
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ construction of a static-dynamic hybrid interface toward stable Zn anodes for aqueous Zn-ion batteries†
Baohua
Liu
a,
Luyan
Yu
a,
Qinghua
Xiao
a,
Shilin
Zhang
 *b,
Guanjie
Li
b,
Kaixin
Ren
a,
Yuxuan
Zhu
a,
Chao
Wang
*a and
Qinghong
Wang
*a
*b,
Guanjie
Li
b,
Kaixin
Ren
a,
Yuxuan
Zhu
a,
Chao
Wang
*a and
Qinghong
Wang
*a
aSchool of Chemistry and Materials Science, Jiangsu Normal University, Xuzhou, Jiangsu 221116, P. R. China. E-mail: wangc@jsnu.edu.cn; wangqh@jsnu.edu.cn
bSchool of Chemical Engineering, Faculty of Sciences, Engineering and Technology, The University of Adelaide, Adelaide, SA 5005, Australia. E-mail: shilin.zhang01@adelaide.edu.au
First published on 6th September 2024
Abstract
Aqueous Zn-ion batteries are promising candidates for next-generation energy storage devices due to the advantages of high safety, low cost and good environmental friendliness. However, the uncontrollable dendrite growth and undesirable side reactions occurring on the Zn anode result in poor cycling stability. Herein, a Lewis base, triethanolamine, is used as the electrolyte additive to construct a hybrid solid-electrolyte interphase layer composed of a static ZnSO4·3Zn(OH)2·4H2O layer and dynamic quaternary ammonium ion adsorption layer. The static SEI layer acts as a physical barrier between the Zn anode and electrolyte, thus effectively suppressing chemical corrosion and the hydrogen evolution reaction. The dynamic layer can not only regulate the ion flux at the interface, but also promote the de-solvation of solvated Zn2+, thus leading to homogenous Zn deposition along the (002) electro-crystallization orientation. As a result, the Zn anode demonstrates an extended cycle life of 2500 h at a current density of 1.0 mA cm−2, with an areal capacity of 1.0 mA h cm−2 and a high coulombic efficiency (CE) of 98.94%. The Zn‖V2O5 cells exhibit a specific capacity of 178.4 mA h g−1 after 500 cycles, indicating both high capacity and robust cycling stability, which are essential for practical applications.
Introduction
The development of large-scale energy storage technology is a significant part of the exploitation and utilization of new clean energy sources. Among various energy storage devices, rechargeable aqueous Zn-ion batteries (ZIBs) are regarded as one of the most promising candidates for large-scale energy storage systems due to their high safety, non-toxicity, and low cost.1,2 Moreover, the metal Zn anode exhibits a high theoretical capacity of 820 mA h g−1 and low electrochemical potential of −0.76 V (vs. SHE), providing ZIBs with high energy density.3,4 In recent years, much effort has been devoted to the exploration of high-performance ZIBs. But their commercialization is still a big challenge, which is largely due to the poor electrochemical performance of Zn anodes. As shown in Fig. 1a, a series of water-parasitic side reactions, such as chemical corrosion and the hydrogen evolution reaction (HER), occur on the surface of the Zn anode, which will cause the passivation of the electrode and consumption of electrolyte, thus resulting in sluggish redox kinetics and quick capacity fading.5,6 Moreover, uncontrolled Zn dendrite growth caused by the inhomogeneous deposition may pierce the separator and lead to a short circuit or even explosion of the cells. 7,8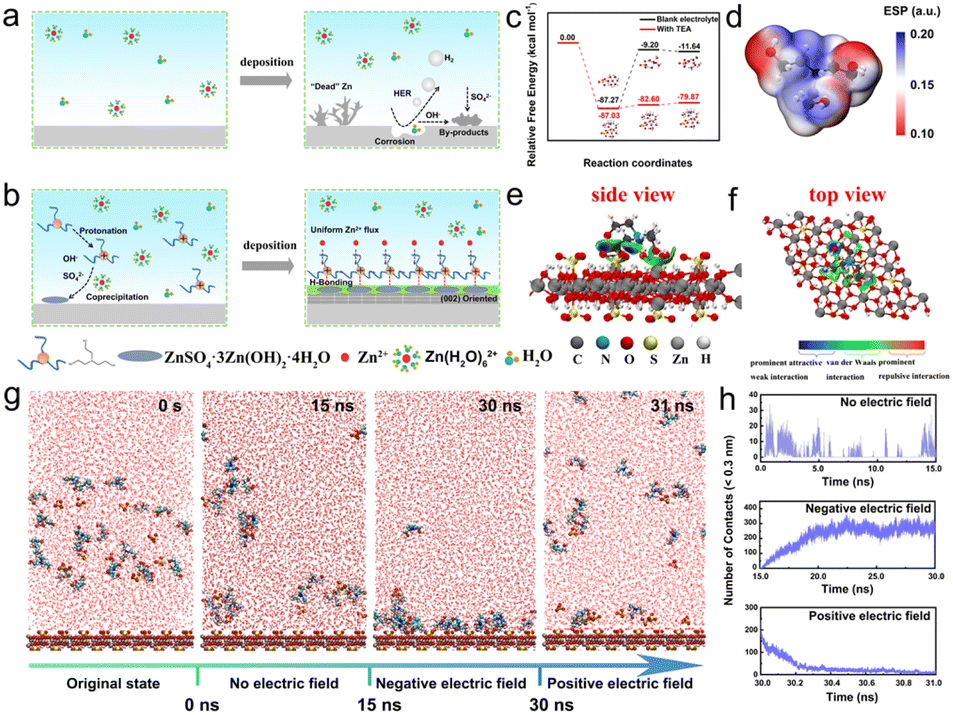 | ||
| Fig. 1 (a) The schematic illustration of the Zn electrodeposition process without a TEA additive. (b) Schematic representation of the formation of an organic–inorganic SEI and its protection effect on the zinc anode. (c) The free energy barrier of the hydrolysis process with or without the TEA additive. (d) Electrostatic potential mapping of the TEAH+ ion. (e and f) The IGMH analysis of the interactions between TEAH+ and the inorganic SEI. (g) MD simulations of the ZHS inorganic layer and electrolyte containing TEAH+ with and without an electric field. (h) The dynamic changes in number of points of contact between the inorganic ZHS layer and organic TEAH+ with and without an electric field. | ||
To solve the above issues, many strategies have been proposed, including controllable synthesis,9,10 artificial surface coatings,11,12 electrolyte engineering,13,14 and advanced separators.15,16 The feasibility of these strategies lies in improving the stability of the solid/electrolyte interface (SEI). Notably, ex situ artificial layers, such as CaCO317 and TiO2,18 possess good ion conductivity and superior anti-corrosion performance due to the structural and chemical stability in aqueous electrolyte. However, they tend to peel off from the Zn anode during the reversible charge/discharge process. Alternatively, employing electrolyte additives, such as small organic molecules,19 inorganic ions and polymers,20,21 leads to high efficiency in regulating the solvation structure and interfacial transportation of Zn2+ for dendrite-free Zn deposition. But it is hard to achieve a uniform and dense SEI to separate the Zn anode from high activity water molecules. Therefore, constructing a stable and multifunctional SEI film that can provide long-term protection for Zn anodes is of great importance.22,23
Herein, a unique hybrid SEI layer composed of a static ZnSO4·3Zn(OH)2·4H2O (ZHS) sedimentary layer and dynamic organic TEAH+ adsorption layer is in situ constructed to achieve a stable Zn anode using a Lewis base, triethanolamine (TEA), as the electrolyte additive (Fig. 1b). The static ZHS layer could effectively hinder the direct contact of water molecules with the Zn anode and inhibit the chemical corrosion and the HER. The dynamic TEAH+ layer driven by the induction of an electric field could regulate the interfacial ion flux and accelerate the desolvation process of hydrated Zn2+, thus leading to dendrite-free Zn deposition. As a result, both the Zn‖Zn symmetric cells and Zn‖V2O5 full cells employing TEA-based electrolyte deliver significantly enhanced electrochemical performance.
Results and discussion
Formation and characterization of the static-dynamic hybrid interface layer
We use TEA molecules, a Lewis base, to construct a static-dynamic hybrid SEI layer on the surface of a Zn anode based on the high protonation ability of amino groups and the complexation properties of hydroxyl groups (–OH) with Zn2+. Initially, density functional theory (DFT) calculations were conducted to elucidate the formation mechanism of the hybrid SEI layer. As illustrated in Fig. 1c, the free energy barrier for the hydrolysis of Zn(H2O)62+ in the TEA-containing electrolyte is calculated to be −79.87 kcal mol−1, which is significantly lower than that in the blank ZnSO4 (ZSO) electrolyte (−11.64 kcal mol−1), indicating the enhanced tendency of the hydrolytic process induced by the protonation of TEA. As a result, ZnSO4·3Zn(OH)2·4H2O (ZHS, JCPDS # 09–0204) is generated, which can be confirmed by using the X-ray diffraction (XRD) pattern of the precipitate obtained from the TEA-based electrolyte (Fig. S2†). The hydrolysis of Zn2+ and protonation of TEA slightly increase the pH of the electrolyte (Fig. S1a and b†), which is helpful for reducing the chemical corrosion of the acidic electrolyte. The pH value of the electrolyte did not obviously change after a certain standing time, indicating that the stability of the electrolyte is good (Fig. S1c and d†). Fig. 1d shows a van der Waals' surface electrostatic potential diagram of TEAH+. As can be seen, the positive charge mainly concentrates in the nitrogen atom (blue atom), which may be beneficial to bind with the generated ZHS. To understand the interaction between TEAH+ and ZHS, an independent gradient model based on Hirshfeld partition (IGMH) analysis was used (Fig. 1e and f). Distinct blue-colored isosurfaces appear between hydrogen in TEAH+ and oxygen in sulfate of ZHS, indicating the formation of hydrogen bonds at the ZHS/TEAH+ interface. The snapshots obtained by molecular dynamics (MD) simulations shown in Fig. 1g reveal that TEAH+ ions tend to aggregate into clusters and randomly disperse across the surface of ZHS, demonstrating the formation of the ZHS/TEAH+ hybrid layer induced by H-bonding. Considering the positively charged nature of TEAH+, the interaction between ZHS and TEAH+ under an applied electric field during the plating process and stripping process was further simulated. The snapshot obtained after applying a negative electric field for 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ps displays a uniform distribution of TEAH+ on the surface of the ZHS layer, indicating that TEAH+ could be absorbed on ZHS to form an inorganic–organic SEI layer during the Zn deposition process. Interestingly, after applying a positive electric field for only 1000 ps, the TEAH+ ions migrate off the surface of the ZHS layer, indicating the disappearance of the organic adsorption layer during the stripping process. The dynamic adsorption and desorption process of TEAH+ ions on the surface of ZHS is further confirmed by the changes in contact frequency (Fig. 1h) and minimum contact distance (Fig. S3†) in different electric field environments. This observation suggests that a unique hybrid SEI layer composed of static ZHS and dynamic TEAH+ could be formed during the plating and stripping process.
000 ps displays a uniform distribution of TEAH+ on the surface of the ZHS layer, indicating that TEAH+ could be absorbed on ZHS to form an inorganic–organic SEI layer during the Zn deposition process. Interestingly, after applying a positive electric field for only 1000 ps, the TEAH+ ions migrate off the surface of the ZHS layer, indicating the disappearance of the organic adsorption layer during the stripping process. The dynamic adsorption and desorption process of TEAH+ ions on the surface of ZHS is further confirmed by the changes in contact frequency (Fig. 1h) and minimum contact distance (Fig. S3†) in different electric field environments. This observation suggests that a unique hybrid SEI layer composed of static ZHS and dynamic TEAH+ could be formed during the plating and stripping process.
The formation of the hybrid SEI layer was experimentally confirmed by XRD, FTIR and XPS measurements. As shown in Fig. S4,† distinct diffraction peaks of ZHS are detected on the surface of the Zn anode after 3 cycles in TEA added electrolyte. Notably, ZHS possesses the same crystal structure as the hydrolysis product (JCPDS # 09–0294), rather than that produced by chemical corrosion (JCPDS # 39–0689) in blank electrolyte, indicating that an inorganic ZHS SEI layer is in situ formed on the Zn anode in the presence of the TEA additive. The XRD measurements demonstrate that the in situ ZHS inorganic SEI could separate the Zn anode from the electrolyte and suppress chemical corrosion. In the FTIR spectrum of the Zn anode (Fig. S5†) after the third cycle in the TEA-base electrolyte, typical characteristic peaks of O–H (3355 cm−1), C–O (1074 cm−1), S–O (1113 cm−1), and –CH2 (2921 cm−1, 2851 cm−1) are observed,24,25 which confirm the adsorption of TEAH+ on the surface of the electrode. To further characterize the chemical composition of the hybrid SEI layer, XPS measurement was conducted on the Zn anode after the third stripping/plating cycle. The survey spectra (Fig. S6†) of the SEI layer show signals corresponding to Zn, S, O, N, and C elements, indicating the existence of TEAH+ and ZHS in the SEI layer. The O 1s spectrum (Fig. 2a) displays peaks for Zn–O (530.45 eV), C–O/O–H (531.70 eV), and S–O (532.50 eV) bonds;11,26,27 the C–C (284.70 eV), C–O (285.72 eV), C–N (288.60 eV), and C–H (283.97 eV) bonds in the C 1s spectrum (Fig. 2b), 8–30 and the N–C (398.57 eV) and N–H (400.38 eV) bonds in the N 1s spectrum (Fig. 2c) further support the successful construction of the ZHS/TEAH+ hybrid SEI layer. 24,31 Additionally, the Zn 2p spectrum shown in Fig. S7† exhibits Zn 2p3/2 (1021.3 eV) and Zn 2p1/2 (1044.5 eV) orbitals of Zn(II), confirming the existence of ZHS.27
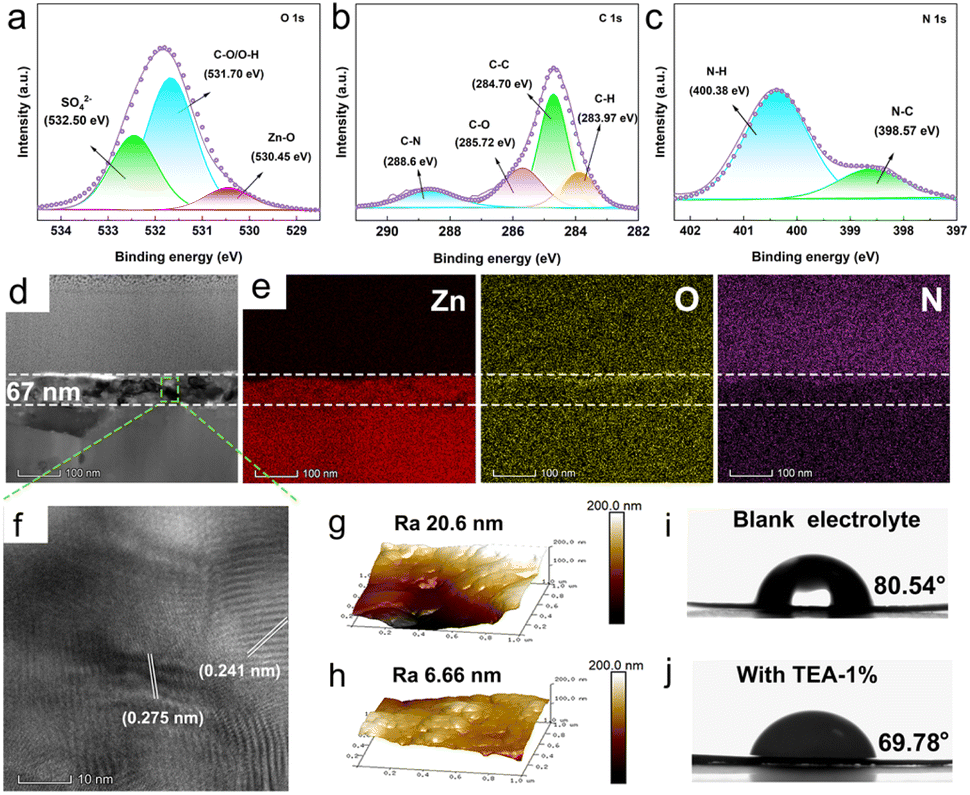 | ||
| Fig. 2 High resolution spectra of (a) O 1s, (b) C 1s and (c) N 1s after the 3rd stripping/plating process in the electrolyte containing TEA. (d) Cross-sectional focused ion beam (FIB)-SEM image (e) with EDS elemental mapping images and (f) TEM image of the Zn anode after the 3rd stripping/plating process with electrolyte containing TEA. AFM after 3 cycles in the (g) blank electrolyte and (h) with TEA electrolyte. The contact angle values of electrolyte on Zn anodes: (i) blank electrolyte and (j) TEA-based electrolyte. | ||
The focused ion beam (FIB)-microscopy technique and atomic force microscopy (AFM) were used to further characterize the hybrid SEI layer. A uniform and dense layer with a thickness of 67 nm was observed with uniform distribution of Zn, O, and N at the interface (Fig. 2d and e). High-resolution transmission electron microscope images reveal clear lattice fringes with spacings of 0.275 and 0.241 nm, corresponding to the planes of ZHS (JCPDS # 09–0294, Fig. 2f). This result is in accordance with the XRD results. AFM images show a smooth SEI layer on the Zn anode in TEA–1.0% electrolyte with a surface roughness of 6.7 nm when compared to the bare Zn surface roughness (20.6 nm) (Fig. 2g and h). Additionally, the bare Zn anode exhibits hydrophobic properties in aqueous electrolyte, with a contact angle as high as 80.54° (Fig. 2i). In contrast, the hybrid SEI layer-modified Zn anode shows a significantly reduced contact angle of 69.78° (Fig. 2j), which will be favorable for reducing the interfacial energy barriers and inducing homogeneous Zn plating.32,33
Effects of the static-dynamic hybrid interface layer on the Zn anode
The electron-rich oxygen in hydroxyl groups can also act as an electron donor and coordinating site when interacting with Zn2+ ions.34 As shown in Fig. 3a, the binding energy between Zn2+ and TEAH+ is calculated to be −1.77 eV, which is higher than that between Zn2+ and H2O (−1.25 eV), suggesting preferential coordination of TEAH+ with Zn2+. Therefore, the dynamic TEAH+ layer at the interface will be favorable for homogenizing the distribution of Zn2+ at the interface and facilitating the desolvation process of solvated Zn2+. As shown in Fig. 3b, the Zn(H2O)62+ group in blank ZSO electrolyte needs high desolvation energies to remove water molecules, which significantly decreases in the presence of the TEA electrolyte additive. Experimentally, the activation energy (Ea) for the desolvation process of solvated Zn2+ is determined from temperature-dependent electrochemical impedance spectrum (EIS) measurements (Fig. 3c and S8†). The Ea for the desolvation process in TEA–1.0% electrolyte is 40.7 kJ mol−1, which is lower than that in pure ZSO electrolyte (50.1 kJ mol−1), indicating enhanced Zn2+ transfer kinetics due to the existence of an inorganic–organic SEI layer. Conversely, during the dissolution process, the rapid migration of TEAH+ coupled with Zn2+ is expected to reduce interfacial ion aggregation and lead to uniform stripping, which will provide a flat configuration for the subsequent deposition process (Fig. S9†). The Zn2+ transference number increases from 0.4 in blank electrolyte to 0.57 in TEA–1.0% electrolyte (Fig. 3d and S10†).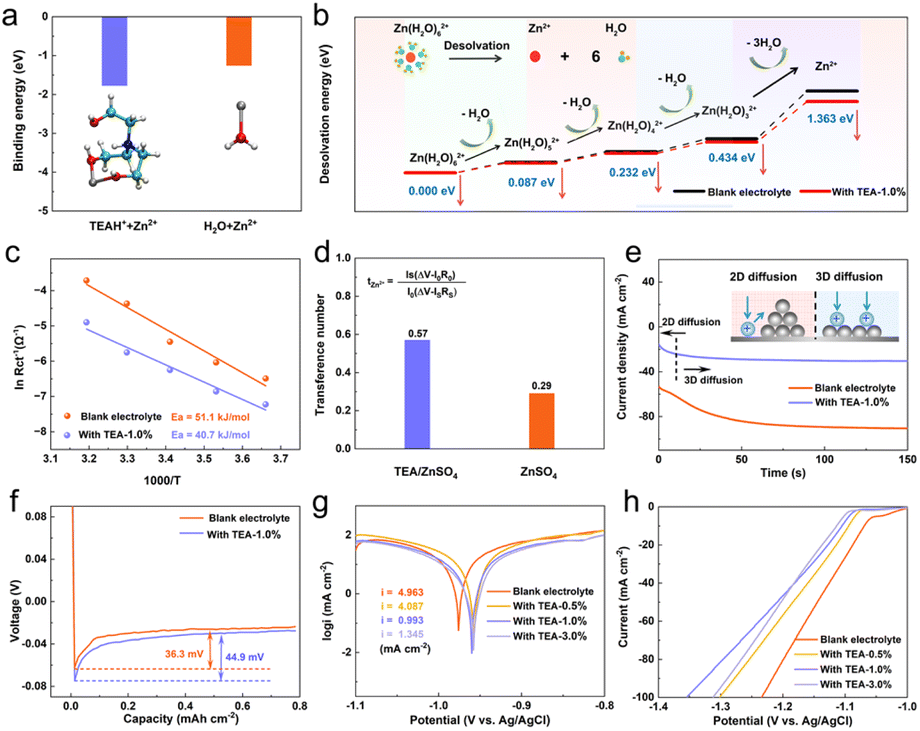 | ||
| Fig. 3 (a) Binding energy of Zn2+ with TEAH+ and H2O, respectively. (b) Desolvation energy values for the removal of H2O molecules on the Zn(H2O)62+ group with or without the TEA additive. (c) The activation energy of Zn2+ diffusion at the interface between the Zn plate and the SEI layer calculated by using the Arrhenius equation. (d) Transference number of Zn2+. (e) Time–current curves of zinc electrodes in different electrolytes. (f) Voltage profiles during Zn2+ ion plating at 1 mA cm−2. (g) Tafel polarization curves in different electrolytes. (h) Linear sweep voltammetry showing the HER potential of Zn electrodes in different electrolytes. | ||
To further understand the impact of this hybrid SEI layer on Zn deposition behavior, chronoamperometry (CA) at a fixed voltage of −150 mV (Fig. 3e) was used. In pure ZSO electrolyte, the rapid current density increase lasts for 50 s, indicating a boost of surface area caused by random nucleation and uneven Zn deposition (in the 2D model). In TEA–1.0% electrolyte, a steady current density is achieved after only 10 s, suggesting an instant nucleation mode promoting dense Zn growth without dendrite formation (in the 3D model). 35,36 The Zn nucleation overpotential in TEA–1.0% electrolyte (44.9 mV) exceeds that in pure ZSO electrolyte (36.3 mV, Fig. 3f). Generally, the larger nucleation overpotential could provide a sufficient driving force for nucleation and help to increase the nucleation rate, thus refining the grain size in the nucleation process and further inducing more uniform Zn deposition.37,38 To confirm the Zn deposition behavior, SEM characterization was conducted on the surface evolution of the Zn anode during plating. As shown in Fig. S11,† Zn dendrites emerge on the Zn anode surface only after 10 minutes of plating in pure ZSO electrolyte, gradually evolving into large Zn plates due to the well-known “tip” effect. In the TEA–1.0% electrolyte, a dense and flat surface was observed throughout the deposition process, indicating effective inhibition of Zn dendrite growth by the hybrid SEI layer.
Chemical corrosion significantly impacts the cycling stability of Zn anodes in aqueous electrolytes, which results in the brittle and loose Zn4SO4(OH)6·xH2O byproduct mainly formed due to the HER, followed by a local pH increase, leading to Zn surface passivation and reduced coulombic efficiency.39–44 The anti-corrosion behavior of Zn anodes in TEA-based electrolytes with varying concentrations was studied using Tafel polarization curve measurements (Fig. 3g). Zn anodes exhibit higher corrosion voltages in TEA–1.0% electrolyte compared to pure ZSO electrolyte, indicating reduced corrosion due to the protection of the inorganic–organic SEI layer. Corrosion current densities of 4.087, 0.993, and 1.345 mA cm−2 were determined for Zn foil in the electrolytes containing 0.5%, 1.0%, and 1.5% TEA, respectively, lower than that in pure ZSO electrolyte (4.963 mA cm−2), suggesting reduced corrosion rates in the presence of the TEA additive. Accordingly, the corrosion inhibition efficiency was calculated to be 77.23% for Zn anodes in TEA-based electrolytes, demonstrating that the in situ formed ZHS/TEAH+ hybrid SEI layer significantly enhances the anti-corrosion resistance of the Zn anode. Furthermore, the Zn anode exhibits a slight increase in charge transfer resistance (Rct) after soaking in TEA–1.0% electrolyte for 7 days. In contrast, the Zn anode soaked in pure ZSO electrolyte for 7 days shows a dramatic increase in Rct by two orders of magnitude, indicating severe chemical corrosion caused by active water and the formation of inert byproducts on the Zn anode surface (Fig. S12†). SEM images and XRD patterns of the immersed Zn anode shown in Fig. S13† further confirm the reduced chemical corrosion in TEA-based electrolyte, demonstrating that the SEI layer acts as a physical barrier protecting Zn metal from corrosion in aqueous electrolytes. The hydrogen evolution current densities in the electrolytes containing 0.5%, 1.0%, and 1.5% TEA are all lower than that in pure ZSO electrolyte, indicating that the organic–inorganic SEI layer inhibits the HER (Fig. 3h). Additionally, pH changes in the electrolytes with and without the TEA additive during the deposition process at a current density of 1 mA cm−2 are shown in Fig. S14.† The pH values of the ZSO electrolyte continue to increase due to the ongoing HER. In contrast, the pH values of the TEA–1.0% electrolyte remain stable, further demonstrating that the HER is inhibited by the TEA additive.
Cycling stability of the Zn anode in the TEA-based electrolyte
The electrochemical performance of Zn anodes in TEA–1.0% and pure ZSO electrolytes is investigated by evaluating symmetric cells with these different electrolytes. The cells using TEA–1.0% electrolyte exhibit a prolonged cycle lifetime of 2500 h with a stable polarization voltage at a current density of 1.0 mA cm−2 with an areal capacity of 1.0 mA h cm−2 (Fig. 4a and b). In contrast, the cell using pure ZSO electrolyte exhibits significant voltage fluctuations after only 100 h and suffers from a short circuit after 180 h. Notably, with a low concentration of TEA in the electrolyte, the symmetric cells maintain stability for only 1280 h, likely because the as-formed SEI is not dense enough to inhibit chemical corrosion and dendrite growth over a long time period (Fig. S15†). Additionally, the symmetric cells using TEA–1.0% electrolyte exhibit a long cycle life of 600 h at a current density of 4 mA cm−2 with an areal capacity of 4 mA h cm−2, about 8 times longer than that in pure ZSO electrolyte (Fig. 4c). The cycling performance is also superior to that of the cells with a single ZHS layer or single TEAH+ SEI layer (Fig. S16†). Moreover, the Zn anode in TEA-based electrolyte exhibits stable polarization curves even at a high current density of 10 mA cm−2 (Fig. S17†). Importantly, at a current density of 1.0 mA cm−2 with an areal capacity of 1.0 mA h cm−2, the Zn//Cu cell in TEA–1.0% electrolyte displays an impressive coulombic efficiency (CE) of 98.94% after 380 cycles, while the Zn‖Cu cell using ZSO electrolyte shows significant CE fluctuations after only 54 cycles (Fig. 4d). The electrochemical performance of Zn anodes in different electrolytes is further investigated for high Zn utilization. At a depth of discharge (DOD) of 50% (Zn foil thickness: 50 μm), the Zn‖Zn cells using TEA–1.0% electrolyte exhibit a lifespan of 250 h at a current density of 15 mA cm−2 for a fixed capacity of 15 mA h cm−2 (Fig. 4e). The superior cycling stability of the Zn anode in our TEA-based electrolyte compared to that in previous studies (Table S1†) can be attributed to the in situ formed hybrid SEI layer with enhanced mechanical strength, boosted ionic transference number, and adequate ionic conductivity.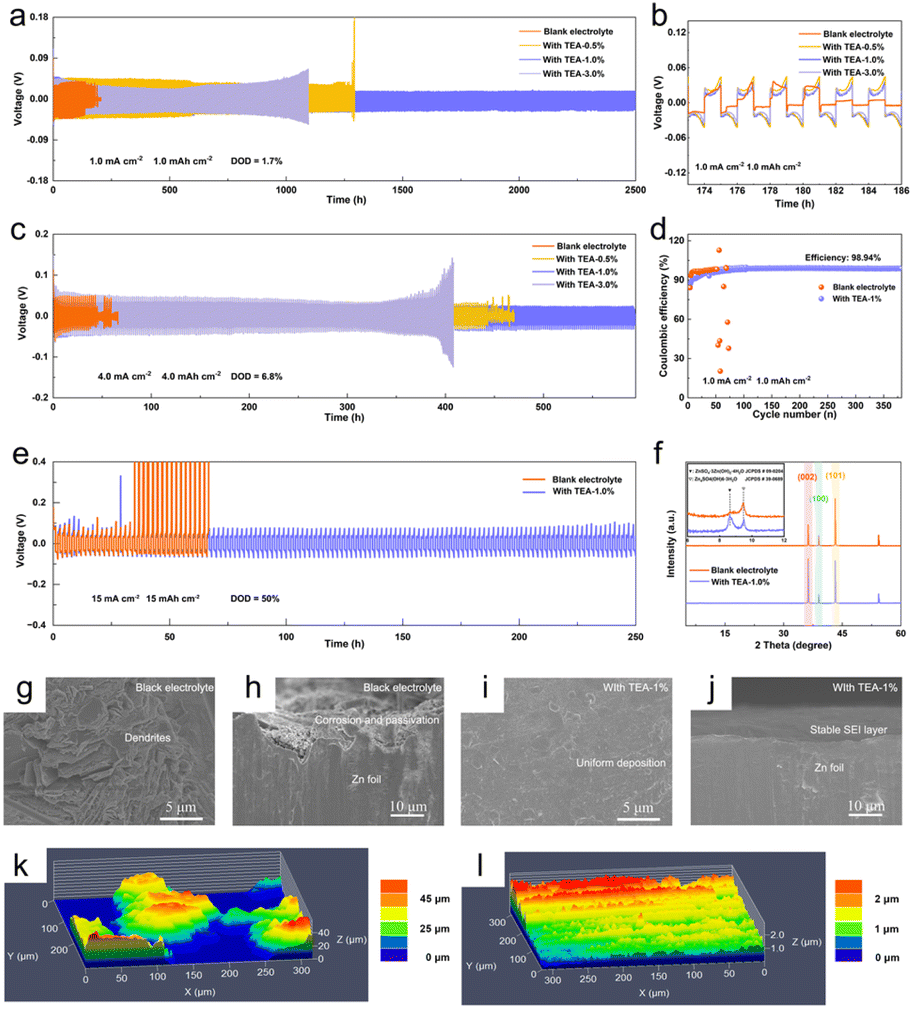 | ||
| Fig. 4 (a) Cycling performance and (b) the enlarged charge–discharge curves of symmetric Zn‖Zn cells at 1.0 mA cm−2 and 1.0 mA h cm−2. (c) Cycling performance of the symmetric cells at 4.0 mA cm−2 and 4.0 mA h cm−2. (d) Coulombic efficiencies of asymmetric Zn‖Cu cells at 1.0 mA cm−2. (e) Cycling performances of symmetric Zn‖Zn cells at a high DOD of 50%. (f) XRD patterns and (g–j) SEM images of the Zn anode after 50 cycles: (g and h) in blank electrolyte and (i and j) in TEA based electrolyte. LCSM images of the Zn anode in (k) blank electrolyte and (l) TEA-based electrolyte after plating for 10 h at a current density of 1.0 mA cm−2. | ||
To assess the long-term protective efficacy of the in situ formed ZHS/TEAH+ hybrid SEI layer, the morphology and composition of the Zn anodes after 50 cycles were analyzed using SEM and XRD. The SEM images reveal that, after cycling in the ZSO electrolyte, the Zn anode surface exhibits numerous irregular aggregates. In contrast, the Zn anode cycled in the TEA–1.0% electrolyte displays a relatively smooth and homogeneous surface, as observed from both top-view and side-view SEM images (Fig. 4g–j). These results support that the hybrid SEI layer can suppress side reactions and inhibit dendrite growth during the long-term charge–discharge process. Furthermore, the EDS mapping shows a uniform distribution of C, O, and N elements, corroborating the sustained influence of TEA on the Zn anode (Fig. S18†). The XRD pattern (Fig. 4f) of the Zn anode cycled in pure ZSO electrolyte shows clear diffraction peaks of the ZHS by-product (JCPDS # 39–0689), while these peaks are less obvious for the Zn anode cycled in TEA–1.0% electrolyte, indicating effective inhibition of side reactions by the hybrid SEI layer. Furthermore, the diffraction peaks of ZHS (JCPDS # 09–0294) are still detected even after 50 cycles, suggesting that the SEI layer exhibits good structural stability and can provide long-term protection for the Zn anode, in good agreement with the EDS results. Additionally, the intensity ratios of the diffraction peaks, I(002)/I(100) and I(002)/I(101), can be utilized to assess the preferred Zn deposition orientation. Compared to the values of 2.02 for I(002)/I(100) and 0.44 for I(002)/I(101) for the Zn anode cycled in pure ZSO electrolyte, these values increased to 4.09 and 1.03 for the Zn anode cycled in TEA–1.0% electrolyte, respectively. This indicates that the in situ constructed hybrid SEI layer also possesses the function of inducing the preferred (002) electro-crystallization orientation, thus facilitating dendrite-free Zn deposition. The laser confocal scanning microscopy (LCSM) results (Fig. 4k and l) show that the surface of the Zn anode cycled in TEA–1.0% electrolyte exhibits uniform Zn deposition at a current density of 1.0 mA cm−2 with a capacity of 10 mA h cm−2. In contrast, the Zn anode cycled in pure ZSO electrolyte exhibits roughness with accumulated protrusions.
Electrochemical performance of the full cells using the TEA-based electrolyte
To assess the practical applicability of the TEA additive, Zn‖V2O5 full batteries were assembled to study electrochemical properties. CV curves of the full cells using both pure ZSO and TEA–1.0% electrolyte exhibit similar redox peaks in the voltage range of 0.2 to 1.6 V at a scan rate of 0.05 mV s−1 (Fig. 5a). Two pairs of charge–discharge plateaus are observed on the galvanostatic charge–discharge curves (Fig. 5b), in good agreement with the CV curves. Positive shifts of anodic peaks and negative shifts of cathodic peaks of the cell using the TEA–1.0% electrolyte imply enhanced electrochemical kinetics. Rate performance (Fig. 5c) examined at the current densities from 0.2 A g−1 to, 0.5, 1.0 and 2.0 A g−1 shows that the Zn‖V2O5 cells using the TEA–1.0% electrolyte deliver high average specific capacities of 284.1, 236.4, 209.3, and 178.0 mA h g−1, respectively, which are higher than those obtained using the pure ZSO electrolyte, indicating significantly promoted rate performance using the TEA–1.0% electrolyte. The long-term cycling performance of the cells was measured at a current density of 2.0 A g−1. As shown in Fig. 5d, the Zn‖V2O5 cells using the TEA–1.0% electrolyte exhibit better reversible cycling stability than those using pure ZSO electrolyte. The specific capacity is maintained 178.4 mA h g−1 after 500 cycles with an average CE value of nearly 100%. These results prove that the in situ formed inorganic–organic hybrid SEI layer ensures high stability and utilization of the Zn anode by inhibiting dendrite growth and suppressing side reactions during the long-term charge/discharge process, indicating the practical potential of the TEA electrolyte additive.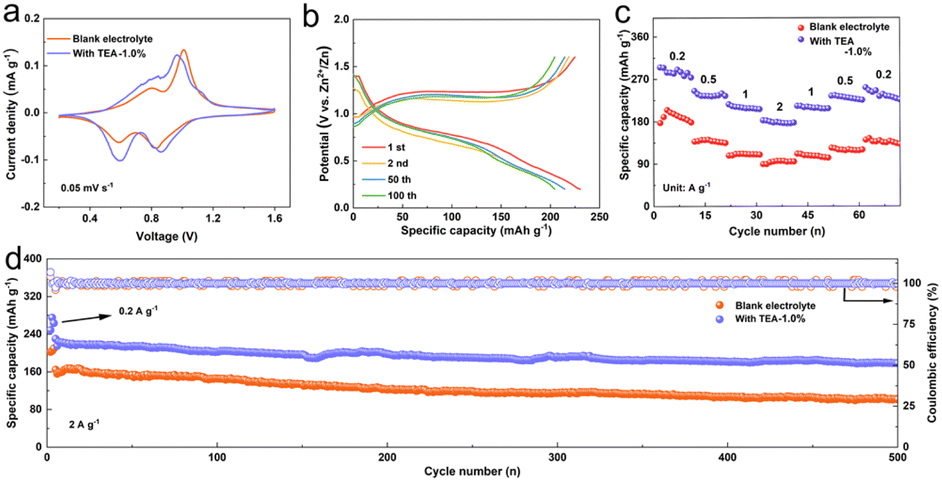 | ||
| Fig. 5 (a) CV curves, (b) the voltage profiles, (c) rate capability and (d) long-term cycling performance of the Zn‖V2O5 batteries in the electrolytes with/without 1.0%-TEA additive. | ||
Conclusions
In summary, a Lewis base, TEA is used as an electrolyte additive to in situ construct a multifunctional ZHS/TEAH+ inorganic–organic hybrid SEI layer on a Zn anode for high-performance ZIBs. MD simulations reveal that TEAH+ could be absorbed on ZHS via strong H-bonds to form a uniform and dense adsorption layer with an electric field during the deposition process. The favorable H-bonding interaction between ZHS and TEAH+ is also verified by DFT calculations. Such a hybrid SEI could not only suppress side reactions on the surface of the Zn anode, but also inhibit dendrite growth by regulating the interfacial ion flux and inducing the preferred (002) electro-crystallization orientation. Therefore, the Zn anode in the electrolyte with the TEA additive demonstrates an improved cycling stability of 2500 h at 1.0 mA cm−2, as well as long cycle life at a DOD of 50%. The Zn‖V2O5 cells also display high specific capacity, long cycle life and good rate performance, indicating the practicality of the TEA additive for high-performance ZIBs. This work provides a facile approach for the design of hybrid SEI layers in aqueous batteries.Data availability
We state that the data supporting this article have been included as part of the ESI.†Author contributions
Q. W. and S. Z. conceived the idea of this study. B. L., L. Y., Q. X., K. R. and G. L. performed the experiments. Q. W., S. Z., and C. W. supervised the conduct of this study. B. L. and Y. Z. provided the first draft of the paper that was first corrected by S. Z., Q. W. and C. W. All authors critically reviewed and revised the manuscript draft and approved the final version for submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Jiangsu Province (no. BK20211352), National Natural Science Foundation of China (no. 22075115, 52472208), Joint Funds of the National Natural Science Foundation of China (no. U2141201), and Natural Science Foundation of Jiangsu Education Committee of China (no. 22KJA430005).Notes and references
- B. Tang, L. Shan, S. Liang and J. Zhou, Energy Environ. Sci., 2019, 12, 3288–3304 RSC.
- X. Zheng, T. Ahmad and W. Chen, Energy Storage Mater., 2021, 39, 365–394 CrossRef.
- C. Nie, G. Wang, D. Wang, M. Wang, X. Gao, Z. Bai, N. Wang, J. Yang, Z. Xing and S. Dou, Adv. Energy Mater., 2023, 13, 2300606 CrossRef CAS.
- Y. Zhu, G. Liang, X. Cui, X. Liu, H. Zhong, C. Zhi and Y. Yang, Energy Environ. Sci., 2024, 17, 369–385 RSC.
- J. Jiang, Z. Li, Z. Pan, S. Wang, Y. Chen, Q. Zhuang, Z. Ju and X. Zhang, Energy Environ. Mater., 2022, 6, e12410 CrossRef.
- J. Chen, W. Zhao, J. Jiang, X. Zhao, S. Zheng, Z. Pan and X. Yang, Energy Storage Mater., 2023, 59, 102767 CrossRef.
- J. Hao, X. Li, X. Zeng, D. Li, J. Mao and Z. Guo, Energy Environ. Sci., 2020, 13, 3917–3949 RSC.
- X. Yang, Z. Dong, G. Weng, Y. Su, J. Huang, H. Chai, Y. Zhang, K. Wu, J. B. Baek, J. Sun, D. Chao, H. Liu, S. Dou and C. Wu, Adv. Energy Mater., 2024, 14, 2401293 CrossRef CAS.
- P. Xue, C. Guo, L. Li, H. Li, D. Luo, L. Tan and Z. Chen, Adv. Mater., 2022, 34, 2110047 CrossRef CAS PubMed.
- S. Wang, Q. Ran, R. Yao, H. Shi, Z. Wen, M. Zhao, X. Lang and Q. Jiang, Nat. Commun., 2020, 11, 1634 CrossRef CAS PubMed.
- J. B. Park, C. Choi, J. H. Park, S. Yu and D. W. Kim, Adv. Energy Mater., 2022, 12, 2202937 CrossRef CAS.
- K. Liu, M. Sun, S. Yang, G. Gan, S. Bu, A. Zhu, D. Lin, T. Zhang, C. Luan, C. Zhi, P. Wang, B. Huang, G. Hong and W. Zhang, Adv. Energy Mater., 2024, 2401479 CrossRef.
- J. Hao, L. Yuan, Y. Zhu, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2022, 34, 2206963 CrossRef CAS PubMed.
- S. Chen, D. Ji, Q. Chen, J. Ma, S. Hou and J. Zhang, Nat. Commun., 2023, 14, 3526 CrossRef CAS PubMed.
- Y. Li, X. Peng, X. Li, H. Duan, S. Xie, L. Dong and F. Kang, Adv. Mater., 2023, 35, 2300019 CrossRef CAS PubMed.
- Y. Zong, H. He, Y. Wang, M. Wu, X. Ren, Z. Bai, N. Wang, X. Ning and S. X. Dou, Adv. Energy Mater., 2023, 13, 2300403 CrossRef CAS.
- L. Kang, M. Cui, F. Jiang, Y. Gao, H. Luo, J. Liu, W. Liang and C. Zhi, Adv. Energy Mater., 2018, 8, 1801090 CrossRef.
- Q. Zhang, J. Luan, X. Huang, Q. Wang, D. Sun, Y. Tang, X. Ji and H. Wang, Nat. Commun., 2020, 11, 3961 CrossRef CAS PubMed.
- K. Qiu, G. Ma, Y. Wang, M. Liu, M. Zhang, X. Li, X. Qu, W. Yuan, X. Nie and N. Zhang, Adv. Funct. Mater., 2024, 34, 2313358 CrossRef CAS.
- Z. Hu, F. Zhang, Y. Zhao, H. Wang, Y. Huang, F. Wu, R. Chen and L. Li, Adv. Mater., 2022, 34, 2203104 CrossRef CAS PubMed.
- C. Huang, J. Mao, S. Li, W. Zhang, X. Wang, Z. Shen, S. Zhang, J. Guo, Y. Xu, Y. Lu and J. Lu, Adv. Funct. Mater., 2024, 34, 2315855 CrossRef CAS.
- M. Liu, W. Yuan, G. Ma, K. Qiu, X. Nie, Y. Liu, S. Shen and N. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304444 CrossRef CAS PubMed.
- S. Liu, J. Mao, W. K. Pang, J. Vongsvivut, X. Zeng, L. Thomsen, Y. Wang, J. Liu, D. Li and Z. Guo, Adv. Funct. Mater., 2021, 31, 2104281 CrossRef CAS.
- H. Tian, J. L. Yang, Y. Deng, W. Tang, R. Liu, C. Xu, P. Han and H. J. Fan, Adv. Energy Mater., 2022, 13, 2202603 CrossRef.
- J. Dong, L. Su, H. Peng, D. Wang, H. Zong, G. Wang and J. Yang, Angew. Chem., Int. Ed., 2024, 63, e202401441 CrossRef CAS PubMed.
- C. Li, R. Kingsbury, A. S. Thind, A. Shyamsunder, T. T. Fister, R. F. Klie, K. A. Persson and L. F. Nazar, Nat. Commun., 2023, 14, 3067 CrossRef CAS PubMed.
- T. Zhou, Y. Mu, L. Chen, D. Li, W. Liu, C. Yang, S. Zhang, Q. Wang, P. Jiang, G. Ge and H. Zhou, Energy Storage Mater., 2022, 45, 777–785 CrossRef.
- Y. Liu, X. Tao, Y. Wang, C. Jiang, C. Ma, O. Sheng, G. Lu and X. W. Lou, Science, 2022, 375, 739–745 CrossRef CAS PubMed.
- K. Ren, M. Li, Q. Wang, B. Liu, C. Sun, B. Yuan, C. Lai, L. Jiao and C. Wang, Nano-Micro Lett., 2024, 16, 117 CrossRef CAS PubMed.
- J. Xu, W. Lv, W. Yang, Y. Jin, Q. Jin, B. Sun, Z. Zhang, T. Wang, L. Zheng, X. Shi, B. Sun and G. Wang, ACS Nano, 2022, 16, 11392–11404 CrossRef CAS PubMed.
- H. Yu, Y. Chen, W. Wei, X. Ji and L. Chen, ACS Nano, 2022, 16, 9736–9747 CrossRef CAS PubMed.
- S. H. Park, S. Y. Byeon, J.-H. Park and C. Kim, ACS Energy Lett., 2021, 6, 3078–3085 CrossRef CAS.
- J. Hao, X. Li, S. Zhang, F. Yang, X. Zeng, S. Zhang, G. Bo, C. Wang and Z. Guo, Adv. Funct. Mater., 2020, 30, 2001263 CrossRef CAS.
- M. C. Han, J. H. Zhang, C. Y. Yu, J. C. Yu, Y. X. Wang, Z. G. Jiang, M. Yao, G. Xie, Z. Z. Yu and J. Qu, Angew. Chem., Int. Ed., 2024, 63, e202403695 CrossRef CAS PubMed.
- Z. Liu, G. Li, M. Xi, Y. Huang, H. Li, H. Jin, J. Ding, S. Zhang, C. Zhang and Z. Guo, Angew. Chem., Int. Ed., 2024, 63, e202319091 CrossRef CAS PubMed.
- G. Li, Z. Zhao, S. Zhang, L. Sun, M. Li, J. A. Yuwono, J. Mao, J. Hao, J. Vongsvivut, L. Xing, C.-X. Zhao and Z. Guo, Nat. Commun., 2023, 14, 6526 CrossRef CAS PubMed.
- A. Pei, G. Zheng, F. Shi, Y. Li and Y. Cui, Nano Lett., 2017, 17, 1132–1139 CrossRef CAS PubMed.
- L. Zhang, J. Huang, H. Guo, L. Ge, Z. Tian, M. Zhang, J. Wang, G. He, T. Liu, J. Hofkens, D. J. L. Brett and F. Lai, Adv. Energy Mater., 2023, 13, 2203790 CrossRef CAS.
- H. Yan, S. Li, J. Zhong and B. Li, Nano-Micro Lett., 2023, 16, 15 CrossRef PubMed.
- W. Zhang, Y. Dai, R. Chen, Z. Xu, J. Li, W. Zong, H. Li, Z. Li, Z. Zhang, J. Zhu, F. Guo, X. Gao, Z. Du, J. Chen, T. Wang, G. He and I. P. Parkin, Angew. Chem., Int. Ed., 2022, 62, e202212695 CrossRef PubMed.
- R. Wang, Q. Ma, L. Zhang, Z. Liu, J. Wan, J. Mao, H. Li, S. Zhang, J. Hao, L. Zhang and C. Zhang, Adv. Energy Mater., 2023, 13, 2302543 CrossRef CAS.
- Y. Liu, F. Li, J. Hao, H. Li, S. Zhang, J. Mao, T. Zhou, R. Wang, L. Zhang and C. Zhang, Adv. Funct. Mater., 2024, 34, 2400517 CrossRef CAS.
- Z. Liu, R. Wang, Y. Gao, S. Zhang, J. Wan, J. Mao, L. Zhang, H. Li, J. Hao, G. Li, L. Zhang and C. Zhang, Adv. Funct. Mater., 2023, 33, 2308463 CrossRef CAS.
- R. Zhang, W. K. Pang, J. Vongsvivut, J. A. Yuwono, G. Li, Y. Lyu, Y. Fan, Y. Zhao, S. Zhang, J. Mao, Q. Cai, S. Liu and Z. Guo, Energy Environ. Sci., 2024, 17, 4569–4581 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05127k |
| This journal is © The Royal Society of Chemistry 2024 |