
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn anthracene-containing crown ether: synthesis, host–guest properties and modulation of solid state luminescence†
Weijie
Zhu‡
*abc,
Bohan
Zhao‡
a,
Shuai
Fang
a,
Huangtianzhi
Zhu
 *a and
Feihe
Huang
*ab
*a and
Feihe
Huang
*ab
aDepartment of Chemistry, Stoddart Institute of Molecular Science, Zhejiang University, Hangzhou 310058, P. R. China. E-mail: htzzhu@zju.edu.cn; fhuang@zju.edu.cn; Fax: (+86) 571-87953189
bZhejiang-Israel Joint Laboratory of Self-Assembling Functional Materials, ZJU-Hangzhou Global Scientific and Technological Innovation Center, Hangzhou, 311215, P. R. China
cSchool of Chemical and Environmental Engineering, Hunan Institute of Technology, Hengyang 421002, P. R. China. E-mail: weijiezhu@zju.edu.cn
First published on 18th September 2024
Abstract
Organic solid state vapochromic materials are of great significance for the development of supramolecular chemistry and materials science. Herein, we synthesize a crown ether derivative (An34C10) containing two anthracene units and construct new crown ether-based vapochromic host–guest co-crystals. Due to the presence of anthracene, An34C10 not only shows good fluorescence properties but also displays mechanochromism. Single crystal structural analysis, powder X-ray diffraction and differential scanning calorimetry experiments demonstrate that the transformation between different stacking modes of An34C10 is responsible for mechanochromism. In addition, An34C10 can complex with 1,2,4,5-tetracyanobenzene (TCNB) to form host–guest complex (An34C10@TCNB) co-crystals. Because organic solvent fuming alters charge-transfer interactions in An34C10@TCNB, the fluorescence of the co-crystals can be turned on and off by 4-methylpyridine and chloroform vapors, respectively, realizing selective detection with opposite emission outputs. Meanwhile, the stimuli-responsive properties of An34C10 and An34C10@TCNB possess good cycling performance. This work provides a new strategy for the construction of organic solid state luminescent materials.
Introduction
Organic solid state vapochromic materials have attracted considerable attention from scientists because of their wide applications in chemical sensors, light-emitting diodes, environmental monitors, and other fields.1–9 In particular, for molecules containing donor–acceptor structures, their luminescence can be regulated by certain gases such as volatile organic compounds (VOCs), leading to functions in smart devices.10–14 Such organic donor–acceptor structures usually require tedious synthesis, and molecules with a large degree of conjugation are commonly less stable.15,16 Therefore, it is urgent to develop new vapochromic materials with simple synthesis and robustness.Crystal engineering provides a feasible way for preparing crystalline functional materials.17–28 Owing to solid state complexation upon crystallization, organic host–guest complex co-crystals are promising candidates for robust luminescent materials.29,30 During co-crystallization, both the donor and acceptor, as the host or guest, respectively, can be included in one system via host–guest binding, avoiding tedious covalent synthesis. Meanwhile, compared with the single-component crystalline system, host–guest complex co-crystals show stimuli-responsiveness, leading to more functions, such as molecular ferroelectrics,31 room-temperature phosphorescence32–35 and fluorescent devices.36,37
As the first generation of macrocycles in supramolecular chemistry, crown ethers have been vigorously studied since they were reported in 1967.38,39 They have been applied in many fields including supramolecular polymers, supramolecular gels, molecular machines, detection and sensing.40–54 In general, guests of crown ethers are cationic compounds, such as alkali metal ions and dialkylammonium salts.55,56 These guests cannot be vaporized under mild conditions, and thus it is difficult to realize vapochromic behaviors based on crown ether-based host–guest complexes. We anticipate that new applications related to vapochromism could be achieved by introducing luminescent crown ethers as hosts.
In this work, we prepared an anthracene-containing crown ether derivative (An34C10) and used it to construct new crown ether-based vapochromic host–guest co-crystals (Fig. 1). Due to the presence of anthracene groups, An34C10 exhibited intense blue fluorescence emission and mechanochromism. When An34C10 was ground vigorously in a mortar for several minutes, the emission color changed from blue to yellow-green. After fuming with organic solvents or heating, the fluorescence of the ground sample recovered to the initial blue emission. Furthermore, as the host is a good electron-donor, 1,2,4,5-tetracyanobenzene (TCNB) was chosen as the acceptor to form host–guest complex (An34C10@TCNB) co-crystals. Although the emission of the co-crystals was quenched by strong charge-transfer (CT), the emission was turned on when the co-crystals were placed in the vapor of 4-methylpyridine, realizing sensing of such vapor with high contrast. Subsequent fuming with chloroform vapor was able to turn off the emission, and thus the material could be recycled. The fluorescence turn-on and turn-off of An34C10@TCNB showed excellent robustness and remained unchanged for five cycles.
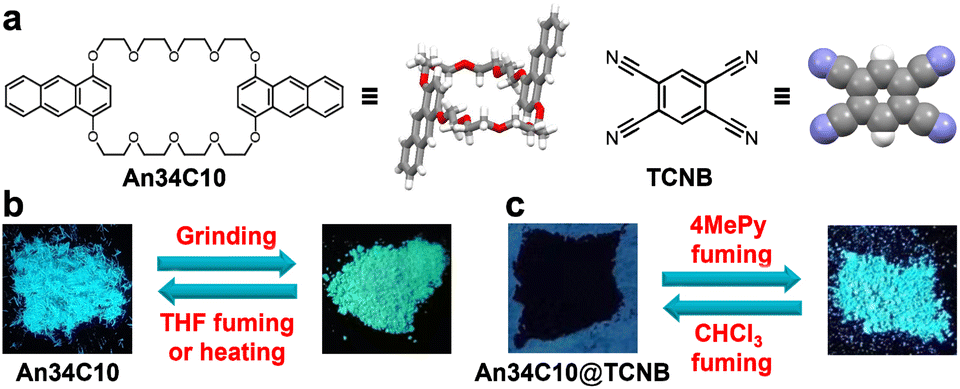 | ||
| Fig. 1 (a) Chemical structures of An34C10 and TCNB. Schematic representations of (b) An34C10 and (c) An34C10@TCNB under different external stimuli. | ||
Results and discussion
The synthetic route to An34C10 is shown in Fig. S1, ESI†. By using 1,4-anthraquinone and tetraethylene glycol as starting materials, the target macrocycle was finally obtained in a yield of 29% after a five-step reaction.57–59 All of the new compounds were confirmed by 1H and 13C nuclear magnetic resonance (NMR) and quadrupole-time of flight (Q-TOF) mass spectroscopies (Fig. S2–S9, ESI†).The photophysical properties of An34C10 were explored in solution. Fig. S10, ESI† exhibits the absorption spectra of An34C10 in different solvents. It can be seen that the absorbance of An34C10 was mainly located between 350 and 450 nm, the characteristic peak of anthracene chromophores.60 When the solvent polarity increased from hexane to DMSO, the absorption wavelength of An34C10 displayed a bathochromic shift of about 7 nm. The emission of An34C10 showed a significant bathochromic shift of 50 nm with the increase of solvent polarity (Fig. S11, ESI†), and the maximum emission peak was red-shifted from 452 nm to 502 nm, indicating that the excited state of An34C10 was easily polarized compared with the ground state.60,61
Anthracene derivatives exhibit photoluminescence not only in solution but also in the solid state.62 We then explored the solid state emissive properties of An34C10. As shown in Fig. 2a, An34C10 appeared as a yellowish powder under daylight and emitted a bright cyan fluorescence under irradiation of 365 nm UV. According to the solid state photoluminescence (PL) spectra (Fig. 2a and b), the maximum emission peak of An34C10 was located at 470 nm with a fluorescence lifetime of 2.96 ns. No long-lived emission was observed. We also determined the fluorescence quantum yield (QY) of An34C10, which was 13%.
 | ||
| Fig. 2 (a) Normalized fluorescence spectra of An34C10 in the solid state. Insets: pictures of An34C10 under daylight and UV illumination. (b) Emission decay of An34C10 in the solid state. (c) Normalized fluorescence spectra of An34C10 in the solid state under different stimulus conditions. (d) The 1931 CIE coordinate diagram of An34C10 before and after grinding. | ||
We next investigated the mechanochromism of An34C10.62,63 As shown in Fig. 1b, when An34C10 crystals were ground in a mortar for 20 minutes, the luminescent color changed from cyan to yellow-green. The maximum emission of An34C10 displayed a bathochromic shift from 470 nm to 498 nm after grinding (Fig. 2c), and the CIE coordinates changed from (0.18, 0.29) to (0.24, 0.44) (Fig. 2d). The emission of the ground sample was restored to its initial state after heating at 100 °C for 5 minutes or fuming with THF for 2 hours. As characterized by the PL spectra (Fig. 2c), the maximum emission recovered to 470 nm upon heating and THF fuming, indicating that the mechanochromism of An34C10 was reversible.
In order to study the mechanism of mechanochromism, we tried to get single crystals of An34C10. Slowly evaporating a saturated ethyl acetate solution of An34C10 at room temperature afforded crystals with bright cyan fluorescence. In the single crystal structure (Fig. 3) of An34C10, the distance between two anthracene units in one An34C10 molecule was 6.532 Å, greatly exceeding the effective distance of aryl stacking.64 Besides, negligible interactions were observed between two adjacent An34C10 molecules, demonstrating a relatively loose packing. Therefore, the pristine crystals of An34C10 exhibited emission from anthracene monomers. We infer that, upon grinding, the packing mode was partially destroyed (vide infra), leading to a relatively dense packing between adjacent anthracene units that further resulted in the bathochromic emission.60–63 Upon heating or fuming, the dense packing mode became loose again, thus the emission recovered to cyan.
 | ||
| Fig. 3 (a) Single crystal structure of An34C10. Carbon atoms are grey, oxygen atoms are red, and hydrogen atoms are white. Crucial distances are marked with dashed lines. One water molecule was encapsulated in the cavity that might come from a trace amount of water in the solvent. Solvent molecules are omitted for clarity. (b) The packing mode of An34C10. Hydrogen atoms and solvents are omitted for clarity. | ||
In addition to the single crystal analysis, we also performed 1H NMR, Fourier transform infrared spectroscopy (FT-IR), powder X-ray diffraction (PXRD) and differential scanning calorimetry (DSC) experiments for An34C10 under different conditions. As can be seen from Fig. S12 and S13, ESI†, the 1H NMR and FT-IR spectra of An34C10 didn't change after grinding, heating or solvent fuming, which indicated good chemical stability under these stimuli. The PXRD patterns revealed that An34C10 in the initial state exhibited strong and sharp diffraction peaks that agreed well with the simulated data derived from the single crystal structure, suggesting a well-ordered molecular arrangement (Fig. S14, ESI†). For the ground sample, however, some of the characteristic peaks disappeared in the PXRD pattern, and the intensity of the diffraction peaks was greatly reduced, indicating that the crystal packing changed from ordered stacking to a disordered state. These analyses implied that the emission change resulted from the altered packing mode of the crystals. In addition, after heating or fuming with THF, the sample exhibited restored PXRD diffraction peaks and intensities, and the patterns were consistent with those of the pristine material, indicating that the molecular arrangement of An34C10 became ordered again.
Then we conducted thermal analysis. DSC curves revealed that the crystalline sample of An34C10 only exhibited an endothermic peak at about 138 °C, belonging to the melting point (Fig. S15, ESI†). No other phase transition was observed. However, ground An34C10 displayed two signals in DSC curves. The first one was an exothermic peak located at about 57 °C, which was attributed to the phase transition process, and the other was a melting peak located at about 138 °C. Heating the ground sample for 5 minutes produced an identical DSC curve to that of the pristine material. A similar result was also obtained with the solvent-fumed sample, which only gave a minor difference at about 69 °C on account of solvent loss. Based on the above analysis, it can be inferred that the transformations between packing modes of the An34C10 crystal are responsible for its reversible mechanochromism.65 Grinding the crystals produced a metastable dense stacking state, rendering the emission bathochromic shifted. Stimuli such as heating or solvent fuming transformed the unstable dense packing state to the thermodynamically stable crystalline state, which restored the initial emission.
Cycling performance is an important parameter to evaluate the reversibility of mechanochromism, so alternative grinding-heating and grinding-solvent fuming experiments were carried out. As expected, the fluorescence of An34C10 could be altered between cyan and yellow-green five times without any performance loss, indicating the excellent reversibility of mechanochromism of An34C10 (Fig. S16 and S17, ESI†).
Considering that An34C10 is electron-rich, we next investigated the construction of donor–acceptor co-crystals with 1,2,4,5-tetracyanobenzene (TCNB) as an electron-deficient guest. We determined the binding stoichiometry of An34C10 and TCNB using the Job's plot, and as expected, the ratio of the host and guest was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S18, ESI†). Next, the host–guest recognition between An34C10 and TCNB in solution was investigated by 1H NMR. As shown in Fig. 4a–c, compared with the spectrum of free TCNB, upfield shifts occurred for the signal related to proton Ha of TCNB in the presence of 1.00 equiv. of An34C10. Meanwhile, the proton resonances H1 and H2 on An34C10 exhibited upfield shifts and H3–H8 shifted downfield upon complexation. These results suggested that TCNB was encapsulated in the cavity of An34C10, allowing the protons on TCNB to be shielded by the electron-rich host. The complexation was also supported by 2D nuclear Overhauser effect spectroscopy (NOESY). Strong correlations between Ha on TCNB and H1 and H2 on An34C10 were observed, and thus the guest was located in the cavity of An34C10 (Fig. S19, ESI†).
:1 (Fig. S18, ESI†). Next, the host–guest recognition between An34C10 and TCNB in solution was investigated by 1H NMR. As shown in Fig. 4a–c, compared with the spectrum of free TCNB, upfield shifts occurred for the signal related to proton Ha of TCNB in the presence of 1.00 equiv. of An34C10. Meanwhile, the proton resonances H1 and H2 on An34C10 exhibited upfield shifts and H3–H8 shifted downfield upon complexation. These results suggested that TCNB was encapsulated in the cavity of An34C10, allowing the protons on TCNB to be shielded by the electron-rich host. The complexation was also supported by 2D nuclear Overhauser effect spectroscopy (NOESY). Strong correlations between Ha on TCNB and H1 and H2 on An34C10 were observed, and thus the guest was located in the cavity of An34C10 (Fig. S19, ESI†).
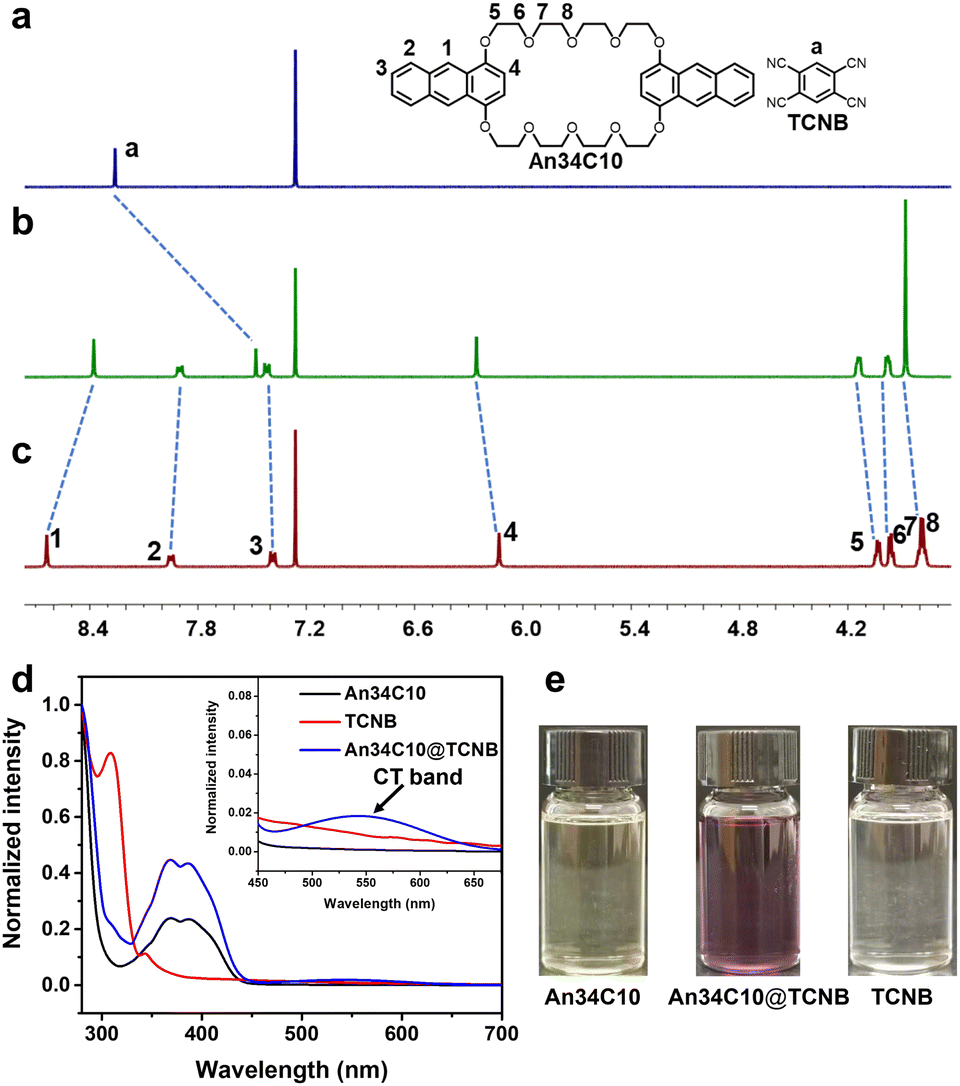 | ||
| Fig. 4 1H NMR spectra (400 MHz, chloroform-d, 298 K): (a) TCNB; (b) 5.00 mM TCNB and 5.00 mM An34C10; (c) An34C10. (d) Normalized UV-vis absorption spectra (CHCl3): An34C10 (1.00 mM); TCNB (1.00 mM); An34C10 and TCNB (1.00 mM, each). (e) Photographs: An34C10 (1.00 mM); TCNB (1.00 mM); An34C10 and TCNB (1.00 mM, each). | ||
We further studied the host–guest complexation between An34C10 and TCNB through UV-vis spectroscopy. As shown in Fig. 4e, after mixing colorless TCNB and the light yellow An34C10 in chloroform, an obvious color change occurred. Different from the UV absorbances of the free host and guest, a new absorption band appeared at about 450–650 nm for the mixed solution ascribed to CT between the host and guest (Fig. 4d). In addition, fluorescence titration experiments were performed to afford the binding constant between An34C10 and TCNB in chloroform. It was found that the color of the solution became darker and the fluorescence intensity decreased during the titration of the guest (Fig. S20, ESI†). The association constant was determined to be (2.34 ± 0.12) × 103 M−1 by non-linear fitting in a 1:1 complexation mode (Fig. S21, ESI†). Meanwhile, the binding affinity of the host–guest complexation was also investigated by 1H NMR titration experiments (Fig. S22, ESI†). And the association constant was determined to be (1.91 ± 0.17) × 103 M−1 (Fig. S23, ESI†). The above results revealed strong intermolecular CT interactions between An34C10 and TCNB in chloroform, which enabled the formation of a stable host–guest complex.
The strong interactions in solution prompted us to explore whether CT existed in the solid state. Evaporating an acetone solution of the host–guest complex afforded dark purple crystals (An34C10@TCNB) suitable for X-ray single crystal diffraction (Fig. 5). In the single crystal structure, TCNB was located in the cavity of An34C10, in line with the above spectroscopic results in solution. The guest was stabilized by multiple C–H⋯N and C–H⋯O hydrogen bonds, where the distances were 2.849 Å, 3.147 Å, 3.255 Å and 3.284 Å, respectively. Additionally, strong aryl stackings between An34C10 and TCNB with distances of 3.427 Å and 3.439 Å evidenced CT between the host and the guest in the co-crystals. PXRD experiments were performed to analyze the stacking mode of co-crystals. As shown in Fig. S24, ESI†, An34C10@TCNB co-crystals showed different PXRD patterns from those of free An34C10 and TCNB, demonstrating that the initial molecular packing modes of single components vanished. In addition, the existence of CT in An34C10@TCNB was also confirmed by FT-IR spectroscopy (Fig. S25, ESI†) and thermogravimetric analysis (TGA) (Fig. S26, ESI†).
 | ||
| Fig. 5 Single crystal structure of An34C10@TCNB: (a) side view; (b) top view. Carbon atoms are grey, oxygen atoms are red, nitrogen atoms are blue, and hydrogen atoms are white. Crucial distances are marked with dashed lines. Solvents are omitted for clarity. (c) The packing mode of An34C10@TCNB. Hydrogen atoms and solvent molecules are omitted for clarity. | ||
Then photophysical properties of the co-crystals were explored to further confirm the existence of intermolecular CT. Fig. 6a displays the solid state UV-vis absorption spectrum of An34C10@TCNB. A new absorbance appeared at 500–700 nm in the co-crystals that could be attributed to intermolecular CT. In addition, the PL spectrum of An34C10@TCNB co-crystals revealed that the host–guest complex was nearly non-emissive (Fig. 6b). This was because the strong intermolecular CT between the host and the guest generated dark states that quenched the emission.66
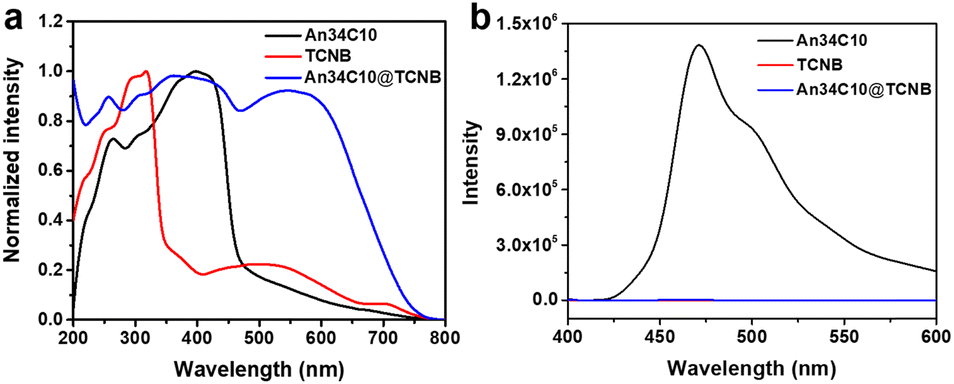 | ||
| Fig. 6 (a) Normalized solid state UV-vis absorption spectra and (b) fluorescence spectra of An34C10, TCNB and An34C10@TCNB. | ||
Intermolecular CT is vulnerable to surrounding environments, we thus envisaged that external stimuli could be applied to restore the emission. After screening VOCs, we found that fuming the co-crystals with 4-methylpyridine (4MePy) vapor was an effective method to turn on the emission. That is, when An34C10@TCNB co-crystals were placed in a saturated atmosphere of 4MePy vapor for 3 days, the fluorescence of the material recovered. The 1H NMR spectrum of a solution of the fumed co-crystals (denoted as An34C10@TCNB-4MePy, Fig. S27, ESI†) illustrated that 4MePy was trapped in the co-crystals. Besides, the proton signal of TCNB showed a slight downfield shift compared with that of An34C10@TCNB (Fig. S28, ESI†), evidencing weakened CT interactions that turned on the emission of the host. Such turn-on emission clearly indicates that the co-crystal is a promising candidate for sensing and detection of pyridyl compounds.
TGA also confirmed the existence of three components (Fig. S29, ESI†). Three plateaus were observed in the thermogravimetric curve of An34C10@TCNB-4MePy. The first plateau implied the desorption of solvent molecules, that is, 4MePy. The amount of 4MePy adsorbed by co-crystals was determined to be 0.89 molecules per host–guest complex, in good agreement with the results of the 1H NMR spectrum. The latter two plateaus corresponded to the decompositions of TCNB and An34C10, respectively. Similarly, the photophysical properties of An34C10@TCNB-4MePy were characterized. The solid state UV-vis spectrum showed a decrease in the CT band compared with that of An34C10@TCNB (Fig. 7a). The PL spectrum of An34C10@TCNB-4MePy was the same as that of An34C10, indicating that the fluorescence came from the luminescence of the macrocycle (Fig. 7b). However, the luminescence intensity was greatly reduced, and the fluorescence quantum yield was determined to be 1.9%. PXRD patterns revealed that after 4MePy fuming, some of the diffraction signals corresponded well to those of the macrocycle (Fig. S30, ESI†). These results confirmed that 4MePy fuming weakened CT interactions in the co-crystal, thus enabling the fluorescence of An34C10 to turn on again. Besides, when An34C10@TCNB was exposed to other common VOC vapors such as toluene, cyclohexane, methylcyclohexane, pyridine, 2-methylpyridine, and 3-methylpyridine at room temperature, no fluorescence was observed (Fig. S31, ESI†). The main diffraction peaks in PXRD patterns and fluorescence spectra of An34C10@TCNB did not change after absorption of these VOCs, which exhibited the selective vapochromic behaviors for 4MePy (Fig. S32 and S33, ESI†).
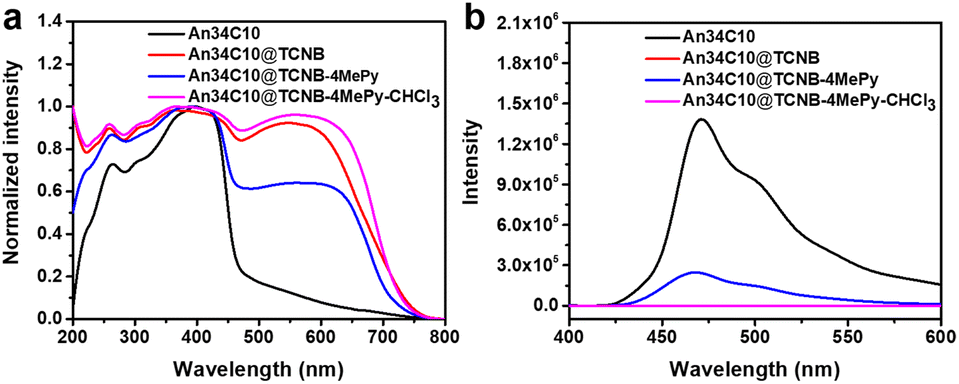 | ||
| Fig. 7 (a) Normalized solid state UV-vis absorption spectra and (b) solid state fluorescence spectra of An34C10, An34C10@TCNB, An34C10@TCNB-4MePy and An34C10@TCNB-4MePy-CHCl3. | ||
Although crystallization of An34C10, TCNB, and 4MePy failed, we still observed that when An34C10@TCNB co-crystals were immersed in a 1.0 mol L−14MePy aqueous solution for 10 days (Fig. S34, ESI†), light yellow cracks appeared on the surfaces of the dark purple crystals. These cracks emitted blue fluorescence under 365 nm UV irradiation, which is consistent with the induced emission in the presence of 4MePy.
Additionally, it was found that when An34C10@TCNB-4MePy was placed in chloroform vapor, the fluorescence of the material was quickly quenched (denoted as An34C10@TCNB-4MePy-CHCl3, Fig. 7b). PXRD experiments (Fig. S30, ESI†) showed that the pattern after fuming with chloroform was consistent with that of An34C10@TCNB, indicating that the crystal structure was restored to the initial CT state. This implied that chloroform could be a competitive guest, whereby 4MePy is released and the co-crystals become non-emissive again. However, under vapors of other haloalkanes and aryl halides, such as tetrachloromethane, 1-iodobutane, 1,4-dibromobutane, chlorobenzene and bromobenzene, the fluorescence of An34C10@TCNB-4MePy remained unchanged (Fig. S35 and S36, ESI†). And the main PXRD diffractions of crystals after vapor exposure were consistent with those before fuming (Fig. S37, ESI†). These results showed that An34C10@TCNB-4MePy displayed a high selectivity for chloroform vapor.
Moreover, we tested the recyclability of the vapochromic behaviors for An34C10@TCNB (Fig. S38, ESI†). As expected, after alternative fuming with 4MePy and chloroform vapors, the fluorescence of An34C10@TCNB could be switched between on and off at least five times without any performance loss, which indicated the good cycling performance of host–guest co-crystals. The on and off switches hold potential in the fabrication of smart luminescent materials with response to pyridyl and alkyl chloride compounds.
Conclusions
In summary, we synthesized a crown ether derivative containing two anthracene units and investigated the host–guest co-crystals based on crown ether for vapochromism. Due to the presence of anthracene, An34C10 not only emitted bright blue fluorescence, but also showed mechanochromic properties, and the fluorescence change could be recovered by heating or organic solvent fuming. Through various experimental analyses, it was found that the mechanochromism of An34C10 was derived from a transition between different stacking structures. In addition, An34C10 complexes with TCNB to form co-crystals. Due to the strong charge-transfer interactions between the host and guest, the fluorescence was quenched. Since solvent fuming would weaken intermolecular charge-transfer interactions, the fluorescence of co-crystals was turned on by fuming with 4MePy. Interestingly, when the emissive co-crystal was further fumed with chloroform vapor, the fluorescence was quenched again, and the structure of co-crystals returned to the initial state. Meanwhile, both the mechanochromism of An34C10 and the fluorescence turn-on and off in An34C10@TCNB displayed good cycling performance. This work not only broadens the application of supramolecular chemistry but also combines a macrocycle with co-crystal engineering, providing a new strategy for the preparation of various stimuli-responsive crystalline materials.Data availability
The crystallographic data for An34C10 and An34C10@TCNB have been deposited at CCDC with deposition numbers 2226677 and 2226678, respectively. The data can be obtained viahttps://www.ccdc.cam.ac.uk/structures. Data for this paper, including synthesis and structural characterizations, are available in the ESI.† The raw data are available from the authors upon reasonable request.Author contributions
W. Zhu, H. Zhu and F. Huang proposed the project and designed the study. W. Zhu and B. Zhao performed the experiments. W. Zhu, S. Fang and H. Zhu analyzed the data. W. Zhu, H. Zhu and F. Huang wrote the manuscript with inputs from all authors. F. Huang directed the project with critical consultation from H. Zhu.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Key Research and Development Program of China (2021YFA0910100), National Natural Science Foundation of China (22035006, 22320102001, and 22350007), Zhejiang Provincial Natural Science Foundation of China (LD21B020001), the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study (SN-ZJU-SIAS-006), and the Leading Innovation Team grant from Department of Science and Technology of Zhejiang Province (2022R01005). We thank The Chemistry Instrumentation Center of Zhejiang University and The Instrumentation and Service Center for Molecular Sciences at Westlake University for the technical support.Notes and references
- X. Zhang, B. Li, Z.-H. Chen and Z.-N. Chen, J. Mater. Chem., 2012, 22, 11427–11441 RSC.
- M. H. Keefe, K. D. Benkstein and J. T. Hupp, Coord. Chem. Rev., 2000, 205, 201–228 CrossRef CAS.
- E. J. Fernández, A. Laguna and J. M. López-De-Luzuriaga, Coord. Chem. Rev., 2005, 249, 1423–1433 CrossRef.
- M. J. Katz, K. Sakai and D. B. Leznoff, Chem. Soc. Rev., 2008, 37, 1884–1895 RSC.
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC.
- L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 CrossRef CAS PubMed.
- Y. Sagara, M. Karman, E. Verde-Sesto, K. Matsuo, Y. Kim, N. Tamaoki and C. Weder, J. Am. Chem. Soc., 2018, 140, 1584–1587 CrossRef CAS PubMed.
- Y. Sagara, H. Traeger, J. Li, Y. Okado, S. Schrettl, N. Tamaoki and C. Weder, J. Am. Chem. Soc., 2021, 143, 5519–5525 CrossRef CAS PubMed.
- S. Thazhathethil, T. Muramatsu, N. Tamaoki, C. Weder and Y. Sagara, Angew. Chem., Int. Ed., 2022, 61, e202209225 CrossRef CAS PubMed.
- M.-S. Yuan, D.-E. Wang, P. Xue, W. Wang, J.-C. Wang, Q. Tu, Z. Liu, Y. Liu, Y. Zhang and J. Wang, Chem. Mater., 2014, 26, 2467–2477 CrossRef CAS.
- S. H. Lim, M. M. Olmstead and A. L. Balch, Chem. Sci., 2013, 4, 311–318 RSC.
- S. Ito, CrystEngComm, 2022, 24, 1112–1126 RSC.
- E. Li, K. Jie, Y. Fang, P. Cai and F. Huang, J. Am. Chem. Soc., 2020, 142, 15560–15568 CrossRef CAS PubMed.
- D. Gentili, M. Gazzano, M. Melucci, D. Jones and M. Cavallini, Chem. Soc. Rev., 2019, 48, 2502–2517 RSC.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- B. Kim, G. Storch, G. Banerjee, B. Q. Mercado, J. Castillo-Lora, G. W. Brudvig, J. M. Mayer and S. J. Miller, J. Am. Chem. Soc., 2017, 139, 15239–15244 CrossRef CAS PubMed.
- G. R. Desiraju, Angew Chem. Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- J. J. Wolff, Angew Chem. Int. Ed. Engl., 1996, 35, 2195–2197 CrossRef CAS.
- H. Yamagishi, H. Sato, A. Hori, Y. Sato, R. Matsuda, K. Kato and T. Aida, Science, 2018, 361, 1242–1246 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- A. G. Slater, M. A. Little, A. Pulido, S. Y. Chong, D. Holden, L. Chen, C. Morgan, X. Wu, G. Cheng, R. Clowes, M. E. Briggs, T. Hasell, K. E. Jelfs, G. M. Day and A. I. Cooper, Nat. Chem., 2017, 9, 17–25 CrossRef CAS PubMed.
- G. Li, Z. Zhou, C. Yuan, Z. Guo, Y. Liu, D. Zhao, K. Liu, J. Zhao, H. Tan and X. Yan, Angew. Chem., Int. Ed., 2020, 59, 10013–10017 CrossRef CAS PubMed.
- K. Gao, Q. Feng, Z. Zhang, R. Zhang, Y. Hou, C. Mu, X. Li and M. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202209958 CrossRef CAS PubMed.
- K. Wang, S. Huang, Y. Zhang, S. Zhao, H. Zhang and Y. Wang, Chem. Sci., 2013, 4, 3288–3293 RSC.
- H. Xia, D. Liu, K. Song and Q. Miao, Chem. Sci., 2011, 2, 2402–2406 RSC.
- Z. Li, Y. Han, F. Nie, M. Liu, H. Zhong and F. Wang, Angew. Chem., Int. Ed., 2021, 60, 8212–8219 CrossRef CAS PubMed.
- B. Li, L. Cui and C. Li, Angew. Chem., Int. Ed., 2020, 59, 22012–22016 CrossRef CAS PubMed.
- M. Wang, Q. Li, E. Li, J. Liu, J. Zhou and F. Huang, Angew. Chem., Int. Ed., 2021, 60, 8115–8120 CrossRef CAS PubMed.
- H. Zhu, L. Chen, B. Sun, M. Wang, H. Li, J. F. Stoddart and F. Huang, Nat. Rev. Chem, 2023, 7, 768–782 CrossRef CAS PubMed.
- L. Sun, Y. Wang, F. Yang, X. Zhang and W. Hu, Adv. Mater., 2019, 31, 1902328–1902349 CrossRef.
- X.-J. Song, T. Zhang, Z.-X. Gu, Z.-X. Zhang, D.-W. Fu, X.-G. Chen, H.-Y. Zhang and R.-G. Xiong, J. Am. Chem. Soc., 2021, 143, 5091–5098 CrossRef PubMed.
- W.-L. Zhou, W. Lin, Y. Chen and Y. Liu, Chem. Sci., 2022, 13, 7976–7989 RSC.
- Z.-Y. Zhang and Y. Liu, Chem. Sci., 2019, 10, 7773–7778 RSC.
- Y. Sun, L. Jiang, L. Liu, Y. Chen, W.-W. Xu, J. Niu, Y. Qin, X. Xu and Y. Liu, Adv. Opt. Mater., 2023, 11, 2300326–2300334 CrossRef CAS.
- Y. Zhao, B. Ding, Z. Huang and X. Ma, Chem. Sci., 2022, 13, 8412–8416 RSC.
- B. Hua, W. Zhou, Z. Yang, Z. Zhang, L. Shao, H. Zhu and F. Huang, J. Am. Chem. Soc., 2018, 140, 15651–15654 CrossRef CAS PubMed.
- B. Hua, C. Zhang, W. Zhou, L. Shao, Z. Wang, L. Wang, H. Zhu and F. Huang, J. Am. Chem. Soc., 2020, 142, 16557–16561 CrossRef CAS PubMed.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 2495–2496 CrossRef CAS.
- R. M. Izatt, Chem. Soc. Rev., 2017, 46, 2380–2384 RSC.
- G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 CrossRef CAS PubMed.
- B. Zheng, F. Wang, S. Dong and F. Huang, Chem. Soc. Rev., 2012, 41, 1621–1636 RSC.
- Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC.
- E. Li, K. Jie, M. Liu, X. Sheng, W. Zhu and F. Huang, Chem. Soc. Rev., 2020, 49, 1517–1544 RSC.
- A. Dhara, A. Dmitrienko, R. N. N. Hussein, A. Sotomayor, B. H. H. Wilson and S. J. J. Loeb, Chem. Sci., 2023, 14, 7215–7220 RSC.
- C.-H. Wang, K.-J. Chen, T.-H. Wu, H.-K. Chang, Y. Tsuchido, Y. Sei, P.-L. Chen and M. Horie, Chem. Sci., 2021, 12, 3871–3875 RSC.
- A. Aster, G. Licari, F. Zinna, E. Brun, T. Kumpulainen, E. Tajkhorshid, J. Lacour and E. Vauthey, Chem. Sci., 2019, 10, 10629–10639 RSC.
- Z. Niu and H. W. Gibson, Chem. Rev., 2009, 109, 6024–6046 CrossRef CAS PubMed.
- M. Lee, R. B. Moore and H. W. Gibson, Macromolecules, 2011, 44, 5987–5993 CrossRef CAS.
- A. Aydogan, D. J. Coady, S. K. Kim, A. Akar, C. W. Bielawski, M. Marquez and J. L. Sessler, Angew. Chem., Int. Ed., 2008, 47, 9648–9652 CrossRef CAS PubMed.
- O. A. Fedorova, E. Y. Chernikova, Y. V. Fedorov, E. N. Gulakova, A. S. Peregudov, K. A. Lyssenko, G. Jonusauskas and L. Isaacs, J. Phys. Chem. B, 2009, 113, 10149–10158 CrossRef CAS PubMed.
- R. Saha, B. Mondal and P. S. Mukherjee, Chem. Rev., 2022, 122, 12244–12307 CrossRef CAS PubMed.
- R. Kumar, A. Sharma, H. Singh, P. Suating, H. S. Kim, K. Sunwoo, I. Shim, B. C. Gibb and J. S. Kim, Chem. Rev., 2019, 119, 9657–9721 CrossRef CAS PubMed.
- D. Zhao, Z. Zhang, J. Zhao, K. Liu, Y. Liu, G. Li, X. Zhang, R. Bai, X. Yang and X. Yan, Angew. Chem., Int. Ed., 2021, 60, 16224–16229 CrossRef CAS PubMed.
- P. Wei, X. Zhang, J. Liu, G.-G. Shan, H. Zhang, J. Qi, W. Zhao, H. H.-Y. Sung, I. D. Williams, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9293–9298 CrossRef CAS PubMed.
- C. Zhang, S. Li, J. Zhang, K. Zhu, N. Li and F. Huang, Org. Lett., 2007, 9, 5553–5556 CrossRef CAS PubMed.
- X. Ji, Y. Yao, J. Li, X. Yan and F. Huang, J. Am. Chem. Soc., 2013, 135, 74–77 CrossRef CAS PubMed.
- S. Murkli, J. Klemm, D. King, P. Y. Zavalij and L. Isaacs, Chem.–Eur. J., 2020, 26, 15249–15258 CrossRef CAS PubMed.
- E. Chirkin, V. Muthusamy, P. Mann, T. Roemer, P. G. Nantermet and D. A. Spiegel, Angew. Chem., Int. Ed., 2017, 56, 13036–13040 CrossRef CAS PubMed.
- Y.-L. Zhao, L. Liu, W. Zhang, C.-H. Sue, Q. Li, O. Š. Miljanić, O. M. Yaghi and J. F. Stoddart, Chem.–Eur. J., 2009, 15, 13356–13380 CrossRef CAS PubMed.
- Q. Dai, J. Zhang, R. Tan, S. Wang, Y. Li, Q. Li and S. Xiao, Mater. Lett., 2016, 164, 239–242 CrossRef CAS.
- R. Li, S. Xiao, Y. Li, Q. Lin, R. Zhang, J. Zhao, C. Yang, K. Zou, D. Li and T. Yi, Chem. Sci., 2014, 5, 3922–3928 RSC.
- K. C. Naeem, A. Subhakumari, S. Varughese and V. C. Nair, J. Mater. Chem. C, 2015, 3, 10225–10231 RSC.
- K. Duraimurugan, M. Harikrishnan, J. Madhavan, A. Siva, S. J. Lee, J. Theerthagiri and M. Y. Choi, Environ. Res., 2021, 194, 110741–110748 CrossRef CAS.
- C. Janiak, J. Chem. Soc. Dalton Trans., 2000, 21, 3885–3896 RSC.
- X. Zhang, Z. Chi, X. Zhou, S. Liu, Y. Zhang and J. Xu, J. Phys. Chem. C, 2012, 116, 23629–23638 CrossRef CAS.
- H. Zhu, J. Liu, Y. Wu, L. Wang, H. Zhang, Q. Li, H. Wang, H. Xing, J. L. Sessler and F. Huang, J. Am. Chem. Soc., 2023, 145, 11130–11139 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra, mass spectra, FT-IR spectra, TGA data, DSC data, PXRD data, crystallographic data, photophysical data, and other materials. CCDC 2226677 and 2226678. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05077k |
| ‡ Weijie Zhu and Bohan Zhao contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |