
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantifying variation in cooperative B–H bond activations using Os(II) and Os(III) κ2-N,S-chelated complexes: same, but different†
Sourav
Gayen
,
Faneesha
Assanar
,
Sampad
Shyamal
,
Dorothy Priyanka
Dorairaj
and
Sundargopal
Ghosh
 *
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: sghosh@iitm.ac.in
First published on 12th September 2024
Abstract
In an effort to investigate small molecule activation by heavier transition metal (TM) based κ2-N,S-chelated species, we have synthesised a series of bis-κ2-1,3-N,S-chelated complexes of osmium, [Os(PPh3)2(κ2-N,S-La/Lb)2], 2a–b, and [Os(PPh3)(La/Lb)(κ2-N,S-La/Lb)2], 3a–b (2a and 3a: La![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) = C7H4NS2; 2b and 3b: Lb = C5H4NS), from the thermolysis of [Os(PPh3)3Cl2], 1, with a potassium salt of heterocyclic ligands La and Lb. The former complexes are diamagnetic in nature, while the EPR spectra, XPS study and density functional theory (DFT) calculations have substantiated the paramagnetic behaviour of 3a–b with a significant spin contribution from non-innocent ligands. These species were engaged in B–H activation of boranes utilizing the combined effect of hemilability and metal–ligand cooperativity (MLC), where 2a–b upon treatment with BH3·SMe2 yielded Os(σ-borate)hydride complexes, [Os(PPh3)2(H){κ3-H,S,S′-BH2(La/Lb)2}], 4a–b (4a: La = C7H4NS2; 4b: Lb = C5H4NS). The formation of 4a–b was emphasized on the basis of possible dual B–H activation that involved a few concerted steps, i.e., (i) cleavage of one hemilabile Os–N bond and cooperative B–H bond activation, (ii) cleavage of another hemilabile Os–N bond and formation of a B–N bond and (iii) activation of another B–H bond. In stark contrast, the paramagnetic bis-κ2-1,3-N,S-chelated species 3a–b manifested diverse activation patterns in the light of the different electronic nature of non-innocent ligands. While the reaction of 3b with borane generated dihydridoborate species, [Os(PPh3)(κ2-N,S-Lb)(κ3-H,H,S′-BH2(OH)C5H4NS)], 5, complex 3a led to the formation of an Os(σ-borate) complex, [Os(PPh3)(κ2-N,S-La)(κ3-H,S,S′-BH2L2a)], trans-6. As the σ-borate entity shows a tendency to adapt to different spatial arrangements around the metal center, we have established a modified synthetic strategy to isolate the cis isomer of 6 that involved the reaction of [Os(PPh3)3Cl2], 1, with NaBH2La2. In a similar fashion, the treatment of [Os(PPh3)3Cl2], 1, with NaBH2Lb2 yielded [Os(PPh3)(κ2-N,S-Lb)(κ3-H,S,S′-BH2L2b)], cis-7. The kinetic and thermodynamic stability of these isomeric species were investigated on the basis of extensive density functional theory (DFT) calculations. Theoretical calculations also provided insightful information on the electronic nature of the species, generated from B–H activations of boranes.
= C7H4NS2; 2b and 3b: Lb = C5H4NS), from the thermolysis of [Os(PPh3)3Cl2], 1, with a potassium salt of heterocyclic ligands La and Lb. The former complexes are diamagnetic in nature, while the EPR spectra, XPS study and density functional theory (DFT) calculations have substantiated the paramagnetic behaviour of 3a–b with a significant spin contribution from non-innocent ligands. These species were engaged in B–H activation of boranes utilizing the combined effect of hemilability and metal–ligand cooperativity (MLC), where 2a–b upon treatment with BH3·SMe2 yielded Os(σ-borate)hydride complexes, [Os(PPh3)2(H){κ3-H,S,S′-BH2(La/Lb)2}], 4a–b (4a: La = C7H4NS2; 4b: Lb = C5H4NS). The formation of 4a–b was emphasized on the basis of possible dual B–H activation that involved a few concerted steps, i.e., (i) cleavage of one hemilabile Os–N bond and cooperative B–H bond activation, (ii) cleavage of another hemilabile Os–N bond and formation of a B–N bond and (iii) activation of another B–H bond. In stark contrast, the paramagnetic bis-κ2-1,3-N,S-chelated species 3a–b manifested diverse activation patterns in the light of the different electronic nature of non-innocent ligands. While the reaction of 3b with borane generated dihydridoborate species, [Os(PPh3)(κ2-N,S-Lb)(κ3-H,H,S′-BH2(OH)C5H4NS)], 5, complex 3a led to the formation of an Os(σ-borate) complex, [Os(PPh3)(κ2-N,S-La)(κ3-H,S,S′-BH2L2a)], trans-6. As the σ-borate entity shows a tendency to adapt to different spatial arrangements around the metal center, we have established a modified synthetic strategy to isolate the cis isomer of 6 that involved the reaction of [Os(PPh3)3Cl2], 1, with NaBH2La2. In a similar fashion, the treatment of [Os(PPh3)3Cl2], 1, with NaBH2Lb2 yielded [Os(PPh3)(κ2-N,S-Lb)(κ3-H,S,S′-BH2L2b)], cis-7. The kinetic and thermodynamic stability of these isomeric species were investigated on the basis of extensive density functional theory (DFT) calculations. Theoretical calculations also provided insightful information on the electronic nature of the species, generated from B–H activations of boranes.
Introduction
Metal–ligand cooperation (MLC), the synergistic interplay between a metal and ligand, plays a dominant role in the catalytic activation of small molecules.1,2 While classical catalysis involves solely metal-based catalytic activity, ligands in cooperative transition metal architectures concertedly participate in the cleavage or formation of inert chemical bonds. In particular, the activation of H–H, C–H, N–H, O–H, B–H and Si–H bonds has been extensively studied utilizing MLC.3–6 Thus, these representatives evolved as privileged catalysts in various catalytic processes, such as hydrogenation, borylation, hydroboration, hydrosilylation, etc.7,8 Conceptually, these species can be better defined as a hybrid molecular architecture by incorporating a transition metal as one of the components in Frustrated Lewis Pairs (FLPs), often termed transition metal frustrated Lewis pairs (TMFLPs).9 The molecular control over the bond activation process can be implemented by tuning the transition metal as well as modulating the steric and electronic properties of ligand scaffolds, thus resulting in a polarized hybrid platform. Based on the metal–ligand combination, these types of polarized systems can be differentiated into two categories; (i) electron-donating metal (LB)–electron-accepting ligand (LA) and (ii) electron-accepting metal (LA)–electron-donating ligand (LB).10In the recent past, significant progress has been made in E–H (E = H, B, Si, C) bond activation utilizing a cooperative design, comprising an electron-accepting metal centre and electron-donating functionality. For example, Oestreich and co-workers extensively studied the heterolytic activation of the E–H bond across the Ru–S bond of a tethered ruthenium thiolate complex that generates a metal hydride and sulphur stabilized E+ cation, I (Chart 1).3b,d,11 As per the reports of the Gessner group, a methandiide derived carbene complex of ruthenium was engaged in the activation of several small molecules, e.g. hydrogen, silanes, phosphines and phosphine oxides, where protonation took place in the carbonic carbon atom (II, Chart 1).3c,12,13 In contrast, a versatile pattern of activation was investigated in the case of complexes of palladium, which successfully cleaved E–H (E= B, Si) bonds to generate hydrido derivatives, III, while treating with Ph3SiH and germanes, possessing high acidity, led to the formation of silyl and germyl complexes, respectively.4a Interestingly, Casey and Clark isolated silyloxy and boroxycyclopentadienyl hydrido–ruthenium complexes, IV, by the heterolytic cleavage of Si–H and B–H bonds, respectively, using dimeric ruthenium species.14 Apart from other small molecules, hydroboranes exhibit peculiarities due to the high electropositivity of boron and the hydridic nature of bound hydrogens. In this context, Drover, Schafer and Love demonstrated the activation of hindered borane, HBCy2, employing the hemilability and joint metal–ligand cooperativity of κ2-N,O chelated phosphoramidate complexes of group-9 TM (V).15 Similarly, Stradiotto reported that a κ2-P,O chelated phosphino enolate complex of ruthenium promoted the insertion of H2BMes by the cleavage of the Ru–O bond, affording a dihydridoborate complex, VI.16 Daly and co-workers demonstrated the electrochemical behaviour of a redox-active ruthenium complex, VII, in which two BH3 units encountered cooperative binding along the Ru–N bond.17 It is worth noting that the field of MLC has recently evolved by introducing heavier main group elements into the ligand scaffold.18 However, the influence of heavier transition metals as cooperative partners in MLC is less explored.
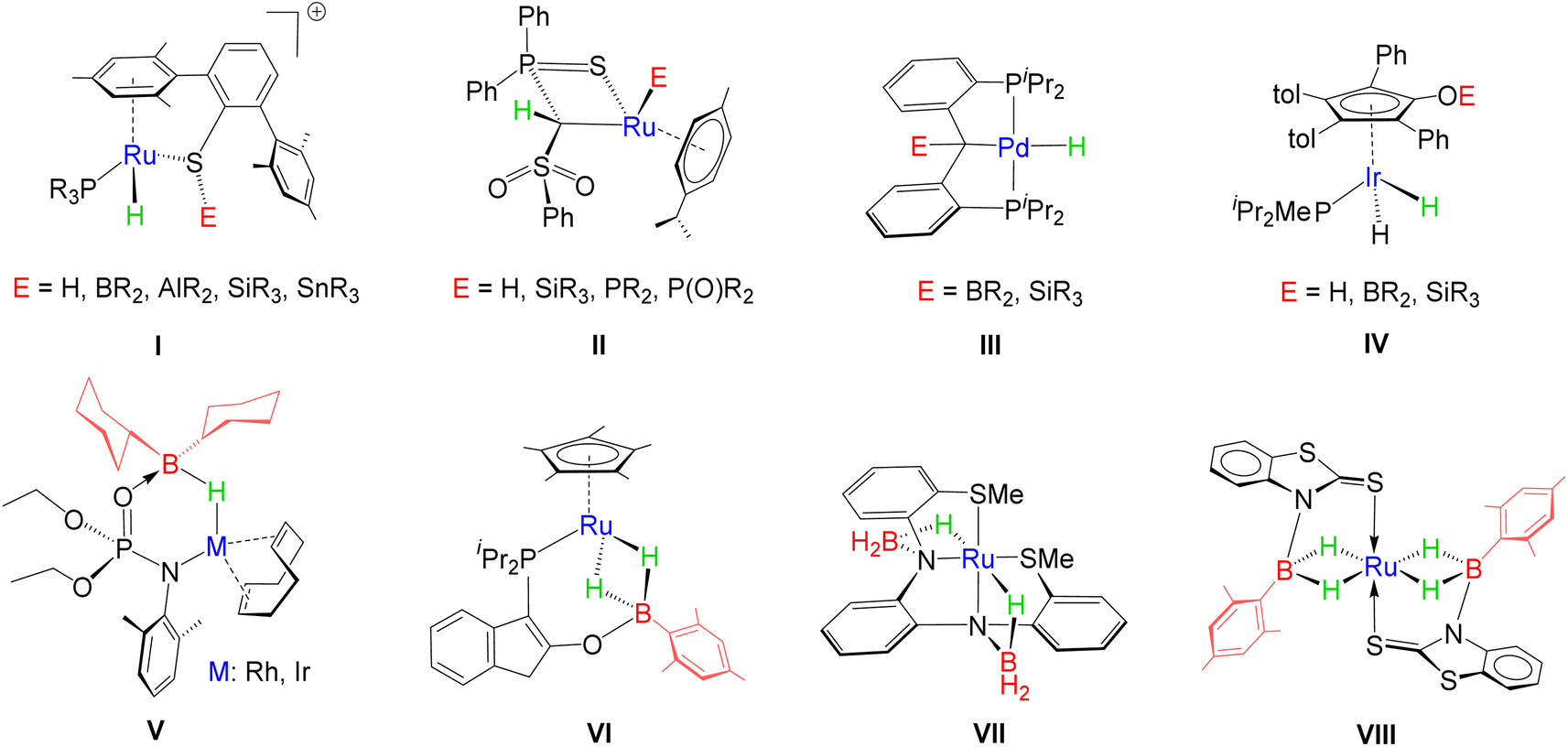 | ||
| Chart 1 Various examples of small molecule activation by TM complexes (I–VIII). (iPr = isopropyl; R = H, alkyl, aryl or alkoxy group). | ||
In the course of our recent studies, we have engaged a series of transition metal based κ2-N,S-chelated complexes in the cooperative activation of various small molecules, such as hydroboranes, hydrosilanes, alkynes and GeCl2.19,20 The combination of hard (N) and soft (S) centres in polydentate ligands offered the activation of E-H (E = B, Si or C) bonds by the cleavage of M–X (X = N, S) bonds. Notably, the activation of boranes yielded TM σ-borate species,19a,b,20a featuring M–H–B interactions. For instance, a redox-active ruthenium system was engaged in unusual dual side B–H activation of H2BMes that resulted in the formation of Ru-bis(dihydridoborate) species, VIII.19b Building on our previous studies, we have sought to explore the activation chemistry of osmium, a heavier group-8 TM based κ2-N,S-chelated species. In this context, we have synthesized two different types of κ2-N,S-chelated osmium complexes, diamagnetic [Os(PPh3)2{κ2-N,S-(La/Lb)}2], 2a–b, and paramagnetic [Os(PPh3)(La/Lb)(κ2-N,S-La/Lb)2], 3a–b (2a and 3a: La = C7H4NS2; 2b and 3b: Lb = C5H4NS). In this article, we report how the incorporation of osmium influences the cooperative activation of boranes.
Results and discussion
Recently, we have synthesized the cis and trans isomers of Ru[(PPh3)2(κ2-N,S-C7H4NS2)2]19d from the reaction of [Ru(PPh3)3 Cl2] with a potassium salt of 2-mercaptobenzothiazole. With a similar objective, we performed the thermolysis of [Os(PPh3)3Cl2], 1, with five equivalents of a potassium salt of two different heterocyclic systems La and Lb (La = C7H4NS2; Lb = C5H4NS). The salt elimination reactions afforded [Os(PPh3)2(κ2-N,S-C7H4NS2)2], 2a (yield = 42%), and [Os(PPh3)2(κ2-N,S-C5H4NS)2], 2b (yield = 46%), respectively (Scheme 1). These complexes were isolated in their purest form by a chromatographic technique and were characterized by multinuclear spectroscopy, mass spectrometry and X-ray diffraction studies. Both species revealed single resonance peaks at δ = −5.5 (2a) and −4.3 (2b) ppm in 31P{1H} NMR, respectively, indicating the presence of a single phosphorus environment. Along with the typical peaks related to the phenyl rings of PPh3, the 1H NMR spectrum showed additional aromatic region peaks in the range of δ = 6.12–7.95 ppm that correspond to heterocyclic ring systems. To get a clear picture, X-ray diffraction analysis was performed on a suitable orange crystal of 2b. As anticipated, the solid state X-ray structure of 2b revealed an octahedral geometry around the Os center, featuring two κ2-1,3-N,S-chelated rings and two triphenylphosphine ligands (Fig. 1). Taking into account that PPh3 units are cis to each other, complex 2b can be designated as cis-[Os(PPh3)2(κ2-N,S-C5H4NS)2]. The molecular ion peak in ESI-MS was in good agreement with the molecular formula. Moreover, in the case of 2a, the intense ion peak at m/z 1049.1018 in mass spectrometry. This data in combination with spectroscopic analysis clearly supported the formation of analogous κ2-1,3-N,S-chelated species, [Os(PPh3)2(κ2-N,S-La)2].
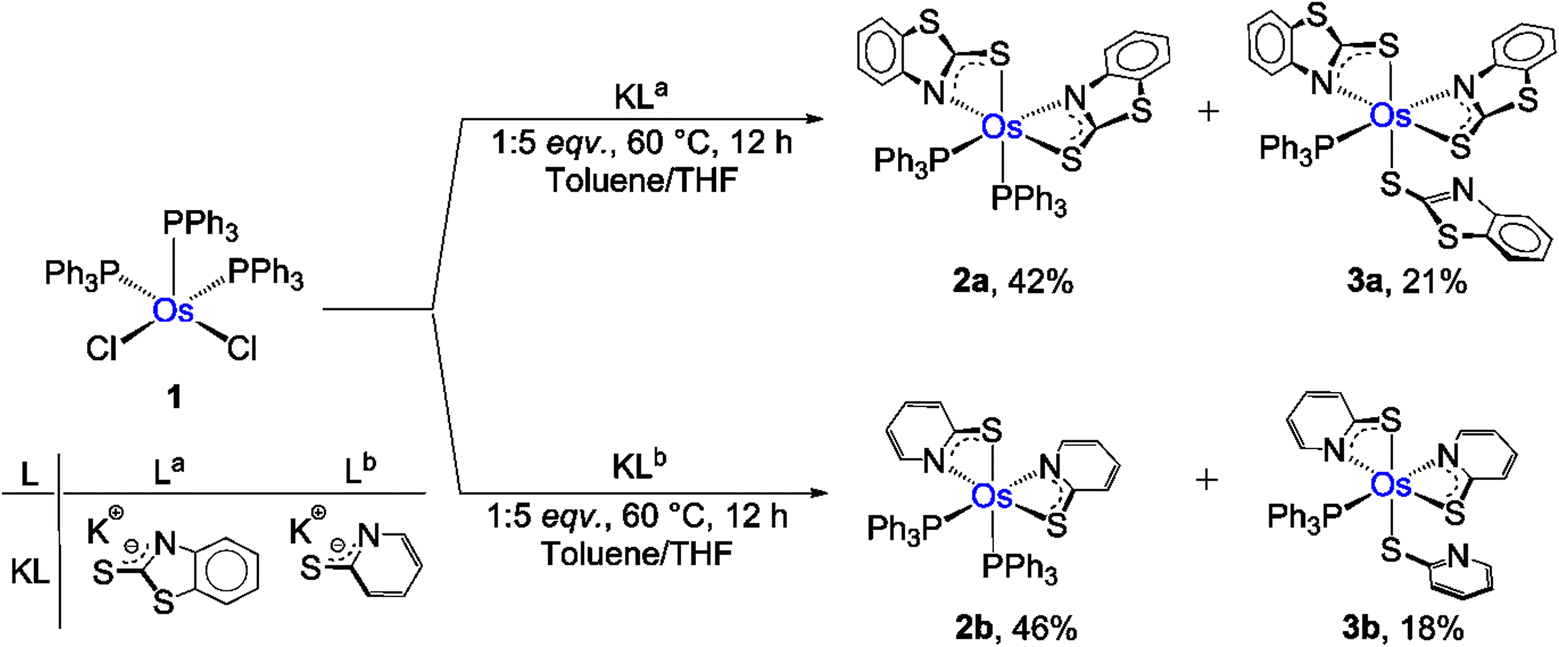 | ||
| Scheme 1 Syntheses of Os(II) and Os(III)-N,S-chelated complexes. | ||
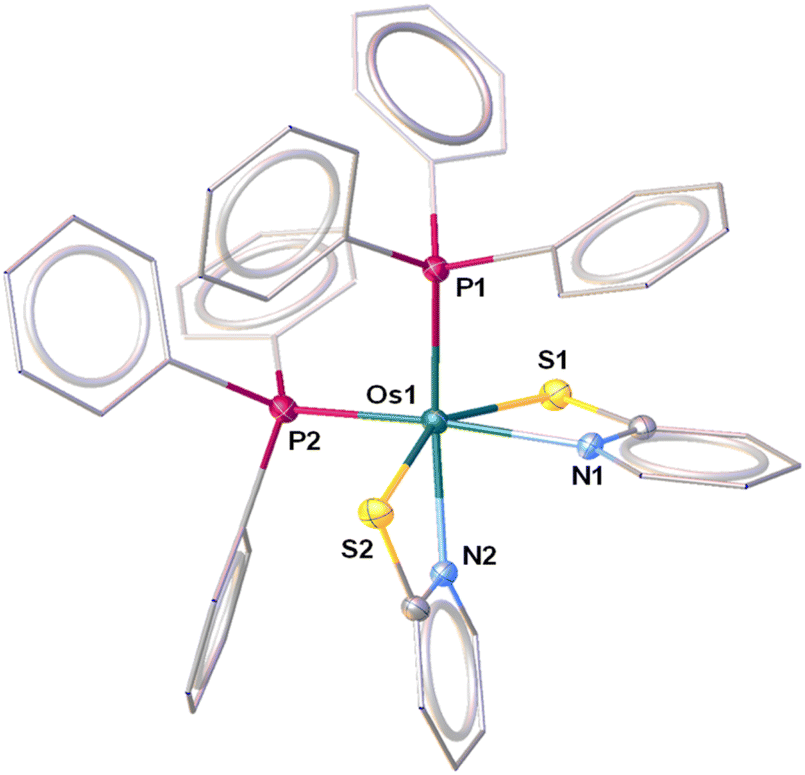 | ||
| Fig. 1 Molecular structure and labelling diagram of 2b. Selected bond lengths (Å) and bond angles (°) of 2b: Os1–N1 2.143(3), Os1–N2 2.134(3), Os1–P1 2.2887(10), Os1–P2 2.2993(10), Os1–S1 2.4318(10), Os1–S2 2.4257(10); S2–Os1–S1 150.98(4), and N2–Os1–N1 80.11(13). | ||
In parallel to the formation of 2a and 2b, the reactions also yielded 3a (yield = 21%) and 3b (yield = 18%), as green and violet solids, respectively. The molecular ion peaks in the ESI-MS spectra of 3a and 3b suggested that these species are analogous to each other with the generalised molecular formula [Os(PPh3)(L)3] (3a: L = La; 3b: L = Lb) (Fig. S10 and S12†). Neither the 1H NMR nor the 31P{1H} NMR spectrum showed any prominent resonances that indirectly indicated their paramagnetic behaviour. Note that, recently, we have isolated paramagnetic Ru(III) species [Ru(PPh3)2(κ2-N,S-C7H4NS2)2(κ1-S-C7H4NS2)] by the aerial oxidation of a Ru(II)–borate complex.19b Similarly, 3a and 3b can be identified as the analogous paramagnetic complexes of Os(III) [Os(Ph3P)2(κ2-N,S-L)2(κ1-S-L)] (L = La or Lb), on the basis of mass spectrometry and preliminary spectroscopic data. Subsequently, the paramagnetic species 3a and 3b were investigated by electron paramagnetic resonance (EPR) spectroscopy and X-ray photoelectron spectroscopy (XPS).
Although the room temperature EPR spectra of both species in dichloromethane solution were ambiguous, the X-band frozen glass EPR spectra (Fig. S14†) displayed signals with g values of 2.157 (3a) and 2.136 (3b). Each spectrum exhibits rhombic features with no hyperfine coupling, presumably due to resolved 14N and 31P nuclei. Similar to analogous Ru species,19bg values differ significantly from those of organic radicals; thus, complexes can be described as a superposition of two resonating forms, i.e., [(L−)OsIII] and [(L˙)OsII]. However, the majority of non-innocent ligand coordinated osmium complexes manifested a significant amount of ligand-based spin.21 Conversely, the Mulliken spin density for the unpaired electron in 3a and 3b was located mostly on the Os atom (3a: +0.67; 3b: +0.71) with a smaller contribution from the monodentate heterocyclic ligand (3a: +0.25; 3b: +0.24), which was consistent with the spin density plot {Fig. 2(a) and (b)}. The molecular orbital analysis also showed that the unpaired electron typically accumulated on the orbital with a significant osmium d-character and a smaller p-character of the sulphur atom of the κ1-S as well as the κ2-N,S-ligand. Therefore, this result provides evidence of a predominantly metal-centred spin with a distinct contribution from the [(L−)OsIII] tautomer. Further, we have used X-ray photoelectron spectroscopy (XPS) to probe the presence of two tautomeric species in 3a–b. Different sets of binding energies of Os 4f7/2 were observed in both species. The binding energy of 50.7 eV is associated with the [(L−)Os(III)] tautomer, while the relatively smaller value of 49.8 eV corresponds to the [(L˙)Os(II)]tautomer {Fig. 2(c) and (d)}. Extensive analysis of the XPS data indicated a greater contribution from the [(L−)Os(III)] tautomer as compared to [(L˙)Os(II)] species. These binding energy values closely resemble those of reported Os(III) and Os(II) species, respectively.22 The characteristic elemental spectra of C, N, S, and P for both complexes were also investigated in a detailed analysis (Fig. S11 and S13†).
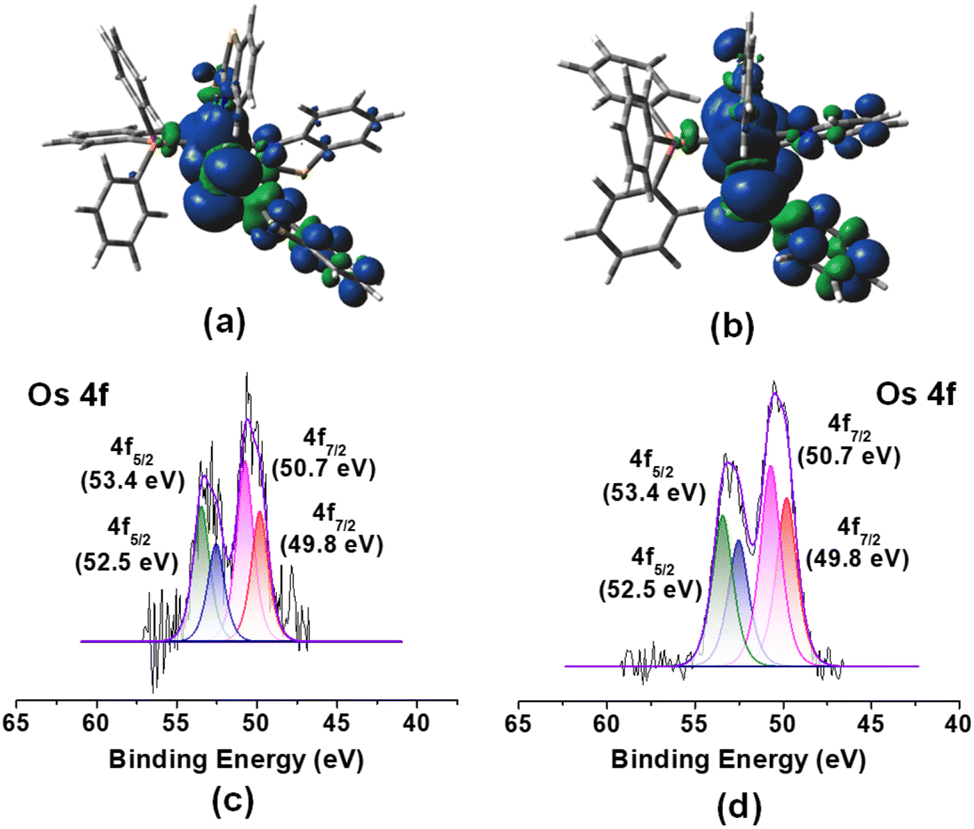 | ||
| Fig. 2 Spin density plot for 3a (a) and 3b (b) (isovalue 0.004 [e bohr−3]1/2); X-ray photoelectron spectroscopy (XPS) spectra of Os 4f7/2 and 4f5/2 for 3a (c) and 3b (d). | ||
An important distinction between these two types of bis-κ2-1,3-N,S-chelated complexes was the presence of a non-chelated κ1-S-L ligand, thereby allowing us to compare the redox and photophysical properties explicitly. To investigate the redox activity of these complexes, we have recorded the cyclic voltammograms of 2a–b in CH2Cl2 and 3a–3b in CH3CN {Fig. 3(a) and (b)}. The redox potential data referenced to the Fc+/Fc couple are summarized in Table S1.† The cyclic voltammogram of 2a yielded two quasi-reversible redox waves at E1/2 = 0.191 V and 0.948 V, while the same appeared at E1/2 = −0.287 V and 0.763 V for 2b. In contrast, three quasi-reversible redox events were observed for 3a in the potential window of 0 V to 1.0 V (Table S1†). The noteworthy observation is that a cathodic quasi-reversible wave at E1/2 = −1.007 V and −1.126 V was recorded for 3a and 3b, respectively.
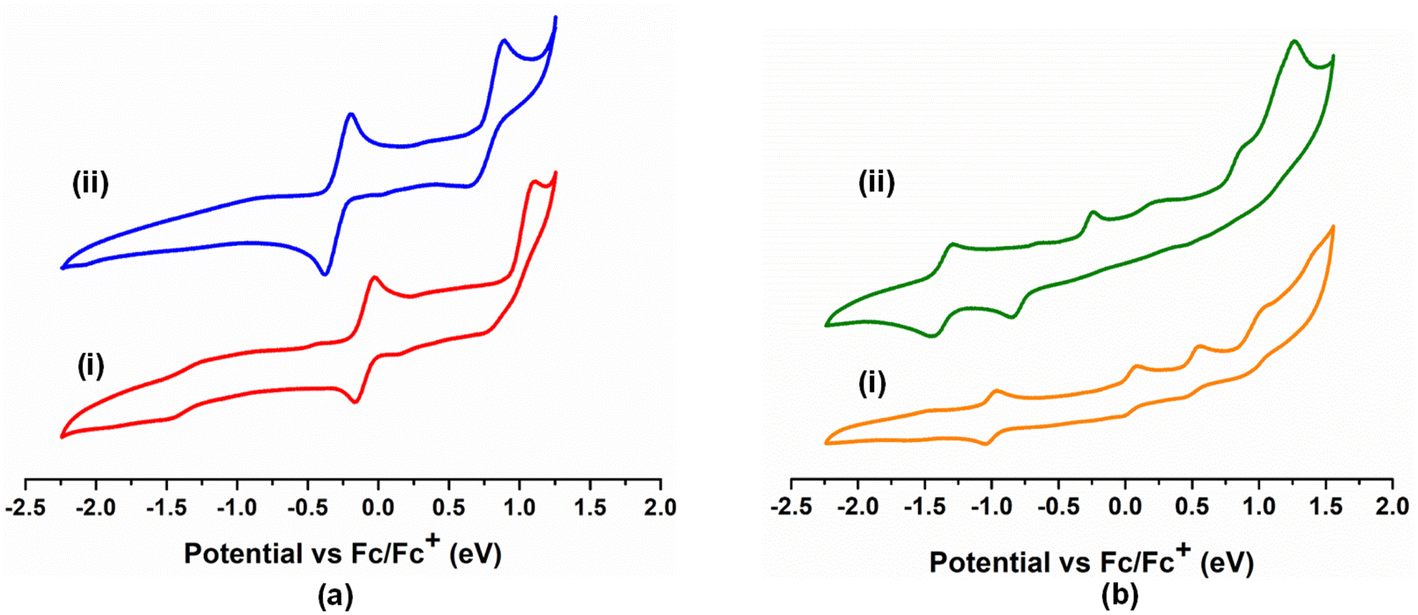 | ||
| Fig. 3 (a) Cyclic voltammograms of 2a (i) and 2b (ii); (b) cyclic voltammograms of 3a (i) and 3b (ii). | ||
Similar kinds of observations and assignments have been investigated for TM complexes containing N, S and P donor ligands. For example, Daly and co-workers demonstrated the redox events of a Ru complex comprising a triaryl non-innocent N2S2 ligand derived from o-phenylenediamine.17b Two reversible redox waves at −0.78 V and −0.28 V correspond to RuII/RuI and RuIII/RuII redox couples, respectively, whereas the irreversible feature at −2.46 V appeared due to reduction of the o-diiminosemiquinone radical. Similarly, Heyduk illustrated that HAT (Hydrogen Atom Transfer) in the non-innocent tridentate [SNS] pincer based square planar nickel complex revealed one irreversible reduction at −2.74 V in addition to a reversible redox event at −0.64 V.23 Mascharak and co-workers reported the cyclic voltammogram of [cis-(dppQ)RuCl2] (dppQ = 1,2-bis-N-[2′ (diphenyl phosphanyl) benzoyl] benzoquinonediimine), wherein a dual ligand centered redox event was observed.24 The ligand-centred reduction of the o-diiminosemiquinone radical to a fully reduced o-phenylenediamine unit was identified as the irreversible wave at −1.35 V, while the reversible wave at −0.28 V was assigned to the second ligand-centred redox event. Nevertheless, the RuIII/RuII redox couple was attributed to the reversible wave at 0.90 V. In this context, [Ru(Ph3P)2(κ2-N,S-C7H4NS2)2(κ1-S-C7H4NS2)], recently reported by us, revealed a reversible wave at 0.25 V associated with the RuII(L˙)/RuII(L−) redox couple.19b Hence, the transition metal complexes derived from non-innocent ligands are known to exhibit rich redox chemistry.25
On the basis of these results, we have tentatively assigned the quasi-reversible feature of 2a at 0.191 V and 0.948 V to the OsIII/OsII and OsIV/OsIII redox couples,23 respectively. A similar trend was also observed for analogous 2b in a slightly deviated electrochemical window, presumably due to the variation in the heterocyclic moiety. Notably, the cathodic quasi-reversible redox waves at −1.007 V (3a) and −1.126 V (3b) correspond to monodentate heterocyclic ligand (La and Lb) centred redox events, i.e., the OsII(L˙)/OsII(L−) couple.23 Additionally, the cyclic voltammograms of 3a and 3b unveiled three sets of quasi-reversible redox waves, which were assigned to OsIII/OsII, OsIV/OsIII and OsIV/OsV redox couples, respectively (Table S1†).23 Thus, it demonstrates the potential of these heterocyclic ligand systems (La and Lb) that can adopt a delocalised state of [(L−)OsIII] and [(L˙)OsII] tautomers upon complexation and display a redox non-innocent nature.
In order to elucidate the photophysical properties associated with these κ2-N,S chelated complexes, the UV-vis absorption spectra of 2a–b and 3a–b were recorded in CH2Cl2. All these species revealed strong absorption in the higher energy region due to the π–π* transition in aromatic rings. Some of the weak absorption peaks were additionally recorded in the window of 300–450 nm, which possibly occurred due to the charge transfer transition (MLCT). Slightly red shifted bands were observed in the case of mercaptobenzothiazolyl congeners. Intriguingly, the absorption features of 2a–b and 3a–b were approximately similar, except for the lower energy region, as illustrated in Fig. 4. The absorption spectrum of 3a displayed a broad band at 650 nm, while the same band of 3b appeared at 580 nm. The origin of these bands was interpreted by TD-DFT calculations in CH2Cl2. The calculated transitions at 666 nm for 3a and 614 nm for 3b correspond to an excitation from β-HOMO-2 to β-LUMO. Thus, the low energy transition can be presumably explained by the contribution of the (L˙)OsII radical, in which the non-chelated radical based ligands get reduced to their anionic form [(L˙)OsII ↔ (L−)OsIII]. A similar phenomenon was also observed in the κ2-N,S chelated Ru(III) species as well as amido ligand based Ru(III) species.25a
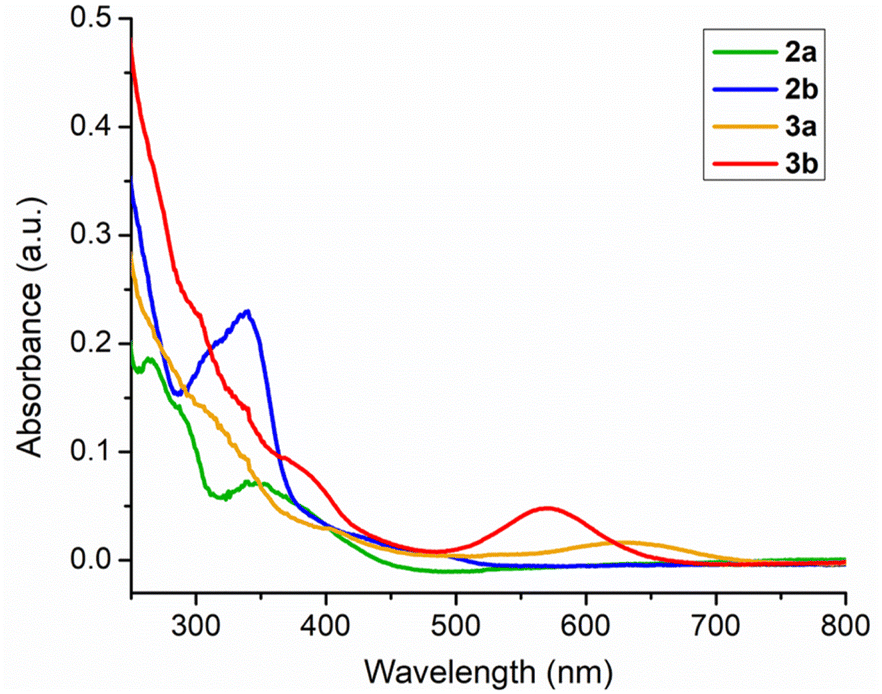 | ||
| Fig. 4 Combined UV-vis spectra of 2a, 2b, 3a and 3b. | ||
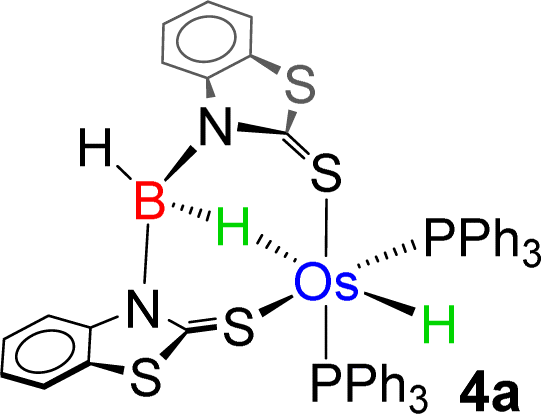
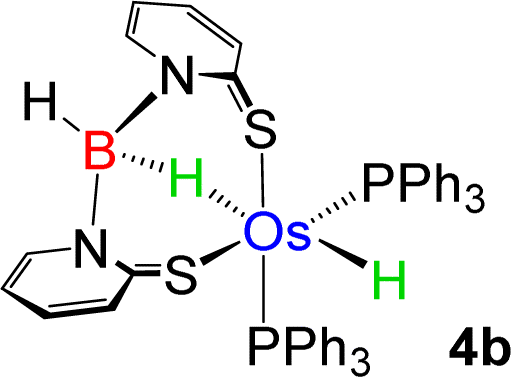
As a part of our ongoing research, we have developed polydentate ligand (N, S) based Ru(II) and Mo(II) complexes, which promoted B–H bond activation of free and bulky boranes.19,20 Based on MLC and hemilability present in κ2-N,S-chelated metallacycles, the M–N bonds exhibit a tendency to capture boranes. Based on our previous studies, we have treated a stoichiometric amount of BH3·SMe2 with 2a and 2b, which led to the formation of 4a and 4b, respectively (Scheme 2). The ESI-MS spectrum of 4a showed isotopic distribution patterns at m/z 1061.1318, which is consistent with the molecular formula [2a + BH2]. The 31P{1H} NMR spectrum disclosed a singlet at δ = 19.2 ppm, which was shifted downfield in comparison to that of 2a. The appearance of a new chemical shift at δ = −3.3 ppm in the 11B{1H} NMR spectrum was attributed to the presence of a single boron environment. In addition to the presence of mercaptobenzothiazolyl and phenyl signals, the room temperature 1H NMR spectra of 4a unveiled two inequivalent hydridic signals at δ = −7.87 and −13.26 ppm. Strikingly, the resonance at δ = −7.87 ppm is broad in nature, while a more shielded signal at δ = −13.26 ppm appeared as an apparent triplet (2JHP = 18.5 Hz). Eventually, the former peak at δ = −7.87 ppm corroborated the interaction of the metal with the B–H entity, resulting in an M–H–B motif. On the other hand, the higher-field signal at δ = −13.26 ppm indirectly confirmed the presence of a metal hydride, which is cis-oriented to the phosphine ligands. Likewise, in 4b, two distinct types of upfield 1H NMR resonances at δ = −9.36 and −12.57 ppm were detected. The 11B{1H} NMR spectrum of 4b also featured a single chemical shift at δ = 10.1 ppm, which was shifted relatively downfield compared to that of 4a. Moreover, ESI-MS revealed an intense ion peak at m/z 936.1506, which was associated with the formulation of C46H38N2S2P2Os. Systematic investigation by NMR spectroscopy along with mass spectrometry clearly suggests that compound 4b is analogous to 4a (see the ESI†). Finally, in order to confirm the spectroscopic assignments, a single-crystal X-ray diffraction study was performed on one of the complexes.
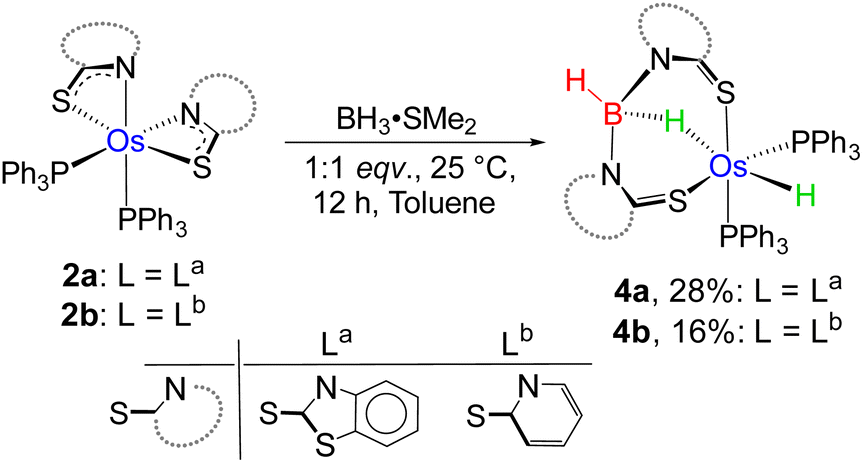 | ||
| Scheme 2 B–H activation by Os(II)–N,S-chelated complexes, 2a–b. | ||
The X-ray diffraction study of 4a was carried out on an orange crystal obtained from hexane layered CH2Cl2 solution. The geometry around the osmium center can be viewed as a distorted octahedral geometry with a void at the axial position. As shown in Fig. 5, the osmium center in the particular octahedral geometry is supported by two homoleptic sulphur atoms of the mercaptobenzothiazolyl entity, two phosphorus atoms of the PPh3 ligand and a bridging hydride of the BH2 moiety. Although, Os–H was not detected in the solid state X-ray diffraction analysis, we believe that a hydrogen atom occupies the vacant site of the octahedron. The presence of Os–H can also be elucidated by the aforementioned sharp hydridic resonance signal at δ = −13.26 ppm in the 1H NMR spectrum, which remained unperturbed on decoupling to 11B (Fig. S18†). The Os1–B1 separation of 2.689 Å is significantly longer than that of various osmium boryl species,26 but still in good agreement with that of reported σ-borate species27,28 of ruthenium.29 Therefore, this can be better emphasized as a δ(B–H) agostic interaction, having primary σ(B–H) as well as secondary interactions through sulphur with metal.27a Indeed, the species 4a and 4b were well identified as the Os(σ-borate)hydride complex, [Os(PPh3)2(H){κ3-H,S,S′-H2B(La/Lb)2}]. Based on previous reports,3b,c,15,16 we believe that the formation of Os(σ-borate)hydride species, 4a–b, presumably occurred via a few concerted steps: (i) cleavage of one hemilabile Os–N bond and cooperative B–H bond activation that eventually formed an Os(II) hydride, (ii) cleavage of another hemilabile Os–N bond and formation of a B–N bond, and (iii) activation of another B–H bond of the BH2L2 unit to form Os(σ-borate) species. Considering this fact, the event can be rationalized as the activation of two B–H bonds in borane.
 | ||
| Fig. 5 Molecular structure and labelling diagram of 4a. Selected bond lengths (Å) and bond angles (°) of 4a: Os1–P2 2.3048(10), Os1–P1 2.3085(11), Os1–S4 2.3675(10), Os1–S2 2.3985(11), Os1–B1 2.689; P2–Os1–P1 97.83(4), and P2–Os1–S4 170.54(4). | ||
Density functional theory (DFT) studies aided us to gain insight into the energetics as well as to differentiate the bonding and electronic properties between 4a and 4b. The changes in the Gibbs free energy (ΔG) of these reactions 3a–b → 4a–b were highly negative, implying the thermodynamic spontaneity of the borane activation reaction. Interestingly, the more negative value of ΔG for 3a → 4a showed the more feasibility of borane activation in the case of the mercaptobenzo-thiazolyl analogue. Further, the second-order perturbation theory suggested a donor–acceptor interaction between σ(B–H) and Os in 4a–b, but a lower stabilization energy was observed for 4a. Natural bonding orbital (NBO) analysis of 4b revealed that the electron population of the B–Hbridging bonding orbital in both species is lower than that of the non-bonding B–H orbital in free borane, clearly indicating the formation of a σ-borate unit through B–H activation. The electron population and Wiberg bond indices (WBI) of the B–Hbridging bonding orbital possess relatively smaller values for 4b in comparison to 4a. This implied an uneven σ-donation from σ(B–H) to the metal center, which presumably occurred due to the presence of different heterocyclic counterparts. As shown in Fig. 6(b), significant bonding interactions along Os–H bonds were also observed in these species, which is consistent with their high WBI values. Furthermore, topological analyses of the electron densities of 4a and 4b were performed using the quantum theory of atoms in molecules (QTAIM). The contour line diagram showed bond critical points (BCPs) between Os–H and B–H bonds in the Os–H–B plane. However, no BCP was located between osmium and boron, which indicated the absence of any strong bonding interaction.
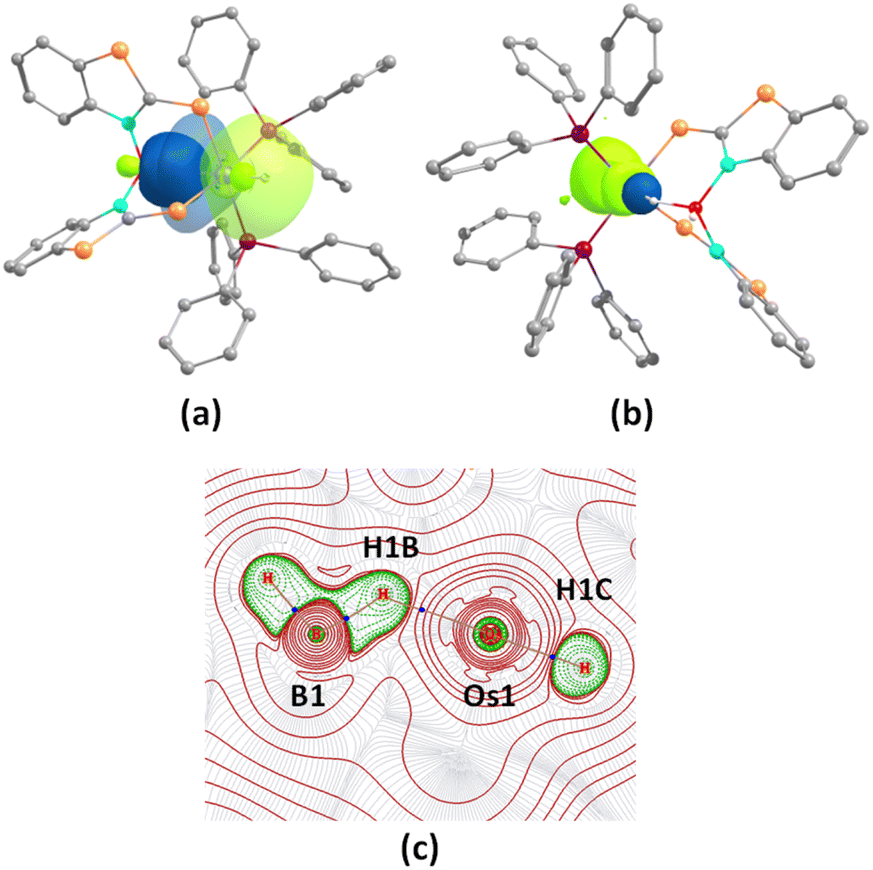 | ||
| Fig. 6 (a) Donor–acceptor interaction between σ(B–H) and the Os center in 4a; (b) Os–H bonding interaction in 4a (isovalue 0.004 [e bohr−3]1/2); (c) contour-line diagram of the Laplacian of the electron density along Os–H–B planes in 4a. | ||
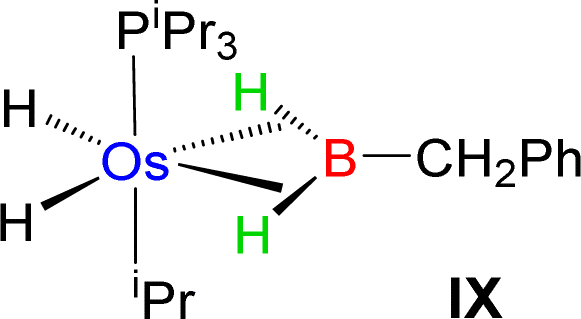
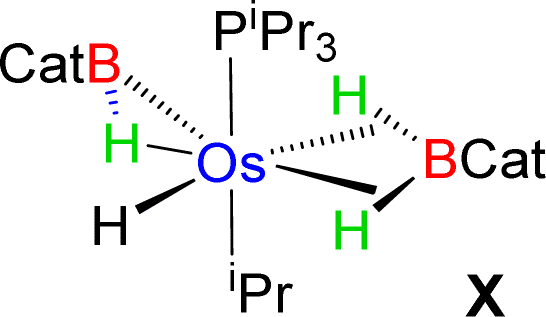
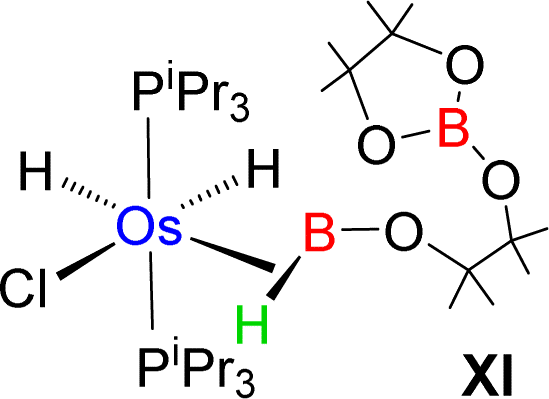
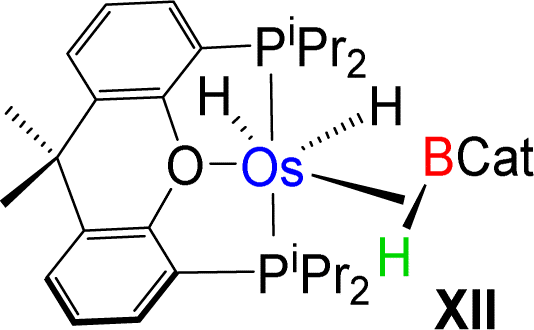
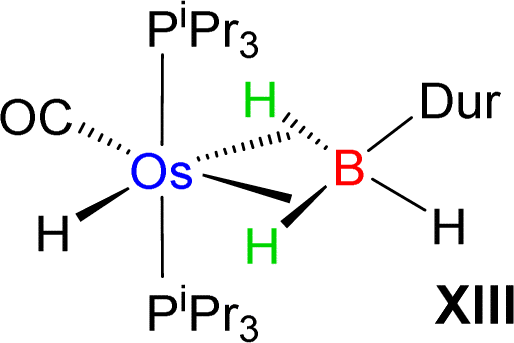
Esteruelas and co-workers30 have extensively studied the structure and bonding of distinct types of osmium σ-borane species, which allowed us to perform a comparative study of 4a–b based on their structural parameters and spectroscopic data (Table 1). The very first structurally characterized bis-σ-borane complex of osmium, [Os(H)2(η2:η2-H2BCH2Ph)(iPr)(PiPr3)],30aIX, was obtained by the successive hydrogenation and hydroboration of a cationic Os–alkylidene complex, [OsH(OH)(≡CPh) (IPr)(PiPr3)] [OTf]. The same group also reported a novel σ(B–H) complex of osmium, [OsH(η3-H2BCat)(η2-HBCat) (PiPr3)2]30b, X, offering η2-coordinated elongated σ-borane and η3-coordinated bis(elongated σ)-dihydridoborate entities. Moreover, the σ(B–H)-borinium derivative,30cXI, exhibited a downfield 11B NMR chemical shift, certainly emerged due to high cationic charge density on borinium boron. Consequently, the Os–B bond length is notably shorter in the η2-BH coordination entity of XI. Further, based on spectroscopic, structural and theoretical points of view, Esteruelas described a novel osmium POP-pincer skeleton stabilized σ-borane complex,30dXII, bearing two hydrides and a σ(B–H) moiety. It is worth noting that the 11B NMR resonance associated with Os(σ-borate) complexes, 4a–b, appeared significantly upfield shifted in comparison to the aforementioned osmium σ-borane complexes (IX–XII). The observation can be well explained on the basis of reduced bonding interaction between the metal and boron in 4a–b, which is also reflected in the longer M–B separation. Apart from a variety of σ-borane complexes of osmium, Braunschweig reported tris-isopropyl based Os dihydridoborate species,31XII. It also shows inequivalent characteristic structural and spectroscopic properties in respect of 4a–b. This fascinating mode of bonding can be described on the basis of the anionic borate nature of the BH2(La/Lb)2 counterpart, which eventually restricted the π-backdonation from the metal to the ligand.27a For this reason, species 4a–b can be considered as one of the newest and most unique additions in the series of osmium σ-complexes.
| Os-(σ-borane/borate) | Spectroscopic parameter | d av.M–B (Å) | |
|---|---|---|---|
| 1H NMRb | 11B NMRb | ||
| a iPr = isopropyl; Cat = catechol. b in ppm. | |||
|
−8.26 | 81.0 | 1.913(4) |
| −9.40 | |||
|
−9.50 | 35.0 | 2.112(4) |
| 2.159(4) | |||
|
−4.0 | 55.0 | 1.899(7) |
| −6.9 | |||
| −16.3 | |||
|
−1.53 | 45.5 | 2.057(4) |
| −5.60 | |||
| −18.80 | |||
|
−5.67 | 21.0 | 2.331(3) |
| −7.07 | |||
|
−7.87 | −3.3 | 2.689 |
| −13.26 | |||
|
−9.36 | 10.1 | — |
| −12.57 | |||
We have observed that complexes, 2a–b, readily undergo a borane activation reaction by ring opening of two hemilabile four-membered OsNCS cycles. Similarly, the paramagnetic species 3a–b are composed of two osma-heterocyclic entities. However, the salient difference between the two types of complexes can be rationalized by the presence of the non-innocent ligand. Transition metal complexes with redox non-innocent ligands in combination with MLC activate BH3 and 9-BBN molecules.8c,32 The isolation of VIII (vide supra, Chart 1) by the B–H activation of the abovementioned κ2-N,S-chelated ruthenium complex, [Ru(Ph3P)2(κ2-N,S-C7H4NS2)2(κ1-S-C7H4NS2)], is one of the prominent examples in this perspective.19b These results encouraged us to investigate the combined effect of non-innocent ligands and heavier transition metals in the B–H bond activation process. Thus, we have explored the reactivity of 3a and 3b with smaller boranes employing different reaction conditions (Scheme 3).
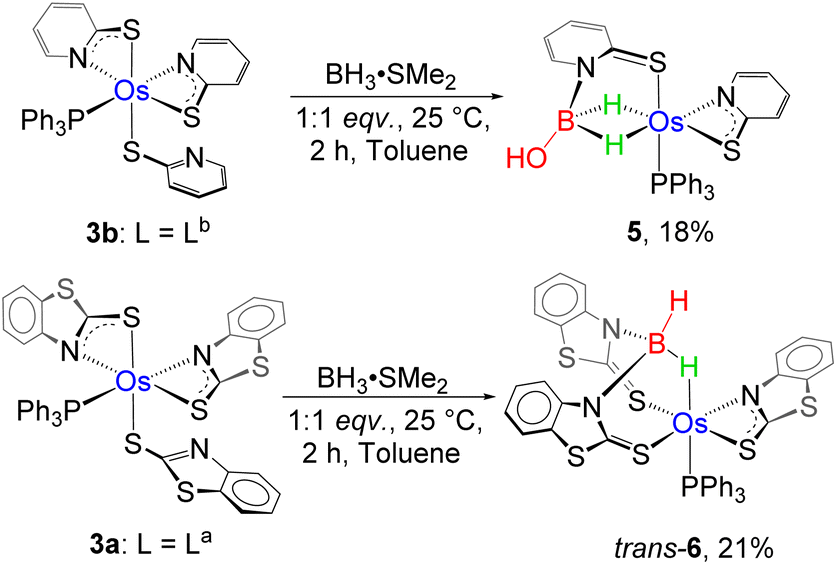 | ||
| Scheme 3 B–H activation by paramagnetic Os(III)-N,S-chelated species, 3a–b. | ||
The stoichiometric addition of BH3·SMe2 into a solution of 3b at room temperature showed a gradual colour change from violet to yellow and yielded 5 in 18% yield along with some air and moisture sensitive products. The complex was further purified using thin layer chromatography and was characterised using different spectroscopic techniques. Unlike 4a–b, the high-field region in the 1H NMR spectrum of 5 disclosed two broad resonances at δ = −13.65 and −13.67 ppm, which indicated the presence of two non-equivalent Os–H–B protons. Interestingly, the resonance signal in the 11B{1H} NMR spectrum appeared relatively downfield shifted at δ = 62.5 ppm. The retention of a single phosphine unit was predicted from the peak at δ = 16.4 ppm in 31P{1H} NMR. Moreover, the ESI-MS spectrum of 5 showed a molecular ion peak at m/z 703.0827 with isotopic distribution patterns. However, a clear account could not be envisaged until the single-crystal X-ray diffraction analysis of 5 was carried out.
The molecular structure of 5 can be designated as the κ2-N,S-chelated osmium(II) dihydridoborate complex, [Os(PPh3)(κ2-N,S-C5H4NS){κ3-H,S,S′-H2B(OH)(C5H4NS)}] (Fig. 7). The formation of the Os(dihydridoborate) unit clearly suggests the insertion of borane across the Os–N bond of one of the hemilabile OsNCS metallacycles, followed by the removal of the κ1-S-C5H4NS ligand. However, we believe that the addition of BH3·SMe2 into a solution of 3b initially led to the formation of a H-substituted Os–dihydridoborate complex, [Os(PPh3)(κ2-N,S-C5H4NS){κ3-H,H,S-H3B(C5H4NS)}], 5′, which further gets converted to 5 upon hydrolysis by a trace amount of water during chromatographic separation. It was further established by the deshielded chemical shift at δ = 3.81 ppm in 1H NMR and significantly low-field 11B{1H} NMR resonance. In this context, Stradiotto isolated a Ru–dihydridoborate complex, [Cp*Ru(κ3-P,H,H-(iPr))P-(C9H6OH2B-Mes)], by the activation of BH2Mes (vide supra, VI, Chart 1).16 Also, a piano-stool Fe–dihydridoborate complex5c was reported by Liang et al. and was generated by cooperative B–H bond activation utilizing [(Cp*L′′Fe)(μ-N2)(FeCp*L′′)] (L′′ = deprotonated N-picoly N-heterocyclic carbene ligand). The Os–B separation of 2.057 Å in 5 is in good agreement with the M–B bond lengths of the abovementioned lighter congener dihydridoborate species.29 Moreover, the same parameter is marginally shorter in comparison to the Ru–B distances of Ru–σ-borane(dihydridoborate) species [Ru(PCy3)2{Bpin(μ-H)2}(H){(μ-H)Bpin}] (2.188(5) Å),33 reported by Sabo-Eteinne, and Ru-bis(dihydridoborate) species, [Ru{(μ-H)2BH (C5H4NS)}2] (2.161(8) Å),29b reported by our group.
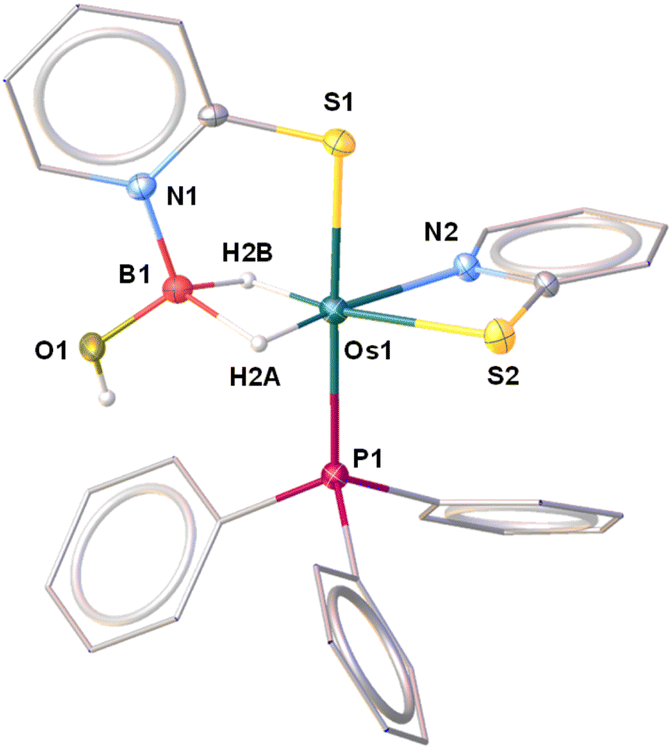 | ||
| Fig. 7 Molecular structure and labelling diagram of 5. Selected bond lengths (Å) and bond angles (°) of 5: Os1–S1 2.3669(9), Os1–S2 2.5045(9), Os1–B1 2.059(4), B1–O1 1.371(5), and B1–Os1–N2 145.20(13). | ||
To get insight into the bonding situations in dihydridoborate coordination, we have carried out DFT calculations on model system 5′. The four-membered cyclic 5′ possessed a BCP along the Os–B bond in addition to two other BCPs along B–H and one Os–H bonds, respectively. In this particular topology, the bond paths were inwardly curved {Fig. 8(b)}, which strongly resembles the situation of diborane (6). The BCPs associated with Os–H and B–H bond paths have significantly large ellipticity, ∈, and considerably smaller negative energy density, H(r) values, thus reflecting the presence of an anisotropic σ(B–H) agostic interaction with metal. In this context, a strong 3c–2e bonding interaction along Os–H–B was observed in the Os–dihydridoborate complex, 5′ {Fig. 8(a)}, in NBO calculation. The computed natural charge on the boron atom further well agreed with a greater charge accumulation in the case of 5′ than that of 4a–b. It clearly addressed the manifestation of a stronger Os–B bonding interaction in the dihydridoborate component than the σ-borate moiety.
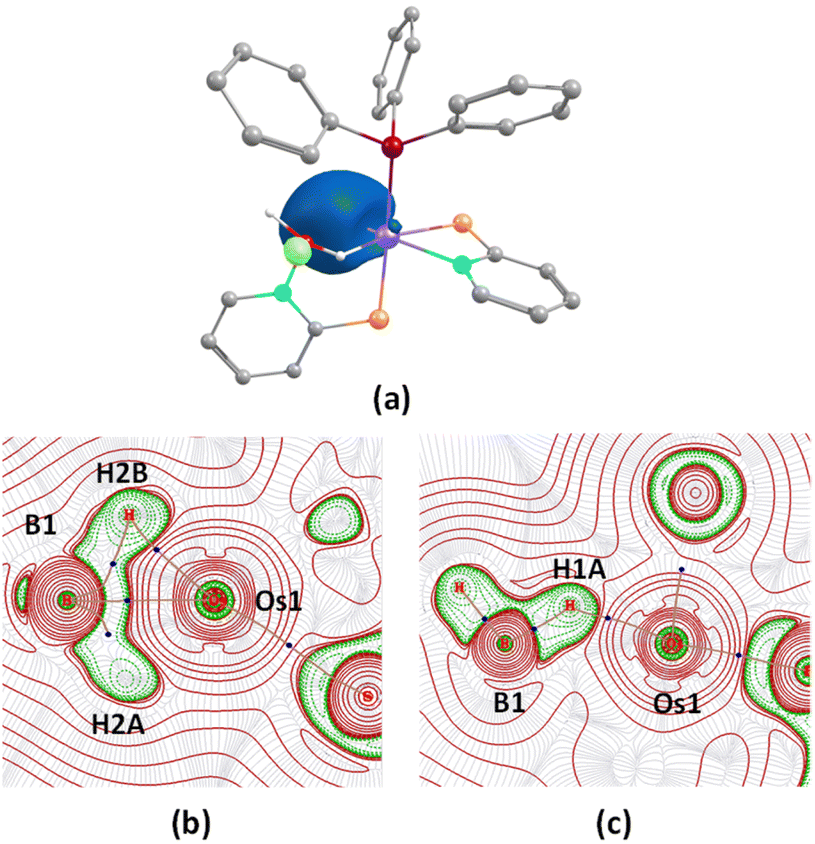 | ||
| Fig. 8 (a) 3c-2e bonding interaction along the Os–H–B plane in 5′ (isovalue 0.004 [e bohr−3]1/2); (b and c) contour-line diagram of the Laplacian of the electron density along the Os–H–B planes in 5′ and trans-6, respectively. | ||
On the other hand, the reaction of 3a with BH3·SMe2 yielded compound 6 as a red crystalline solid in 15% yield. The single peak at δ = 9.1 ppm in the 31P{1H} NMR spectrum of 6 suggested the retention of the phosphine unit of 3a. The 11B{1H} NMR spectrum showed a sharp peak at δ = −3.7 ppm, which was observed considerably in the upfield region relative to 5. Along with the signal corresponding to the aromatic hydrogens, a high-field broad resonance was also detected at δ = −4.56 ppm in the 1H NMR spectrum of 6, which probably emerged due to the presence of Os–H–B. In addition to Os–H–B, another broad signal at δ = 5.15 ppm in 1H NMR resolved upon decoupling to boron, seemingly appeared due to the presence of terminal B–H. Furthermore, ESI-MS exhibited a molecular-ion peak at m/z 965.0099 with an isotopic distribution pattern, which is consistent with the molecular formula C39H29N3S6OsPB. Although the spectroscopic assessment proposed the probable activation of the B–H bond, a precise explanation could not be envisaged until the single-crystal X-ray diffraction investigation was performed.
As shown in Fig. 9(left), the molecular structure of 6 can be identified as a κ2-N,S-chelated osmium(II) mercaptobenzo–thiazolyl borate complex. Similar to 4a, the distorted octahedral geometry around the Os center in 6 is composed of [H2BL2]− in κ3-(H,S,S′) coordination mode, featuring comparable separation between metal and boron. Although we were unable to find any proper mechanistic evidence behind the formation of 6, we assumed that the reaction progressed via two steps. The first step involved the capture of the BH2 bond after two Os–N bond cleavages, which resulted in a σ-borate counterpart. Then, the chelation of the κ1-S-La ligand through Os–N bond formation led to the generation of an osma-heterocycle (OsNCS) in the second step. Based on the trans orientation of the hydride ligand with respect to phosphine, we have designated the species as trans-6. Moreover, tris-homoleptic mercaptobenzothiazolyl sulphur moieties were coordinated to the osmium center in a meridional fashion. Thus, 6 can be well described as trans-mer-[Os (PPh3)(κ2-N,S-C7H4NS2){κ3-H,S,S′-H2B(C7H4NS2)2}], which is analogous to [Ru(PPh3)(κ2-N,S-C7H4NS2){κ3-H,S,S′-H2B(C7H4NS2)2}],19c isolated from the reaction of [(η6-p-cymene)RuCl2PPh3] with Na[H2B(C7H4NS2)2]. Further, QTAIM analysis revealed that bond critical points (BCPs) were detected along Os–H and B–H bonds in the Os–H–B plane of trans-6 {Fig. 8(c)}. In contrast to 5, relatively lower ellipticity, ∈ values are observed for Os–H and B–H bonds. Subsequently, NBO analysis also addressed the distinct type of bonding scenario in trans-6. Rather than the 3c–2e bonding interaction, Os(σ-borate) species displayed a donor–acceptor interaction, in which the filled σ(B–H) orbital donates electron density to an empty metal orbital.
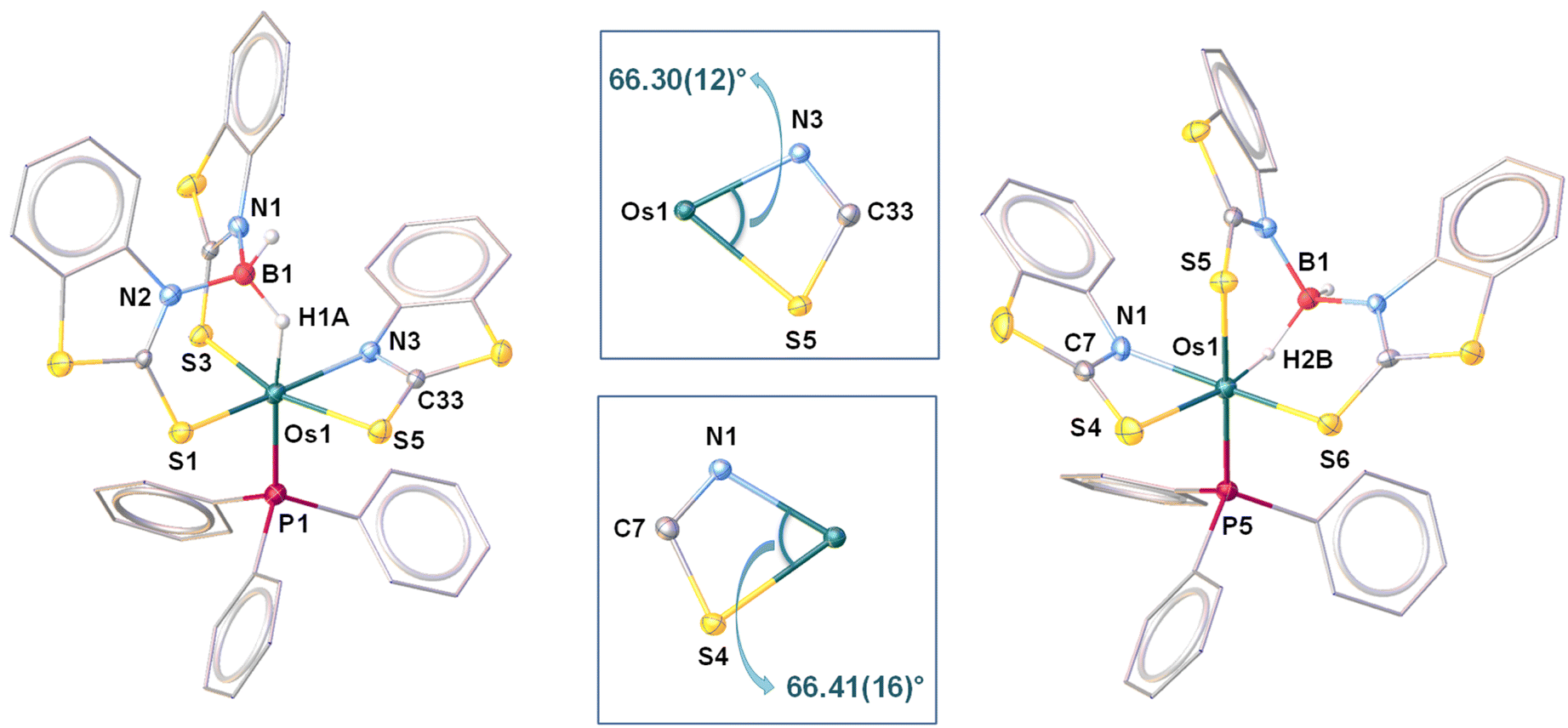 | ||
| Fig. 9 Molecular structure and labelling diagram of trans-6 (left) and cis-6 (right). Selected bond lengths (Å) and bond angles (°) of trans-6: Os1–B1 2.712, Os1–N3 2.127(4), Os1–S5 2.4816(15), and Os1–S1 2.3162(15); N3–Os1–S5 66.30(12). Selected bond lengths (Å) and bond angles (°) of cis-6: Os1–B1 2.497(8), Os1–N1 2.138(5), Os1–S4 2.4741(18), and Os1–S5 2.4852(17); N1–Os1–S4 66.41(16). | ||
The coordination chemistry of TM σ-borate complexes has become a significant research topic due to their ability to adopt versatile coordination geometries around metal centres. Recently, our group has carried out an explicit structural investigation on several stereoisomers of κ2-N,S-chelated Ru(II) mercaptobenzothiazolyl borate complexes.19d The formation of trans-6, therefore, tempted us to investigate the efficient synthetic pathway for the synthesis of its cis analogue, i.e. cis-6. Thus, we have carried out the thermolysis of [Os(PPh3)3Cl2], 1, with two equivalents of [bis-(2-mercapto-benzothiazolyl)borate], which led to the formation of cis-6 along with 2a, 3a and trans-6 (Scheme 4).
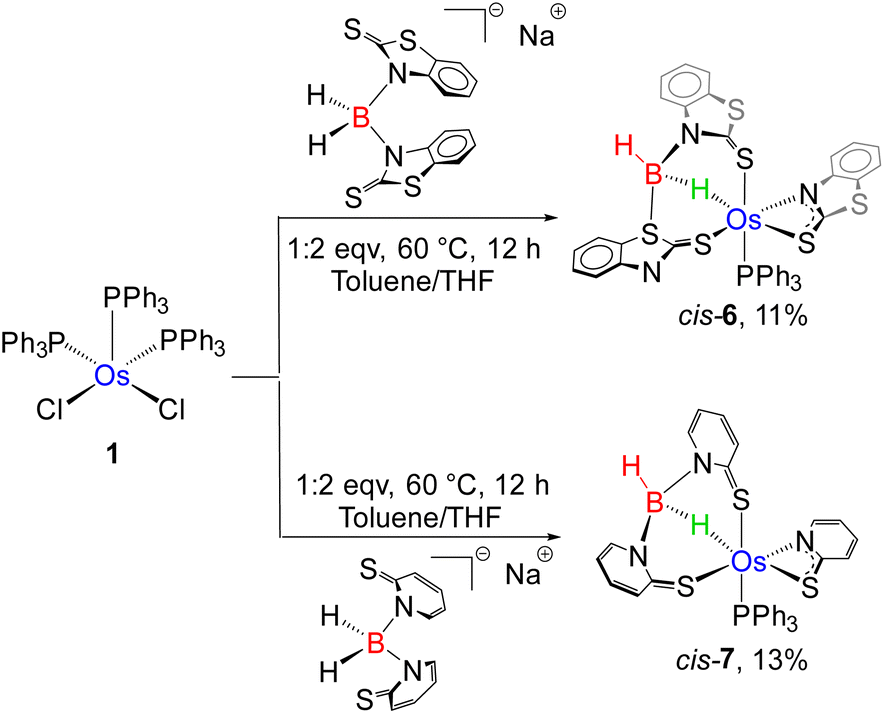 | ||
| Scheme 4 Modified syntheses of isomeric Os(σ-borate) species, cis-6 and cis-7. | ||
The solid-state structure revealed a distorted octahedral geometry, in which three sulphur atoms are present on one of the triangular faces {Fig. 9(right)}. Unlike trans-6, the bridging hydride was placed cis to the phosphine ligand. As a result, it can be described as cis-6, more specifically cis-fac-6. Both of the isomers exhibited comparable bite angle values for the four-membered chelating osmacycles, which suggested that the borate counterpart has no significant steric influence on the coordination of the bidentate mercaptobenzothiazolyl ligand. In this connection, the hydridic chemical shift of cis-6 at δ = −13.83 ppm related to Os–H–B appeared in the relatively upfield region in comparison to trans-6. The trans-isomer was composed of a good σ-donor phosphine ligand trans to the hydride, whereas the electronegative S atom occupied the trans site of the hydride in the cis isomer. The electron donation from the B–H bond to the metal centre is expected to be more, where sulphur is the trans atom. Therefore, the hydridic character of Os–H–B is greater in cis-6 in comparison to trans-6. However, the 11B{1H} NMR resonance of both isomers remained unperturbed instead of adapting different spatial arrangements.
The effect of structural variation on the thermodynamic and kinetic stability of these isomeric species was analysed by means of DFT studies. The structural parameters of the optimized geometries agreed well with those obtained from the X-ray diffraction data. However, the Os–B separation in cis-6 was slightly elongated compared to that found in the solid-state structure (Table S2†). The relative thermodynamic stability of trans-6 can be described on the basis of ground-state energy calculations. The energy difference between these two isomers is 1.2 kcal mol−1. On the other hand, molecular orbital (MO) analysis suggested that the HOMOs of both isomers were mainly localized on the d-orbitals of osmium centres with a weak π-bonding interaction along the CS bond. Meanwhile, a delocalized electron density over the heterocyclic ring of the σ-borate unit was observed in LUMOs. The energy gap between the HOMO and LUMO, ΔEHOMO–LUMO, of cis-6 is quite higher than that of trans-6, which presumably indicated the greater kinetic stability of the cis-isomer (Fig. 10). Therefore, trans-6 is a thermodynamically controlled product, while the formation of cis-6 is seemingly kinetically favoured.
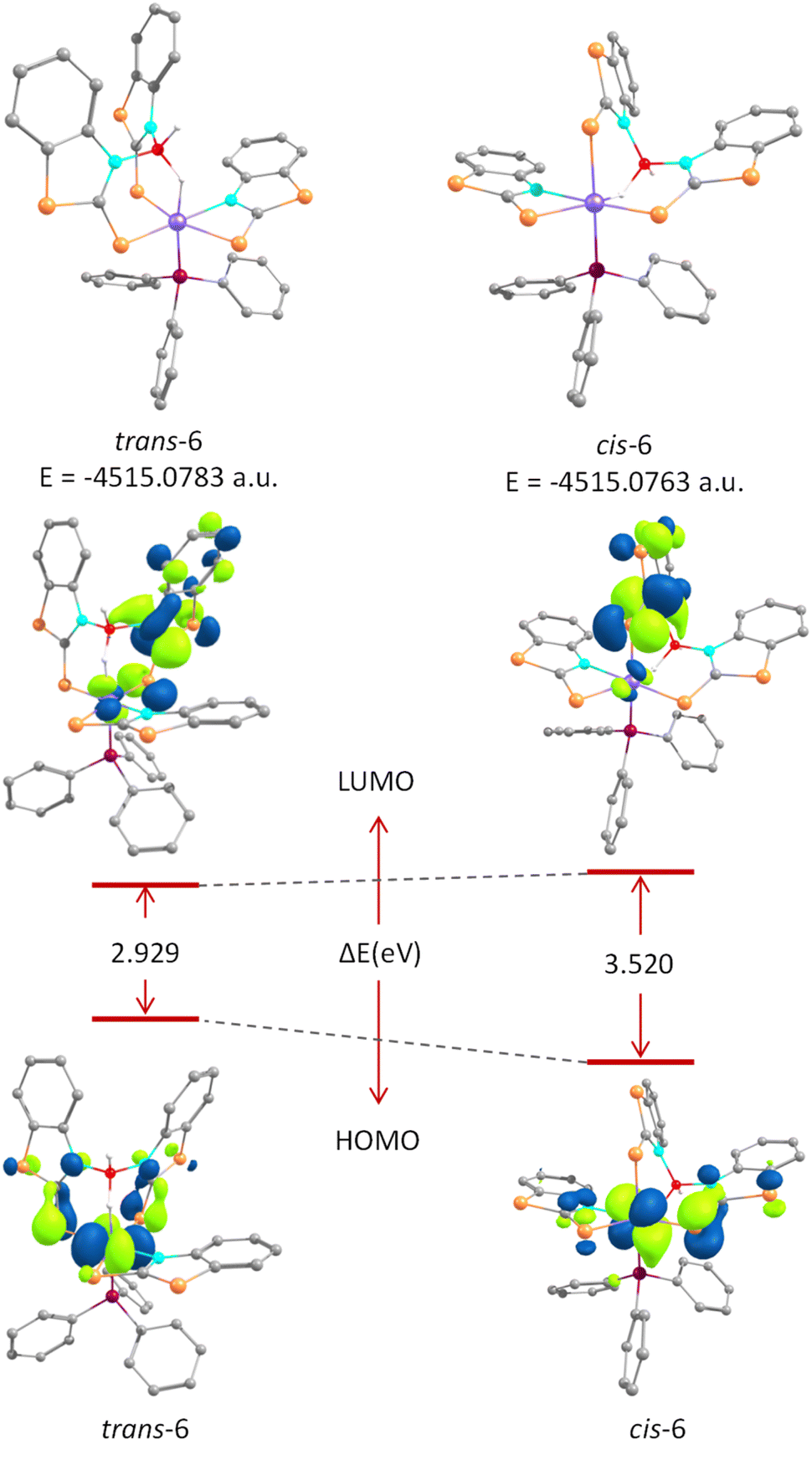 | ||
| Fig. 10 Top: ground-state energies of trans-6 (left) and cis-6 (right). Bottom: HOMO–LUMO energy gap between trans-6 and cis-6. | ||
In stark contrast, we were unable to isolate the mercaptopyridinyl analogue of trans-6 or cis-6, while the borane activation was carried out using [Os(Ph3P)(κ2-N,S-C5H4NS)2(κ1-S-C5H4NS)], 3b. Thus, we have imitated the methodology that was employed to synthesise cis-6, which is the treatment of 1 with 2 equivalents of NaBH2Lb2. Indeed, the reaction led to the formation of 7 along with the formation of 2b, 3b, 4b and 5. The 1H NMR chemical shift related to Os–H–B of 7 (δ = −12.46 ppm) was in the upfield region compared to that of analogous trans-6 (δ = −4.60 ppm), but closely resembles that of cis-6 (δ = −13.81 ppm). The single chemical shift at δ = 13.9 ppm in the 11B{1H} NMR of 7 was associated with the tetra-coordinated boron atom. The boron centre in all these σ-borate complexes exhibited a diverse electronic nature, predominately due to the presence of different heterocyclic entities and various possible geometrical orientations. Indeed, the solid-state X-ray structure, shown in Fig. S1,† revealed that the hydrides of the σ(B–H) moiety and PPh3 ligand were placed in the axial and equatorial positions of the octahedron, respectively. This confirmed that the geometry of 7 can be considered as a cis isomer. Moreover, three homoleptic sulphur atoms occupied three vertices of one of the triangular faces. Thus, the latter complex can be better designated as cis-fac-[Os(PPh3)(κ2-N,S-C5H4NS){κ3-H,S,S′-H2B(C5H4NS)2}]. The Os–B bond distance of 2.497 Å was in good accord with analogous cis-6, but evidently shorter than that of trans-6. The bite angle (N–Os–S) of the four membered metallacycle in cis-7 is akin to those of the analogous isomeric species.
Conclusions
In summary, we have developed a series of osmium bis-κ2-N,S-chelated complexes featuring hemilabile osma-heterocycles that illustrate their susceptibility towards activation of B–H bonds in free boranes. Subsequently, we have investigated the redox, photochemical and electronic behaviour to differentiate between various diamagnetic and paramagnetic bis-κ2-N,S-chelated species. Cooperative activation of free boranes by diamagnetic complexes led to the formation of Os(σ-borate)hydride complexes through dual non-identical B–H bond activation. In contrast, two redox-active paramagnetic bis κ2-N,S-chelated species possessing different non-innocent heterocyclic ligands unveiled different patterns of activation. The capture of the B–H bond by the cleavage of the Os–N bond in hemilabile osma-heterocycles yielded dihydridoborate and σ-borate complexes of osmium, respectively. To investigate the influence of spatial arrangements on the stability and bonding of isomeric species, we have further isolated several isomeric Os(σ-borate) complexes. In the course of our study, combined experimental and theoretical studies show that a combination of electronic behaviour and metal–ligand cooperativity significantly influences the activation of borane.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
S. Gayen, F. Assanar, S. Shyamal and D. P. Dorairaj have executed the experimental synthesis, characterization and analysis of the data. S. Gayen has conducted the theoretical calculations. All authors have contributed to the preparation of the manuscript. S. Ghosh has supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thankfully acknowledge the support from SERB-DST (Grant No. CRG/2023/000189), New Delhi, India. S. G. thanks CSIR, F. A and S. S. thank UGC, and D. P. D thanks IIT Madras for research fellowships. The computational facility of IIT Madras is gratefully acknowledged.Notes and references
- (a) R. Peters, Cooperative Catalysis, Wiley-VCH, Weinheim, 2015 CrossRef; (b) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS; (c) R. H. Morris, Acc. Chem. Res., 2015, 48, 1494–1502 CrossRef CAS.
- (a) M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC; (b) H. Li, T. P. Goncalves, D. Lupp and K.-W. Huang, ACS Catal., 2019, 9, 1619–1629 CrossRef CAS.
- (a) T. Higashi, S. Kusumoto and K. Nozaki, Chem. Rev., 2019, 119, 10393–10402 CrossRef CAS; (b) L. Omann, C. D. F. Konigs, H. F. T. Klare and M. Oestreich, Acc. Chem. Res., 2017, 50, 1258–1269 CrossRef CAS PubMed; (c) K.-S. Feichtner and V. H. Gessner, Chem. Commun., 2018, 54, 6540–6553 RSC; (d) F. Forster and M. Oestreich, Metal-Ligand Cooperative Si–H Bond Activation in Organosilicon Chemistry – Novel Approaches and Reactions, ed. T. Hiyama, and M. Oestreich, Wiley-VCH, Weinheim, 2019, pp. 115–130 Search PubMed.
- (a) C. C. Comanescu and V. M. Iluc, Chem. Commun., 2016, 52, 9048–9051 RSC; (b) C. C. Comanescu and V. M. Iluc, Polyhedron, 2018, 143, 176–183 CrossRef CAS; (c) J. Y. Corey, Chem. Rev., 2016, 116, 11291–11435 CrossRef CAS PubMed; (d) M. K. Bisai, V. Sharma, R. G. Gonnade and S. S. Sen, Organometallics, 2021, 40(13), 2133–2138 CrossRef CAS.
- (a) J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2015, 137, 12808–12814 CrossRef CAS PubMed; (b) C. Erken, A. Kaithal, S. Sen, T. Weyhermuller, M. Holscher, C. Werle and W. Leitner, Nat. Commun., 2018, 9, 4521 CrossRef PubMed; (c) Q. Liang, H. A. Garcia Mayerstein and D. Song, Organometallics, 2023, 42, 816–824 CrossRef CAS.
- (a) S. P. Cronin, J. M. Strain, M. S. Mashuta, J. M. Spurgeon, R. M. Buchanan and C. A. Grapperhaus, Inorg. Chem., 2020, 59, 4835–4841 CrossRef CAS PubMed; (b) R. Stichauer, A. Helmers, J. Bremer, M. Rohdenburg, A. Wark, E. Lork and M. Vogt, Organometallics, 2017, 36, 839–848 CrossRef CAS; (c) I. Heuermann, B. Heitmann, R. Stichauer, D. Duvinage and M. Vogt, Organometallics, 2019, 38, 1787–1799 CrossRef CAS; (d) R. Stichauer and M. Vogt, Organometallics, 2018, 37, 3639–3643 CrossRef CAS.
- (a) J. W. Nugent, M. Garcia-Melchor and A. R. Fout, Organometallics, 2020, 39, 2917–2927 CrossRef CAS; (b) X. Zhai, M. Pang, L. Feng, J. Jia, C.-H. Tung and W. Wang, Chem. Sci., 2021, 12, 2885–2889 RSC; (c) J. V. Obligacion and P. J. Chirik, ACS Catal., 2017, 7, 4366–4371 CrossRef CAS PubMed; (d) S.-F. Hou, J.-Y. Chen, M. Xue, M. Jia, X. Zhai, R.-Z. Liao, C.-H. Tung and W. Wang, ACS Catal., 2020, 10, 380–390 CrossRef CAS.
- (a) M. Ito, M. Itazaki and H. Nakazawa, Inorg. Chem., 2017, 56, 13709–13714 CrossRef CAS PubMed; (b) J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2015, 137, 12808–12814 CrossRef CAS PubMed; (c) H. Song, K. Ye, P. Geng, X. Han, R. Liao, C.-H. Tung and W. Wang, ACS Catal., 2017, 7, 7709–7717 CrossRef CAS.
- (a) S. R. Flynn and D. F. Wass, ACS Catal., 2013, 3, 2574–2581 CrossRef CAS; (b) A. R. Jupp and D. W. Stephan, Trends Chem., 2019, 1(1), 35–48 CrossRef CAS; (c) E. R. M. Habraken, A. R. Jupp, M. B. Brands, M. Nieger, A. W. Ehlers and J. C. Slootweg, Eur. J. Inorg. Chem., 2019, 2436–2442 CrossRef CAS PubMed; (d) J. Bauer, H. Braunschweig and R. D. Dewhurst, Chem. Rev., 2012, 112, 4329–4346 CrossRef CAS PubMed.
- B. Chatterjee, W.-C. Chang, S. Jena and C. Werle, ACS Catal., 2020, 10, 14024–14055 CrossRef CAS.
- (a) A. Lefranc, Z.-W. Qu, S. Grimme and M. Oestreich, Chem.–Eur. J., 2016, 22, 10009–10016 CrossRef CAS; (b) T. Stahl, K. Mether, Y. Ohki, K. Tatsumi and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 10978–10981 CrossRef CAS PubMed; (c) F. Forster, T. T. Metsanen, E. Irran, P. Hrobarik and M. Oestreich, J. Am. Chem. Soc., 2017, 139, 16334–16342 CrossRef CAS PubMed; (d) H. F. T. Klare, M. Oestreich, J.-i. Ito, H. Nishiyama, Y. Ohki and K. Tatsumi, J. Am. Chem. Soc., 2011, 133, 3312–3315 CrossRef CAS PubMed; (e) F. Forster, V. M. R. Lopez and M. Oestreich, J. Am. Chem. Soc., 2018, 140, 1259–1262 CrossRef CAS PubMed.
- (a) J. Weismann, R. Waterman and V. H. Gessner, Chem.–Eur. J., 2016, 22, 3846–3855 CrossRef CAS PubMed; (b) J. Weismann, L. T. Scharf and V. H. Gessner, Organometallics, 2016, 35, 2507–2515 CrossRef CAS.
- L. T. Scharf, J. Weismann, K.-S. Feichtner, F. Lindl and V. H. Gessner, Chem.–Eur. J., 2018, 24, 3439–3443 CrossRef CAS PubMed.
- (a) C. P. Casey, S. W. Singer, D. R. Powell, R. K. Hayashi and M. Kavana, J. Am. Chem. Soc., 2001, 123, 1090–1100 CrossRef CAS PubMed; (b) L. Korean-Selfridge, H. N. Londino, J. K. Vellucci, B. J. Simmons, C. P. Casey and T. B. Clark, Organometallics, 2009, 28, 2085–2090 CrossRef.
- (a) M. W. Drover, L. L. Schafer and J. A. Love, Angew. Chem., Int. Ed., 2016, 55, 3181–3186 CrossRef CAS; (b) M. W. Drover, E. G. Bowes, J. A. Love and L. L. Schafer, Organometallics, 2017, 36, 331–341 CrossRef CAS.
- M. A. Rankin, K. D. Hesp, G. Schatte, R. McDonald and M. Stradiotto, Dalton Trans., 2009, 4756–4765 RSC.
- (a) G. Durgaprasad, J. A. Luna, K. D. Spielvogel, C. Haas, S. K. Shaw and S. R. Daly, Organometallics, 2017, 36, 4020–4031 CrossRef CAS; (b) K. D. Spielvogel, J. A. Luna, S. M. Loria, L. P. Weisburn, N. C. Stumme, M. R. Ringenberg, G. Durgaprasad, J. M. Keith, S. K. Shaw and S. R. Daly, Inorg. Chem., 2020, 59, 10845–10853 CrossRef CAS PubMed.
- B. Pan, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Organometallics, 2011, 30, 5560–5563 CrossRef CAS.
- (a) Md. Zafar, R. Ramalakshmi, A. Ahmad, P. K. S. Antharjanam, S. Bontemps, S. Sabo-Etienne and S. Ghosh, Inorg. Chem., 2021, 60, 1183–1194 CrossRef CAS PubMed; (b) M. Zafar, A. Ahmad, S. Saha, R. Rongala, T. Roisnel and S. Ghosh, Chem. Sci., 2022, 13, 8567–8575 RSC; (c) M. Zafar, R. Ramalakshmi, K. Pathak, A. Ahmad, T. Roisnel and S. Ghosh, Chem.–Eur. J., 2019, 25, 13537–13546 CrossRef CAS PubMed; (d) A. Ahmad, S. Saha, M. Zafar, T. Roisnel, P. Ghosh and S. Ghosh, Eur. J. Org Chem., 2023, 989, e202201283 CrossRef.
- (a) S. Saha, A. Haridas, F. Assanar, C. Bansal, P. K. S. Antharjanam and S. Ghosh, Dalton Trans., 2022, 51, 4806–4813 RSC; (b) S. Saha, F. Assanar, A. Ahmad, A. Bains, S. Nagendran and S. Ghosh, Organometallics, 2024, 43, 718–725 CrossRef CAS.
- (a) S. Ye, B. Sarkar, C. Duboc, J. Fiedler and W. Kaim, Inorg. Chem., 2005, 44(8), 2843–2847 CrossRef CAS PubMed; (b) M. K. Biswas, S. C. Patra, A. N. Maity, S. C. Ke, N. D. Adhikary and P. Ghosh, Inorg. Chem., 2012, 51, 6687–6699 CrossRef CAS PubMed.
- (a) F. Chen, G. –F. Wang, Y. –Z. Li, X. –T. Chen and Z. –L. Xue, Inorg. Chem. Commun., 2012, 21, 88–91 CrossRef CAS; (b) D. O'Hare, J. C. Green, T. P. Chadwick and J. S. Miller, Organometallics, 1988, 7, 1335–1342 CrossRef; (c) P. G. Gass man, J. W. Mickelson and J. R. Sowa Jr, J. Am. Chem. Soc., 1992, 114, 6942–6944 CrossRef CAS.
- K. E. Rosenkoetter, M. K. Wojnar, B. J. Charette, J. W. Ziller and A. F. Heyduk, Inorg. Chem., 2018, 57, 9728–9737 CrossRef CAS PubMed.
- N. L. Fry, M. J. Rose, C. Nyitray and P. K. Mascharak, Inorg. Chem., 2008, 47, 11604–11610 CrossRef CAS PubMed.
- (a) S. Kundu, D. Dutta, S. Maity, T. Weyhermuller and P. Ghosh, Inorg. Chem., 2018, 57, 11948–11960 CrossRef CAS PubMed; (b) N. P. V. Leest, M. A. Tepaske, J. H. Oudsen, B. Venderbosch, N. R. Rietdijk, M. A. Siegler, M. Tromp, J. I. V. D. Vlugt and B. D. Bruin, J. Am. Chem. Soc., 2020, 142, 552–563 CrossRef PubMed; (c) H. Masui, A. B. P. Lever and P. R. Auburn, Inorg. Chem., 1991, 30, 2402–2410 CrossRef CAS; (d) S. D. J. McKinnon, B. O. Patrick, A. B. P. Lever and R. G. Hicks, Chem. Commun., 2010, 46, 773–775 RSC.
- (a) M. A. Esteruelas, I. Fernandez, A. M. Lopez, M. Mora and E. Onate, Organometallics, 2012, 31, 4646–4649 CrossRef CAS; (b) M. L. Buil, M. A. Esteruelas, K. Garces and E. Onate, J. Am. Chem. Soc., 2011, 133, 2250–2263 CrossRef CAS PubMed.
- (a) K. Saha, D. K. Roy, R. D. Dewhurst, S. Ghosh and H. Braunschweig, Acc. Chem. Res., 2021, 54, 1260–1273 CrossRef CAS PubMed; (b) K. Pathak, S. Mishra, C. Nandi, S. Saha and S. Ghosh, Inorg. Chem., 2023, 62, 160–169 CrossRef CAS PubMed.
- (a) M. R. St.-J. Foreman, A. F. Hill, G. R. Owen, A. J. P. White and D. J. Williams, Organometallics, 2003, 22, 4446–4450 CrossRef CAS; (b) G. R. Owen, Chem. Soc. Rev., 2012, 41, 3535–3546 RSC; (c) R. J. Abernethy, A. F. Hill, N. Tshabang, A. C. Willis and R. D. Young, Organometallics, 2009, 28, 488–492 CrossRef CAS; (d) M. Jiménez-Tenorio, M. C. Puerta and P. Valerga, Organometallics, 2009, 28, 2787–2798 CrossRef.
- (a) R. Ramalakshmi, K. Saha, D. K. Roy, B. Varghese, A. K. Phukan and S. Ghosh, Chem.–Eur. J., 2015, 21, 17191–17195 CrossRef CAS PubMed; (b) K. Pathak, S. Gayen, S. Saha, C. Nandi, S. Mishra and S. Ghosh, Chem.–Eur. J., 2022, 28, e202104393 CrossRef CAS PubMed; (c) S. Gayen, S. Shyamal, S. Mohapatra, P. K. S. Antharjanam and S. Ghosh, Chem.–Eur. J., 2023, 30, e202302362 CrossRef PubMed.
- (a) M. L. Buil, J. J. F. Cardo, M. A. Esteruelas, I. Fernandez and E. Onate, Organometallics, 2015, 34, 547–550 CrossRef CAS; (b) J. C. Babon, M. A. Esteruelas, I. Fernandez, A. M. Lopez and E. Onate, Inorg. Chem., 2018, 57, 4482–4491 CrossRef CAS PubMed; (c) M. A. Esteruelas, F. J. Fernandez-Alvarez, A. M. Lopez, M. Mora and E. Onate, J. Am. Chem. Soc., 2010, 132(16), 5600–5601 CrossRef CAS PubMed; (d) M. A. Esteruelas, I. Fernandez, C. García-Yebra, J. Martín and E. Onate, Organometallics, 2017, 36(12), 2298–2307 CrossRef CAS.
- N. Arnold, S. Mozo, U. Paul, U. Radius and H. Braunschweig, Organometallics, 2015, 34, 5709–5715 CrossRef CAS.
- V. Lyaskovskyy and B. Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- V. Montiel-Palma, M. Lumbierres, B. Donnadieu, S. Sabo-Etienne and B. Chaudret, J. Am. Chem. Soc., 2002, 124, 5624–5625 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2353030 (for 2b), 2353031 (for 4a), 2353032 (for 5), 2353033 (for trans-6), 2353034 (for cis-6) and 2353035 (for cis-7) contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05092d |
| This journal is © The Royal Society of Chemistry 2024 |