
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn endo-functionalized molecular cage for selective potentiometric determination of creatinine†
Yu
Lu‡
a,
Song-Meng
Wang‡
b,
Sui-Sui
He‡
a,
Qicheng
Huang‡
b,
Cheng-Da
Zhao
ac,
Shan
Yu
c,
Wei
Jiang§
 b,
Huan
Yao
*a,
Li-Li
Wang
*a and
Liu-Pan
Yang
*a
b,
Huan
Yao
*a,
Li-Li
Wang
*a and
Liu-Pan
Yang
*a
aSchool of Pharmaceutical Science, Hengyang Medical School, University of South China, Hengyang, Hunan 421001, China. E-mail: yaoh@usc.edu.cn; wangll@usc.edu.cn; yanglp@usc.edu.cn
bDepartment of Chemistry, Southern University of Science and Technology, Xueyuan Blvd 1088, Shenzhen, 518055, China
cThe Affiliated Nanhua Hospital, University of South China, Hengyang, Hunan 421001, China
First published on 22nd August 2024
Abstract
Potentiometric ion-selective electrodes (ISEs), which rely on selective and lipophilic ionophores, are commonly employed in clinical diagnostics. However, there are very limited specific ionophores for the detection of creatinine, a critical biomarker for renal function assessment. In the present research, we designed and synthesized an endo-functionalized cage, which is able to selectively bind the creatininium cation (Ka = 8.6 × 105 M−1) through the formation of multiple C–H⋯O and N–H⋯N hydrogen bonds and cation⋯π interactions. ISEs prepared with this host show a Nernstian response to creatinine and exhibit excellent selectivity and a low detection limit of 0.95 μM. In addition, the creatinine levels in urine or plasma samples determined by our sensor are consistent with those analyzed using enzymatic assay on a Cobas c702. The method is simple, fast and accurate, and amenable to clinical detection of creatinine levels.
Introduction
Kidney diseases are common clinical conditions and increasingly recognized global public health concerns.1 Currently, creatinine level measurement is a routine clinical test for diagnosing kidney disease and monitoring kidney function.2 Creatinine is a metabolic byproduct of phosphocreatine in the muscles, mainly cleared through glomerular filtration in the kidneys and excreted in urine during blood circulation.3 Normally, the serum creatinine level in the body remains relatively stable. However, when there is kidney dysfunction, creatinine accumulates in the bloodstream and thus leads to an increase in serum creatinine levels. Therefore, the determination of creatinine levels in plasma and urine is the most commonly used and fundamental indicator of kidney function in clinical practice.4 The creatinine test results' accuracy significantly impacts kidney function assessment, choice of treatment plans, treatment effectiveness monitoring, and prognosis prediction. This diagnostic method has been the gold standard for evaluation and management of chronic kidney disease for a century.5 Therefore, the development of rapid, efficient, user-friendly, and highly accurate creatinine testing methods is essential for the diagnosis and treatment of related diseases.6In 1886, Max Jaffé discovered that creatinine could react with picric acid in an alkaline solution, forming a measurable chromogenic compound using photometric techniques.7 The Jaffe reaction has been employed for creatinine detection for more than a century, but it is easily affected by proteins, glucose, bilirubin, and some drugs.8 Since 1980, enzymatic assays have been developed to enhance the specificity of creatinine measurement, and these approaches are increasingly employed in clinical detection settings.9 Besides, other bio-sensing methods have also been developed in recent years. However, these detection methods still suffer from some limitations, which hinder their practical application.10 Ion-selective electrodes (ISEs), as potentiometric-based sensors, have been commonly employed in clinical detection techniques benefitting from the development of artificial ionophores.11 However, the application of this technique for creatinine detection is extremely rare, primarily due to the scarcity of suitable ionophores.12 From 2016, several elegant applications of aryl-extended calix[4]pyrrole as the ionophores in the potentiometric determination of creatinine levels in serum and urine were reported by Ballester and co-workers.13 To date, this class of synthetic ionophores still stands alone as the only one capable of achieving accurate creatinine detection in biological fluids. Herein, developing affordable macrocyclic ionophores for creatinine detection remains of great clinical significance.
The creatininium cation has a pKa value of 4.8, thus the cationic species will be predominant at pH values below 4.8 (Fig. 1a). To achieve sensitive and selective creatinine sensing by ISEs, a tailored ionophore with strong and selective binding affinity for the creatininium cation is necessary. To fulfill this requirement, an ideal host should possess certain characteristics. Firstly, it should have a well-defined cavity that exhibits shape and charge complementarity with creatininium cations to ensure binding affinity. In addition, the decoration of appropriate binding sites inside the host cavity will enhance the selectivity. In recent years, the “endo-functionalized cavity” strategy has emerged as an effective approach for enhancing selectivity in molecular recognition.14 This strategy involves incorporating specific functional groups within the interior of a host, leading to exceptional selectivity in recognizing target molecules and further promising exciting advancements in spectroscopic analysis,15 biomedicine,16 and materials science.17 In this study, we reported a molecular cage that followed the concept of “endo-functionalized cavity” (Fig. 1a). The host exhibits high selectivity for binding creatininium cations, with a binding constant of 8.6 × 105 M−1, placing it at the forefront among synthetic hosts for creatininium cations. By utilizing the cage as an ionophore, we developed an ISE sensor capable of accurate determination of creatinine levels. This electrode demonstrated superiority in terms of selectivity, sensitivity, and other critical parameters, enabling accurate measurement of creatinine levels in both urine and serum samples.
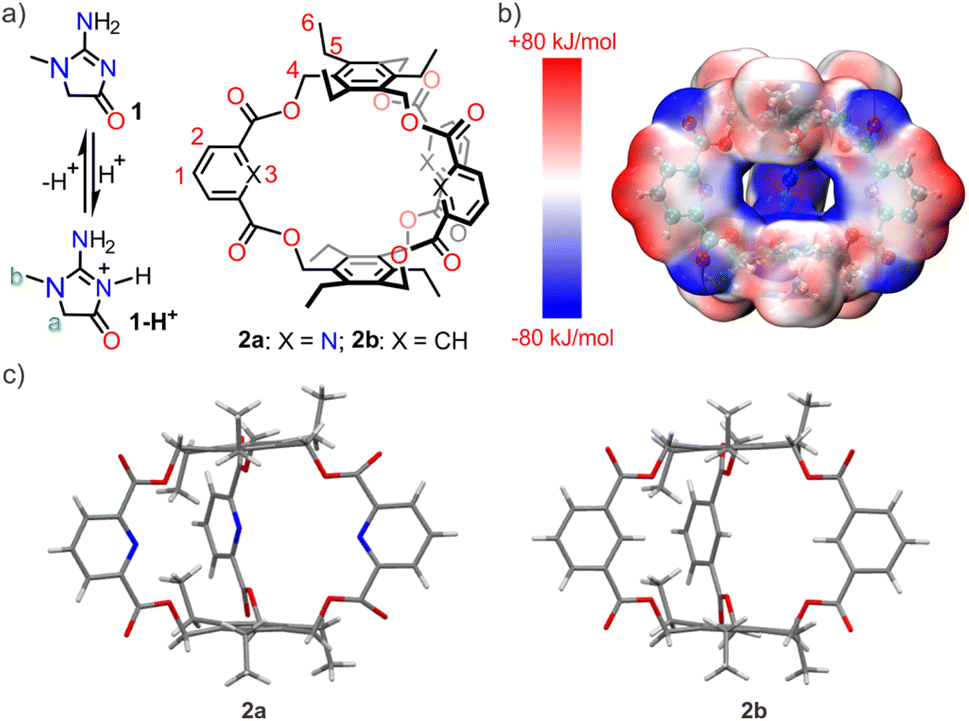 | ||
| Fig. 1 (a) Chemical structures of creatinine (1) and its protonated form (1-H+), and two cages 2a and 2b; (b) electrostatic potential surface of 2a calculated and visualized by Multiwfn 3.8 (dev) and VMD 1.9.3 programs after optimizing with DFT (ωB97XD/6-311+G(d,p)) in a vacuum; (c) X-ray single crystal structures of 2a and 2b. | ||
Results and discussion
Design and synthesis
Synthetic hosts that exhibit selective recognition abilities towards creatinine or its cationic species are extremely rare in the literature.13,18 In this study, we designed and synthesized an endo-functionalized cage (2a) tailored to the structural characteristics of the creatininium cation. The molecular cage exhibits the following features in its structure: (a) triethylbenzene is chosen as the top and bottom planes, creating an electron-rich environment in the cavity to match the charge of the creatininium cation. The calculated electrostatic potential surface (Fig. 1b) suggests that the cavity of 2a is rather electron-rich, providing a favorable electrostatic milieu capable of accommodating the electron-deficient creatininium cation. (b) The preorganization of triethylbenzene due to steric gearing19 may facilitate the synthesis, as a similar amide-linked cage (Anslyn's cage) for recognizing anions was synthesized in surprisingly high yield (40%).20 (c) Pyridine is selected as the sidewalls because the inwardly directed nitrogen atoms may serve as hydrogen bonding receptors. This not only enhances the binding strength but also ensures selectivity for the creatininium cation. (d) By incorporating relatively rigid ester groups as linkers, it is possible to create a pre-organized three-dimensional cavity, thereby avoiding the unfavorable effects of entropy during binding processes. (e) In comparison to pyridine, 2,6-ester substituted pyridine exhibits a lower pKa value, decreasing from 5.2 to approximately 2.0,21 which ensures that the endo-functionalized nitrogen-atoms remain deprotonated at lower pH values, thus they are able to act as hydrogen-bonding receptors. (f) The ester linker may get involved in binding when the oxygen atoms are inwardly directed.Ester-bridged cage 2a was synthesized from commercially available pyridine-2,6-dicarboxylic acid and 1,3,5-tris(bromomethyl)-2,4,6-triethylbenzene using carbonate-promoted reactions similar to those employed for ester naphthocage synthesis.22 By optimizing reaction conditions by changing the reaction time and choice of carbonate, the highest isolated yield for 2a reached 23% (Table S1,† entry 1) with K2CO3 as the base and a reaction time of 4 hours. This result suggests that the template effect of the potassium cation likely plays a significant role in the formation of 2a. For comparison, cage 2b featuring benzene sidewalls was synthesized using a similar procedure. Notably, its yield reached 42% with Na2CO3 as the base (Table S2,† entry 5), again benefiting from the template effect of alkali metal ions. The structure of 2a and 2b was characterized by NMR spectroscopy and HRMS spectrometry. The X-ray diffraction structure analysis reveals that the two hosts exhibit similar well-defined cavity structures (Fig. 1c). The primary difference lies in the atoms pointing towards the interior cavity, which may lead to differences in recognition performance.
Guest-binding properties
The working principle of ISEs is based on the analyte's distribution equilibrium at the membrane interface and the liquid phase. Therefore, the binding ability of the ionophores to the target analyte determines the usability of the ISE. In this regard, it is crucial for the molecular cage to effectively and selectively bind with the protonated species of creatinine for it to function as an ionophore in a creatinine ISE. As a result, we initially investigated the binding interaction between the molecular cages and creatinine ion through NMR and isothermal titration calorimetry (ITC). For the NMR and ITC experiments, the noncoordinating tetrakis[3,5-bis(trifluoromethyl)phenyl]borate anion (BArF−) was employed as the counterion for the creatininium cation in order to maintain solubility in non-polar solvents such as CH2Cl2. The binding of 2a with the creatininium cation can be evidenced through analysis of the 1H NMR spectra (Fig. 2a–c and S1†). When host 2a and the guest were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in CD2Cl2, significant chemical shifts of the NMR peaks on both the guest and the host were observed. Notably, the methyl protons b of creatininium exhibited an upfield shift of 0.63 ppm, indicating that the guest is encapsulated inside the host cavity. It should be noted that the methylene protons a on the guest displayed much smaller upfield shifts, which may be attributed to the formation of C–H⋯O hydrogen bonds with the ester linker and thus the shielding effect by the host cavity was balanced. Control experiments (Fig. S1†) employing excess guest or host support a slow exchange on the NMR timescale. No free guest or host was detected, indicating highly stable binding (Ka > 9.0 × 104 M−1). The binding affinity of the creatininium cation with cage 2a was then determined using ITC titrations, and the corresponding thermodynamic parameters, ΔH° and TΔS°, were also obtained (Fig. S2†). The association constant was found to be 8.6 × 105 M−1, mainly driven by enthalpic contributions with a slightly favorable entropic contribution. This association constant is the highest reported for synthetic hosts to the creatininium cation.
:1 ratio in CD2Cl2, significant chemical shifts of the NMR peaks on both the guest and the host were observed. Notably, the methyl protons b of creatininium exhibited an upfield shift of 0.63 ppm, indicating that the guest is encapsulated inside the host cavity. It should be noted that the methylene protons a on the guest displayed much smaller upfield shifts, which may be attributed to the formation of C–H⋯O hydrogen bonds with the ester linker and thus the shielding effect by the host cavity was balanced. Control experiments (Fig. S1†) employing excess guest or host support a slow exchange on the NMR timescale. No free guest or host was detected, indicating highly stable binding (Ka > 9.0 × 104 M−1). The binding affinity of the creatininium cation with cage 2a was then determined using ITC titrations, and the corresponding thermodynamic parameters, ΔH° and TΔS°, were also obtained (Fig. S2†). The association constant was found to be 8.6 × 105 M−1, mainly driven by enthalpic contributions with a slightly favorable entropic contribution. This association constant is the highest reported for synthetic hosts to the creatininium cation.
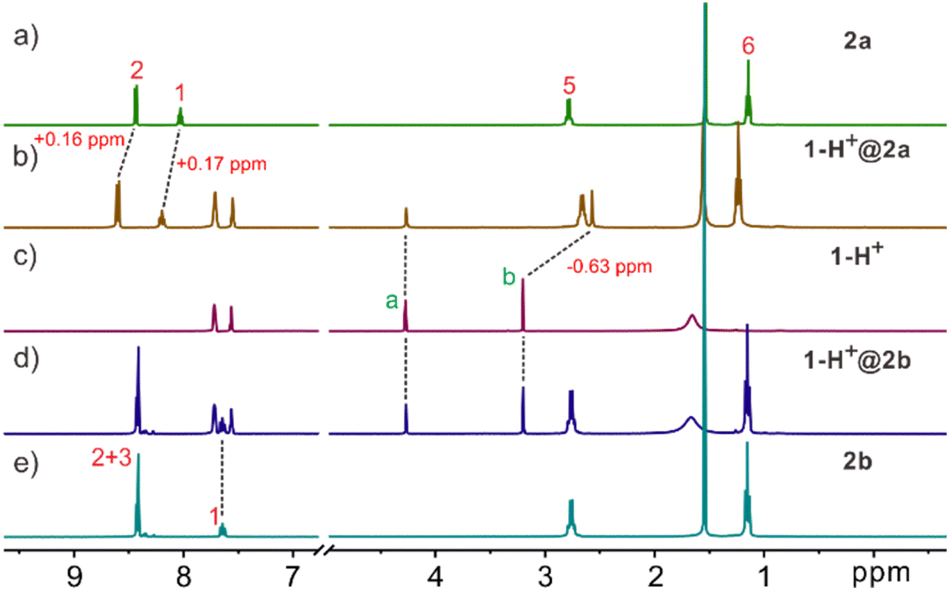 | ||
| Fig. 2 Partial 1H NMR spectra (500 MHz, CD2Cl2, 2.0 mM, 25 °C) of (a) 2a, (b) a 1:1 mixture of 2a and 1-H+, (c) 1-H+, (d) a 1:1 mixture of 2b and 1-H+, and (e) 2b. | ||
However, 1H-NMR spectra suggest that there is no obvious binding interaction between the creatininium cation and the molecular cage 2b with benzene as the sidewalls (Fig. 2c–e and S3†). We also synthesized Anslyn's cage according to ref. 20b and investigated its interaction with the creatininium cation by 1H NMR. The results showed that the interaction between the Anslyn's cage and creatininium cation is very weak (Fig. S4†). These results implied that the endo-functionalized sites play an indispensable role in the binding process, and subtle variations of endo-functionalized sites result in significant differences in recognition capabilities. This phenomenon parallels selective recognition by biological receptors, where a single atom difference can lead to substantial changes in recognition behavior, thereby influencing function.23 Additionally, through comparing the binding with possible interfering substances or analogs such as tetramethyl ammonium (core structure of choline and acetylcholine, Fig. S5†), N-phenylacetamide (an aromatic compound with an H-bond donor, Fig. S6†), and 5-N-propyluracil (a uracil analog, Fig. S7†), it was discovered that 2a exhibits very high selectivity in recognizing the creatininium cation.
To reveal the binding geometry between the endo-functionalized cage and the creatininium cation, the complex structures in a vacuum (Fig. 3) and in water (Fig. S8†) were calculated using density functional theory (DFT) at the ωB97XD/6-311+G(d,p) level of theory.24 The results indicated that both the energy-minimized structures in the vacuum and in water share a similar sandwich-like structure where the creatininium cation is encased between two aromatic planes. The peripheral hydrogen atoms are stabilized by the endo-functionalized sidewalls through the formation of multiple intramolecular N–H⋯N or C–H⋯O hydrogen bonds, with bond lengths ranging from 2.1 Å to 2.4 Å. Visual analysis of intermolecular interactions (independent gradient models based on Hirshfeld partition of molecular density, IGMH)25 further supports the presence of multiple hydrogen bonds, and cation⋯π interactions are also involved in the binding. Therefore, the combination of charge and size complementation, along with the cooperative effects of these weak interactions, results in a highly selective interaction between the endo-functionalized cage and the creatininium cation. For comparison, the energy-minimized structure the complex structure of 2b with the creatininium cation (Fig. S9†) indicates that 2b cannot form multiple effective hydrogen bonds with the guest as effectively as 2a does. Meanwhile, the packing coefficient of the 2a complex is closer to the ideal 55% (Fig. S10 and S11 and Table S3†),26 which further supports the superior binding of creatininium cations within the cavity of 2a.
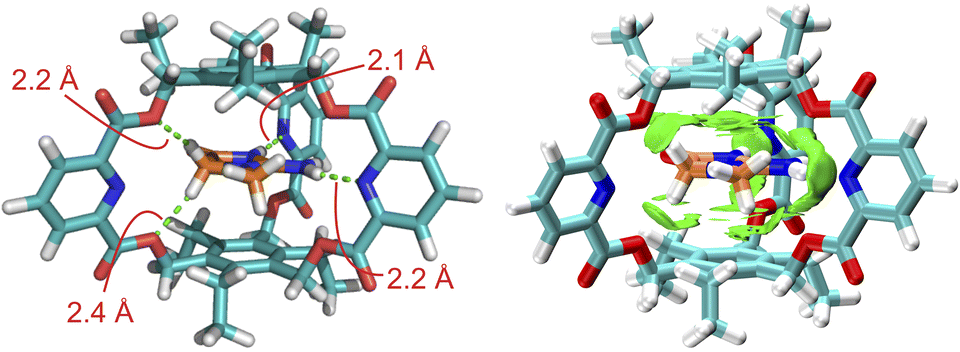 | ||
| Fig. 3 Energy-minimized structures and visual analysis of intermolecular interactions (IGMH) of 1-H+@2a in a vacuum obtained by DFT (wB97XD/6-31G(d)) calculations and visualized by Multiwfn 3.8 (dev) and VMD 1.9.3 programs. The dotted lines indicate the hydrogen bonds. | ||
Fabrication and analytical performance of membrane sensors
ISEs are frequently fabricated by incorporating PVC-based electrode membranes doped with plasticizers, ion exchangers and ionophores. Ion exchangers and ionophores are responsible for augmenting selectivity, whereby ionophores should exhibit a superior binding affinity towards the targeted analyte, subsequently lowering the free energy required for the analyte's permeation across the aqueous membrane interface. In this study, we prepared three membrane electrodes to investigate the performance of endo-functionalized cages as ionophores for creatinine. The electrode membrane preparation method was adopted from previous literature,27 employing PVC as the polymer matrix, ortho-nitrophenyl octyl ether (o-NPOE) as the plasticizer, and potassium tetrakis[3,5-bis(trifluoromethyl)phenyl] borate as the ion exchanger. 2a or 2b was used as the ionophore, with a “blank” sensor serving as a control without any ionophores. To determine the most suitable creatinine ionophore, Bu4N+ was employed as a reference compound given its wide application in potentiometry for characterizing ionophores owing to its high hydrophobicity.28 The ionophore that exhibited the optimal response could be identified by comparing the responses of the creatininium cation from different ionophores. Fig. 4a depicts the potential differences, measured in terms of electromotive force (EMF) values, between the ion-selective electrodes (ionophore-free, containing 2a, and containing 2b) for the creatininium cation relative to Bu4N+. The ion-selective electrode containing 2a demonstrated the smallest potential difference (54 mV) compared to the electrode containing 2b (422 mV) and the ionophore-free electrode (441 mV). This result confirms that 2a possesses a stronger affinity for creatinine than 2b.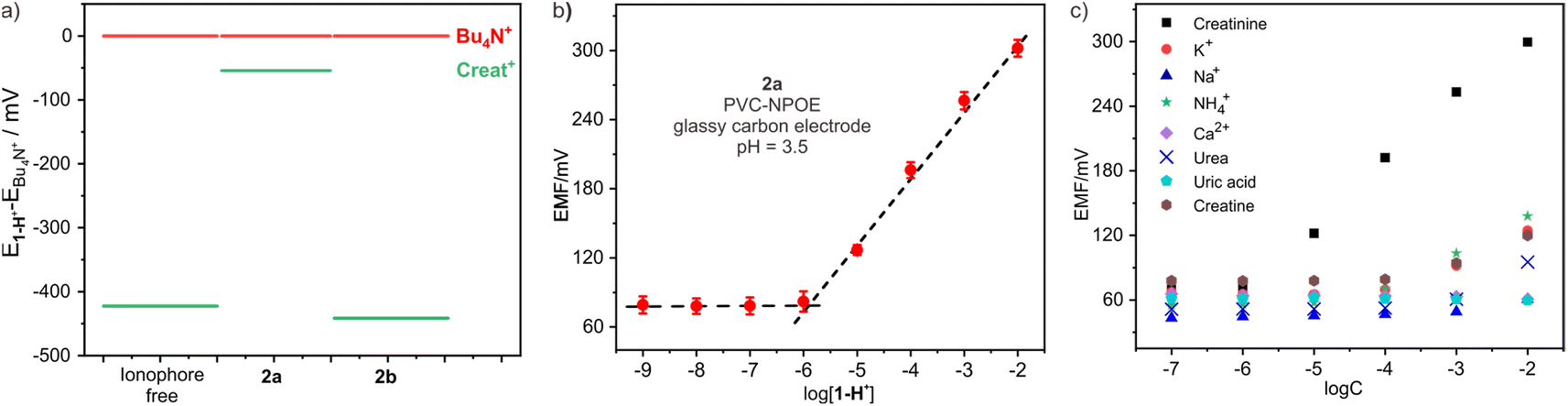 | ||
| Fig. 4 (a) The EMF values of ISEs containing diverse ionophores exposure to 1.0 mM creatininium cation subtracted the corresponding EMF value exposure to 1.0 mM Bu4N+; (b) calibration curves of the 2a-based electrode to 1-H+; (c) the selectivity for 1-H+ over Na+, K+, NH42+, Ca2+, Mg2+, urea, uric acid, and creatine of the electrodes containing 2a. | ||
After determining the optimal ionophore, we optimized the selection of plasticizer, electrode and working pH. The results revealed that o-NPOE exhibited significantly higher sensitivity as a plasticizer compared to dioctyl sebacate (Fig. S13†). Furthermore, the sensitivity of the glassy carbon electrode was noticeably superior to that of the gold and platinum electrodes (Fig. S14†). During the pH optimization process, we observed a potential response to pH changes in the buffer solution, with the maximum electromotive force occurring at pH 3.5 (Fig. S15†). Therefore, the optimal working pH was determined to be 3.5. For the subsequent experiments, a 10 mM acetic acid/sodium acetate buffer with a pH of 3.5 was used.
Fig. 4b illustrates the measured EMF response at different creatinine concentrations for the electrode doped with ionophore 2a. The limit of detection (LOD) for the electrode was determined to be 0.95 μM, which is comparable to the reported value.13a The calibration curve displayed a linear relationship, with a slope of 58.2 mV per decade, consistent with the expected Nernstian slope and even slightly better than that of Ballester's sensor.13a The linear range is from 1.0 μM up to 10 mM, which fully covers the concentration range of creatinine in bodily fluids, enabling reliable analysis of real biological samples such as urine and blood. In contrast, the sensor without ionophore or 2b-based sensor both shows lower sensitivity (Fig. S16†). To ensure the reliability and longevity of ISEs, ionophores must exhibit hydrophobic properties to prevent undesired leakage from the electrode membrane into the sample solution. Host 2a exhibits exceptional hydrophobicity, thus the electrode potentials in a solution containing 0.1 mM creatinine were very stable over time, indicating the absence of any leakage of the ionophore from the electrode membrane (Fig. S17†).
Interference studies
One obstacle in the accurate determination of creatinine levels in real samples through potentiometric measurements is to avoid interference of cations commonly seen in biological samples with high concentrations (Na+, K+, NH4+ and Ca2+). This is why only very limited creatinine potentiometric sensors are suitable for practical applications. To address this issue, we evaluated the influence of these common cations on the sensor and calculated their selectivity coefficients. As shown in Fig. 4c, the sensor containing 2a showed excellent selectivity against these cations. The stability constant of 2a with creatininium cations in the electrode membrane in the buffer medium was determined to be (4.9 ± 0.1) × 106 M−1 through the Stefanova–Mokrov method.29 The selectivity coefficients of the 2a-based electrode for creatininium over other cations were all over 3.0 (Table S4†), meaning that it is nearly 1000 times more selective for creatininium over other cations. Additionally, the presence of high concentrations of neutral substances such as urea, uric acid, creatinine, and glucose in urine or blood can also significantly compromise the accuracy of detecting authentic samples during testing, thus we also calculated the selectivity coefficients of these compounds, finding that they were also lower by more than three orders of magnitude compared to creatininium (Table S4†). When compared with Ballester's sensor,13 we have found that our sensor has selectivity parameters that are essentially comparable to or even better than those of his sensor (Table S5†). This indicates that our sensor holds promising potential for real sample detection applications.Analysis of real samples
To accurately measure creatinine levels in real samples, it is essential to have a sensor with high selectivity, as well as the ability to resist non-specific interference, such as biological fouling caused by the adhesion of matrix components on the electrode membrane. This can lead to serious signal drift and permanent electrode damage if left unchecked. Non-specific interference significantly affects the accuracy of creatinine level detection. To overcome this, common methods include diluting the sample before measurement. In our study, we utilized this technique to investigate the measurement of creatinine in actual urine samples. We first measured the creatinine content of a real urine sample using a standard enzymatic method using a Cobas c702, which yielded a result of 21.3 mM. We then diluted the sample three times with a pH 3.5 buffer, using dilutions of 1:10, 1:50, and 1:100. Between each sample, a 1.0 mM creatinine standard solution was measured to check the accuracy of the sensor. The obtained potential time trajectory showed that the signal from our sensor was relatively stable under different dilutions, with a drift of less than 2 mV min−1, and a high recovery rate of up to nearly 100% (Fig. 5a). Moreover, the signal from the 1.0 mM creatinine standard indicated that the sensor's response did not change during the experiment, demonstrating that the accuracy of the sensor is ensured when continuously testing different samples. Subsequently, we used the dilution method to measure real biological samples.
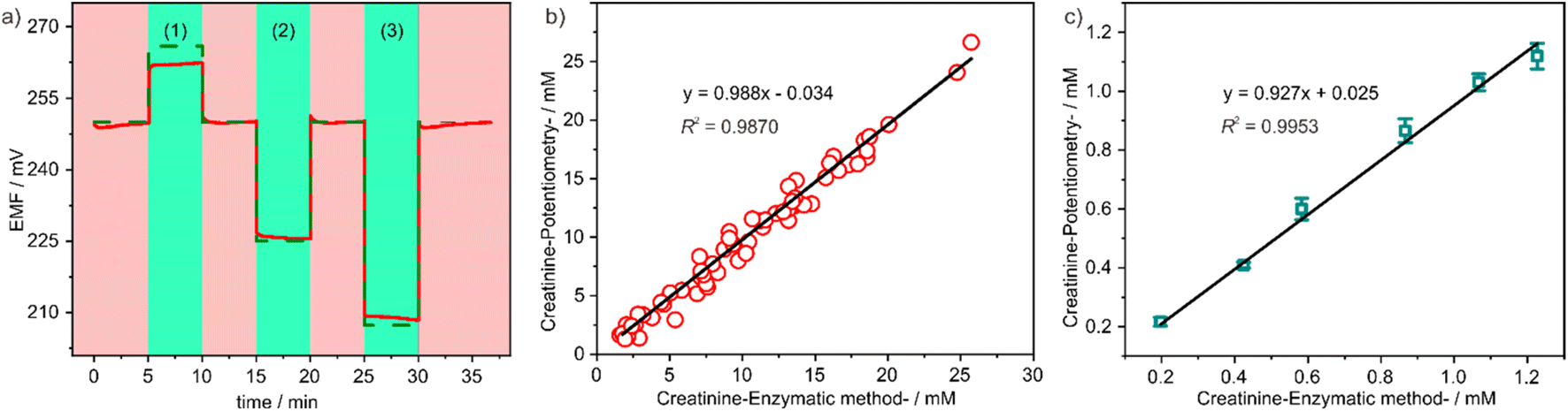 | ||
| Fig. 5 (a) Expected (dashed line) and experimental (full line) potentiometric response obtained for 1:10 (1), 1:50 (2) and 1:100 (3) dilutions of urine (red) for the sensor. The baseline in between samples (green) corresponds to a 1.0 mM creatinine standard; creatinine values obtained in several urine ((b), N = 60) and serum ((c), N = 6) samples with the creatinine ion-selective electrode compared with the standard colorimetric standard method – the enzymatic method. | ||
In order to assess the accuracy of the sensor in real sample detection, a total of 60 urine samples and 6 serum samples were randomly collected at the Affiliated Nanhua Hospital, University of South China. The creatinine levels of these samples were firstly quantified using a Cobas c702 instrument. The urine samples exhibited creatinine concentrations ranging from 2.0 mM to 28 mM, while the serum samples displayed creatinine concentrations ranging from 0.2 mM to 1.2 mM. Consequently, the urine samples were diluted 100-fold and the serum samples were diluted 10-fold before measurement using our potentiometric sensor. The obtained measurement results were subsequently compared with those obtained from the Cobas c702 instrument. As illustrated in Fig. 5b and c, a strong linear correlation was observed, with a slope close to 1 and an intercept close to 0. This finding signifies the effectiveness of our method in accurately analysing creatinine levels in actual biological samples.30
Conclusions
In summary, we have designed and synthesized an endo-functionalized molecular cage to target the structural features of creatinine cations. The cage consists of benzene rings as the upper and lower planes, connected by ester bonds to three pyridine side walls, forming a three-dimensional cavity with an electron-rich environment. This molecular cage not only exhibits a precise charge and shape complementarity with the creatinine cation but also provides multiple hydrogen bonding sites inside the cavity to ensure strong and selective binding with the guest. The binding affinity between the cage and creatinine cations reached a remarkable value of 8.6 × 105 M−1.The subtle variations at endo-functionalized sites yield significant differences in recognition capabilities, which is reminiscent of the selective recognition seen in biological receptors. A mere single-atom difference can greatly alter recognition behavior and thus impact biological functions.23 This study further highlights the efficacy of the endo-functionalized macrocyclic design concept in the selective recognition of relevant disease biomarkers.
In application, we employed this molecular cage to fabricate an ion-selective electrode for creatinine detection, which exhibited comparable or even superior performance in terms of detection range, sensitivity, and selectivity compared to Ballester's sensor, which is currently the only ISE sensor capable of accurately measuring creatinine levels in real biological samples. Furthermore, our sensor successfully detected creatinine levels in urine and plasma, exhibiting excellent agreement with the enzymatic methods from the Cobas c702 instrument. Notably, our ionophore offers an easy synthesis route and the proposed detection method is convenient and precise, representing a highly promising approach for clinical creatinine measurement.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
W. Jiang and L.-P. Yang conceived the project with contributions from L.-L. Wang and H. Yao. Y. Lu, Q.-C. Huang, C.-D. Zhao and S. Yu performed the experiments with help from S.-S. He. S.-M. Wang performed the DFT calculations. L.-P. Yang, L.-L. Wang and H. Yao wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
In memory of Prof. Wei Jiang, who initiated this research but sadly passed away on December 25th, 2022. This research was financially supported by the National Natural Science Foundation of China (No. 21801125, 22174059 and 22201128), Hunan Provincial Natural Science Foundation of China (No. 2022JJ40363 and 2022JJ40365), the Young Science and Technology Innovation Program of Hunan Province (No. 2022RC1230), and China Post-doctoral Science Foundation (No. 2022M721542). We are grateful for the technical support from SUSTech-CRF and the Center for Computational Science.Notes and references
- (a) R. J. Glassock, D. G. Warnock and P. Delanaye, Nat. Rev. Nephrol., 2017, 13, 104–114 CrossRef CAS PubMed; (b) E. F. Carne, Nat. Rev. Nephrol., 2020, 16, 251 CrossRef PubMed; (c) P. Cockwell and L. Fisher, Lancet, 2020, 395, 662–664 CrossRef PubMed.
- (a) M. Wyss and R. Kaddurah-Daouk, Physiol. Rev., 2000, 80, 1107–1213 CrossRef CAS PubMed; (b) S. Hallan, A. Åsberg, M. Lindberg and H. Johnsen, Am. J. Kidney Dis., 2004, 44, 84–93 CrossRef PubMed; (c) R. Cánovas, M. Cuartero and G. A. Crespo, Biosens. Bioelectron., 2019, 130, 110–124 CrossRef PubMed.
- (a) K. Kashani, M. H. Rosner and M. Ostermann, Eur. J. Intern. Med., 2020, 72, 9–14 CrossRef CAS PubMed; (b) L. Kazak and P. Cohen, Nat. Rev. Endocrinol., 2020, 16, 421–436 CrossRef PubMed.
- A. S. Levey, S. M. Titan, N. R. Powe, J. Coresh and L. A. Inker, Clin. J. Am. Soc. Nephrol., 2020, 15, 1203–1212 CrossRef CAS PubMed.
- A. S. Levey, C. Becker and L. A. Inker, J. Am. Med. Assoc., 2015, 313, 837–846 CrossRef CAS PubMed.
- E. Mohabbati-Kalejahi, V. Azimirad, M. Bahrami and A. Ganbari, Talanta, 2012, 97, 1–8 CrossRef CAS PubMed.
- M. Jaffe, Z. Physiol. Chem., 1886, 10, 391–400 Search PubMed.
- (a) K. Spencer, Ann. Clin. Biochem., 1986, 23, 1–25 CrossRef CAS PubMed; (b) J. A. Weber and A. P. van Zanten, Clin. Chem., 1991, 37, 695–700 CrossRef CAS PubMed.
- (a) N. Greenberg, W. L. Roberts, L. M. Bachmann, E. C. Wright, R. N. Dalton, J. J. Zakowski and W. G. Miller, Clin. Chem., 2012, 58, 391–401 CrossRef CAS PubMed; (b) E. J. Erlandsen and E. Randers, Scand. J. Clin. Lab. Invest., 2018, 78, 490–495 CrossRef PubMed.
- (a) R. Cánovas, M. Cuartero and G. A. Crespo, Biosens. Bioelectron., 2019, 130, 110–124 CrossRef PubMed; (b) C. S. Pundir, P. Kumar and R. Jaiwal, Biosens. Bioelectron., 2019, 126, 707–724 CrossRef CAS PubMed.
- (a) E. Bakker, P. Buhlmann and E. Pretsch, Chem. Rev., 1997, 97, 3083–3132 CrossRef CAS PubMed; (b) E. Zdrachek and E. Bakker, Anal. Chem., 2019, 91, 2–26 CrossRef CAS PubMed; (c) R. Hein, P. D. Beer and J. J. Davis, Chem. Rev., 2020, 120, 1888–1935 CrossRef CAS PubMed; (d) Y. Shao, Y. Ying and J. Ping, Chem. Soc. Rev., 2020, 49, 4405–4465 RSC; (e) E. Zdrachek and E. Bakker, Anal. Chem., 2021, 93, 72–102 CrossRef CAS PubMed.
- P. M. Kelly, R. Kataky, D. Parker and A. F. Patti, J. Chem. Soc., Perkin Trans. 2, 1995, 1955–1963 RSC.
- (a) T. Guinovart, D. Hernández-Alonso, L. Adriaenssens, P. Blondeau, M. Martínez-Belmonte, F. X. Rius, F. J. Andrade and P. Ballester, Angew. Chem., Int. Ed., 2016, 55, 2435–2440 CrossRef CAS PubMed; (b) T. Guinovart, D. Hernández-Alonso, L. Adriaenssens, P. l. Blondeau, F. X. Rius, P. Ballester and F. J. Andrade, Biosens. Bioelectron., 2017, 87, 587–592 CrossRef CAS PubMed; (c) M. M. Erenas, I. Ortiz-Gómez, I. de Orbe-Payá, D. Hernández-Alonso, P. Ballester, P. Blondeau, F. J. Andrade, A. Salinas-Castillo and L. F. Capitán-Vallvey, ACS Sens., 2019, 4, 421–426 CrossRef CAS PubMed; (d) A. F. Sierra, D. Hernández-Alonso, M. A. Romero, J. A. González-Delgado, U. Pischel and P. Ballester, J. Am. Chem. Soc., 2020, 142, 4276–4284 CrossRef CAS PubMed; (e) L. Escobar, Q. Sun and P. Ballester, Acc. Chem. Res., 2023, 56, 500–513 CrossRef CAS PubMed; (f) A. Corba, A. F. Sierra, P. Blondeau, B. Giussani, J. Riu, P. Ballester and F. J. Andrade, Talanta, 2022, 246, 123473 CrossRef CAS PubMed.
- (a) L. Adriaenssens and P. Ballester, Chem. Soc. Rev., 2013, 42, 3261–3277 RSC; (b) L.-P. Yang, X. Wang, H. Yao and W. Jiang, Acc. Chem. Res., 2020, 53, 198–208 CrossRef CAS PubMed; (c) A. P. Davis, Chem. Soc. Rev., 2020, 49, 2531–2545 RSC; (d) C.-D. Zhao, H. Yao, S.-Y. Li, F. Du, L.-L. Wang and L.-P. Yang, Chin. Chem. Lett., 2023, 35, 108879 CrossRef.
- (a) L.-L. Wang, Z. Chen, W.-E. Liu, H. Ke, S.-H. Wang and W. Jiang, J. Am. Chem. Soc., 2017, 139, 8436–8439 CrossRef CAS PubMed; (b) L.-L. Wang, M. Quan, T.-L. Yang, Z. Chen and W. Jiang, Angew. Chem., Int. Ed., 2020, 59, 23817–23824 CrossRef CAS PubMed.
- Y.-L. Ma, C. Sun, Z. Li, Z. Wang, J. Wei, Q. Cheng, L.-S. Zheng, X.-Y. Chang, K. Li, R. Wang and W. Jiang, CCS Chem., 2021, 4, 1977–1989 CrossRef.
- (a) H. Ke, L.-P. Yang, M. Xie, Z. Chen, H. Yao and W. Jiang, Nat. Chem., 2019, 11, 470–477 CrossRef CAS PubMed; (b) L.-P. Yang, H. Ke, H. Yao and W. Jiang, Angew. Chem., Int. Ed., 2021, 60, 21404–21411 CrossRef CAS PubMed; (c) J.-N. Jin, X.-R. Yang, Y.-F. Wang, L.-M. Zhao, L.-P. Yang, L. Huang and W. Jiang, Angew. Chem., Int. Ed., 2021, 60, 21404–21411 CrossRef PubMed.
- T. W. Bell, Z. Hou, Y. Luo, M. G. B. Drew, E. Chapoteau, B. P. Czech and A. Kumar, Science, 1995, 269, 671–674 CrossRef CAS PubMed.
- G. Hennrich and E. V. Anslyn, Chem.–Eur. J., 2002, 8, 2219–2224 Search PubMed.
- (a) A. P. Bisson, V. M. Lynch, M.-K. C. Monahan and E. V. Anslyn, Angew. Chem., Int. Ed., 1997, 36, 2340–2342 CrossRef CAS; (b) J. C. Lauer, A. S. Bhat, C. Barwig, N. Fritz, T. Kirschbaum, F. Rominger and M. Mastalerz, Chem.–Eur. J., 2022, 28, e202201527 CrossRef CAS.
- K. C. Ong, B. Douglas and R. A. Robinson, J. Chem. Eng. Data, 1966, 11, 574–576 CrossRef CAS.
- H. Chai, L.-P. Yang, H. Ke, X.-Y. Pang and W. Jiang, Chem. Commun., 2018, 54, 7677–7680 RSC.
- T. D. H. Bugg, G. D. Wright, S. Dutka-Malen, M. Arthur, P. Courvalin and C. T. Walsh, Biochemistry, 1991, 30, 10408–10415 CrossRef CAS PubMed.
- (a) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS; (b) J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- (a) T. Lu and Q. Chen, J. Comput. Chem., 2022, 43, 539–555 CrossRef CAS PubMed; (b) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- S. Mecozzi and J. Rebek Jr, Chem.–Eur. J., 1998, 4, 1016–1022 CrossRef CAS.
- Q. Chen, L.-P. Yang, D.-H. Li, J. Zhai, W. Jiang and X. Xie, Sens. Actuators, B, 2021, 326, 128836 CrossRef CAS.
- A. Spath and B. Konig, Beilstein J. Org. Chem., 2010, 6, 32 Search PubMed.
- (a) Y. Mi and E. Bakker, Anal. Chem., 1999, 71, 5279–5287 CrossRef CAS PubMed; (b) M. M. Shultz, O. K. Stefanova, S. B. Mokrov and K. N. Mikhelson, Anal. Chem., 2002, 74, 510–517 CrossRef CAS.
- One may wonder about the LOD of our sensor in the real urine and serum samples. However, considering the inherent variability of creatinine levels in blood and urine, precise lower detection limits are challenging to ascertain. Nevertheless, experiments using real samples diluted 100-fold for urine and 10-fold for blood consistently yield reliable detection results, suggesting that detection limits in blood and urine should not exceed 20 μM, which could be adequate for the analysis of real biological samples.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2323488 and 2360598. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04950k |
| ‡ These authors contributed equally. |
| § Deceased (2022.12). |
| This journal is © The Royal Society of Chemistry 2024 |