
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLysosome–targeting chimeras containing an endocytic signaling motif trigger endocytosis and lysosomal degradation of cell-surface proteins†
Tong
Fang
,
Zhenting
Zheng
,
Na
Li
,
Yishu
Zhang
,
Jing
Ma
,
Chengyu
Yun
and
Xiaoqing
Cai
 *
*
School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, China. E-mail: caixq7@mail.sysu.edu
First published on 1st October 2024
Abstract
Lysosome–targeting degradation technologies have emerged as a promising therapeutic strategy for the selective depletion of target extracellular and cell-surface proteins by harnessing a cell-surface effector protein such as lysosome–targeting receptors (LTRs) or transmembrane E3 ligases that direct lysosomal degradation. We recently developed a lysosome–targeting degradation platform termed signal-mediated lysosome-targeting chimeras (SignalTACs) that functions independently of an LTR or E3 ligase; these are engineered fusion proteins comprising a target binder, a cell-penetrating peptide (CPP), and a lysosomal sorting signal motif (P1). Herein, we present the next-generation SignalTACs containing a single endocytic signal that bypasses the need for a CPP. We demonstrate that the fusion with a 10-amino acid endocytic signaling peptide (P3) derived from the cation-independent mannose-6-phosphate receptor (CI-M6PR) induces robust internalization and lysosomal degradation of the target protein. The P3-based SignalTAC exhibited enhanced antitumor efficacy compared to the parent antibody. We envision that the fusion of the endocytic signaling peptide P3 to a target binder may allow the construction of an effective degrader for membrane-associated targets. Furthermore, mechanistic studies identified different drivers for the activities of the P3- and P1-based SignalTACs, which is expected to provide crucial insights toward the harnessing of the intrinsic signaling pathways to direct protein trafficking and degradation.
Introduction
Targeted protein degradation (TPD) has attracted great attention as a novel paradigm for biological research and the development of new therapeutics.1–3 Technologies that hijack the ubiquitin-proteasome system (UPS), including proteolysis-targeting chimeras (PROTACs) and molecular glues, have demonstrated great therapeutic potential in clinical practice.4–7 The emergence of novel TPD technologies that exploit the endocytosis–lysosome system has expanded the scope of TPD to include targets such as extracellular and cell-surface proteins; examples of such technologies include the lysosome–targeting chimeras (LYTACs),8–12 antibody-based PROTACs (AbTACs),13 cytokine receptor-targeting chimeras (KineTACs),14 and others.15–26 These novel techniques exploit the central waste disposal system of cells and present promising approaches for tackling proteins associated with specific diseases.The vast majority of the lysosome–targeting degradation platforms hitherto employed rely on the recruitment of a cell-surface effector protein that directs lysosomal degradation of the target protein; a lysosome–targeting receptor (LTR) or transmembrane E3 ubiquitin ligase are typically employed as effectors.25,27 Extracellular protein degradation uses bifunctional molecules, such as antibody–glycan conjugates, bispecific antibodies, and small molecules, which bind simultaneously to a protein of interest (POI) and a lysosome-directed protein effector to form a ternary complex and trigger the lysosomal trafficking of POI. Despite the great advances these new platforms have made, there remain significant challenges to address. First, the degradation efficacy of such strategies is highly dependent on the expression of a lysosome-directed protein effector, which is usually not ubiquitously expressed on all cell or tissue types, thus limiting the degradation on the desired site of action. In addition, LTR-based degraders compete with endogenous protein ligands that are present in excess, which can limit the access of degraders to LTRs. Lastly, a high-affinity binder to the effector protein with KD values ranging from picomolar to low nanomolar is typically needed to ascertain efficient degradation.11,27
Lysosomes can be accessed via the biosynthetic or endocytic pathway through intrinsic protein sorting codes and the cellular decoding machinery. We have previously reported a lysosome–targeting signaling tag (R9-NPGY) composed of a nona-arginine (R9) cell-penetrating peptide linked with the lysosomal sorting sequence (NPGY) from the low-density lipoprotein receptor (LDLR). The linking of this tag to a nanobody induced robust endocytosis and lysosomal trafficking of the nanobody.28 Subsequently, the Chen group generated an elegant degradation method termed GlueTAC, involving the enzymatic linking of this tag (R9-NPGY) with a covalently engineered nanobody to enable the lysosomal degradation of the captured POI.17 We have independently developed the degradation platform signal-mediated lysosome–targeting chimeras (SignalTACs) that is entirely genetically encoded and harnesses the intrinsic lysosomal sorting signals to promote the lysosomal trafficking and degradation of the target membrane POI (Fig. 1A).22 The first-generation SignalTACs consist of three modular components: a target binder (such as a monoclonal antibody or mAb, nanobody, and peptide), a CPP, and a dieucine-based sorting signal motif from the cation-independent mannose-6-phosphate receptor (CI-M6PR).29,30 A short CPP (R4K) was required to increase the cytosolic delivery of this signaling motif, which in turn enhanced the lysosomal degradation of the target membrane POI. Despite intense research efforts and the proven therapeutic potential of CPPs, significant concerns remain regarding their stability, cell-type specificity, and potential systemic toxicity.31–35
 | ||
| Fig. 1 The construction and internalization of next-generation SignalTACs containing a tyrosine-based endocytosis signal. (A) Schematic representation of the lysosomal targeting degradation of membrane proteins mediated by SignalTACs and the construction of two types of SignalTACs. (B) Electrostatic potential properties of the tyrosine-based endocytic sorting motif P3 and the dileucine-based lysosomal sorting motif P1. (C) The mean fluorescence intensity of SKOV3 after treatment with 100 nM TAMRA-labeled Tz, Tz-P1, Tz-P2-P1, Tz-P3, or Tz-P2-P3 for 12 h as determined by flow cytometry. (D) Confocal microscopy images of SKOV3 cells incubated with 100 nM TAMRA-labeled Tz, Tz-P2-P1, Tz-P3, or Tz-P2-P3 for 24 h. (E) The mean fluorescence intensity of the MDA-MB-231 cells after treatment with 100 nM TAMRA-labeled Az, Az-P1, Az-P2-P1, Az-P3, or Az-P2-P3 for 24 h as determined by flow cytometry. Scale bars indicate 20 μm. Data are shown as mean ± s.e.m from three independent experiments. Statistical analysis was determined by one-way ANOVA with Tukey's post hoc test. NS, no significance, *P < 0.05, **P < 0.01, ****P < 0.0001. | ||
The M6PR-dependent pathway represents the classic route of transport of lysosomal proteins to the lysosome. CI-M6PR is one of the lysosomal shuttling receptors that has been harnessed for the design of the LYTAC degradation platforms.8,12,24 CI-M6PR is chiefly located in the trans-Golgi network (TGN) and endosomes with only a small fraction (∼10%) expressed on the cell surface.36,37 CI-M6PR regulates the sorting and trafficking of proteins to the lysosomal compartments via two distinct pathways. In the first pathway, CI-M6PR plays an important intracellular role by delivering the newly synthesized mannose-6-phosphate (M6P)-modified acid hydrolases along the biosynthetic pathway to the lysosomes.29,37 In the second pathway, the cell-surface CI-M6PR functions as a receptor for various extracellular ligands; this, in turn, can trigger a series of cellular processes such as the proteolytic activation of proteins, growth and development, as well as receptor internalization.30,38,39 These two distinct pathways are regulated via two different cytoplasmic sorting signal motifs of CI-M6PR. A dileucine-based sorting signal is chiefly responsible for the transport of lysosomal enzymes along the TGN to the lysosomes via the biosynthetic pathway.40,41 By contrast, the internalization of cell-surface CI-M6PR is believed to be mediated by a tyrosine-based signal.42–44 The dileucine-based motif has been exploited for the construction of the first-generation SignalTACs; however, whether the tyrosine-based signal tag can be incorporated in a target binder for promoting the internalization and lysosomal trafficking of the bound POI remains to be explored. Furthermore, the molecular events and pathway(s) employed for the intracellular trafficking of SignalTACs containing these two distinct signaling motifs remain unclear.
In the current work, we sought to exploit the tyrosine-based endocytic signaling motif from CI-M6PR for the construction of next-generation SignalTACs (Fig. 1A). We demonstrated that the fusion of this linear internalization motif with the target binder induces the internalization and degradation of the target protein without the need for a CPP motif. In addition, we evaluated the antitumor efficacy of the next-generation SignalTACs so generated. Lastly, we performed a mechanistic study to elucidate the underlying molecular events and factors essential for SignalTAC-mediated lysosomal trafficking and degradation of the target POI.
Results and discussion
Design and validation of next-generation SignalTACs containing a tyrosine-based internalization motif P3
The sequence YKYSKV spanning the positions 24–29 of the cytoplasmic tail of CI-M6PR has been recognized as the signal motif that facilitates the rapid internalization of the cell-surface CI-M6PR,45 this activity is believed to be predominantly mediated by four carboxy-terminal amino acid residues, particularly Tyr26 and Val29.46 The mechanism underlying the role of this small linear motif in the backbone of the cytosolic tail of CI-M6PR as a signal for cargo recruitment remains unclear, as does the issue of whether the binding of M6P changes the conformation of the protein to display the sorting motif, thereby triggering the process of endocytosis. We have previously shown that a longer sequence that contains the key sorting motif secures the critical structural elements for allowing the molecular events.22 Therefore, we chose a longer sequence containing the YSKV motif (SYKYSKVNKE) that spans the positions 23–32 of the cytoplasmic tail of CI-M6PR and is referred to as P3 for simplicity, for the construction of the next-generation SignalTACs.First, we observed considerable differences between the chemical properties of the tyrosine-based signaling motif P3 and the dileucine-based signaling motif (SFHDDSDEDLLHI) referred to hereafter as P1. The P3 motif appears to be a cationic peptide containing cationic (K3, K6, N8, and K9) and polar residues, whereas the P1 motif is featured by a cluster of acidic residues, notably multiple Asp residues (D4, D5, D7, and D9). Therefore, we performed an analysis of the surface electrostatic properties by calculating the electrostatic potential of the peptide surface (Fig. 1B); as expected, the electrostatic potential of the surfaces of P3 and P1 were strikingly different. Presumably, the anionic surface charge of the dileucine-based signal P1 impedes the interaction of the signaling motif with the negatively charged cell membrane; this, in turn, may explain the need for a CPP motif containing five positively charged residues for promoting the lysosomal trafficking and degradation activities of the first-generation SignalTACs. Therefore, we proposed that the cationic surface charge of P3 may enhance the cell penetrability and therefore, facilitate the trafficking of the target binder into the cytosol.
We hypothesized that the P3 signaling motif could be incorporated in a target binder to allow the delivery of the target POI to the lysosomes for degradation. As a proof of concept, we fused the P3 signaling motif to the C-termini of the light and heavy chains of a monoclonal antibody (mAb). We subsequently examined whether the SignalTACs facilitate the internalization and degradation of the two membrane proteins human epidermal growth factor receptor 2 (HER2) and programmed death-ligand 1 (PD-L1). HER2 is a well-established oncogenic driver, as also an internalization-impaired receptor owing to the lack of a natural ligand.47–50 PD-L1 is an important immune checkpoint protein that suppresses T cell-mediated cytotoxicity.51 Accordingly, we incorporated the P3 signaling motif in the C-termini of both the heavy and light chains of trastuzumab (Tz),52 a United States Food and Drug Administration (FDA)-approved anti-HER2 antibody, to generate SignalTAC Tz-P3. A (G4S)3 linker was inserted before this signaling motif to enhance the flexibility of the signaling motif. We have previously demonstrated that the insertion of a short CPP before the signaling motif promotes the internalization and lysosomal trafficking.22,28 The sequence R4K (P2) with minimum overall positive charge was optimized and fused with the P1 signaling motif to generate the first-generation SignalTAC.22 To evaluate whether the insertion of a positively charged CPP improves the internalization, we inserted P2 before the P3 motif to yield Tz-P2-P3. Similarly, we also incorporated the sequences P3 and P2-P3 into atezolizumab53 (Az), an anti-PD-L1 antibody approved by the FDA, to generate two anti-PD-L1 SignalTACs termed Az-P3 and Az-P2-P3 (Fig. 1A). The abovementioned four antibody–peptide fusions, Tz-P3, Tz-P2-P3, Az-P3, and Az-P2-P3, were expressed in 293-F cells and characterized by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), high-performance liquid chromatography (HPLC), and electrospray ionization mass spectrometry (ESI-MS; ESI Fig. S1–S12†). Additionally, we conducted an evaluation of the chemical stability of Tz-P3 at both 25 °C and 40 °C after incubation for 12 h and 60 h. SDS-PAGE and SEC-HPLC analyses confirmed that there was no aggregation or degradation resulting from the introduction of the P3 motif (ESI Table S1 and Fig. S13–S16†).
Subsequently, we examined whether the P3-based SignalTACs (Tz-P3 and Tz-P2-P3) are internalized and transported to the lysosomes. We also included two P1-based HER2-targeting SignalTACs (Tz-P1 as Tz-P2-P1) for the purpose of comparison. Accordingly, we employed the human cancer cell line SKOV3 with high levels of HER2 expression and treated the cells with Tz, Tz-P1, Tz-P2-P1, Tz-P3, and Tz-P2-P3. Flow cytometry analysis revealed that all the antibody–peptide fusions displayed significantly higher internalization activities compared to Tz (Fig. 1C). The P3-based SignalTACs (Tz-P3 and Tz-P2-P3) demonstrated the highest potential for internalization followed by Tz-P2-P1, and then Tz-P1. To our surprise, the internalization of the CPP-containing Tz-P2-P3 was only slightly more efficient than that of Tz-P3. In other words, the insertion of CPP did not significantly impact the internalization of P3-based SignalTACs. To further verify the internalization and lysosomal trafficking of Tz-P3 and Tz-P2-P3, confocal microscopy study was carried out, which confirmed that Tz-P3 and Tz-P2-P3 exhibited comparable potential for internalization and lysosomal trafficking (Fig. 1D). Internalization was not observed by treatment of HER2-negative cells with Tz-P3 or Tz-P2-P3 (ESI Fig. S18†), indicating that the internalization is HER2 dependent. Lastly, SignalTACs were constructed using P1 and P3 as the signaling motifs and an HER2-targeting nanobody (Nb) as the target binder. To further validate the superiority of P3 motif, we also incorporated the endocytic signal54 (P4) derived from the asialoglycoprotein receptor (ASGPR), a liver-specific LTR employed for lysosome-targeted protein degradation.10 Flow cytometry study confirmed that the P3 motif exhibited a superior ability to promote the internalization of Nb compared to both P1 and P4 (ESI Fig. S17†).
We also compared the internalization activities of the P3-based anti-PD-L1 SignalTACs (Az-P3 and Az-P2-P3) and P1-based anti-PD-L1 SignalTACs (Az-P1 and Az-P2-P1). Consistent with the above results, Az-P3 and Az-P2-P3 exhibited similar potential for internalization, followed by Az-P2-P1, Az-P1, and finally, Az (Fig. 1E and ESI Fig. S19†). Neither Az-P3 nor Az-P2-P3 could be uptaken by PD-L1-negative cells (ESI Fig. S20†). Taken together, these data suggest that P3 is a strong endocytic signaling motif that can be exploited to direct robust internalization of the target binder.
P3-based SignalTACs are potent degraders that promote lysosomal degradation of membrane proteins
We subsequently evaluated whether the P3-based SignalTACs can induce the degradation of the target POI. First, we sought to compare the degradation of HER2 mediated by all the four SignalTACs. Accordingly, we treated SKOV3 cells with Tz-P1, Tz-P2-P1, Tz-P3, and Tz-P2-P3, and assessed the level of HER2 by western blot analysis. Treatment with Tz-P3 resulted in the degradation of >75% of HER2 in SKOV3 cells, which is similar to the degradation observed with Tz-P2-P1 and Tz-P2-P3 treatment (Fig. 2A). We also evaluated the degradation of HER2 in cell lines SKBR3 and NCI-N87; Tz-P2-P3 and Tz-P3 exhibited comparable HER2 degradation efficiency (Fig. 2A). We then evaluated the degradation of HER2 using flow cytometry. Consistent with the above results, the treatment of SKOV3 cells with Tz-P3 resulted in the degradation >75% of HER2 (Fig. 2B). We also carried out confocal microscopy imaging to confirm the substantial depletion of HER2 on the surface of cells (Fig. 2C). | ||
| Fig. 2 P3-based SignalTACs mediate the degradation of target proteins. (A) Western blot analysis of HER2 degradation after treatment with 100 nM Tz and indicated SignalTACs for 48 h in SKOV3, SKBR3, and NCI-N87 cells. (B) Degradation of cell-surface HER2 in SKOV3 cells as determined by flow cytometry after treatment with 100 nM Tz or Tz-P3 for 48 h. (C) Visualization of HER2 degradation in SKOV3 cells by confocal microscopy after treatment with 100 nM Tz or Tz-P3 for 48 h. (D) Western blot analysis of PD-L1 degradation after treatment with 100 nM Az and indicated SignalTACs for 48 h in MDA-MB-231, A431, or HCC-1954 cells. (E) Time course of degradation of PD-L1 in MDA-MB-231 cells incubated with 100 nM Az-P3 or Az for 3, 6, 12, 24, and 48 h. (F) Dose–response curve for PD-L1 degradation in MDA-MB-231 cells incubated with 1 nM, 10 nM, 50 nM, 100 nM, and 200 nM Az-P3 or Az for 48 h. (G) & (H) Western blot analysis of PD-L1 degradation in MDA-MB-231 cells treated with 100 nM Az-P3 in the presence or absence of 50 μM chloroquine, 0.1 mg/mL leupeptin, or 20 μM MG132 for 24 h. (I) Western blot analysis of HER2 degradation in SKOV3 cells treated with 100 nM Tz-P3 in the presence or absence of 20 μM chloroquine or 20 μM MG132 for 48 h. Results are representative of three (A–I) or four (E and F) independent experiments. Scale bars indicate 20 μm. Data are shown as means ± s.e.m. Statistical analysis was determined by one-way ANOVA with Tukey's post hoc test. ****P < 0.0001. | ||
We subsequently evaluated the potential of P3-based SignalTAC to degrade another membrane protein, PD-L1, by treating PD-L1-positive cells with 100 nM Az, Az-P2, Az-P2-P1, Az-P3, and Az-P2-P3. Western blot analysis revealed that the two P3-based SignalTACs (Az-P3 and Az-P2-P3) resulted in comparable downregulation of PD-L1 (Fig. 2D). Confocal microscopy imaging further confirmed the considerable degradation of PD-L1 in the breast cancer cell line MDA-MB-231 upon treatment with Az-P3 and Az-P2-P3 (ESI Fig. S21†). Next, we evaluated the efficiency of Az-P3-mediated depletion of cell-surface PD-L1 in MDA-MB-231 cells through western blotting, considering both time and dose factors (ESI Fig. S22 and S23†). The degradation of PD-L1 by Tz-P3 was found to be time-dependent, with maximum degradation attained at 48 h (Fig. 2E). Moreover, Az-P3 degraded PD-L1 in a dose-dependent manner, with maximum degradation achieved at a concentration of 100 nM (Fig. 2F). Overall, these data reveal that the fusion of the internalization motif P3 to Tz or Az is sufficient to induce efficient degradation of the target POIs.
Lastly, we set to verify whether the P3-based SignalTAC-mediated degradation of target POIs occurred via the lysosomal pathway. We first treated MDA-MB-231 cells with Az-P3 in the presence or absence of the lysosome inhibitors chloroquine or leupeptin and the proteasome inhibitor MG132. The treatment with chloroquine or leupeptin, but not MG132, inhibited the degradation of PD-L1, indicating the involvement of the lysosomes but not the proteosome in SignalTAC-mediated PD-L1 degradation (Fig. 2G and H). Moreover, we incubated SKOV3 cells with Tz-P3 in the presence or absence of chloroquine or MG132. Again, the pretreatment of chloroquine inhibited the degradation of HER2, while MG132 treatment failed to exert any effect (Fig. 2I). Together, these results indicate that the P3-based SignalTACs mediate target degradation in a lysosome-dependent manner.
P3-based SignalTACs exhibited enhanced antitumor activity
We subsequently investigated whether P3-based SignalTACs exhibit enhanced antitumor activities compared to their parent antibodies. First, we carried out an EdU assay to determine the efficacy of Tz-P3 in inhibiting the proliferation of SKOV3 cells. Tz-P3 was substantially more effective at inhibiting cell proliferation than Tz (Fig. 3A). In addition, the treatment of SKOV3 cells with various concentrations of Tz-P3 showed significant inhibition of colony formation and growth, further confirming the superior antiproliferation activity of Tz-P3 (ESI Fig. S24†). Encouraged by these results, we next evaluated the antitumor activities of P3-based SignalTACs using the NCI–N87 xenograft mouse model. When the tumors reached a size of ∼500 mm3, we intraperitoneally injected Tz, Tz-P3, and PBS into the tumor-bearing mice (n = 4 per group) every four days at a dose of 10 mg kg−1 (Fig. 3B). We measured the tumor size and body weight every two days. Significant weight loss was not observed during this process, suggesting the biosafety of the P3-based SignalTACs (ESI Fig. S25†). The tumor growth was substantially suppressed by Tz-P3 treatment compared to Tz or PBS treatment (Fig. 3C). At the end of the study, tumor tissues were dissected and weighted (Fig. 3D and E). The rates of tumor inhibition upon the administration of Tz and Tz-P3 were calculated to be approximately 24% and 76% relative to that of PBS treatment, respectively (Fig. 3F). Furthermore, immunohistochemistry (IHC) analysis indicated that P3-based SignalTACs significantly depleted HER2 levels in tumor tissues (Fig. 3G). Taken together, these results demonstrate that P3-based SignalTACs effectively suppress tumor growth without any substantial toxicity, suggesting that the SignalTAC-mediated degradation strategy may have potential therapeutic applications.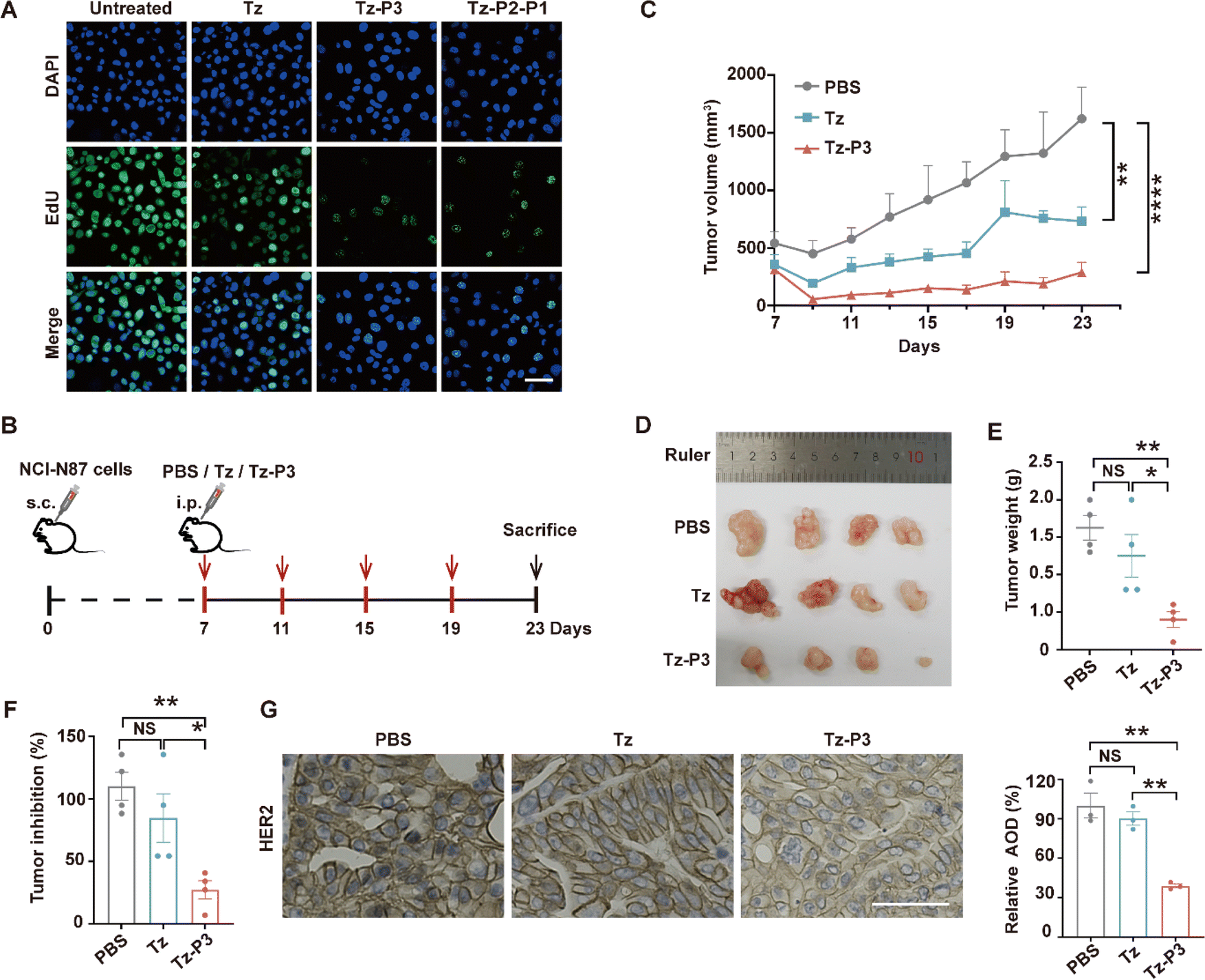 | ||
| Fig. 3 P3-SignalTACs exhibit enhanced anti-tumor activity. (A) Analysis of proliferation by EdU assay in SKBR3 after treatment with 100 nM Tz or Tz-P3 for 48 h. (B) Schematic illustration of the in vivo study for therapeutic efficacy in an NCI-N87 xenograft mouse model. (C) Evaluation of tumor size after treatment of PBS, Tz, or Tz-P3 every 2 days (n = 4 mice/group). (D) Photographs of dissected tumor tissues after different treatment at day 23. (E) Comparison of the tumor weight after tumor dissection. (F) Tumor inhibitory rates calculated based on the weight of dissected tumor tissues. (G) Representative immunohistochemical staining of HER2 expression on tumor tissues. Scale bars indicate 50 μm. Data are shown as means ± s.e.m. (n = 4). Statistical analysis was determined by one-way ANOVA with Tukey's post hoc test. NS, no significance, *P < 0.05, **P < 0.01, ****P < 0.0001. | ||
P3-based SignalTACs mediate target degradation via caveolae-dependent endocytosis
To obtain a better understanding of the mechanisms driving the degradation of the target POIs, we first investigated the involvement of the endocytosis pathway in P3-based SignalTAC-mediated degradation. We preincubated SKOV3 cells with inhibitors of clathrin-mediated endocytosis (chlorpromazine, CPZ), caveolae/lipid raft-dependent endocytosis (nystatin), and macropinocytosis (5-[N-ethyl-N-isopropyl]amiloride, EIPA), followed by incubation with Tz-P3. Western blot analysis demonstrated that treatment with nystatin substantially inhibited HER2 degradation, unlike CPZ which failed to impact the process (Fig. 4A). The addition of EIPA moderately inhibited HER2 degradation. Surprisingly, these data suggest that the degradation activity of P3-based SignalTACs is dependent on caveolae but not clathrin. By contrast, our previous study showed that clathrin plays a key role in P1-based SignalTAC-mediated target degradation.22 Subsequently, we carried out confocal microscopy to verify the cellular uptake of Tz-P3 in the presence of the abovementioned endocytosis inhibitors; the quantitative analysis confirmed caveolae-dependent endocytosis as the key pathway of uptake (Fig. 4B). Lastly, we performed immunofluorescence staining for clathrin and caveolae to visualize the two corresponding distinct endocytosis pathways for the uptake of Tz-P3 and Tz-P2-P1 (Fig. 4C and D); the results further confirmed that Tz-P3 internalization occurred via caveolae-mediated endocytosis while the uptake of Tz-P2-P1 predominantly occurred via clathrin-mediated endocytosis. | ||
| Fig. 4 The endocytosis mechanism of SignalTACs. (A) Western blot analysis of HER2 degradation in SKOV3 cells incubated with 100 nM Tz-P3 in the presence or absence of 10 μg/mL EIPA, Nystatin, or CPZ for 48 h. (B) Confocal microscopy images of SKOV3 cells incubated with 100 nM Tz-P3 in the presence or absence of 10 μg/mL chlorpromazine, 10 μg/mL Nystatin or 10 μg/mL EIPA for 24 h. (C) Confocal microscopy images of clathrin in SKOV3 cells incubated with 100 nM Tz, Tz-P2-P1, or Tz-P3 for 6 h. (D) Confocal microscopy images of caveolin in SKOV3 cells incubated with 100 nM Tz, Tz-P2-P1, or Tz-P3 for 6 h. Scale bars indicate 20 μm. Data are shown as means ± s.e.m. Statistical analysis was determined by one-way ANOVA with Tukey's post hoc test. NS, no significance, *P < 0.05, **P < 0.01, ****P < 0.0001. | ||
The activity of P3- and P1-based SignalTACs are dependent on different endocytic adaptor proteins
Protein sorting signals are known to interact with endocytic adaptor proteins peripherally associated with the cytosolic face of the cell membrane; these proteins serve as a link between the endocytic machinery and the cargo proteins and promote endocytosis.55 Therefore, we probed the roles of two main families of endocytic adaptor proteins; these include adaptor proteins complexes (AP-1, AP-2, and AP-3) and Golgi-located, γ ear-containing, ADP-ribosylation factor-binding proteins (GGA1, GGA2, and GGA3). Toward this end, we employed the RNA interference approach to knock down the expression of particular adaptor proteins (ESI Fig. S26†). Accordingly, we incubated SKOV3 cells with Tz-P3 or Tz-P2-P1 in conjunction with siRNAs targeting AP-1 (AP1B1), AP-2 (AP2S1), AP-3 (AP3S1), GGA1, GGA2, or GGA3. Western blot analysis revealed that the knockdown of AP-1 and AP-3 disrupted Tz-P3-mediated HER2 degradation, suggesting that the degradation of HER2 could be dependent on the functions of AP-1 and AP-3 (Fig. 5A and B). Interestingly, the knockdown of AP-2 and the members of the GGA family (GGA1, GGA2, and GGA3) did not exert a significant impact on the Tz-P3-mediated degradation of HER2, suggesting that AP2 and the GGA family may not play a role in Tz-P3-mediated protein degradation. By contrast, the knockdown of AP-2, AP-3, and the members of the GGA family (GGA1, GGA2, and GGA3) exerted a significant impact on Tz-P2-P1 activity (Fig. 5C and D). AP-1, which appeared to be essential for Tz-P3 activity, did not exert a critical impact on the activity of Tz-P2-P1. Interestingly, only AP-3 appeared to play an essential role for the activities of both Tz-P3 and Tz-P2-P1. We carried out confocal microscopy to visualize the internalization and lysosomal trafficking of both Tz-P3 and Tz-P2-P1 in SKOV3 cells pretreated with various siRNAs targeting the subunits of AP complexes and members of the GGA family (ESI Fig. S27†). The result confirmed that the activities of P3-based SignalTACs are dependent only on AP-1 and AP-3, while those of the P1-based SignalTACs require AP-2, AP-3, GGA1, GGA2, and GGA3.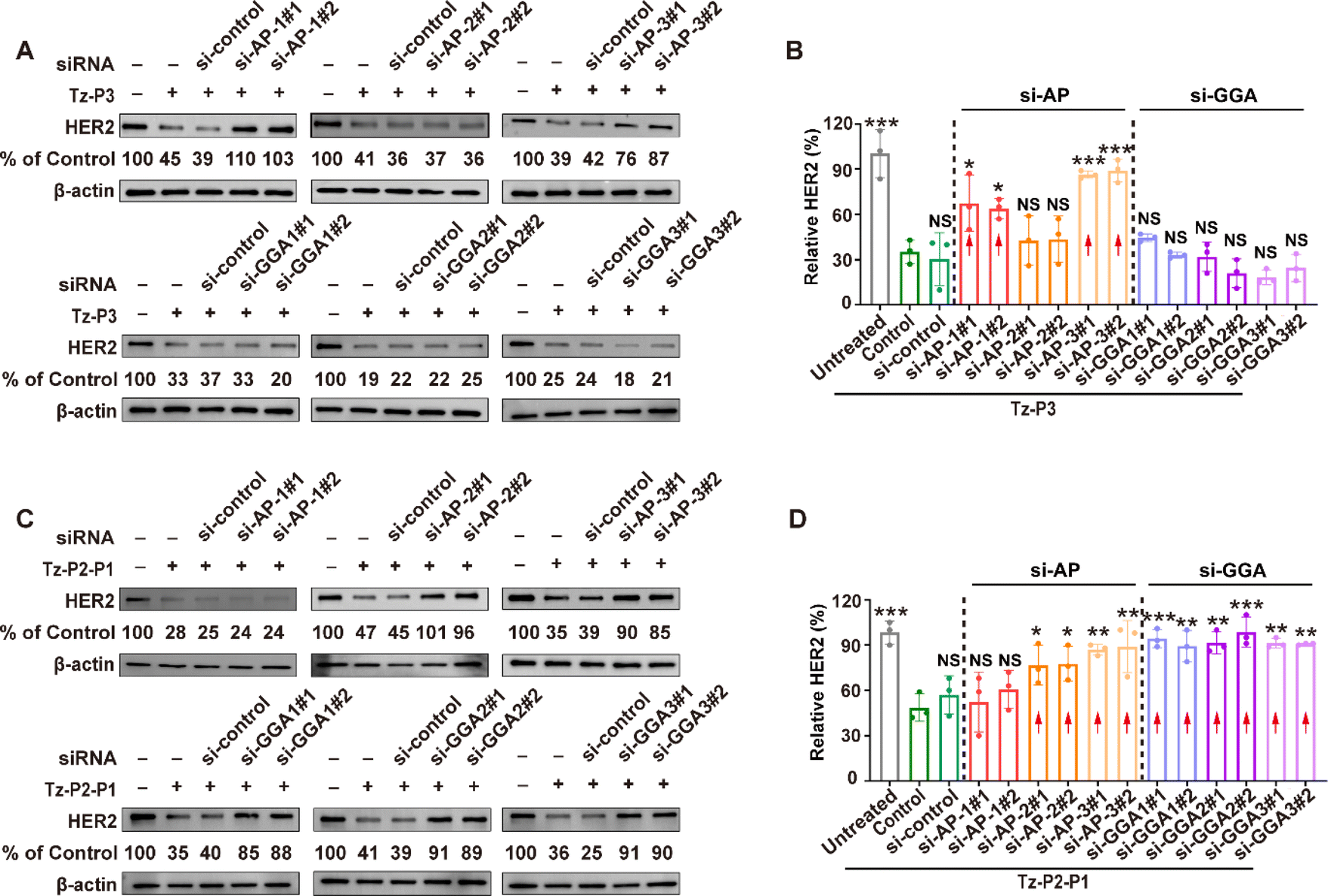 | ||
| Fig. 5 Effect of endocytic adaptor proteins on SignalTAC activity. (A) Western blot analysis of HER2 degradation after siRNA knockdown of AP-1, AP-2, or AP-3 followed by 48 h of treatment with 100 nM Tz-P3 in SKOV3 cells. (B) Quantitative analysis of relative HER2 as shown in (A). (C) Western blot analysis of HER2 degradation after siRNA knockdown of GGA1, GGA2, or GGA3 followed by 48 h of treatment with 100 nM Tz-P2-P1 in SKOV3 cells. (D) Quantitative analysis of relative HER2 as shown in (C). Results are representative of three independent experiments. Data are shown as means ± s.e.m. Statistical analysis was determined by one-way ANOVA with Tukey's post hoc test. NS, no significance, *P < 0.05, **P < 0.01, ***P < 0.001 as compared to the Tz-P3 control for (B), and as compared to the Tz-P2-P1 control for (D). | ||
Taken together, the SignalTACs containing P3 and P1 motifs interact with different components of the cellular decoding machinery to trigger the lysosomal sorting process. The activity of Tz-P3 requires only AP-1 and AP-3, while the functions of Tz-P2-P1 rely on AP-2, AP-3, and GGA1–3. Notably, AP-2, which plays a key role in the initiation of clathrin-mediated endocytosis,56,57 appears to be essential for the activity of Tz-P2-P1, but not Tz-P3; this result further confirms that the endocytosis of Tz-P3 is clathrin-independent unlike that of Tz-P2-P1, which is dependent on clathrin. AP3, which has been previously reported to play a role in both clathrin-dependent and clathrin-independent transport mechanisms,58 is shown to play a critical role in the activities of both Tz-P3 and Tz-P2-P1. Interestingly, AP-1, which is also known as a clathrin-associated adaptor, seems to be essential for the endocytosis and activity of Tz-P3. The observed role of the GGAs in the functions of P1 motif is also supported by previous studies, which elucidated the crystal structures of the VHS domain of the GGA proteins complexed with the dileucine sorting peptide from M6PR.59,60
P3- and P1-based SignalTACs employ different pathways of intracellular trafficking
Given the critical roles of the AP complexes and GGA1–3 for the activity of SignalTACs, we subsequently investigated the dynamic intracellular movement of the P3- and P1-based SignalTACs. We examined the colocalization of Tz-P3 or Tz-P2-P1 with the early endosome (Rab5), late endosome (Rab7), Golgi apparatus (Golgi Tracker Green), and the recycling endosome (Rab11). Accordingly, we treated SKOV3 cells with the above markers following incubation with Tz-P3 or Tz-P2-P1 for 6, 12, and 24 h. As shown in Fig. 6A and B, Tz-P3 colocalized with both early and late endosomes (EE and LE, respectively) after 6 h, but chiefly with LE at 12 h, which was maintained at 24 h; the colocalization of Tz-P3 with EE decreased with increasing time of incubation. Tz-P3 exhibited only marginal colocalization with the Golgi apparatus, which was maintained at the three time points. The subcellular distribution of Tz-P2-P1 over time was found to be different from that of Tz-P3 (Fig. 6C and D). At the time point of 6 h, Tz-P2-P1 was largely colocalized with EE and the Golgi apparatus, but only marginally with LE. However, the colocalization of Tz-P2-P1 with the Golgi apparatus decreased significantly at 12 h, which was maintained at 24 h. Taken together, these data suggest that the Golgi apparatus plays a role in the lysosomal trafficking of Tz-P2-P1. Moreover, an increased colocalization with LE at 12 and 24 h suggests the transport of Tz-P2-P1 from the EE and Golgi apparatus to LE. Notably, colocalization of either Tz-P3 or Tz-P2-P1 with the recycling endosome (RE) was not observed, indicating that the RE pathway may not be involved in the lysosomal trafficking of SignalTACs (ESI Fig. S28†). Subsequently, the colocalization with Golgi apparatus was examined at 2, 4, 6, 8, 10, and 12 h following 2 h of incubation of the cells with Tz-P3 or Tz-P2-P1 (Fig. 6E); significant colocalization of Tz-P3 with the Golgi apparatus was not observed over time, indicating that Tz-P3 is mainly delivered to the lysosomes via the endo-lysosomal pathway. By contrast, a marked colocalization of Tz-P2-P1 with the TGN was observed after 2 h that reached a peak at 6 and 8 h, suggesting the sorting of Tz-P2-P1 to the TGN for further transport to the LE. | ||
| Fig. 6 The intracellular trafficking study of SignalTACs. (A) Visualization of Rab5, Rab7 or Golgi in SKOV3 cells by confocal microscopy after treatment with 100 nM Tz-P3 labeled with TAMRA for 6 h, 12 h or 24 h. (B) The statistical results of colocalization factor (Pearson's R value) in (A). (C) Visualization of Rab5, Rab7 or Golgi in SKOV3 cells by confocal microscopy after treatment with 100 nM Tz-P2-P1 labeled with TAMRA for 6 h, 12 h or 24 h. (D) The statistical results of colocalization factor (Pearson's R value) in (C). (E) Confocal images showing the colocalization between Golgi with Tz-P3 or Tz-P2-P1 in SKOV3 cells for 2, 4, 6, 8, 10 or 12 h after treatment with 100 nM TAMRA-labeled Tz-P3 or Tz-P2-P1 for 2 h. The intensity profiles of SignalTACs and Golgi along the white line are plotted in the right panels. Scale bars indicate 10 μm. Data are shown as means ± s.e.m. (n = 3). Statistical analysis was determined by one-way ANOVA with Tukey's post hoc test. NS, not significance, *P < 0.05, **P < 0.01, ****P < 0.0001. | ||
Collectively, these findings support the notion that the internalization and lysosomal trafficking of Tz-P3 occurs via a direct route while that of Tz-P2-P1 may occur via the conventional endo-lysosomal pathway as well as an indirect route of transport from the TGN to the endosomes (Fig. 7). The data from our mechanistic studies further support the merits of employing the endocytic signaling motif P3 for the construction of next-generation SignalTACs. Further investigation is still necessary to elucidate the detailed trafficking pathways and identify other possible molecular players that drive targeted membrane protein degradation by SignalTACs.
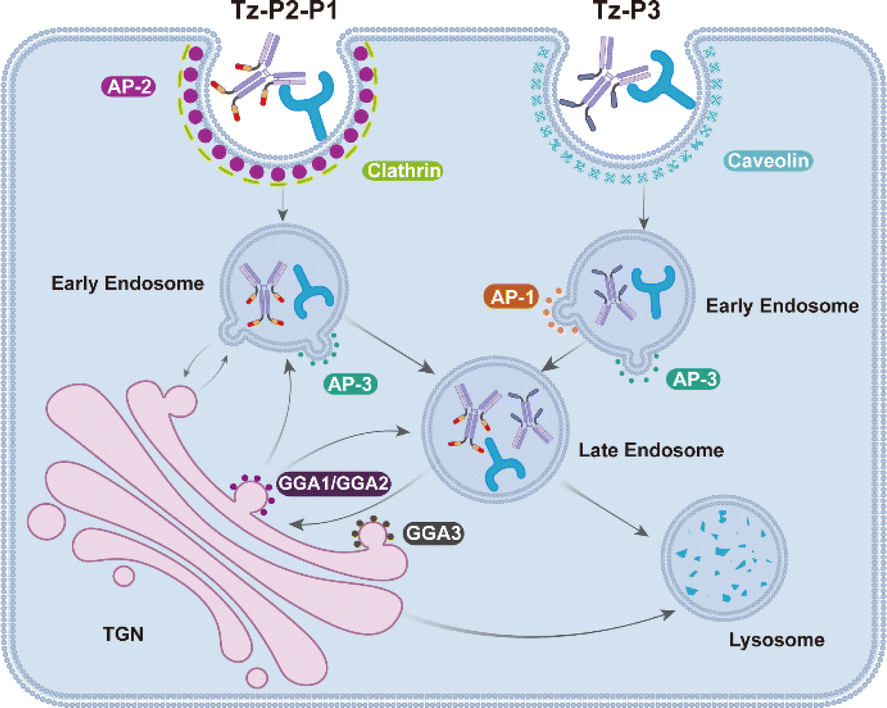 | ||
| Fig. 7 Schematic representation showing the endocytosis and trafficking route of Tz-P3 and Tz-P2-P1. | ||
Conclusions
In summary, we present the development of the next-generation SignalTAC degradation platform by exploiting tyrosine-based endocytic signals and the intrinsic protein sorting machinery. We demonstrated that the fusion of a single 10-amino acid signaling peptide P3 induces robust internalization and lysosomal degradation of the target protein. The P3-based SignalTAC exhibited enhanced antitumor efficacy compared to the parent antibody. We envision that the fusion of this endocytic signaling peptide P3 to a target-binding moiety such as an antibody, nanobody, peptide, or small molecule may allow the construction of an effective degrader for membrane-associated targets. Lastly, mechanistic studies identified different drivers for the activities of the P3- and P1-based SignalTACs, which is likely to provide crucial insights toward the harnessing of intrinsic signaling pathways to direct protein trafficking and degradation. Efforts are underway to construct SignalTACs targeting additional pathogenic proteins to further validate the potential of this platform.Data availability
The data supporting this article have been included in the ESI.†Author contributions
X. C. conceived and designed the project, and provided the supervision. T. F. prepared and purified all antibodies and conjugates. N. L and Y. Z. performed HPLC analysis and mass spectral characterization. T. F., Z. Z., N. L, J. M. and Y. Z. carried out the in vitro activity experiments. T. F. performed the mechanism study. T. F., Y. Z., C. Y., N. L. and Z. Z. worked collaboratively on the in vivo study. X. C. wrote the manuscript with input from all other authors.Conflicts of interest
The authors declare no competing interest.Acknowledgements
This study was funded by the National Natural Science Foundation of China (82173723 to X. C.) and Guangdong Basic and Applied Basic Research Foundation (2024A1515010432 and 2022A1515011964 to X. C.).References
- M. Békés, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef PubMed.
- P. P. Chamberlain and L. G. Hamann, Nat. Chem. Biol., 2019, 15, 937–944 CrossRef PubMed.
- X. Li and Y. Song, J. Hematol. Oncol., 2020, 13, 50 CrossRef.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef.
- G. E. Winter, D. L. Buckley, J. Paulk, J. M. Roberts, A. Souza, S. Dhe-Paganon and J. E. Bradner, Science, 2015, 348, 1376–1381 CrossRef PubMed.
- M. Schapira, M. F. Calabrese, A. N. Bullock and C. M. Crews, Nat. Rev. Drug Discovery, 2019, 18, 949–963 CrossRef PubMed.
- A. C. Lai and C. M. Crews, Nat. Rev. Drug Discovery, 2017, 16, 101–114 CrossRef.
- S. M. Banik, K. Pedram, S. Wisnovsky, G. Ahn, N. M. Riley and C. R. Bertozzi, Nature, 2020, 584, 291–297 CrossRef PubMed.
- Y. Zhou, P. Teng, N. T. Montgomery, X. Li and W. Tang, ACS Cent. Sci., 2021, 7, 499–506 CrossRef.
- G. Ahn, S. M. Banik, C. L. Miller, N. M. Riley, J. R. Cochran and C. R. Bertozzi, Nat. Chem. Biol., 2021, 17, 937–946 CrossRef.
- J. T. Bagdanoff, T. M. Smith, M. Allan, P. O'Donnell, Z. Nguyen, E. A. Moore, J. Baird, S. Wang, V. Subramanian, B. Tigani, D. O. Nettleton, L. G. Monovich, I. Lewis, A. N. Flyer, B. Granda, J. W. Blankenship, D. Barnes-Seeman and K. B. Clairmont, Cell Chem. Biol., 2023, 30, 97–109.e109 CrossRef.
- G. Ahn, N. M. Riley, R. A. Kamber, S. Wisnovsky, S. Moncayo von Hase, M. C. Bassik, S. M. Banik and C. R. Bertozzi, Science, 2023, 382, eadf6249 CrossRef.
- A. D. Cotton, D. P. Nguyen, J. A. Gramespacher, I. B. Seiple and J. A. Wells, J. Am. Chem. Soc., 2021, 143, 593–598 CrossRef.
- K. Pance, J. A. Gramespacher, J. R. Byrnes, F. Salangsang, J. C. Serrano, A. D. Cotton, V. Steri and J. A. Wells, Nat. Biotechnol., 2023, 41, 273–281 CrossRef.
- Y. Miao, Q. Gao, M. Mao, C. Zhang, L. Yang, Y. Yang and D. Han, Angew. Chem., Int. Ed. Engl., 2021, 60, 11267–11271 CrossRef.
- D. F. Caianiello, M. Zhang, J. D. Ray, R. A. Howell, J. C. Swartzel, E. M. J. Branham, E. Chirkin, V. R. Sabbasani, A. Z. Gong, D. M. McDonald, V. Muthusamy and D. A. Spiegel, Nat. Chem. Biol., 2021, 17, 947–953 CrossRef.
- H. Zhang, Y. Han, Y. Yang, F. Lin, K. Li, L. Kong, H. Liu, Y. Dang, J. Lin and P. R. Chen, J. Am. Chem. Soc., 2021, 143, 16377–16382 CrossRef PubMed.
- H. Marei, W. K. Tsai, Y. S. Kee, K. Ruiz, J. He, C. Cox, T. Sun, S. Penikalapati, P. Dwivedi, M. Choi, D. Kan, P. Saenz-Lopez, K. Dorighi, P. Zhang, Y. T. Kschonsak, N. Kljavin, D. Amin, I. Kim, A. G. Mancini, T. Nguyen, C. Wang, E. Janezic, A. Doan, E. Mai, H. Xi, C. Gu, M. Heinlein, B. Biehs, J. Wu, I. Lehoux, S. Harris, L. Comps-Agrar, D. Seshasayee, F. J. de Sauvage, M. Grimmer, J. Li, N. J. Agard and E. M. F. de Sousa, Nature, 2022, 610, 182–189 CrossRef.
- J. Zheng, W. He, J. Li, X. Feng, Y. Li, B. Cheng, Y. Zhou, M. Li, K. Liu, X. Shao, J. Zhang, H. Li, L. Chen and L. Fang, J. Am. Chem. Soc., 2022, 144, 21831–21836 CrossRef PubMed.
- C. Zhu, W. Wang, Y. Wang, Y. Zhang and J. Li, Angew. Chem., Int. Ed. Engl., 2023, 62, e202300694 CrossRef.
- Y. Wu, B. Lin, Y. Lu, L. Li, K. Deng, S. Zhang, H. Zhang, C. Yang and Z. Zhu, Angew. Chem., Int. Ed. Engl., 2023, 62, e202218106 CrossRef.
- J. Yu, H. Li, T. Fang, C. Yun, X. Liu, J. Xu, X. Jiang and X. Cai, J. Am. Chem. Soc., 2023, 145, 19107–19119 CrossRef PubMed.
- E. Loppinet, H. A. Besser, C. E. Lee, W. Zhang, B. Cui and C. Khosla, J. Am. Chem. Soc., 2023, 145, 18705–18710 CrossRef.
- B. Zhang, R. K. Brahma, L. Zhu, J. Feng, S. Hu, L. Qian, S. Du, S. Q. Yao and J. Ge, J. Am. Chem. Soc., 2023, 145, 24272–24283 CrossRef PubMed.
- X. Chen, Y. Zhou, Y. Zhao and W. Tang, Trends Pharmacol. Sci., 2023, 44, 762–775 CrossRef.
- R. A. Howell, S. Wang, M. Khambete, D. M. McDonald and D. A. Spiegel, J. Am. Chem. Soc., 2024, 146, 16404–16411 CrossRef CAS.
- J. A. Wells and K. Kumru, Nat. Rev. Drug Discovery, 2024, 23, 126–140 CrossRef PubMed.
- Y. Han, Y. Da, M. Yu, Y. Cheng, X. Wang, J. Xiong, G. Guo, Y. Li, X. Jiang and X. Cai, Org. Biomol. Chem., 2020, 18, 3229–3233 RSC.
- P. Ghosh, N. M. Dahms and S. Kornfeld, Nat. Rev. Mol. Cell Biol., 2003, 4, 202–212 CrossRef PubMed.
- C. Gauthier, K. El Cheikh, I. Basile, M. Daurat, E. Morère, M. Garcia, M. Maynadier, A. Morère and M. Gary-Bobo, J. Controlled Release, 2024, 365, 759–772 CrossRef PubMed.
- M. Zorko, S. Jones and Ü. Langel, Adv. Drug Delivery Rev., 2022, 180, 114044 CrossRef.
- H. Derakhshankhah and S. Jafari, Biomed. Pharmacother., 2018, 108, 1090–1096 CrossRef.
- S. H. Nam, J. Park and H. Koo, Arch. Pharmacal Res., 2023, 46, 18–34 CrossRef CAS PubMed.
- J. Wu, S. Roesger, N. Jones, C. J. Hu and S. D. Li, J. Controlled Release, 2024, 366, 864–878 CrossRef CAS PubMed.
- D. Raucher and J. S. Ryu, Trends Mol. Med., 2015, 21, 560–570 CrossRef CAS.
- T. Braulke, C. Gartung, A. Hasilik and K. von Figura, J. Cell Biol., 1987, 104, 1735–1742 CrossRef CAS.
- H. J. Geuze, W. Stoorvogel, G. J. Strous, J. W. Slot, J. E. Bleekemolen and I. Mellman, J. Cell Biol., 1988, 107, 2491–2501 CrossRef CAS.
- I. Martin-Kleiner and K. Gall Troselj, Cancer Lett., 2010, 289, 11–22 CrossRef CAS.
- T. Takeda, M. Komatsu, F. Chiwaki, R. Komatsuzaki, K. Nakamura, K. Tsuji, Y. Kobayashi, E. Tominaga, M. Ono, K. Banno, D. Aoki and H. Sasaki, Cell Death Dis., 2019, 10, 876 CrossRef CAS.
- H. J. Chen, J. Remmler, J. C. Delaney, D. J. Messner and P. Lobel, J. Biol. Chem., 1993, 268, 22338–22346 CrossRef CAS.
- H. J. Chen, J. Yuan and P. Lobel, J. Biol. Chem., 1997, 272, 7003–7012 CrossRef CAS.
- P. Lobel, K. Fujimoto, R. D. Ye, G. Griffiths and S. Kornfeld, Cell, 1989, 57, 787–796 CrossRef CAS.
- K. F. Johnson and S. Kornfeld, J. Cell Biol., 1992, 119, 249–257 CrossRef CAS PubMed.
- M. Jadot, W. M. Canfield, W. Gregory and S. Kornfeld, J. Biol. Chem., 1992, 267, 11069–11077 CrossRef CAS.
- T. Braulke and J. S. Bonifacino, Biochim. Biophys. Acta, 2009, 1793, 605–614 CrossRef CAS PubMed.
- W. M. Canfield, K. F. Johnson, R. D. Ye, W. Gregory and S. Kornfeld, J. Biol. Chem., 1991, 266, 5682–5688 CrossRef CAS PubMed.
- J. Yu, T. Fang, C. Yun, X. Liu and X. Cai, Front. Mol. Biosci., 2022, 9, 847835 CrossRef CAS.
- A. Sorkin and L. K. Goh, Exp. Cell Res., 2009, 315, 683–696 CrossRef.
- C. D. Austin, A. M. De Mazière, P. I. Pisacane, S. M. van Dijk, C. Eigenbrot, M. X. Sliwkowski, J. Klumperman and R. H. Scheller, Mol. Biol. Cell, 2004, 15, 5268–5282 CrossRef PubMed.
- K. Roepstorff, L. Grøvdal, M. Grandal, M. Lerdrup and B. van Deurs, Histochem. Cell Biol., 2008, 129, 563–578 CrossRef PubMed.
- D. Escors, M. Gato-Cañas, M. Zuazo, H. Arasanz, M. J. García-Granda, R. Vera and G. Kochan, Signal Transduction Targeted Ther., 2018, 3, 26 CrossRef.
- S. M. Swain, M. Shastry and E. Hamilton, Nat. Rev. Drug Discovery, 2023, 22, 101–126 CrossRef PubMed.
- Y. M. Ning, D. Suzman, V. E. Maher, L. Zhang, S. Tang, T. Ricks, T. Palmby, W. Fu, Q. Liu, K. B. Goldberg, G. Kim and R. Pazdur, Oncologist, 2017, 22, 743–749 CrossRef PubMed.
- I. Geffen, C. Fuhrer, B. Leitinger, M. Weiss, K. Huggel, G. Griffiths and M. Spiess, J. Biol. Chem., 1993, 268, 20772–20777 CrossRef CAS.
- J. Gruenberg, Nat. Rev. Mol. Cell Biol., 2001, 2, 721–730 CrossRef CAS.
- J. Z. Rappoport, S. M. Simon and A. Benmerah, Traffic, 2004, 5, 327–337 CrossRef CAS PubMed.
- L. M. Traub, J. Cell Biol., 2003, 163, 203–208 CrossRef CAS PubMed.
- A. Pompa, F. De Marchis, M. T. Pallotta, Y. Benitez-Alfonso, A. Jones, K. Schipper, K. Moreau, V. Žárský, G. P. Di Sansebastiano and M. Bellucci, Int. J. Mol. Sci., 2017, 18, 703 CrossRef PubMed.
- S. Misra, R. Puertollano, Y. Kato, J. S. Bonifacino and J. H. Hurley, Nature, 2002, 415, 933–937 CrossRef CAS.
- T. Shiba, H. Takatsu, T. Nogi, N. Matsugaki, M. Kawasaki, N. Igarashi, M. Suzuki, R. Kato, T. Earnest, K. Nakayama and S. Wakatsuki, Nature, 2002, 415, 937–941 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05093b |
| This journal is © The Royal Society of Chemistry 2024 |