04833D
(Edge Article)
Chem. Sci., 2024,
15, 19745-19751
Wiring proton gradients for energy conversion†
Received
20th July 2024
, Accepted 7th November 2024
First published on 8th November 2024
Abstract
Light-switchable buffer solutions based on merocyanine photoacids can be used as efficient photoenergy harvesting systems. Varying the solvation environment of merocyanine photoacids in water–methanol mixtures allows one to carefully tune their photoacidity, relaxation kinetics, and solubility, opening up the possibility to install persistent pH gradients of approximately 4 pH units under 500 nm light. When interfaced between two electrodes and exposed to asymmetric light irradiation, these solutions can be photoactivated precisely both in space and time, generating open circuit voltages as high as 240 mV that can last hours under steady-state irradiation – an outcome that is akin the peak performance of biological transmembrane proton pumps.
Introduction
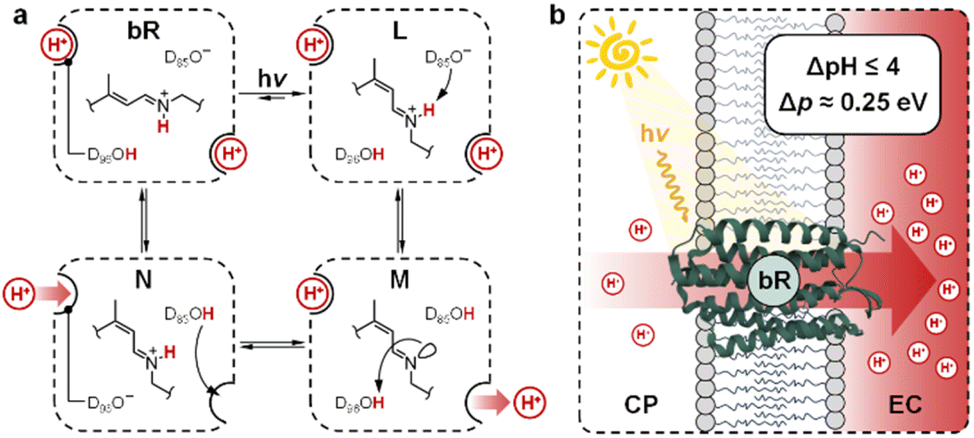
Transmembrane proton gradients are pivotal in regulating the activity of bacteria, animals, and plants cells. They constitute the natural expedient through which external energy can be efficiently transduced into adenosine triphosphate (ATP), the energy currency of life.1 In chloroplasts, for example, energy transduction originates from water splitting and proton-coupled electron transfer reactions, which harvest light energy generating a proton motive force (Δp) across the thylakoid membrane that, in turn, activates ATP-synthase through chemiosmosis.2 In some prokaryotic organisms such as Haloarchaea, however, chemiosmotic coupling3 does not rely upon an electron transport chain. Their energy harvesting is based instead on bacteriorhodopsin (bR), a light-driven proton pump complex composed of a retinal molecule covalently bound to bacteriopsin through a protonated Schiff base linkage.4 Over the past decades, bR has become a model system for light-driven proton transport and its molecular mechanism has been elucidated by a tremendous amount of crystallographic and spectroscopic studies.5 Its photocycle6 comprises the sequential formation of several optically-distinct states, four of which (referred to as bR and intermediate species L, M, and N) are evoked to better highlight the coupling between the retinal molecular switch and the spatially-oriented release and uptake of a proton (Fig. 1a).7 In the resting state (bR), the retinal Schiff base resides in its all-trans conformation (pKa = 13.3 (ref. 8)), with adjacent aspartic acid residues D96 (pKa > 12 (ref. 9)) and D85 (pKa < 3 (ref. 10)) in their protonated and deprotonated state, respectively. Visible light absorption triggers trans-to-cis isomerization of the retinal Schiff base, which results in the formation of intermediate species L; the resulting change in protein conformation is associated with pKa shifts that allow selective proton transfer from the retinal Schiff base to D85 to give intermediate M, which subsequently expels a proton into the extracellular (EC) environment.11 At this stage, the proton affinity difference observed between D96 (pKa = 7.1) and the retinal Schiff base (pKa = 8.2) enables reprotonation of the latter to give intermediate N.12 In the final stage, D85 donates its proton to the water cluster located near the EC surface, while D96 uptakes a proton from the cytoplasmatic (CP) medium prior to back-isomerization of the chromophore.11 Overall, this cycle of conformationally-induced proton transfer reactions drives the vectorial transport of protons from the CP to the EC space, generating a Δp ranging from 0.2 to 0.3 eV.13 In other words, this means that bR can harvest and transduce light energy into proton concentration differences of up to four orders of magnitude,14 before being inhibited by the so-called back-pressure effect15 – i.e., the Δp it generates works against the active transfer of additional protons (Fig. 1b). Interestingly, this effect is regarded to as a general control mechanism in membrane bioenergetics16 and explains why proton concentration gradients (ΔpH) in biological systems can hardly exceed the four pH units.17
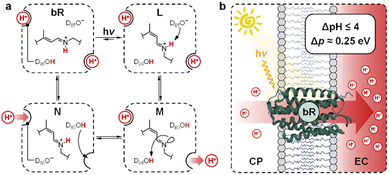 |
| | Fig. 1 (a) Simplified view of bR photocycle highlighting the coupling of retinal Schiff base isomerization with proton transfer reactions. (b) Illustration of the vectorial proton translocation mediated by bR in Halobacteria and corresponding peak performance. | |
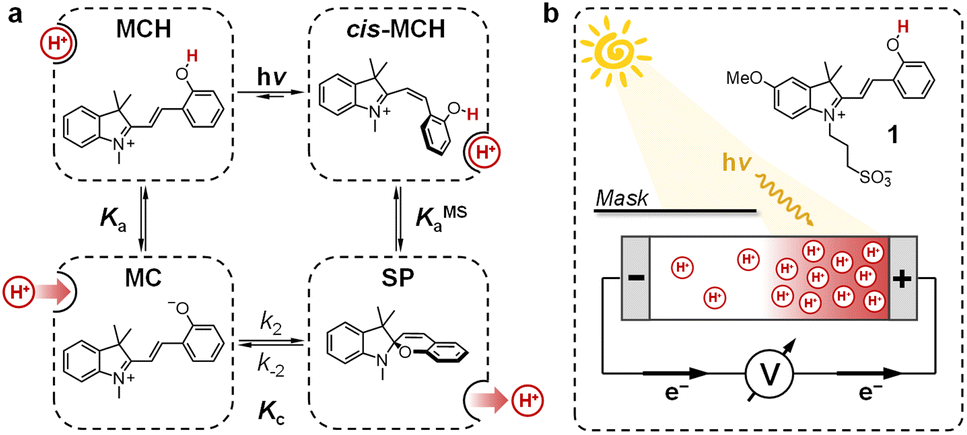
In recent years, chemists have succeeded in mimicking autotrophs' photosynthesis,18 and made groundbreaking achievements in the design and operation of artificial molecular ratchets19 including pumps20 and motors,21 and membrane transport systems.22 Recently, we23 and others24 have shown that merocyanine photoacids (MCHs) are molecular switches able to transduce visible light into persistent and reversible drops of pH. As in the case of bR, the trans-to-cis isomerization of MCHs is accompanied by a significant pKa downshift, which can result in proton transfer to solvent molecules25 or suitable proton acceptors.26 Specifically, the thermodynamics and kinetics of their proton release/uptake in water can be described by the four-state cyclic model depicted in Fig. 2a.27 Under dark conditions, dissociation (Ka) of open protonated form (MCH) is followed by isomerization (Kc) of the open deprotonated form (MC) into the corresponding closed form (SP) – i.e., the ground state of MCHs comprises a three-component equilibrium system featuring an apparent acidity constant that can be formulated as KGSa = Ka(1 + Kc). Yet, under visible light irradiation, photoproduct cis-MCH forms a photostationary state with SP characterized by an apparent acidity constant (KMSa) that is almost four orders of magnitude higher than that of the ground state. The quantum yield of isomerization, however, does not appear to be a limiting factor for bulk pH switching as are the photoacidity and the solubility of MCHs.24b Herein, we report on a strategy to rationally augment both the photoacidity28 (which is defined as Π = pKGSa − pKMSa) and solubility of MCHs in aqueous environments. It will be shown that MCHs can (i) transduce 500 nm light into proton concentration gradients of about 4 pH units and (ii) act as energy harvesters to produce persistent open-circuit voltages (VOC) of up to 240 mV (Fig. 2b) – a result that strongly resembles the peak performance of bR in Halobacteria (see Fig. 1b).
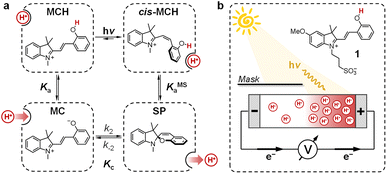 |
| | Fig. 2 (a) Four-state model describing MCHs' operation in aqueous environments: visible light-triggered isomerization (upper-right path) contrasts with thermal relaxation (bottom-left path), where the SP ring-opening is the rate-determining step. Substituents are omitted for the sake of clarity. (b) Working principle of the energy harvesting device employed in this study: asymmetric light irradiation of a tube-shaped cell filled with a solution of compound 1 triggers the formation of a persistent proton concentration gradient (ΔpH) across the cell corresponding to an open circuit voltage, VOC (mV) = 0.2·T·ΔpH (see below). | |
Results and discussion
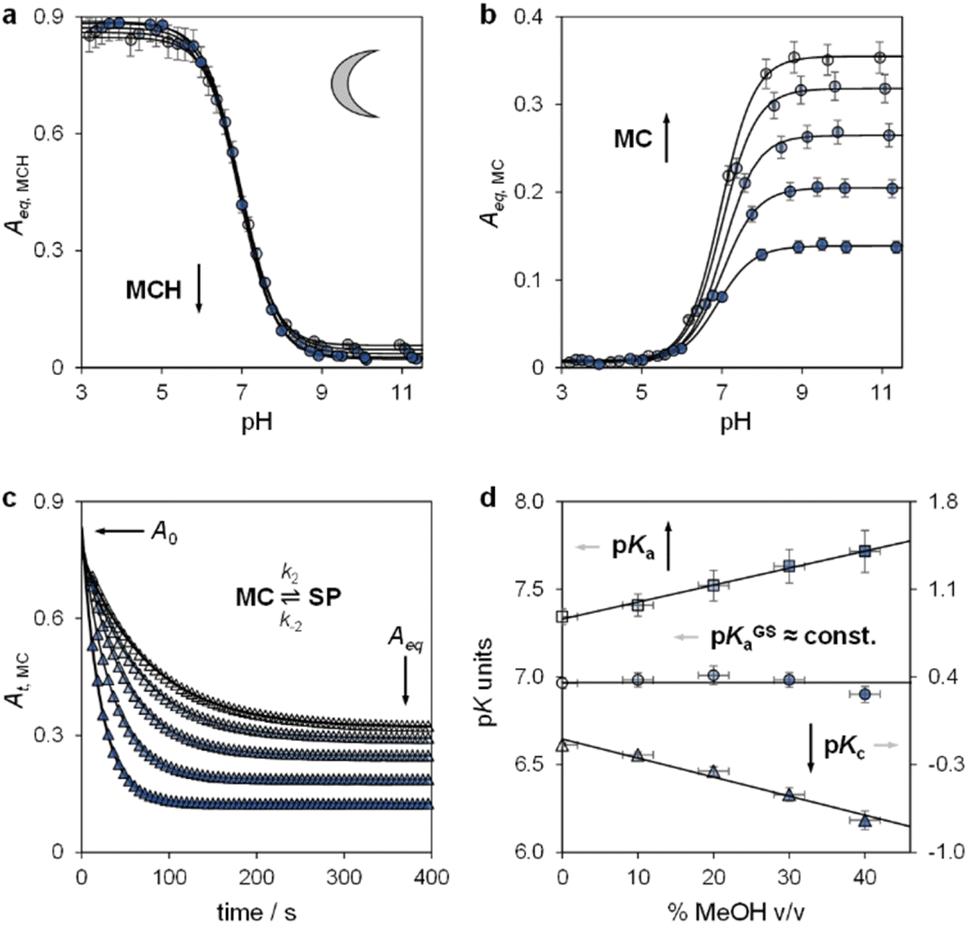
Lukatskaya and co-workers29 have recently shown that the solubility and the hydrolytic stability of MCHs in aqueous binary mixtures can be regulated by changing the solvent composition. The effect of tuning the solvation environment on the photoacidity of MCHs, however, remains unexplored. We thus decided to contribute in this direction by dissecting the (photo)chemical properties of compound 1 (Fig. 2b, top right corner) in water–methanol30 mixtures. We decided to test compound 1 because, compared to the parent compound introduced by Liao and coworkers,31 it bears a methoxy group on the indolenine side which makes it more hydrolytically stable.25a As for the water co-solvent, we opted for methanol (MeOH) for three main reasons, which are: (i) MCHs are more soluble in MeOH than water by at least an order of magnitude,32 (ii) MeOH may stabilize MCHs towards hydrolysis, possibly increasing the robustness of the photoswitch,33 and (iii) accurate pH readings of aqueous MeOH mixtures up to 40% v/v can be easily made by calibrating the glass electrode with standard aqueous buffers.34 In practical terms, this last consideration represents a point of convenience, as it means that comparative evaluation of pKa values can be done without changing the standard state of reference.35 First, we focused on the effect of the solvation environment on the ground state acidity constant. pKGSa values of compound 1 in aqueous MeOH mixtures were determined spectrophotometrically. Samples at different pH values were prepared by adding 1 into potassium phosphate buffers at the desired solvent composition, and left equilibrating at 25 °C in the dark for 15 minutes prior to acquisition – all spectra and corresponding elaborations are given in the ESI (see Fig. S1†). Absorbance profiles of both the MCH and the MC forms as a function of the pH (Fig. 3a and b) were used to determine the corresponding ground state acidity constant as described previously.27 We found that the pKGSa of compound 1 remains constant irrespective of solvent composition (pKGSa = 6.97 ± 0.04). This is quite surprising if one considers that phenols in aqueous MeOH mixtures have lower acidity constants (i.e., higher pKa values) as the MeOH content increases (see below). On the contrary, however, the maximum absorption of both the MCH and the MC forms did increase linearly by 10 and 20 nm, respectively, passing from 0 to 40% MeOH v/v (see Fig. S2†). These bathochromic shifts may indicate a loss of charge delocalization, with the MC form gradually acquiring a more quinoidal character.36 This observation, together with the progressive weakening of the MC band at high pH (Fig. 3b), strongly suggests an increasing tendency of the system towards ring closing.37 To confirm whether the equilibrium distribution shifts towards the SP form, we examined the MC ⇌ SP isomerization at high pH, monitoring the decay of the MC form kinetically at varying solvent composition (Fig. 3c). The rate constant of both the forward (k2) and the backward (k−2) reaction were calculated considering the best fit to a first-order decay, whereas Kc was determined by dividing the loss of absorbance (A0 − Aeq) for the plateau value reached at equilibrium (Aeq) (see Fig. S3†). We found that k−2 remained constant within the experimental error while k2 gradually increased, resulting in a 5-fold increase of Kc passing from 0 to 40% MeOH v/v – i.e., indicating a preference of the system for the SP form in less polar solvent media. The acidity constant of the phenol moiety can thus be extrapolated through the inverse function of pKGSa (pKa = pKGSa − p(1 + Kc)), and plotted together with pKGSa and pKc for comparison (Fig. 3d). Interestingly, the obtained linear increase in pKa looks very similar to those observed for substituted phenols38 (e.g., 4-nitrophenol, see Fig. S4†), and sums up the big picture emerging from these initial studies: within the range of solvent composition tested, the ground state acidity constant of compound 1 remains constant on account of a mutual increase of both pKa and Kc.
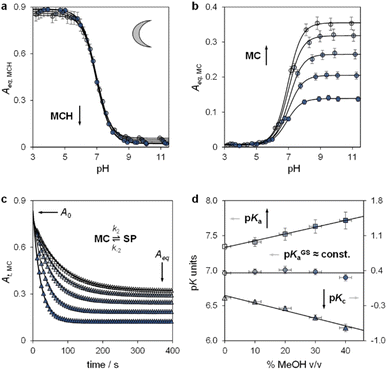 |
| | Fig. 3 The ground state acid–base properties as a function of the solvent composition. Shades of colour represent the effective% MeOH v/v at which each experiment was carried out, as summarized in plot (d). Absorbance profiles of MCH (a) and MC (b) at equilibrium obtained as a function; each solid black line represents the best fit to eqn (S1).† (c) Kinetic profiles of MC equilibration at high pH; solid black lines represent the best fits to a first-order decay model (eqn (S2), see ESI for more details†). (d) Obtained trends of pKGSa, pKa, and pKc as a function of % MeOH v/v. Experimental conditions: [1] = 29 ± 1 μM, [phosphate buffers] = 20 mM, T = 25 °C. | |
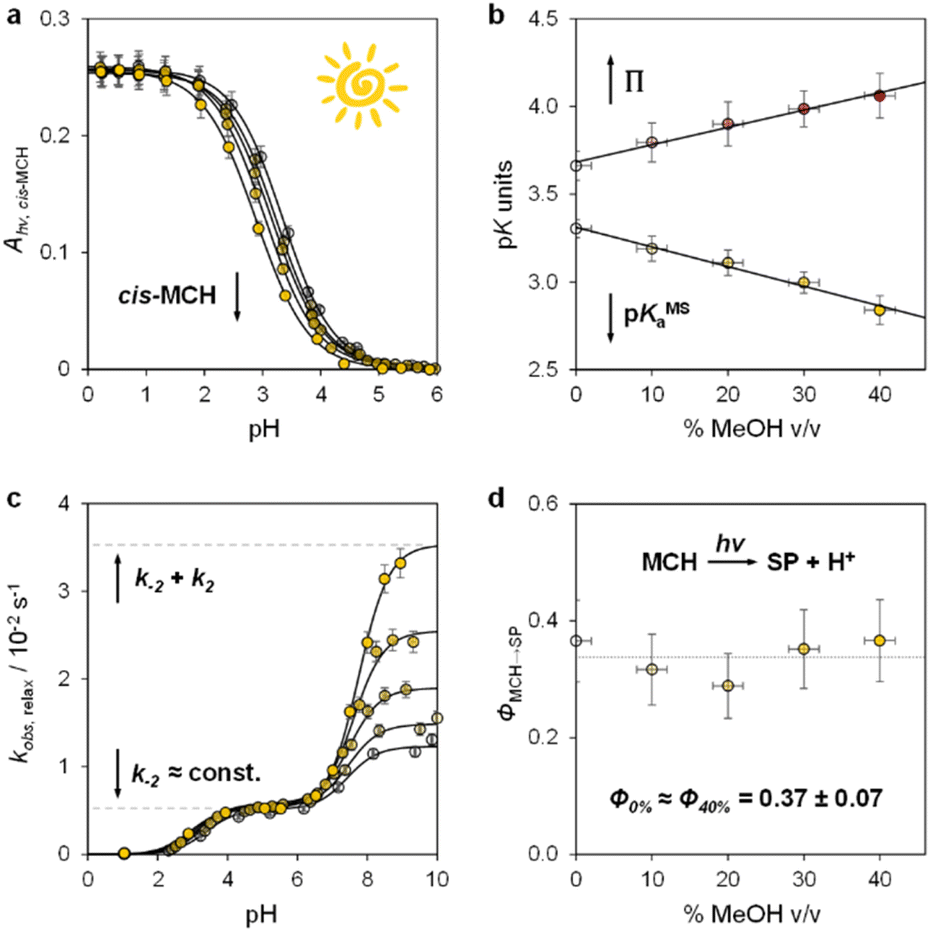
Having elucidated the ground state properties, we investigated the acid–base features of the metastable state. pKMSa values in aqueous MeOH mixtures were determined spectrophotometrically under continuous light irradiation. Samples at different pH values were prepared by adding 1 into potassium phosphate buffers or HCl solutions (below pH 4) at the solvent composition, and left under 500 nm light during acquisition till photostationary conditions were reached – pKMSa were obtained as described above – see Fig. 4a and S5† for all spectra and elaborations. We found that the pKMSa of compound 1 linearly decreased as a function of the MeOH content, resulting in a parallel improvement of the photoacidity from Π = 3.7 to 4.1 pKa units (Fig. 4b). To complement the thermodynamic data, we decided to examine the kinetics of relaxation after light irradiation, monitoring the recovery of the MCH form as a function of the pH. In all cases, the observed rate constant of relaxation gradually increased with pH following double-sigmoidal behaviours (Fig. 4c), indicating that the four-state model cycle evoked to describe the operation of compound 1 in water27 remains valid also in binary aqueous MeOH mixtures. Importantly, the optimized parameters resulting from fitting are in line with the set of kinetic and thermodynamic constants determined above (see Fig. S6† for more details), confirming that the crucial effect of adding MeOH to the system is the decrease of the kinetic barrier leading to the SP form – i.e., the increase of the forward rate constant of the MC-to-SP reaction, k2. We then examined the kinetics of photoisomerization and hydrolysis, monitoring the decay of the MCH form upon light irradiation and under dark conditions, respectively. Inspection of the obtained isomerization profiles39 at pH 4.5 using mild 500 nm light (see Fig. S6†) allowed us to calculate the quantum yield of the MCH-to-SP reaction, which, in line with k−2, was found to remain constant within the experimental error (Φ0% ≈ Φ40% = 0.37 ± 0.07, Fig. 4d). This value closely matches the one we determined previously through pH jump studies in water (0.40 ± 0.03).25a As for the hydrolysis, we studied the depletion of the MCH form over time at different pH values. In all cases, we obtained similar bell-shaped profiles displaying a maximum first-order rate constant of hydrolysis of 4·10−4 min−1, which corresponds to a half-life of about 29 hours at 25 °C (see Fig. S7†). In other words, this means that: (i) the mechanism of hydrolysis of compound 1 does not change up to 40% MeOH v/v, and (ii) compound 1 remains three times more stable than the parent compound lacking the methoxy group.
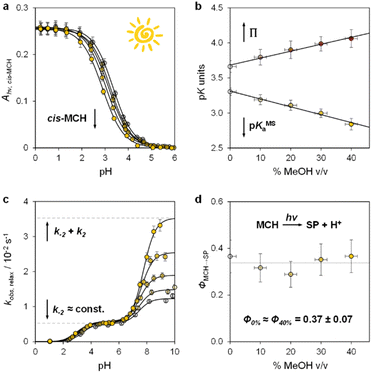 |
| | Fig. 4 The metastable-state acid–base properties as a function of the solvent composition. Shades of colour represent the effective % MeOH v/v at which each experiment was carried out, as summarized in plots (c) and (d). (a) Absorbance profiles of cis-MCH under continuous light irradiation (500 nm, 90 mW) obtained as a function of the pH; each solid black line represents the best fit to eqn (S1).† (b) Obtained trends for pKMSa and Π as a function of % MeOH v/v. (c) Kinetic profiles of relaxation as a function of the pH; each solid black line represents the best fit to eqn (S3).† (d) Quantum yield of the MCH-to-SP reaction. Experimental conditions: [1] = 29 ± 1 μM, [phosphate buffers] = 20 mM or standard HCl solutions, T = 25 °C. | |
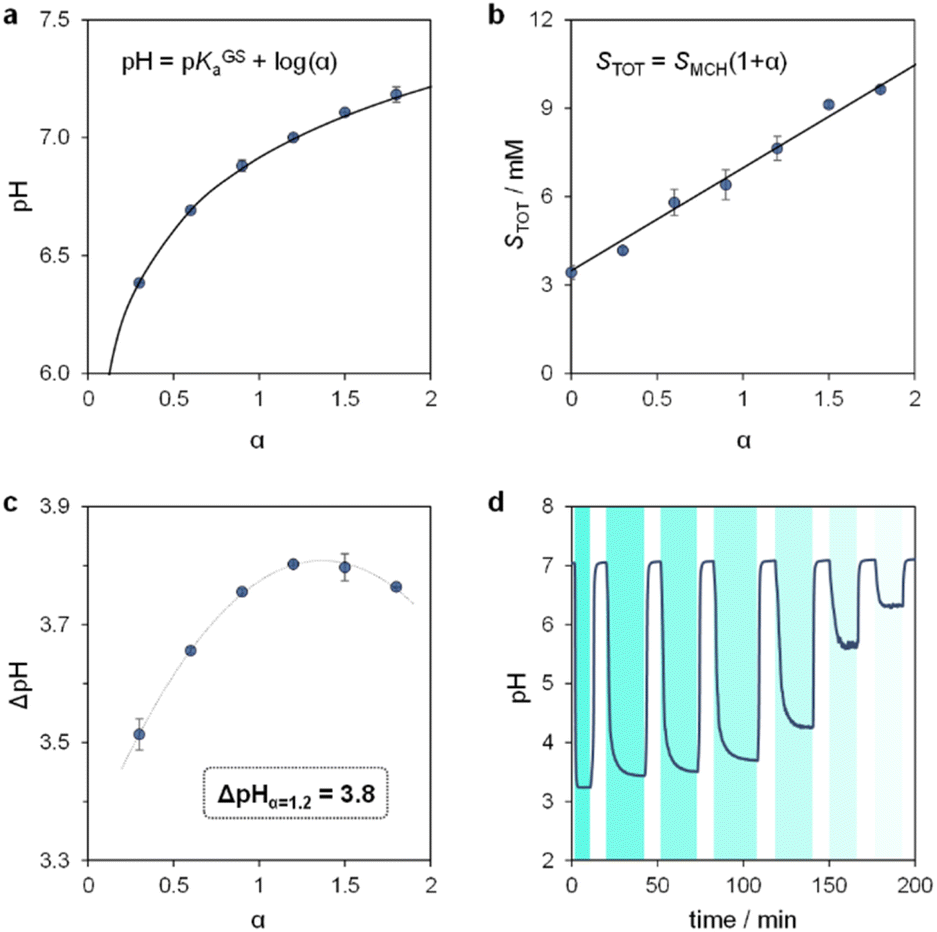
We proceeded to examine the features of light-switchable buffers in 40% MeOH v/v. Saturated mixtures at increasing pH values were prepared by adding compound 1 to standard HCl or NaOH solutions, under stirring and 25 °C till stabilization of the pH. The initial run is required to determine the solubility of the MCH form (SMCH), which is then used to calculate the equivalents of NaOH (α) to be added in the following solubility experiments (see Section 10 of the ESI†). We found that the pH values of the resulting saturated solutions are in line with the Henderson–Hasselbalch equation (Fig. 5a), whereas the total solubility of 1 (STOT) linearly increases as a function of α, with SMCH remaining constant at a value of 3.4 ± 0.2 mM regardless of pH (Fig. 5b). Notably, this saturation concentration is approximately one order of magnitude higher than that in pure water (0.37 mM,25a), and resulted in 500 nm light-triggered ΔpH that are almost one pH unit larger (from pH 7.0 to 3.2) when α = 1.2 (see Fig. S9† and 5c). The optimized buffer solution also shows good robustness and tunability, as it can be programmed repetitively for more than three consecutive hours to obtain ΔpH spanning from 3.8 to 0.8 pH units (Fig. 5d). Overall, these results demonstrate a straightforward strategy to significantly enhance the (photo)chemical properties of MCHs post-synthetically.
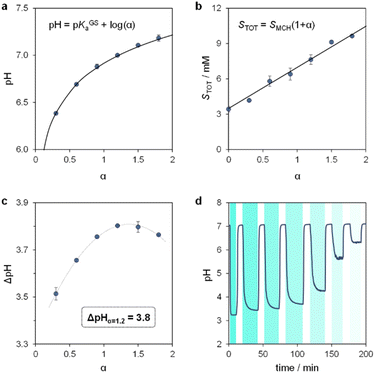 |
| | Fig. 5 The light-switchable buffer characteristics in 40% MeOH v/v. pH (a) and total concentration (b) of the resulting buffer solutions under dark conditions as a function of α, the solid black line represents the best fit to the corresponding model equation. (c) Values of ΔpH obtained under intense light irradiation as a function of α. (d) Tuning of ΔpH by modulation of the light intensity. Experimental conditions: T = 25 °C, 500 nm LED-light irradiance, (c) 55.6; (d) 55.6, 22.0, 21.4, 20.1, 18.8, 17.8, 17.2 mW cm−2 from left to right, respectively (see ESI† for more details about the photochemical setup); the time intervals under light irradiation are highlighted by cyan-coloured areas. | |
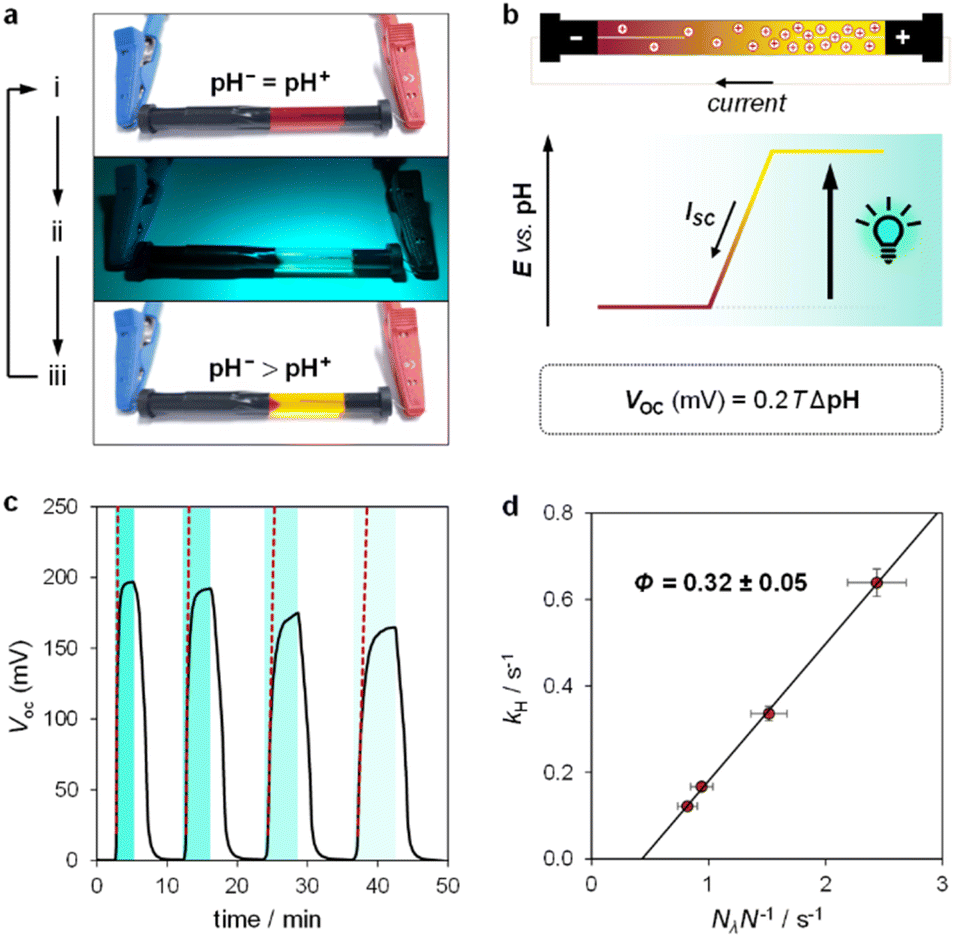
Inspired by the recent work of Kim and co-workers,40 the possibility of harnessing light-switchable buffer solutions for photoenergy harvesting was finally assessed with a cylindrical electrochemical cell, which is nothing but a 5 cm glass tube capped with two silicone rubber caps each bearing a coaxially-inserted Pt wire (see Section 11.1 of the ESI†). The cell was filled with the previously optimized buffer solution and subjected to chronopotentiometry and chronoamperometry studies during asymmetric light irradiation (Fig. 6a). Under these working conditions, although other types of reversible photoacids or photobases may be effective in producing useful work through excited-state proton transfer (ESPT) mechanisms,41 our light-switchable buffer is expected to support in the metastable chemical polarization of the Pt electrode under light irradiation (Fig. 6b). Compared to excited-state photoacids, compound 1 more effectively transduces visible light energy into differences in protonic chemical potential, which in turn generates differences in electrochemical potential between the Pt electrodes:
| | 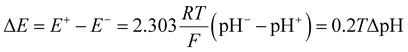 | (1) |
where Δ
E is the cell potential,
R is the gas constant,
T is the temperature,
F is the Faraday constant, while pH
− and pH
+ represent the pH values of the solution around the negative and positive electrode, respectively –
i.e., in the dark and in photochemical equilibrium (
Fig. 6b).
42 We measured the
VOC of our cell over a range of irradiance at 500 nm from 4.7 to 14.0 mW cm
−2. The cell demonstrated effective performance, delivering a voltage close to 200 mV (
Fig. 6c). To clearly confirm that the observed voltage is due to light-triggered proton release, we examined the initial rate change of voltage (d
VOC/d
t) as a function of the photon flux. Theoretically, assuming that the pH near the negative electrode remains constant, the time derivative of the cell potential correlates with the observed rate constant of proton release (
kH) as follows:
| | 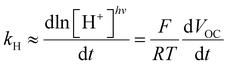 | (2) |
where [H
+]
hv represents the proton concentration experienced solely by the positive electrode (see Section 11.2 ESI for more details
†). The rate constant of proton release calculated in this manner increases linearly with the photon flux (
Nλ) normalized by the total number of molecules in solution (
N), yielding a quantum yield
Φ = 0.32 ± 0.05 (
Fig. 6d). This value aligns with that obtained from UV-Vis isomerization kinetics (see
Fig. 4d) and from pH jump experiments (see Fig. S11
†), demonstrating that the behaviour of our cell is governed by the photochemical properties of MCHs.
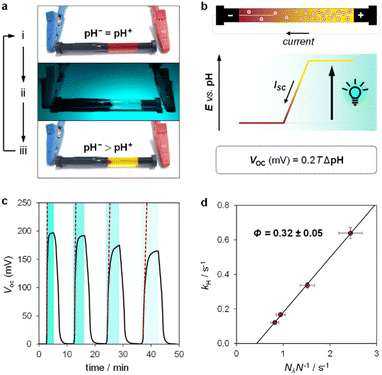 |
| | Fig. 6 Validation of the photoenergy harvesting mechanism. (a) Representative pictures of our cylindrically-shaped electrochemical cell filled with the light-switchable buffer before (i), during (ii), and after (iii) asymmetric 500 nm LED light irradiation. (b) Proposed working mechanism highlighting the correlation of the pH gradient (ΔpH = pH− − pH+) with the open-circuit voltage (VOC) and the short-circuit current (ISC). (c) Chronopotentiometry experiment over four light/dark cycles (i → ii → iii) at different irradiances; dotted red lines represent the initial rate change of voltage. (d) Observed rate constant of proton release as a function of the photon flux normalized by the total number of molecules in solution (Nλ/N). Experimental conditions: [1] = 7.6 ± 0.4 mM in 40% MeOH v/v, α = 1.2, T = 22 °C, 500 nm LED-light irradiance: 14.0, 8.7, 5.4, and 4.7 mW cm−2 from left to right, respectively; the time intervals under light irradiation are highlighted by cyan-coloured areas. | |
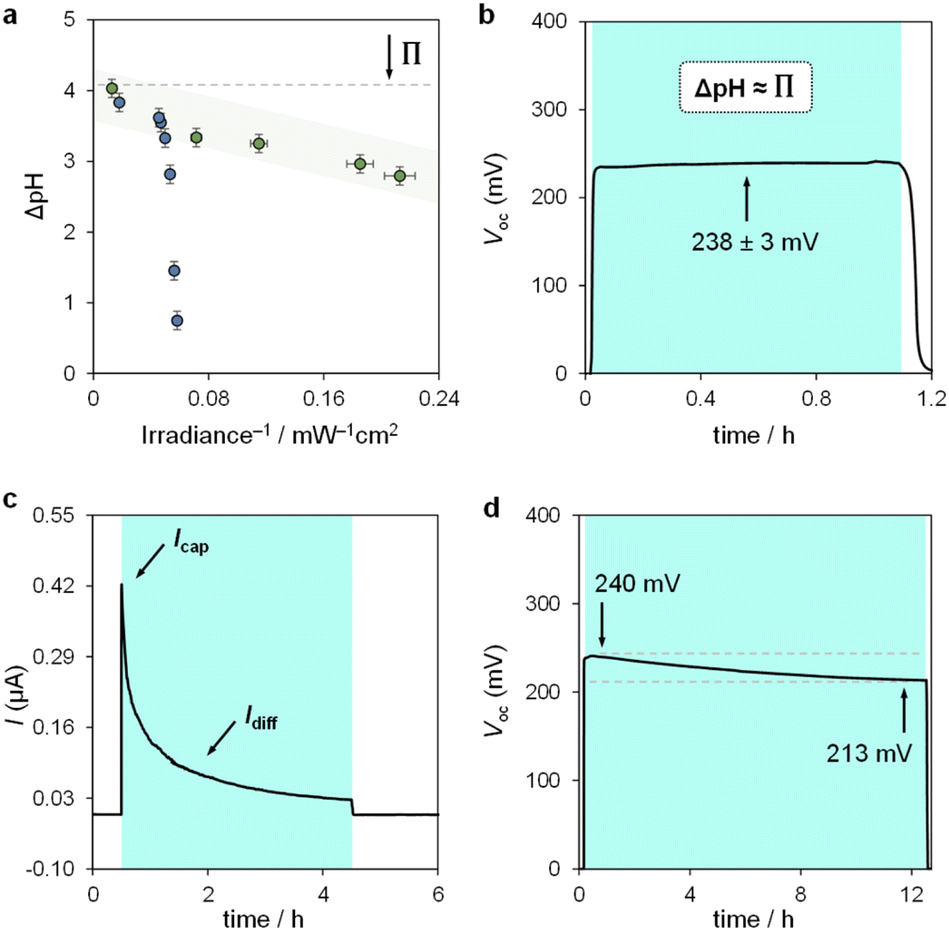
We used the reverse function of the cell potential (eqn (1)) to determine the effective pH gradient installed across the cell (Fig. 7a). Interestingly, compared to the pH jump experiments, we found that similar ΔpH values could be obtained with relatively lower irradiance. For example, a gradient of 2.8 pH units was obtained with an irradiance four times lower (4.7 vs. 18.8 mW cm−2). This is expected, given that the surface area-to-volume ratio (A/V) of the solution under light irradiation in the cell is four times greater than that in the photoreactor used for pH jumps (see Section 11.3 of the ESI†). When the photon flux exceeds the total number of molecules in solution (Nλ > N), however, ΔpH scales linearly with the inverse of irradiance (see the light green region in Fig. 7a). The intercept of this linear regime aligns with the photoacidity of compound 1 in 40% MeOH v/v (Π = 4.07 ± 0.13, see above), representing the maximum ΔpH achievable by the system. This observation can be formulated in terms of VOC as follows:
| | 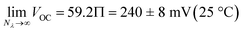 | (3) |
to illustrate the theoretical limit of our approach with regard to the cell potential. Within the framework of sunlight-to-protonic energy conversion, Ardo and colleagues have recently demonstrated that comparable thresholds can be achieved using excited-state photoacids exhibiting (i) high water solubility (1 M), (ii) excellent photoacidity characteristics (Δp
Ka ≈ 8), and (iii) near-optimal sunlight absorption over a very short distance (10 nm).
43 Compounds of this type, however, do not exist yet. In our case, we hypothesized that higher cell potentials could be obtained by increasing the photon flux. We repeated
VOC measurements under a radiant flux per unit area matching that of the AM1.5G standard solar spectrum
44 at 500 nm (75 ± 4 mW cm
−2). In this setup, the
VOC reached a stable value close to the theoretical limit predicted by
eqn (3), maintaining 238 ± 3 mV over one hour (
Fig. 7b). This
VOC corresponds to a pH gradient of approximately 4 pH units at 22 °C. The stability of
VOC suggests that a steady-state condition was achieved, where the ratio of forward and backward ion fluxes across the dark/light interface remains constant, supported by continuous light irradiation.
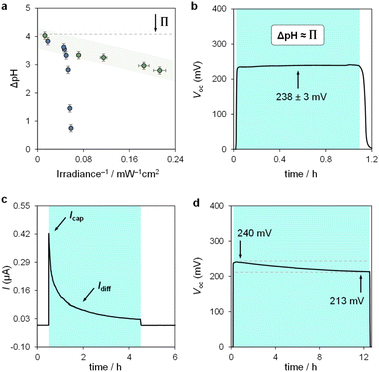 |
| | Fig. 7 Sunlight-to-protonic energy conversion. (a) Proton concentration gradient (ΔpH) as a function of the inverse of irradiance obtained with (i) the photochemical apparatus for pH jumps studies (blue circles) and (ii) the electrochemical cell under asymmetric light irradiation (green circles). Chronopotentiometry (b and d) and chronoamperometry (c) experiments over one cycle (i → ii → iii) at 75 mW cm−2. Experimental conditions: [1] = 7.6 ± 0.4 mM in 40% MeOH v/v, α = 1.2, T = 22 °C; the time intervals under light irradiation are highlighted by cyan-coloured areas. | |
In chronoamperometry experiments measuring short-circuit current (Fig. 7c), we verified that current generation depends directly on illumination. Photodissociation of compound 1 is essential for charge generation, as no current was detected in the absence of light. Under asymmetric light irradiation, the cell exhibited an immediate current peak of 0.42 μA, which decayed to a stable level of 0.03 μA after 4 hours. These results suggest two main current components: a non-faradaic capacitive current (Icap), and a diffusion-driven current (Idiff). The initial rise is attributed to the rapid formation of an electric double layer at the illuminated electrode due to proton dissociation, supported by the corresponding dVOC/dt burst (see Fig. S12†). The gradual decrease in current reflects a reduction in the magnitude of ion migration fluxes over time, rather than a change in their ratio, as the system reaches a steady state. The stable VOC (dVOC/dt = 0) further suggests that a full diffusion front does not propagate across the cell, with local ion migration balancing the charge near the illuminated electrode.
Comparative evaluation of different buffer solutions showed that both Icap and Idiff are higher in the optimized 7.6 mM aqueous methanol buffer than in a 0.7 mM,25a aqueous buffer (see Fig. S13a†). The higher ion concentration in 40% MeOH v/v supports enhanced ion flux under illumination, while the increased viscosity of the binary solvent system45 reduces proton mobility, slowing down current decay and extending capacitive retention. A 0.7 mM buffer solution in 40% MeOH v/v also showed a higher current than the same concentration in water (see Fig. S13b†).
Finally, we assessed the robustness of the system by subjecting the cell to extended operation (Fig. 7d). The VOC remained stable at around 240 mV, decreasing by 30 mV over 12 hours of continuous illumination. This drop aligns with the chemical stability of compound 1 near its pKGSa, where irreversible hydrolysis (τ1/2 ≈ 1 day, see Fig. S7†) gradually reduces the photoactive species in solution and thus the ΔpH over time. Overall, these electrochemical results highlight the advantages of using MCHs over excited-state photoacids in sunlight-to-protonic energy conversion, identifying the A/V ratio as a key parameter for optimizing system performance.
Conclusions
In conclusion, we have demonstrated that the (photo)chemical performance of compound 1 can be precisely tuned in aqueous MeOH mixtures. Specifically, we found that the pKGSa remains constant irrespective of solvation environment on account of a mutual increase of both pKa and Kc. On the contrary, pKMSa linearly decreases as a function of the MeOH content, resulting in a simultaneous increase in photoacidity from 3.6 to 4.1 pK units. The reason for this enhancement accounts to a decrease in the kinetic barrier of isomerization leading to the closed spiro form, SP. The quantum yield and the chemical stability of the system, however, were found not to increase with solvent composition as does the solubility, which dramatically increases by one order of magnitude passing from 0% to 40% MeOH v/v. As a result, photoactive buffer solutions whose pH can be set from 7 to 3 using 500 nm light can be made in a straightforward manner. These solutions can be precisely activated both in space and time, as witnessed by their implementation as liquid-state photoenergy harvesting systems. The results presented here are expected to open new horizons for applying MCHs in energy-related technologies.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
C. P. and D. J. K. conceived the project. C. B. synthesized and purified compound 1. X. D., C. B., and C. P. performed UV-Vis and pH jump analyses. X. D. performed the electrochemical experiments. All authors contributed to data elaboration and interpretation. C. P. and D. J. K. wrote the manuscript and all authors commented on it.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Swiss National Science Foundation (SNSF “Ambizione” PZ00P2_180008), and funding from the Italian Ministry of Education, Universities and Research (MUR project no. PGR19CX3UP). D. J. K. acknowledges the support from the Australian Research Council (DE210101618).
Notes and references
- N. Lane, BioEssays, 2017, 39, 1600217 CrossRef PubMed.
-
(a) P. Mitchell, Nature, 1961, 191, 144–148 CrossRef CAS PubMed;
(b) A. Hahn, J. Vonck, D. J. Mills, T. Meier and W. Kühlbrandt, Science, 2018, 360, eaat4318 CrossRef PubMed.
- P. Mitchell, Bioenergetics, 1972, 3, 5–24 CrossRef CAS PubMed.
- D. Oesterhelt, Angew Chem. Int. Ed. Engl., 1976, 15, 17–24 CrossRef CAS PubMed.
- J. P. Lanyi, Annu. Rev. Physiol., 2004, 66, 665–688 CrossRef CAS PubMed.
- J. P. Lanyi, Biochim. Biophys. Acta, 2006, 1757, 1012–1018 CrossRef CAS PubMed.
-
(a) W. Kühlbrandt, Nature, 2000, 406, 569–570 CrossRef PubMed;
(b) A. Royant, K. Edman, T. Ursby, E. Pebay-Peyroula, E. M. Landau and R. Neutze, Nature, 2000, 406, 645–648 CrossRef CAS PubMed;
(c) H. J. Sass, G. Büldt, R. Gessenich, D. Hehn, D. Neff, R. Schlesinger, J. Berendzen and P. Ormos, Nature, 2000, 406, 649–653 CrossRef CAS PubMed;
(d) S. Subramaniam and R. Henderson, Nature, 2000, 406, 653–657 CrossRef CAS PubMed.
- M. Sheves, A. Albeck, H. Friedman and M. Ottolenghi, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 3262–3266 CrossRef CAS PubMed.
- C. Zscherp, R. Schlesinger, J. Tittor, D. Oesterhelt and J. Heberle, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 5498–5503 CrossRef CAS PubMed.
- S. P. Balashov, E. S. Imasheva, R. Govindjee and T. G. Ebrey, Biophys. J., 1996, 70, 473–481 CrossRef CAS PubMed.
- J. P. Lanyi, J. Struct. Biol., 1998, 124, 164–178 CrossRef CAS PubMed.
- L. S. Brown and J. K. Lanyi, Proc. Natl. Acad. Sci. USA, 1996, 93, 1731–1734 CrossRef CAS PubMed.
- H. V. Westerhoff and Z. Dancsházy, Trends Biochem. Sci., 1984, 9, 112–117 CrossRef CAS.
- T. Kouyama, A. N. Kouyama and A. Ikegami, Biophys. J., 1987, 51, 839–841 CrossRef CAS PubMed.
- N. Calimet and G. M. Ullmann, J. Mol. Biol., 2004, 339, 571–589 CrossRef CAS PubMed.
- B. J. van Rotterdam, H. V. Westerhoff, R. W. Visschers, D. A. Bloch, K. J. Hellingwerf, M. R. Jones and W. Crielaard, Eur. J. Biochem., 2001, 268, 958–970 CrossRef CAS PubMed.
- T. P. Silverstein, J. Bioenerg. Biomembr., 2014, 46, 229–241 CrossRef CAS PubMed.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
-
(a) S. Borsley, D. A. Leigh and B. M. W. Roberts, Angew. Chem., Int. Ed., 2024, 63, e202400495 CrossRef CAS PubMed;
(b) S. Borsley, ChemSystemsChem, 2024, 6, e202400004 CrossRef CAS.
-
(a) K. Xiao, L. Chen, R. Chen, T. Heil, S. D. Cruz Lemus, F. Fan, L. Wen, L. Jiang and M. Antonietti, Nat. Commun., 2019, 10, 74 CrossRef CAS PubMed;
(b) Y. Qiu, Y. Feng, Q.-H. Guo, R. D. Astumian and J. F. Stoddart, Chem, 2020, 6, 195–1977 Search PubMed.
- S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC.
-
(a) G. Steinberg-Yfrach, P. A. Liddell, S.-C. Hung, A. L. Moore, D. Gust and T. A. Moore, Nature, 1997, 385, 239–241 CrossRef CAS;
(b) I. M. Bennett, H. M. Vanegas Farfano, F. Bogani, A. Primak, P. A. Liddell, L. Otero, L. Sereno, J. J. Silber, A. L. Moore, T. A. Moore and D. Gust, Nature, 2002, 420, 398–401 CrossRef CAS PubMed;
(c) T. G. Johnson and M. J. Langton, J. Am. Chem. Soc., 2023, 145, 27167–27184 CrossRef CAS PubMed.
- C. Berton and C. Pezzato, Eur. J. Org Chem., 2023, 26, e202300070 CrossRef CAS.
-
(a) Y. Liao, Acc. Chem. Res., 2017, 50, 1956–1964 CrossRef CAS PubMed;
(b) L. Wimberger, S. K. K. Prasad, M. D. Peeks, J. Andréasson, T. W. Schmidt and J. E. Beves, J. Am. Chem. Soc., 2021, 143, 20758–20768 CrossRef CAS PubMed;
(c) C. Kaiser, T. Halbritter, A. Heckel and J. Wachtveitl, Chem. – Eur. J., 2021, 27, 9160–9173 CrossRef CAS PubMed;
(d) Y. Liao, Phys. Chem. Chem. Phys., 2022, 24, 4116–4124 RSC.
-
(a) C. Berton, D. M. Busiello, S. Zamuner, R. Scopelliti, F. Fadaei-Tirani, K. Severin and C. Pezzato, Angew. Chem., Int. Ed., 2021, 60, 21737–21740 CrossRef CAS PubMed;
(b) L. Wimberger, J. Andréasson and J. Beves, Chem. Commun., 2022, 58, 5610–5613 RSC.
-
(a) S. Jansze, G. Cecot and K. Severin, Chem. Sci., 2018, 9, 4253–4257 RSC;
(b) R.-J. Li, C. Pezzato, C. Berton and K. Severin, Chem. Sci., 2021, 12, 4981–4984 RSC.
- C. Berton, D. M. Busiello, S. Zamuner, E. Solari, R. Scopelliti, F. Fadaei-Tirani, K. Severin and C. Pezzato, Chem. Sci., 2020, 11, 8457–8468 RSC.
- V. J. Périllat, C. Berton and C. Pezzato, Mater. Today Chem., 2022, 25, 100918 CrossRef.
- A. de Vries, K. Golozinina, M. Reiter, M. Salanne and M. R. Lukatskaya, Chem. Mater., 2024, 36, 1308–1317 CrossRef CAS PubMed.
- J.-C. Soetens and P. A. Bopp, J. Phys. Chem. B, 2015, 119, 8593–8599 CrossRef CAS PubMed.
- Z. Shi, P. Peng, D. Strohecker and Y. Liao, J. Am. Chem. Soc., 2011, 133, 14699–14703 CrossRef CAS PubMed.
- J. Liu, W. Tang, L. Sheng, Z. Du, T. Zhang, X. Su and S. X. Zhang, Chem.–Asian J., 2019, 14, 438–445 CrossRef CAS PubMed.
- L. A. Tatum, J. T. Foy and I. Aprahamian, J. Am. Chem. Soc., 2014, 136, 17438–17441 CrossRef CAS PubMed.
- I. Canals, J. A. Portal, E. Bosch and M. Rosés, Anal. Chem., 2000, 72, 1802–1809 CrossRef CAS PubMed.
- R. G. Bates, M. Paabo and R. A. Robinson, J. Phys. Chem., 1963, 67, 1833–1838 CrossRef CAS.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- Y. Shiraishi, M. Itoh and T. Hirai, Phys. Chem. Chem. Phys., 2010, 12, 13737–13745 RSC.
- M. Rosés, F. Rived and E. Bosch, J. Chromatogr. A, 2000, 867, 45–56 CrossRef PubMed.
- T. Halbritter, C. Kaiser, J. Wachtveitl and A. Heckel, J. Org. Chem., 2017, 82, 8040–8047 CrossRef CAS PubMed.
- J. Bae, H. Lim, J. Ahn, Y. H. Kim, M. S. Kim and I.-D. Kim, Adv. Mater., 2022, 34, 2201734 CrossRef CAS PubMed.
- A. Yucknovsky, Y. Shlosberg, N. Adir and N. Amdursky, Angew. Chem., Int. Ed., 2023, 62, e202301541 CrossRef CAS PubMed.
-
A. J. Bard, L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Incorporated, Hoboken, 2000 Search PubMed.
-
(a) S. Luo, W. White, J. M. Cardon and S. Ardo, Energy Environ. Sci., 2021, 14, 4961–4978 RSC;
(b) L. Schulte, W. White, L. A. Renna and S. Ardo, Joule, 2021, 5, 2380–2394 CrossRef CAS;
(c) G. S. Phun, R. Bhide and S. Ardo, Energy Environ. Sci., 2023, 16, 4593–4611 RSC.
- C. A. Gueymard, D. Myers and K. Emery, Sol. Energy, 2002, 73(6), 443–467 CrossRef.
- J. W. Thompson, T. J. Kaiser and J. W. Jorgenson, J. Chromatogr. A, 2006, 1134, 201–209 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04833d |
| ‡ Present address: Department of Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland. |
| § Present address: Laboratory for Macromolecular and Organic Chemistry (MOC), Department of Chemical Sciences, University of Padova, via Marzolo 1, 35131 Padova, Italy. Email: E-mail: cristian.pezzato@unipd.it. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Cesare
Berton‡
a,
Cesare
Berton‡