
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEasy access to amphiphilic nitrogenous block copolymers via switchable catalysis†
Xue
Liang
 a,
Jiachen
Lv
a,
Hongru
Qiang
a,
Jiahui
Li
a,
Wenli
Wang
*a,
Jianzhong
Du
*bcd and
Yunqing
Zhu
*a
a,
Jiachen
Lv
a,
Hongru
Qiang
a,
Jiahui
Li
a,
Wenli
Wang
*a,
Jianzhong
Du
*bcd and
Yunqing
Zhu
*a
aDepartment of Polymeric Materials, School of Materials Science and Engineering, Tongji University, 4800 Caoan Road, Shanghai 201804, China. E-mail: 1019zhuyq@tongji.edu.cn
bDepartment of Gynaecology and Obstetrics, Shanghai Key Laboratory of Anesthesiology and Brain Functional Modulation, Clinical Research Center for Anesthesiology and Perioperative Medicine, Translational Research Institute of Brain and Brain-Like Intelligence, Shanghai Fourth People's Hospital, School of Medicine, Tongji University, Shanghai 200434, China. E-mail: jzdu@tongji.edu.cn
cKey Laboratory of Advanced Civil Engineering Materials of Ministry of Education, School of Materials Science and Engineering, Tongji University, 4800 Caoan Road, Shanghai 201804, China
dSchool of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China
First published on 15th October 2024
Abstract
A key challenge in polymer synthesis is to develop new methods that enable block copolymers to be prepared from mixed monomer feedstock. The emerging switchable polymerization catalysis can generate block copolymers with well-defined structures and tunable properties from monomer mixtures. However, constrained by the reactivity of monomers and the incompatibility of different polymerization mechanisms, this method is usually confined to oxygenated monomers. In this work, the switchable polymerization was successfully applied to nitrogenous monomers for the first time, achieving the efficient copolymerization of N-substituted N-carboxyanhydrides (NNCAs) with epoxides and cyclic anhydrides. This leads to easy access towards amphiphilic nitrogenous copolymers, such as polyester-b-polypeptoids. Density functional theory calculations demonstrated that the reaction of cyclic anhydrides with the alkoxide terminal is thermodynamically more favorable than that of NNCAs. Characterization, using nuclear magnetic resonance spectroscopy, size exclusion chromatography and in situ infrared spectroscopy, has confirmed the well-defined block structure of the obtained copolymers. This switchable polymerization strategy is applicable to a range of monomer mixtures with different oxygenated monomers and NNCAs, providing a highly efficient synthetic route towards nitrogenous block copolymers. Most importantly, the easily accessed amphiphilic polyester-b-polypeptoids demonstrated excellent anti-protein adsorption capabilities and barely any cytotoxicity, showing great potential in the field of biomedicine.
Introduction
Block copolymers, consisting of polymer blocks with different properties, can form ordered structures at the nanoscale by precisely controlling the composition and sequence of blocks.1–3 This imparts novel characteristics to the materials, such as stimulus responsiveness, self-healing capability, and biocompatibility.4–6 Additionally, due to the presence of heteroatoms, such as oxygen, nitrogen, sulfur and silicon, in the main chain, heterochain block polymers exhibit excellent thermal stability, good mechanical properties, biocompatibility, and degradability.7–9 Therefore, they are widely used as the thermoplastic elastomers, drug delivery vehicles, membranes, lithography materials, and mesoporous materials.10Switchable polymerization based on ring-opening polymerization (ROP) and ring-opening copolymerization (ROCOP) can construct block copolymers with complex chain structures in a ‘one-pot/one-step’ process from monomer mixtures, using a single catalytic system.9,11–16 Usually, a spontaneous switch between different catalysis cycles occurs after the complete consumption of the first monomer/monomer pairs. Therefore, compared to the traditional ‘two-pot/two-step’ and ‘one-pot/two-step’ methods for making block copolymers, switchable polymerization can easily reach well-defined block copolymers, while avoiding complex and costly modification/purification procedures. Thus, it is simple and highly industrially valuable.17–22 However, due to the strict limitations of monomer reactivity ratios and selectivity of the initiators/catalysts, it is very challenging to synthesize block copolymers with highly diverse structures and properties, using the switchable polymerization methods.22–25 Currently, switchable polymerization is mainly applicable to oxygenated monomers, such as epoxides, cyclic anhydrides, lactones, O-carboxyanhydrides, as well as CO2 or COS. This leads to the limited formation of oxygenated blocks, including polyethers, poly(thio)esters, or poly(thio)carbonates.20 Few studies have reported the one-pot synthesis of nitrogen- or sulfur-containing block copolymers via switchable polymerization. For example, polyester-b-poly(ester-alt-thioester)s,26 poly(episulfide-alt-isothiocyanate)-b-polythioethers,27 polyether-b-poly(ester amide)s28 or poly(ester amide)-b-polyesters.29 Therefore, expanding the monomer scope beyond oxygenated ones, and developing block copolymers with diverse structures and properties are the key issues to be addressed for switchable polymerization.
Polypeptoids are an important class of nitrogenous polymers, and the ROP of N-substituted N-carboxyanhydrides (NNCAs) or N-substituted N-thiocarboxyanhydrides (NNTAs) using primary amine initiators is the primary method for synthesizing polypeptoids.30–33 Recently, new catalysts have been developed for preparing polypeptoids with complex structures or high molecular weights, such as transition metals34 and carboxylic acids.35 Owing to the excellent biocompatibility and tunable structures of polypeptoids, a wide range of polypeptoid-based block copolymers—such as polypeptoid-b-polypeptides, polyether-b-polypeptoids, and polyester-b-polypeptoids—have been synthesized using various methods. These methods, based on the ROP of NNCA or NNTA, include sequential polymerization, the use of macroinitiators or bifunctional initiators, and chemical ligation strategies.33,36–39 Furthermore, diverse morphologies of nano-assemblies can be prepared using polymerization-induced self-assembly (PISA) or crystallization-driven self-assembly (CDSA) for various pharmaceutical and biomedical applications.40–45 However, the synthesis of these polypeptoid-based block copolymers still relies on multi-step procedures, which including multiple feedings, as well as separation and purification of the initial block polymer. These time-consuming procedures significantly hinder their industrialization and clinical application. Therefore, it is of great value to develop switchable polymerization method, enabling easy access towards polypeptoid-based nitrogenous block copolymers.
During the switchable catalysis of oxygenated monomers, hydroxyl/alkoxide groups are usually able to initiate different monomers, ensuring successful propagation and switching. However, the relatively weak nucleophilicity of these groups makes them less prone to initiating the ROP of NNCAs.46 This issue restricts the adaptability of switchable polymerization on to NNCA monomers. To address this issue, in this work, we applied the triethylborane/bis(triphenylphosphine)iminium chloride (TEB/PPNCl) catalytic system to a mixed monomer feedstock of NNCAs, epoxides and cyclic anhydrides. This led to the successful switchable polymerization, resulting in polyester-b-polypeptoids with controlled molecular weight, narrow molecular weight distribution, and a rich diversity of structures (Fig. 1). It is discovered that TEB can activate not only the NNCA monomer, but also the alkoxide groups, enabling successful ROP of NNCAs once the anhydrides are completely consumed. TEB/PPNCl catalysis system shows high selectivity in monomers and excellent control for two polymerization cycles during the ‘one-pot/one-step’ process. Furthermore, the obtained amphiphilic polyester-b-polypeptoids can easily self-assemble into nanoparticles with controlled morphologies, which further exhibit excellent anti-protein adsorption capabilities and barely no cytotoxicity for potential applications in biomedicine.
 | ||
| Fig. 1 One-pot synthesis of polyester-b-polypeptoids via the switchable polymerization of NNCAs, epoxides and cyclic anhydrides, using TEB/PPNCl. | ||
Results and discussion
ROP of NNCA using the TEB/PPNCl catalytic system
Before studying switchable polymerization of epoxides/cyclic anhydrides/NNCAs, the TEB/PPNCl catalyzed ROCOP of styrene oxide (SO)/phthalic anhydride (PA) and ROP of NNCA were conducted. According to previous reports, the TEB/PPNCl is an effective catalyst for ROCOP of epoxides/cyclic anhydrides with good control.13,16 However, this remains unknown for the ROP of NNCA. Therefore, to verify the ability of the TEB/PPNCl catalytic system to facilitate the ROP of NNCA, two NNCA monomers, [sarcosine N-carboxyanhydrides (Sar-NCA) and N-ethyl N-carboxyanhydrides (Et-NNCA)], were synthesized and polymerized at 50 °C in SO/MeCN solution (1/1, v/v) (Fig. S1–S3,†Table 1 entries 1 and 2). The isolated polysarcosine (PSar) and poly(N-ethyl glycine) (PNEG) were analyzed by the 1H NMR spectroscopy, and the peaks (4.0–4.5 ppm) ascribed to PSar and PNEG were observed (Fig. S3†), indicating that ROP of NNCA occurred successfully. The PSar showed a number-average molecular weight (Mn) of 5.1 kg mol−1 and a low dispersity (Đ = 1.11). These results indicate that the TEB/PPNCl catalytic system can facilitate the controlled ROP of NNCA to produce polypeptoids.| Entry | NNCAs | Epoxides | [NNCA]/[PA]/[TEB]/[PPNCl] | Time (h) | M n,theo (kg mol−1) | M n,SEC (kg mol−1) | Đ |
|---|---|---|---|---|---|---|---|
| a Reactions were conducted at 50 °C in 1.2 mL of epoxide/MeCN (1/1, v/v). Both NNCAs and PA have been completely converted. b Determined by 1H NMR spectra, Mn,theo = M(NNCA) × ([NNCA]0/[PPNCl]0) × conv.(NNCA) + M(epoxide + PA) × ([PA]0/[PPNCl]0) × conv.(PA). c Determined by SEC in DMF, calibrated with polystyrene standards. Representative 1H and 1H DOSY NMR spectra, and SEC curves are in Fig. S21–S30. | |||||||
| 1 | Sar-NCA | CHO | 40/0/3/1 | 3 | 2.8 | 5.1 | 1.11 |
| 2 | Et-NNCA | CHO | 40/0/3/1 | 3 | 3.4 | 5.6 | 1.11 |
| 3 | — | SO | 0/25/3/1 | 48 | 6.7 | 4.9 | 1.08 |
| 4 | — | SO | 0/50/3/1 | 48 | 13.4 | 14.1 | 1.14 |
| 5 | Sar-NCA | SO | 40/50/3/1 | 48 | 16.1 | 11.4 | 1.14 |
| 6 | Sar-NCA | PO | 40/50/3/1 | 48 | 13.0 | 11.3 | 1.24 |
| 7 | Sar-NCA | VPO | 40/50/3/1 | 48 | 13.6 | 11.5 | 1.23 |
| 8 | Sar-NCA | AGE | 40/50/3/1 | 48 | 15.8 | 15.4 | 1.15 |
| 9 | Et-NNCA | SO | 40/50/3/1 | 48 | 16.8 | 4.6 | 1.14 |
| 10 | Et-NNCA | PO | 40/50/3/1 | 48 | 13.7 | 7.2 | 1.05 |
| 11 | Et-NNCA | VPO | 40/50/3/1 | 48 | 14.3 | 3.2 | 1.24 |
| 12 | Et-NNCA | AGE | 40/50/3/1 | 48 | 16.5 | 3.7 | 1.05 |
Next, the in situ ATR-IR spectroscopy was further employed to investigate the initiation process and assess the effects of TEB and PPNCl on the ROP of NNCA (Fig. S4–S9†). It is found that the presence of epoxide is crucial in the ROP of NNCA catalyzed by TEB/PPNCl. The chloride anion generated by PPNCl most likely acts as an effective initiator, leading to the ring-opening of one epoxide molecule and forming an alkoxide intermediate with α-Cl.16 With the presence of TEB, the alkoxide intermediate then further nucleophilically attacked the C5 carbonyl position of NNCA, effectively initiating the controlled ROP of NNCA. These results suggest that the controlled ROP of NNCA is achieved only with the combined use of TEB, PPNCl, and epoxide. In addition, the ROCOP of SO and PA was successfully conducted (Fig. S10–S11,†Table 1 entries 3 and 4).
Furthermore, matrix-assisted laser desorption ionization time-of-flight mass spectroscopy (MALDI-TOF MS) was employed to confirm the structure and terminal functional group of the P(SO-alt-PA) and PSar. As shown in Fig. 2A, a series of perfectly alternating polyester chains were observed. In the TEB/PPNCl catalyzed ROP, the chain growth is typically initiated by alkoxide species, which are generated by the nucleophilic attack of Cl− on epoxide monomers.13,16 However, the MALDI-TOF spectrum indicates that the P(SO-alt-PA) chain ends primarily consist of α,ω-dihydroxy, with no evidence of Cl-terminated chain ends (Fig. 2A). This phenomenon is most likely due to commercial TEB being available in THF solution, which may contain a trace amount of water. These water molecules can react with the epoxide monomers to form diols, acting as the chain transfer agents, leading to the predominant α,ω-dihydroxy chain structure.13,47,48 Furthermore, a series of PSar chains, capped by α,ω-diamine groups, were also observed (Fig. 2B), which further indicated that the diol most likely acted as chain transfer agents to afford the telechelic structures.
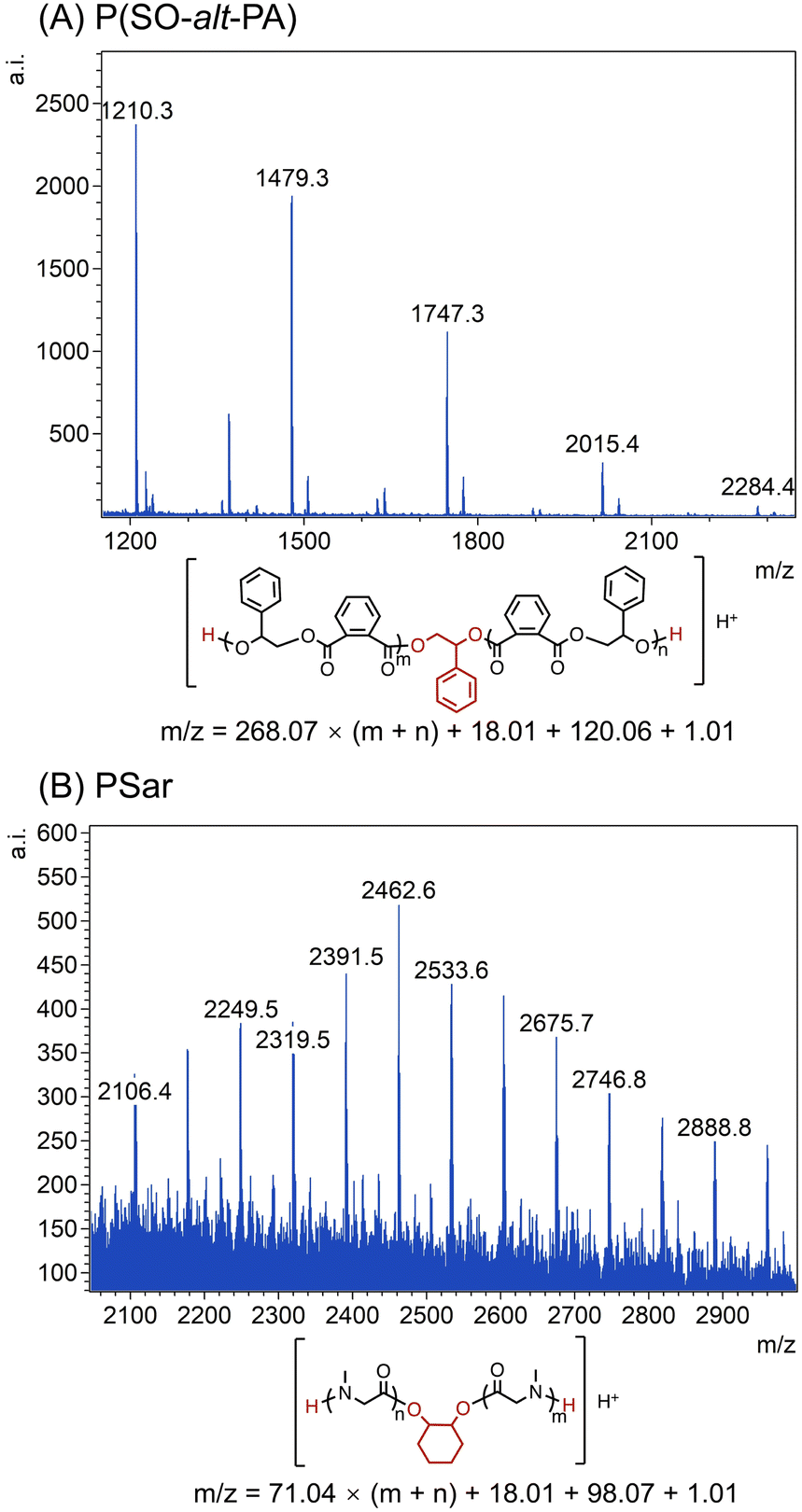
Switchable polymerization of SO/PA/Sar-NCA
The copolymerization of SO/PA/Sar-NCA monomer mixture using TEB/PPNCl as the catalytic system was investigated (Table 1 entry 5). The reaction, with a relative molar ratio of [SO]/[PA]/[Sar-NCA]/[TEB]/[PPNCl] = 210/50/40/3/1, was stopped after 48 h and showed complete consumption of PA (ca. 8.0 ppm) and Sar-NCA (4.1 ppm and 3.1 ppm) (Fig. S12†). The characteristic peaks of PSar are observed in the regions of 3.9–4.3 ppm (e) and 2.9–3.2 ppm (f), corresponding to the methylene and methyl groups of the PSar repeating units, respectively (Fig. 3A). Meanwhile, the signal peak b for the methine of the P(SO-alt-PA) repeating unit is observed at 6.0–6.4 ppm (Fig. 3A). The repeat unit ratio of P(SO-alt-PA) block to PSar block is ca. 1/0.75. It is worth noting that the ring-opening process of Sar-NCA produces CO2 as a byproduct. The TEB/PPNCl has been demonstrated to effectively catalyze the ROCOP of epoxides with CO2, leading to the formation of polycarbonates.49 However, there was no evidence for any polycarbonate (5.5–6.0 ppm) formation in this polymerization process (Fig. S12†).14 It is hypothesized that the copolymerization of epoxides and CO2 could not occur due to the insufficient CO2 pressure. Meanwhile, no polyether (ca. 4.5 ppm) was observed, indicating that the excess epoxides did not undergo homo-polymerization. The peaks at 4.0 ppm and 3.0 ppm are associated with the sarcosine anhydride, a cyclic byproduct formed during the ROP of Sar-NCA. This byproduct is possibly formed through an intramolecular “backbiting” or depolymerization reactions of PSar.50,51 SEC results indicate that the P(SO-alt-PA)-b-PSar copolymers have an Mn of approximately 11.4 kg mol−1, exhibiting a monomodal distribution and a low dispersity (Đ = 1.14) (Fig. S13† and Table 1 entry 5). These results demonstrate that both the ROCOP of SO/PA and the ROP of Sar-NCA can occur in one pot, enabling easy access towards P(SO-alt-PA)-b-PSar copolymers.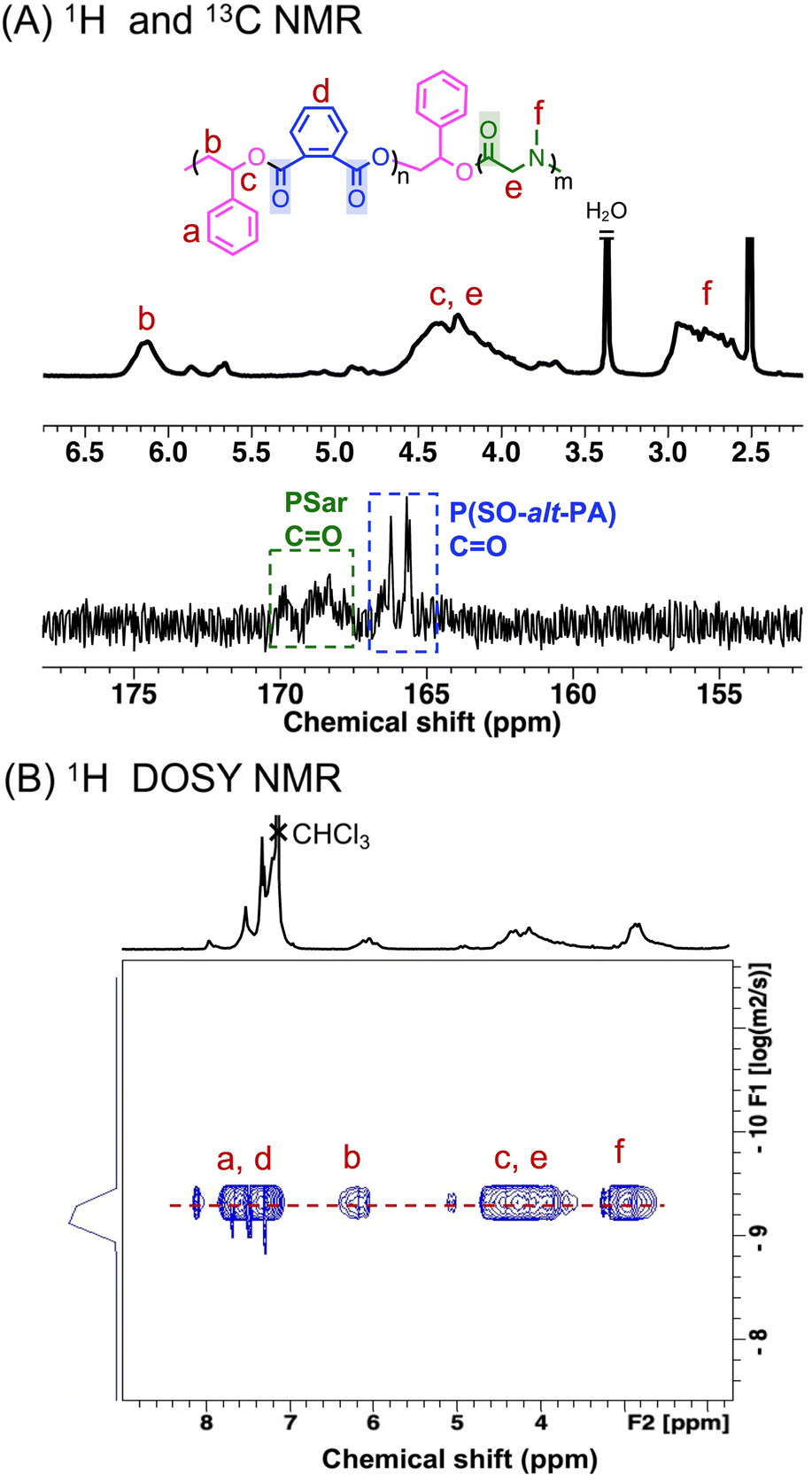 | ||
| Fig. 3 (A) The enlarged 1H NMR and 13C NMR spectra of P(SO-alt-PA)-b-PSar (in DMSO-d6). (B) The 1H DOSY NMR spectrum of P(SO-alt-PA)-b-PSar (in CDCl3). | ||
To further confirm the block structure and linkage units of the P(SO-alt-PA)-b-PSar copolymers, detailed characterization was performed using 1H, 13C, 1H–1H correlation spectroscopy (1H–1H COSY), 1H–13C heteronuclear single quantum coherence (1H–13C HSQC), and 1H diffusion ordered spectroscopy (1H DOSY NMR). As shown in the Fig. S14,† the characteristic peaks of both P(SO-alt-PA) and PSar were observed in the P(SO-alt-PA)-b-PSar copolymer, indicating that the copolymer contains both P(SO-alt-PA) and PSar blocks. New signal appeared at 5.5–6.0 ppm, which is presumed to be the linkage units between P(SO-alt-PA) and PSar. The assignment of the new signal peak was further confirmed by 1H–1H COSY NMR as two sets of coupled cross-peaks were observed (Fig. S16A†). Specifically, H1 and H2 were assigned to the signals of the P(SO-alt-PA) repeating unit, H3 and H4 were assigned to the additional SO unit at the junction between the P(SO-alt-PA) and PSar blocks. In addition, in the P(SO-alt-PA)-b-PSar copolymer, the carbonyl region shows two major peaks with 165.7–166.3 ppm corresponding to the carbonyl of P(SO-alt-PA),52 and 168.0–169.9 ppm corresponding to the carbonyl of PSar (Fig. 3A and S15†).53 Furthermore, the junction unit between P(SO-alt-PA) and PSar blocks at [5.9 ppm (H3), 78.0 ppm (C3)] was identified by the 1H–13C HSQC NMR spectrum (Fig. S16B†). The observation of peak q in the 1H NMR spectrum, in conjunction with the absence of corresponding coupling signals in the 1H–13C HSQC spectrum, suggests that peak q is associated with the hydrogen atoms at the secondary amine terminus [−N(CH3)H] of the copolymers (Fig. S16B†).
Furthermore, the 1H DOSY NMR spectrum of P(SO-alt-PA)-b-PSar copolymers showed a single diffusion coefficient for all resonances (Fig. 3B), indicating that P(SO-alt-PA) and PSar are covalently linked. In contrast, a blend of the P(SO-alt-PA) with PSar shows different diffusion coefficients (Fig. S17†). These findings confirm that P(SO-alt-PA)-b-PSar copolymers with well-defined block structures can be synthesized using TEB/PPNCl from mixed monomer feedstock containing epoxides, cyclic anhydrides, and NNCAs. To the best of our knowledge, this is the first successful synthesis of polyester-b-poly(α-amino acid) copolymers through switchable polymerization.
In situ monitoring of the terpolymerization process
The copolymerization progress was monitored by in situ ATR-IR spectroscopy and 1H NMR spectroscopy. Prior to the monitoring, characteristic IR absorption peaks were identified first. As shown in Fig. S18,† the absorption peaks at 1853 cm−1 and 1784 cm−1 correspond to the carbonyl group (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of both Sar-NCA and PA. Fortunately, the peak at 981 cm−1 is characteristic for Sar-NCA and can be used to monitor its conversion (Fig. S19A†). The absorption peak at 1674 cm−1 is attributed to the PSar, while the peak at 1729 cm−1 belongs to P(SO-alt-PA). Therefore, the absorptions at 981, 1852, 1729, and 1674 cm−1, which correspond to Sar-NCA consumption, PA/Sar-NCA consumption, P(SO-alt-PA) formation, and PSar formation, respectively, were selected. However, the absorptions of P(SO-alt-PA) is overlapped by the PSar (Fig. S18†).
O) of both Sar-NCA and PA. Fortunately, the peak at 981 cm−1 is characteristic for Sar-NCA and can be used to monitor its conversion (Fig. S19A†). The absorption peak at 1674 cm−1 is attributed to the PSar, while the peak at 1729 cm−1 belongs to P(SO-alt-PA). Therefore, the absorptions at 981, 1852, 1729, and 1674 cm−1, which correspond to Sar-NCA consumption, PA/Sar-NCA consumption, P(SO-alt-PA) formation, and PSar formation, respectively, were selected. However, the absorptions of P(SO-alt-PA) is overlapped by the PSar (Fig. S18†).
Initially, the PA/Sar-NCA resonance showed a decrease (1852 cm−1, purple ◆) in intensity and there were increases in the resonance assigned to P(SO-alt-PA) (1729 cm−1, yellow, ●) (Fig. 4). Over this time period, there was no significant change in the absorption peak intensity at 981 cm−1 (Sar-NCA, blue, ▼), indicating that the Sar-NCA does not polymerize during this stage. Once the PA was completely consumed, the intensity of Sar-NCA started to decrease and the intensity of PSar (1674 cm−1, red, ▲) gradually increased. These in situ ATR-IR results confirmed that the copolymerization of PA and SO occurred first. Furthermore, the terpolymerization was also monitored by 1H NMR at regular intervals (Fig. S20†). During the first stage (0–4 h), the signals of PA (7.8–8.0 ppm) decreased and the intensity of peaks ascribed to P(SO-alt-PA) (6.0–6.4 ppm) increased gradually. Meanwhile, the peaks of Sar-NCA kept constant. These results further confirm that the ROCOP of SO and PA occurred first, with the ROP of Sar-NCA being inactive. After the complete consumption of PA, the polymerization switched to the second stage, demonstrating chemoselectivity.
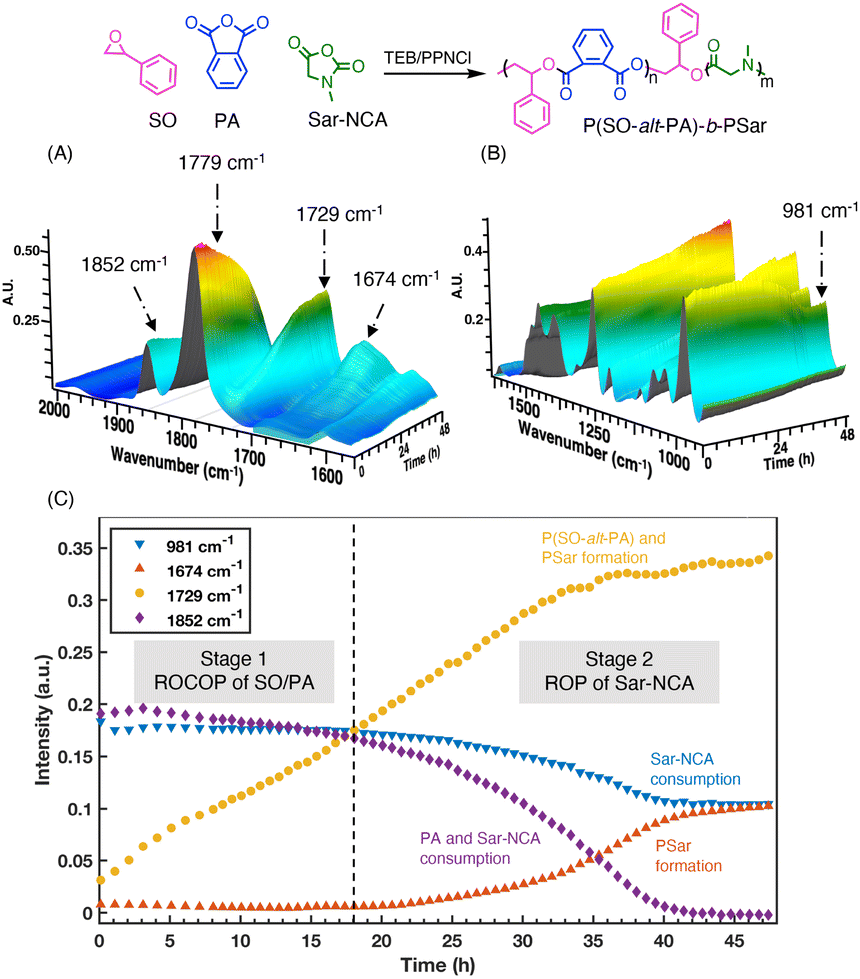 | ||
| Fig. 4 In situ ATR-IR spectroscopy monitoring of the terpolymerization process of SO/PA/Sar-NCA: (A) 2000–1590 cm−1, (B) 1625–900 cm−1. (C) Plots of monomer conversion versus time as monitored by in situ ATR-IR spectroscopy. The reaction was performed in 1.2 mL SO/MeCN (1/1, v/v) at 50 °C with [SO]/[PA]/[Sar-NCA]/[TEB]/[PPNCl] = 210/50/40/3/1. | ||
Monomer adaptability
To verify the generality of TEB/PPNCl-catalyzed switchable polymerization in directly producing various polyester-b-polypeptoid copolymers, the copolymerizations of PA with various epoxides, including SO, propylene oxide (PO), vinyl propylene oxide (VPO), and allyl glycidyl ether (AGE) as well as NNCAs (Sar-NCA and Et-NNCA), were conducted. A series of well-defined polyester-b-polypeptoid copolymers were successfully constructed with high chemoselectivity, confirmed by NMR and SEC analyses (Table 1 entries 5–12, and Fig. S21–S30†). All 1H DOSY NMR spectra exhibited single diffusion coefficients, suggesting that the polyester and polypeptoid blocks are connected to each other. For the VPO or AGE based copolymers (entries 7, 8, 11 and 12), the vinyl groups in the resulting block copolymers can be utilized for post-polymerization modifications.54,55 It is noteworthy that for polyester-b-PNEG copolymers (Table 1 entries 9–12), the Mn values measured by SEC were not in consistent with the theoretical values. This could be due to the potential interaction between the polypeptoid chains and the SEC columns, leading to underestimated molecular weights. In addition, the tendency of 1,4-diethylpiperazine-2,5-dione formation during the ROP of Et-NNCA, due to intramolecular “backbiting”,56–58 may also contribute to this issue (Fig. S28–S30†).Mechanism study of the switchable polymerization by DFT calculations
DFT calculations were conducted using PO, PA, and Sar-NCA as model monomers (Fig. 5). The initiation process of TEB/PPNCl-catalyzed epoxide/cyclic anhydride ROCOP has been reported previously, where epoxide is first activated by TEB and subsequently undergoes ring-opening through a nucleophilic attack by the Cl− anion of PPNCl to generate alkoxide intermediate IM1.13,16 The chain transfer procedure due to the in situ formed diols was not involved in the calculation for simplifying the computational procedure. In addition, as the DFT calculations were primarily focused on investigating the switching process, PO was selected as the epoxide for simplicity. From IM1, the energy barriers for the chain-end alkoxide terminal to attack PA and Sar-NCA was calculated. In the PA pathway (blue line), IM1 attacks the carbonyl of PA, leading to PA ring-opening through intermediate TS1, with an energy barrier of 34.3 kcal mol−1, forming the carboxylate anion intermediate IM4, which is stabilized by TEB. In the Sar-NCA pathway (red line), IM1 attacks the C5 carbonyl of Sar-NCA, passing through TS2 with an energy barrier of 39.7 kcal mol−1 to form the carbamate intermediate IM6. Subsequently, another Sar-NCA molecule approaches and reacts to form IM7. The decarboxylation of intermediate IM7 and the nucleophilic attack of the secondary amine terminal on the carbonyl of Sar-NCA proceed in a concerted manner through TS3, with an activation energy barrier of 40.1 kcal mol−1, ultimately yielding intermediate IM8. During the ROP of Sar-NCA, the calculations suggest that TEB stabilizes the chain end, ensuring the successful removal of CO2. As the energy barrier for PA insertion at the alkoxide terminal (34.3 kcal mol−1) is lower than that for Sar-NCA (39.7 kcal mol−1), the cyclic anhydride/epoxide ROCOP pathway is thermodynamically more favorable than the ROP pathway of Sar-NCA.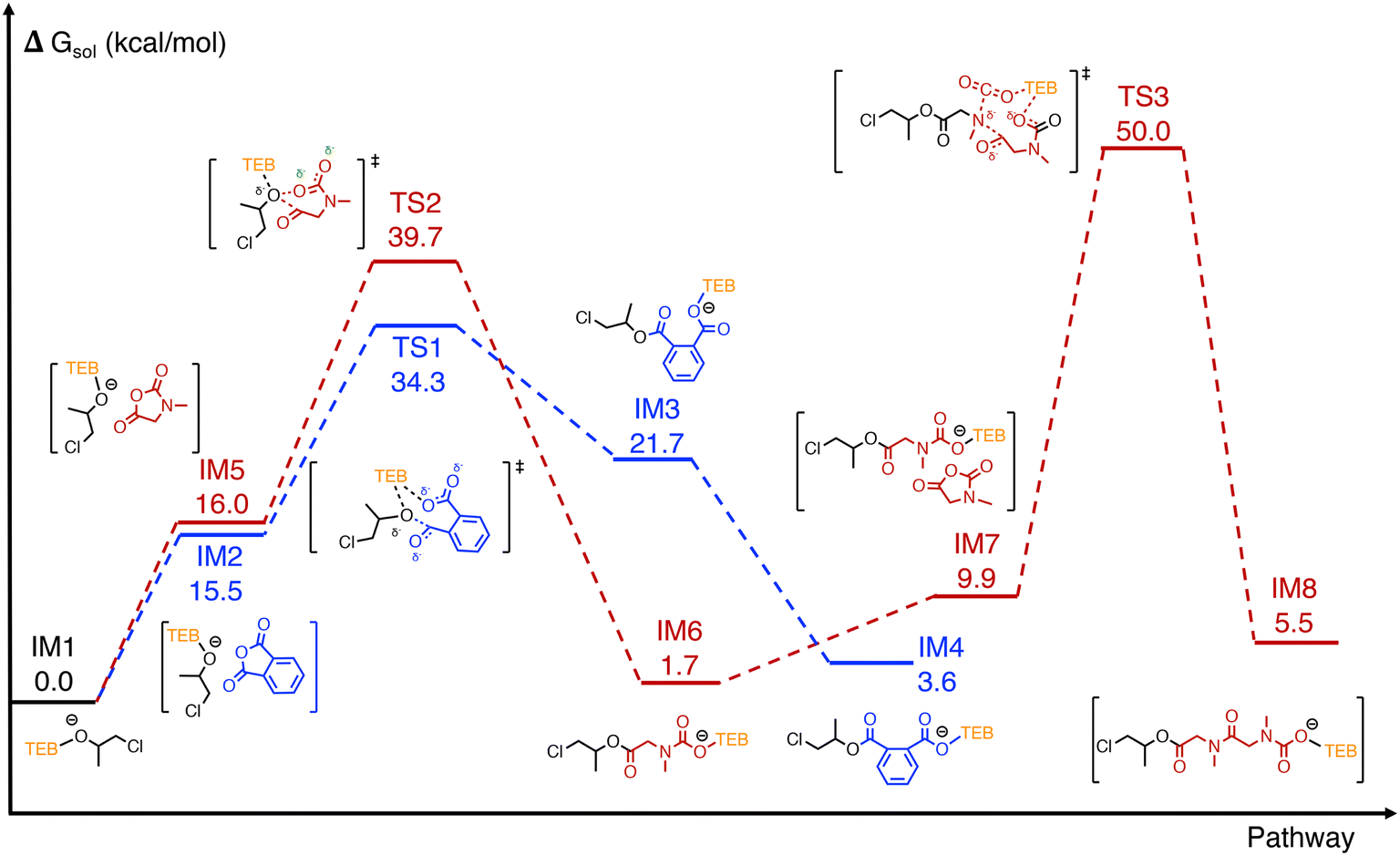 | ||
| Fig. 5 DFT-calculated free energy profiles for activation and initiation in PO/PA ROCOP (blue line), Sar-NCA ROP (red line), including key intermediates and transition states. The detailed three-dimensional structures of the optimized key transition states can be found in Fig. S31 of the ESI.† | ||
Based on the DFT calculations and experimental results, a plausible mechanism for the switchable polymerization was proposed. In the TEB/PPNCl-catalyzed ROCOP of epoxides and cyclic anhydrides, TEB typically functions as an activator, while PPNCl serves as an initiator, producing an alternating polyester with an α-Cl/ω-OH chain structure.13,16 As shown in Fig. 6, once initiated, the insertion of the cyclic anhydride into the alkoxide end is thermodynamically more favorable than the insertion of the NNCA monomer. Therefore, the terminal alkoxide species preferentially reacts with the cyclic anhydride, forming a carboxylate anion intermediate. This intermediate continues to react with the epoxide, generating a new alkoxide species. The continuous alternating insertion of cyclic anhydrides and epoxides eventually forms an alternating polyester, until the cyclic anhydride is completely consumed. Subsequently, due to the presence of excess epoxide, the polymer chain ends revert to alkoxide species. Then, without the competition from PA, NNCA can finally react with the terminal alkoxide species, forming a urethane intermediate. At this stage, the insertion of new NNCA monomers and the decarboxylation of the urethane intermediate occur simultaneously, generating a secondary amine species, which further initiates NNCA. This cyclic process of NNCA insertion and CO2 elimination continues, ultimately forming the polypeptoid block. TEB not only activates the epoxide but also forms an “ate-complex” with the polymer chain-end, thus stabilizing the chain-end (Fig. 6).13,14 This stabilization could weaken the basicity of the terminal alkoxide species, preventing the formation of a highly basic species that could lead to the deprotonation of NNCA.59,60 Additionally, the mild alkalinity of the TEB/PPNCl pairs ensures that excessive epoxides do not undergo homopolymerization throughout the process.15,61
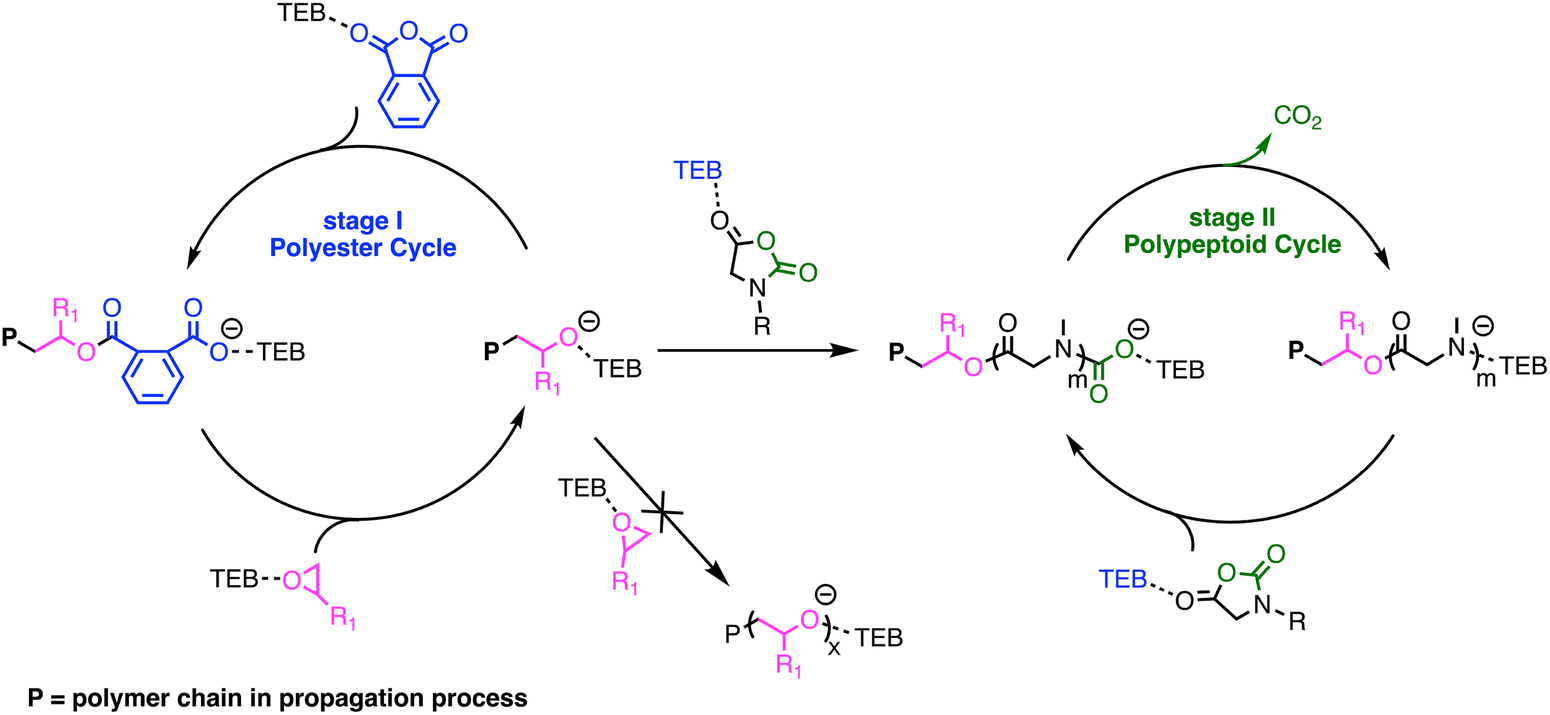 | ||
| Fig. 6 Plausible mechanism of TEB/PPNCl catalyzed switchable polymerization of epoxides/cyclic anhydrides/NNCAs. | ||
Cytotoxicity and anti-protein adsorption properties of copolymers
Using the switchable polymerization of epoxides/cyclic anhydrides/NNCAs, a series of amphiphilic polyester-b-polypeptoid copolymers were successfully synthesized. In these copolymers, the polyester blocks serve as the hydrophobic blocks, while the polypeptoid blocks act as the hydrophilic blocks. Notably, PSar is considered as a potential alternative to PEG. The PSar-based block copolymers exhibit excellent biocompatibility and low immunogenicity, making them suitable for biomedical application.62,63 The amphiphilic polyester-b-polypeptoid copolymers can self-assemble into nanoparticles in aqueous solution. Consequently, the cytotoxicity and anti-protein adsorption properties of P(SO-alt-PA)-b-PSar as a representative example were further investigated.As illustrated in Fig. 7A, TEM showed that P(SO-alt-PA)-b-PSar self-assembled into spherical nanoparticles in aqueous solution. DLS results revealed that the intensity-averaged hydrodynamic diameter (Dh) of the nanoparticles remained ca. 460 nm, with a narrow size distribution (PD = 0.10). Additionally, a negative ζ-potential value of −33.6 mV was observed (Fig. 7A). Furthermore, the cytotoxicity of P(SO-alt-PA)-b-PSar nanoparticles to mouse fibroblast (L929) cells was assessed using a cell counting kit-8 (CCK-8) assay. As shown in Fig. 7B, after 24 h of incubation with different nanoparticle concentrations (from 0.063 to 1.000 mg mL−1), P(SO-alt-PA)-b-PSar nanoparticles exhibited negligible toxicity at concentrations ranging from 0.063 to 0.500 mg mL−1. Even at the highest concentration of 1.000 mg mL−1, the cell viability remained ≥80%.
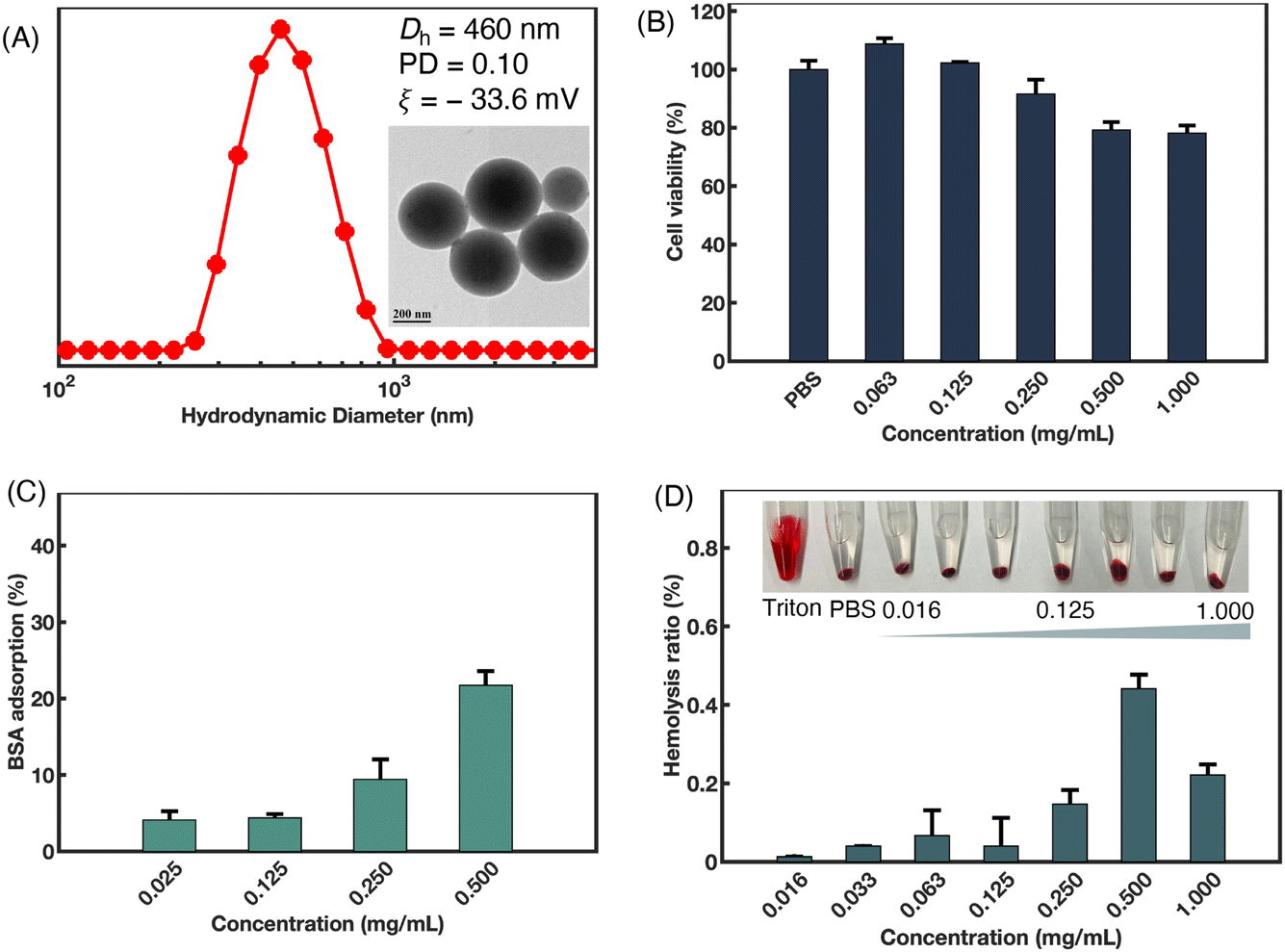 | ||
| Fig. 7 Synthesis and investigation on the properties of P(SO-alt-PA)-b-PSar self-assembled nanoparticles. (A) DLS result and TEM image in aqueous solution at 25 °C. Scale bar in the TEM image: 200 nm. (B) Relative cell viability (%) of P(SO-alt-PA)-b-PSar nanoparticles for L929 cells after 24 h of incubation at 37 °C. (C) BSA adsorption (%) after incubation at 37 °C for 24 h. (D) Hydrolysis ratio (%) with P(SO-alt-PA)-b-PSar nanoparticles after incubation at 37 °C for 1 h. | ||
Protein adsorption on the surface of materials in serum can significantly affect the therapeutic efficacy of drugs. The primary factors influencing protein adsorption include the hydrophobic/hydrophilic ratio of the material surface, the balance of positive and negative charges, and the structural stability of the proteins.64,65 Studies have shown that PSar resists the adhesion of proteins and mouse fibroblasts, which is attributed to its high hydrophilicity, lack of hydrogen bond donor characteristics, and the non-ionic nature of the methylated amide nitrogen atoms.66–68 To assess the protein repelling ability of the P(SO-alt-PA)-b-PSar nanoparticles, Bovine Serum Albumin (BSA) was used as a model protein. BSA (0.375 mg mL−1) was incubated with various concentrations of P(SO-alt-PA)-b-PSar nanoparticles for 24 h. The mixtures were isolated by centrifugation. The liquid supernatant was then taken out, and its UV-vis absorbance at 280 nm (the characteristic absorbance of BSA) was measured. The adsorption levels of BSA are quantitatively displayed in Fig. 7C. The P(SO-alt-PA)-b-PSar exhibited a maximum BSA adsorption of 22% over a concentration range of 0.025 to 0.500 mg mL−1, indicating a certain degree of anti-protein adsorption capability.
Additionally, the blood compatibility of P(SO-alt-PA)-b-PSar nanoparticles was evaluated using an in vitro hemolysis assay. Nanoparticle dispersions at different concentrations (ranging from 0.160 to 1.000 mg mL−1) were directly mixed with a suspension of red blood cells. After 1 h of incubation, the mixtures were centrifuged, and the absorbance of the supernatants was measured using a microplate reader. As depicted in Fig. 7D, all tested concentrations of P(SO-alt-PA)-b-PSar nanoparticles exhibited no hemolysis, while significant hemolysis was observed in the positive control group (Triton). The hemolysis ratio remained below 0.5%, even at the highest concentration of 1.0 mg mL−1. These results demonstrate that the synthesized polyester-b-polypeptoid copolymers have excellent anti-protein adsorption capabilities and barely no cytotoxicity, highlighting their promising potential for biomedical applications.
Conclusion
In summary, we propose an easy access to amphiphilic nitrogenous block copolymers via the ‘one-pot/one-step’ switchable catalysis. The ROCOP of epoxides and cyclic anhydrides proceeds preferentially until the cyclic anhydride is completely consumed, after which the ROP of NNCA initiates. DFT calculations indicate that the reaction of PA with the alkoxide terminal is thermodynamically more favorable than that of Sar-NCA. This switchable polymerization strategy exhibits high selectivity across various epoxides and NNCAs, which is promising and profound for industrial development. By tuning monomer types, block structures, and side chain functionalities, a variety of amphiphilic copolymers with diverse structures and functionalities were efficiently synthesized, suggesting potential applications in more complex multicomponent systems in the future. Additionally, the obtained amphiphilic P(SO-alt-PA)-b-PSar copolymers can self-assemble into nanoparticles, demonstrating excellent anti-protein adsorption capabilities and barely no cytotoxicity for potential applications in biomedicine. Most importantly, this methodology is expected to open new horizons beyond oxygenated monomers for switchable polymerization.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
X. Liang, W. Wang, J. Du and Y. Zhu conceived the idea, proposed the strategy, designed the experiments, evaluated the data, and wrote the manuscript together. X. Liang performed the most of the experiments. J. Lv conducted the DFT calculations. X. Liang, J. Li and H. Qiang participated in polymerization experiments and related characterization analysis. All the authors proofread the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (22175131, 22335005, and 21925505), the National Key R&D Program of China (2022YFC2402900), Innovation Program of Shanghai Municipal Education Commission (2023ZKZD28), Tongcheng Youth Research and Development Fund (CPCIF-RA-0104), and the Interdisciplinary Collaborative Research Project of Tongji University (2023-2-YB-03). J. Du is a recipient of National Science Fund for Distinguished Young Scholars.References
- J. Jennings, G. He, S. M. Howdle and P. B. Zetterlund, Chem. Soc. Rev., 2016, 45, 5055–5084 RSC
.
- H. Dau, G. R. Jones, E. Tsogtgerel, D. Nguyen, A. Keyes, Y.-S. Liu, H. Rauf, E. Ordonez, V. Puchelle, H. Basbug Alhan, C. Zhao and E. Harth, Chem. Rev., 2022, 122, 14471–14553 CrossRef CAS PubMed
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed
- Y. Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC
- C. Li, Q. Li, Y. V. Kaneti, D. Hou, Y. Yamauchi and Y. Mai, Chem. Soc. Rev., 2020, 49, 4681–4736 RSC
- C. C. M. Sproncken, P. Liu, J. Monney, W. S. Fall, C. Pierucci, P. B. V. Scholten, B. Van Bueren, M. Penedo, G. E. Fantner, H. H. Wensink, U. Steiner, C. Weder, N. Bruns, M. Mayer and A. Ianiro, Nature, 2024, 630, 866–871 CrossRef CAS PubMed
- A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2022, 61, e202104495 CrossRef CAS PubMed
- C. Zhang, X. Zhang and X. Zhang, Sci. China: Chem., 2020, 63, 1807–1814 CrossRef CAS
- X. Geng, Z. Liu, C. Zhang and X. Zhang, Macromolecules, 2023, 56, 4649–4657 CrossRef CAS
- H. Feng, M. Dolejsi, N. Zhu, S. Yim, W. Loo, P. Ma, C. Zhou, G. S. W. Craig, W. Chen, L. Wan, R. Ruiz, J. J. De Pablo, S. J. Rowan and P. F. Nealey, Nat. Mater., 2022, 21, 1426–1433 CrossRef CAS PubMed
- C. A. L. Lidston, S. M. Severson, B. A. Abel and G. W. Coates, ACS Catal., 2022, 12, 11037–11070 CrossRef CAS
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC
- S. Ye, W. Wang, J. Liang, S. Wang, M. Xiao and Y. Meng, ACS Sustainable Chem. Eng., 2020, 8, 17860–17867 CrossRef CAS
- J. Tang, M. Li, X. Wang and Y. Tao, Angew. Chem., Int. Ed., 2022, 134, e202115465 CrossRef
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed
- J. Zhang, L. Wang, S. Liu, X. Kang and Z. Li, Macromolecules, 2021, 54, 763–772 CrossRef CAS
- N. Zhu, X. Hu, Z. Fang and K. Guo, Prog. Polym. Sci., 2021, 117, 101397 CrossRef CAS
- Y. Jia, Z. Sun, C. Hu and X. Pang, ChemPlusChem, 2022, 87, e202200220 CrossRef CAS PubMed
- A. C. Deacy, G. L. Gregory, G. S. Sulley, T. T. D. Chen and C. K. Williams, J. Am. Chem. Soc., 2021, 143, 10021–10040 CrossRef CAS PubMed
- C. Hu, X. Pang and X. Chen, Macromolecules, 2022, 55, 1879–1893 CrossRef CAS
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS PubMed
- T. Stößer, T. T. D. Chen, Y. Zhu and C. K. Williams, Philos. Trans. R. Soc., A, 2018, 376, 20170066 CrossRef PubMed
- D. Ryzhakov, G. Printz, B. Jacques, S. Messaoudi, F. Dumas, S. Dagorne and F. Le Bideau, Polym. Chem., 2021, 12, 2932–2946 RSC
- C. K. Williams and K. Nozaki, Inorg. Chem., 2020, 59, 957–959 CrossRef CAS PubMed
- A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Chem. Rev., 2016, 116, 1969–1992 CrossRef CAS PubMed
- X.-L. Chen, B. Wang, D.-P. Song, L. Pan and Y.-S. Li, Macromolecules, 2022, 55, 1153–1164 CrossRef CAS
- X. F. Zhu, G. W. Yang, R. Xie and G. P. Wu, Angew. Chem., Int. Ed., 2021, 61, e202115189 CrossRef PubMed
- J. Xu and N. Hadjichristidis, Angew. Chem., Int. Ed., 2021, 60, 6949–6954 CrossRef CAS PubMed
- J. Xu, X. Wang and N. Hadjichristidis, Nat. Commun., 2021, 12, 7124 CrossRef CAS PubMed
- A. Rasines Mazo, S. Allison-Logan, F. Karimi, N. J.-A. Chan, W. Qiu, W. Duan, N. M. O'Brien-Simpson and G. G. Qiao, Chem. Soc. Rev., 2020, 49, 4737–4834 RSC
- Z. L. Li, B. Cai, W. C. Yang and C. L. Chen, Chem. Rev., 2021, 121, 14031–14087 CrossRef CAS PubMed
- A. M. Clapperton, J. Babi and H. Tran, ACS Polym. Au, 2022, 2, 417–429 CrossRef CAS PubMed
- A. Birke, J. Ling and M. Barz, Prog. Polym. Sci., 2018, 81, 163–208 CrossRef CAS
- Z. S. Clauss and J. R. Kramer, ACS Appl. Mater. Interfaces, 2022, 14, 22781–22789 Search PubMed
- S. Wang, M. Y. Lu, S. K. Wan, C. Y. Lyu, Z. Y. Tian, K. Liu and H. Lu, J. Am. Chem. Soc., 2024, 146, 5678–5692 CrossRef CAS PubMed
- B. Yu, B. S. Chang, W. S. Loo, S. Dhuey, P. O'Reilly, P. D. Ashby, M. D. Connolly, G. Tikhomirov, R. N. Zuckermann and R. Ruiz, ACS Nano, 2024, 18, 7411–7423 CrossRef CAS PubMed
- C. A. R. Picken, O. Buensoz, P. D. Price, C. Fidge, L. Points and M. P. Shaver, Chem. Sci., 2023, 14, 12926–12940 Search PubMed
- M. F. Kabil, H. M. E. Azzazy and M. Nasr, Int. J. Pharm., 2024, 653, 123871 Search PubMed
- P. Zhou, T. Shen and J. Ling, J. Polym. Sci., 2021, 59, 2946–2958 Search PubMed
- N. S. Jiang, T. Y. Yu, O. A. Darvish, S. Qian, I. K. M. Tsengam, V. John and D. H. Zhang, Macromolecules, 2019, 52, 8867–8877 Search PubMed
- R. Y. Li, Y. Zhao, M. Lin and J. Sun, Chin. J. Chem., 2023, 41, 2305–2310 CrossRef CAS
- L. Y. Kang, A. Chao, M. Zhang, T. Y. Yu, J. Wang, Q. Wang, H. H. Yu, N. S. Jiang and D. H. Zhang, J. Am. Chem. Soc., 2021, 143, 5890–5902 CrossRef CAS PubMed
- Z. W. Wang, M. Lin, C. Bonduelle, R. Y. Li, Z. K. Shi, C. H. Zhu, S. Lecommandoux, Z. B. Li and J. Sun, Biomacromolecules, 2020, 21, 3411–3419 CrossRef CAS PubMed
- Z. K. Shi, Y. H. Wei, C. H. Zhu, J. Sun and Z. B. Li, Macromolecules, 2018, 51, 6344–6351 Search PubMed
- S. Varlas, P. G. Georgiou, P. Bilalis, J. R. Jones, N. Hadjichristidis and R. K. O'Reilly, Biomacromolecules, 2018, 19, 4453–4462 Search PubMed
- A. Ma, X. Yu, M. Liao, W. Liu, S. Xuan and Z. Zhang, Macromol. Rapid Commun., 2023, 44, 2200301 CrossRef CAS PubMed
- J. Liang, S. Ye, W. Wang, C. Fan, S. Wang, D. Han, W. Liu, Y. Cui, L. Hao, M. Xiao and Y. Meng, J. CO2 Util., 2021, 49, 101558 Search PubMed
- S. Zhu, Y. Wang, W. Ding, X. Zhou, Y. Liao and X. Xie, Polym. Chem., 2020, 11, 1691–1695 RSC
- V. K. Chidara, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, Macromolecules, 2021, 54, 2711–2719 CrossRef CAS
- S. H. Lahasky, W. K. Serem, L. Guo, J. C. Garno and D. Zhang, Macromolecules, 2011, 44, 9063–9074 Search PubMed
- C. Fetsch, A. Grossmann, L. Holz, J. F. Nawroth and R. Luxenhofer, Macromolecules, 2011, 44, 6746–6758 Search PubMed
- E. Hosseini Nejad, A. Paoniasari, C. E. Koning and R. Duchateau, Polym. Chem., 2012, 3, 1308 Search PubMed
- S. Cui, X. Wang, Z. Li, Q. Zhang, W. Wu, J. Liu, H. Wu, C. Chen and K. Guo, Macromol. Rapid Commun., 2014, 35, 1954–1959 CrossRef CAS PubMed
- D. J. Darensbourg and F. T. Tsai, Macromolecules, 2014, 47, 3806–3813 Search PubMed
- N. Yi, T. T. D. Chen, J. Unruangsri, Y. Zhu and C. K. Williams, Chem. Sci., 2019, 10, 9974–9980 Search PubMed
- K. Chen, Y. Wu, X. Wu, M. Zhou, R. Zhou, J. Wang, X. Xiao, Y. Yuan and R. Liu, Polym. Chem., 2022, 13, 420–426 RSC
- L. Guo, S. H. Lahasky, K. Ghale and D. Zhang, J. Am. Chem. Soc., 2012, 134, 9163–9171 CrossRef CAS PubMed
- B. Zheng, T. Bai, X. Tao, H. Schlaad and J. Ling, Biomacromolecules, 2018, 19, 4263–4269 CrossRef CAS PubMed
- Y. M. Wu, M. Zhou, K. Chen, S. Chen, X. M. A. Xiao, Z. M. Ji, J. C. Zou and R. H. Liu, Chin. Chem. Lett., 2021, 32, 1675–1678 CrossRef CAS
- P. Salas-Ambrosio, A. Tronnet, M. Since, S. Bourgeade-Delmas, J. L. Stigliani, A. Vax, S. Lecommandoux, B. Dupuy, P. Verhaeghe and C. Bonduelle, J. Am. Chem. Soc., 2021, 143, 3697–3702 CrossRef CAS PubMed
- C. Zhang, H. Wu, Y. Li, J. Yang and X. Zhang, Nat. Commun., 2018, 9, 2137 CrossRef PubMed
- E. J. L. Stéen, J. T. Jørgensen, K. Johann, K. Nørregaard, B. Sohr, D. Svatunek, A. Birke, V. Shalgunov, P. E. Edem, R. Rossin, C. Seidl, F. Schmid, M. S. Robillard, J. L. Kristensen, H. Mikula, M. Barz, A. Kjær and M. M. Herth, ACS Nano, 2020, 14, 568–584 CrossRef PubMed
- Y. Hu, Y. Hou, H. Wang and H. Lu, Bioconjugate Chem., 2018, 29, 2232–2238 CrossRef CAS PubMed
- M. Rabe, D. Verdes and S. Seeger, Adv. Colloid Interface Sci., 2011, 162, 87–106 CrossRef CAS PubMed
- X. Lu, P. Xu, H.-M. Ding, Y.-S. Yu, D. Huo and Y.-Q. Ma, Nat. Commun., 2019, 10, 4520 CrossRef PubMed
- M. Schneider, C. Fetsch, I. Amin, R. Jordan and R. Luxenhofer, Langmuir, 2013, 29, 6983–6988 CrossRef CAS PubMed
- M. Schneider, Z. Tang, M. Richter, C. Marschelke, P. Förster, E. Wegener, I. Amin, H. Zimmermann, D. Scharnweber, H. G. Braun, R. Luxenhofer and R. Jordan, Macromol. Biosci., 2016, 16, 75–81 CrossRef CAS PubMed
- E. Ostuni, R. G. Chapman, R. E. Holmlin, S. Takayama and G. M. Whitesides, Langmuir, 2001, 17, 5605–5620 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05047a |
| This journal is © The Royal Society of Chemistry 2024 |