
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCation migration in layered oxide cathodes for sodium-ion batteries: fundamental failure mechanisms and practical modulation strategies
Zhuang-Chun
Jian†
 ab,
Jun-Xu
Guo†
ab,
Yi-Feng
Liu
b,
Yan-Fang
Zhu
*ab,
Jingqiang
Wang
*b and
Yao
Xiao
*ab
ab,
Jun-Xu
Guo†
ab,
Yi-Feng
Liu
b,
Yan-Fang
Zhu
*ab,
Jingqiang
Wang
*b and
Yao
Xiao
*ab
aCollege of Chemistry and Materials Engineering, Wenzhou University, Wenzhou 325035, P. R. China. E-mail: yanfangzhu@wzu.edu.cn; xiaoyao@wzu.edu.cn
bWenzhou Key Laboratory of Sodium-Ion Batteries, Wenzhou University Technology Innovation Institute for Carbon Neutralization, Wenzhou, 325035, P. R. China. E-mail: jqwangchi@hotmail.com
First published on 21st October 2024
Abstract
Sodium-ion batteries (SIBs) are regarded as competitive candidates for the next generation of electrochemical energy storage (EES) systems due to their low cost and abundant sodium resources. Layered oxide cathodes have attracted much interest owing to their simple preparation process, high specific capacity and environmental friendliness. However, undesired cation migration during electrochemical reactions can lead to irreversible phase transitions and structural degradation of layered oxide cathode materials, resulting in a sharp decrease in specific capacity and energy density. Therefore, in order to find effective strategies to suppress cation migration, the fundamental failure mechanism of layered oxides and the practical approaches to solve this key scientific issue are thoroughly investigated, and herein the history and current status of developments in this field are also reviewed. Elemental doping and structural design can directionally modify the electronic structure, energy band structure and electronic density of states in layered oxides and enhance cation migration barriers, which benefits the improvement of electrochemical performance and structural stability during the whole sodiation/desodiation process. The summary and prospects of inhibiting cation migration in layered oxides provide insights into the development of advanced cathode materials with high energy density and excellent structural stability for the commercialization of SIBs.
 Zhuang-Chun Jian | Zhuang-Chun Jian is currently a postgraduate student at College of Chemical and Materials Engineering, Wenzhou University. His research interest mainly focuses on the structures of novel functional materials and their relationship with properties and performances, especially on improving the performance of high energy density Na-based layered oxide cathode materials. |
 Jun-Xu Guo | Jun-Xu Guo is a postgraduate student at the College of Chemical and Materials Engineering, Wenzhou University. His main research focuses on advanced electrode materials for sodium-ion batteries, mainly focusing on the development of low-cost and high-energy density layered oxide cathode materials. |
 Yan-Fang Zhu | Yan-Fang Zhu works at the Institute of Carbon Neutralization, College of Chemistry and Materials Engineering, Wenzhou University. She received her PhD degree from the University of Wollongong, Australia. She is engaged in the research of cathode materials for sodium-ion batteries and related new energy storage devices, mainly focusing on the development and commercialization of layered oxide cathode materials. |
 Jingqiang Wang | Jingqiang Wang received his Ph.D. degree from the Department of Nano Convergence Engineering, Jeonbuk National University, Korea, in 2023. He is currently a postdoctoral researcher at the College of Chemistry and Chemical Engineering of Inner Mongolia University and the Carbon Neutralization Technology Innovation Research Institute of Wenzhou University, China. His research focuses on layered oxide cathode materials for the development of sodium-ion batteries for energy conversion and storage. |
 Yao Xiao | Yao Xiao is a professor and deputy dean at the Institute for Carbon Neutralization, College of Chemistry and Materials Engineering, Wenzhou University. He received his PhD (2019) from Sichuan University. He was a visiting scholar at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS), from 2016 to 2019. He was a postdoctoral fellow at the School of Materials Science and Engineering, Nanyang Technological University, Singapore. At present, he serves as managing editor for Carbon Neutralization (Wiley) and as an Outstanding Young Editorial Board Member of Carbon Energy (Wiley). His research focuses on layered oxide cathodes for sodium-ion batteries. |
1. Introduction
Global climate change, caused partly by the release of greenhouse gases, including carbon dioxide from the burning of fossil fuels, is having a significant impact on both human society and the natural environment.1–6 Therefore, reducing carbon emissions and accelerating the transition to low-carbon and zero-carbon energy sources have become urgent priorities. Secondary rechargeable battery systems, which are less environmentally constrained than other energy supply systems, are expected to be used commercially on a large scale.7–13 Among most secondary rechargeable batteries, lithium-ion batteries (LIBs) have found widespread applications in digital products, automobiles, and other application areas on account of their excellent cycling stability and high efficiency.14–27 However, as the demand for LIBs is increasing, the limited and unevenly distributed lithium resources are facing shortages, which lead to price and cost increases.28,29 Belonging to the same main group, the element sodium has similar physiochemical properties to those of lithium.Sodium-ion batteries (SIBs), as an immensely prospective form of energy storage, show enormous potential in applications in renewable energy storage, electric vehicles, mobile devices, and other fields.30–33 Compared to LIBs, SIBs have garnered increasing attention due to the plentiful availability of sodium resources and lower costs.34–40 Currently, the pivotal point for commercialization is to develop advanced SIB cathode materials with high cycling stability and energy density. SIB cathode materials primarily encompass a diverse array of compounds, such as layered oxides that exhibit a distinct layered structure, Prussian blue analogues renowned for their unique coordination chemistry, polyanionic compounds characterized by their complex anionic frameworks, and organic compounds that offer a wide range of chemical and functional properties.41–47 The layered oxide cathode materials have the merits of high theoretical capacity, simple preparation methods, and low costs.48–51 Therefore, it is promising to apply layered oxides as cathodes for SIBs, yet many challenges are still waiting to be overcome for their large-scale application. For example, when layered transition metal oxides come into contact with air, they are prone to react with water and carbon dioxide in the air, resulting in the emergence of electrochemically inert compounds on the surface. Such interactions have the potential to alter the interface of the active material, leading to compromised battery efficiency.52–54 The structure of layered oxides undergoes irreversible phase transitions during charge and discharge cycles, which results in lattice distortion, deformation, or even destruction.55–58 Additionally, the electrolyte may undergo degradation reactions after a long cycle, bringing about the formation of a robust solid electrolyte interface (SEI) layer and sodium metal dendrites.59–62 These critical issues can restrict ion transport and reduce electron conductivity, resulting in decreased battery performance and reduced cycling stability.
One major but often overlooked issue is the irreversible cation migration occurring amidst the transition metal (TM) layers and Na layers, which can trigger structural instability and affect the properties of materials. Cation migration is an important phenomenon that manifests itself across diverse batteries, including LIBs, SIBs, and perovskite battery configurations, especially within the cathode materials.63–66 The migration of TM in lithium layered oxide cathodes, exemplified by the typical Li/Ni disorder, was initially investigated. Unfavorable TM migration in LIBs is often attributed to the approximate ionic dimensions of Li+ (0.76 Å) and Ni2+ (0.69 Å) ions.67–72 However, due to the significantly larger ionic radius of Na+ (1.02 Å), the migration of TM ions in SIBs differs from that in LIBs. Despite the significantly larger ionic radius of Na+ compared to TM ions, cation migration still occurs for specific structures and under specific conditions, and can be influenced by various factors such as material structure, defects, charge balance, and others.73–75 Cation migration is nothing more than a minor change between the TM and Na layers. However, it can drastically alter the intrinsic properties and structure of layered oxide materials. With the advancement of characterization and analysis techniques, more and more studies have demonstrated that the migration of TM to the Na layer during charging and discharging can lead to harmful structural recombination, which causes structural degradation, irreversible phase transition, and rapid reduction of the capacity/energy density, and consequent cathode material failure. In this regard, the mechanisms of microstructure variation and efficient modulation strategies should be more systematically investigated and comprehended.
Herein, the research history of cation migration within layered oxide cathode materials for SIBs, specific migration phenomena, the fundamental mechanisms leading to cathode materials failure, strategies to mitigate irreversible transitions, and advanced characterization techniques for detecting such migrations are comprehensively reviewed. The advances and findings of related research will provide guidance for designing stable structures and proposing valid strategies for layered oxide cathodes for SIBs. It is firmly believed that the summarized in depth understanding of the detailed evolution and related reactions in the microstructure involved in cation migration can promote the research and advancement of stable cathodes with high energy and a stable structure for commercial application.
2. The history of cation migration exploration
As shown in Fig. 1, the typical NaxTMO2 (0 < x≤ 1, TM = 3d transition metal element) structure of the layered oxide cathode material for SIBs is alternately composed of sodium layers and TM layers.76–80 The TM layers can be arranged in different ways along the c-axis, resulting in a variety of possibilities for the layered structure phases.81–84 According to the difference in the occupying mode of Na+ ions and the stacking mode of oxygen layers in TM layers, the layered oxide can be divided into two categories: P2 and O3, P is the initial letter of the word prismatic, which means that the sodium ion occupies a trigonal site in the interlayer, and O is the initial letter of the word octahedral, which means that the sodium ion occupies an octahedral site in the interlayer.85–87 The numbers 2 and 3 of P2 and O3 representing the counts of TM layers exhibiting varying patterns of oxygen layer configurations which is presented per unit cell.88–91 The TM ions with smaller ionic radii and Na+ ions with larger radii make it relatively easy for the TM ions to migrate to the Na layer.92–94 TM migration was found to be originated from the formation of a Na-free layer when sodium ions were extracted, and the Na-free layer was derived from a random desodiation process.95 For the random desodiation process, the TM ions in two adjacent TM layers would be homogeneously oxidized and then the Na near the oxidized TM ions would be de-inserted from the same Na layer, which would subsequently result in the formation of a Na-free layer and TM migration.96 Moreover, cation migration typically encompasses the relocation of TM ions, as these ions play a crucial role in the course of charging and discharging cycles. TM ion migration can be caused by various factors, including ion concentration in the electrolyte, temperature, and the charge/discharge rate of batteries. The cation migration can lead to issues such as structural degradation, decreased conductivity and irreversible phase transitions that worsen the electrochemical performance and consequently lead to failure of the cathode material.97–99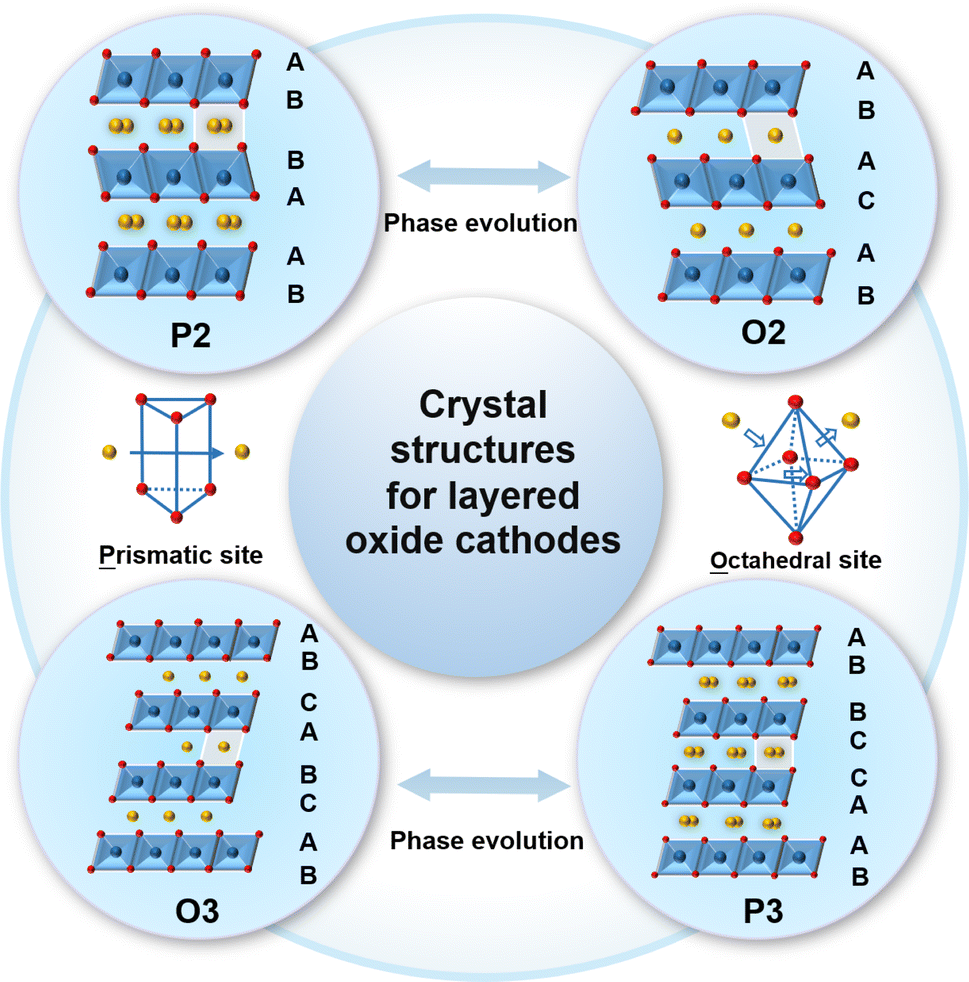 | ||
| Fig. 1 Schematic of the crystal structures for sodium layered oxide cathodes. | ||
To deeply understand cation migration, the advanced research about the concept proposing, scientific conjecture and corresponding observation results of cation migration are summarized in Fig. 2. The concept of cation migration was proposed by MAAZA et al. in 1983 and the host structure NaTiO2 transformed to NaxTiO2 as a result of the migration of certain Ti ions from TM slabs into the intervening space.100 From the discharge curves, the reversible electrochemical behavior can only be observed within a sodium content of 0.7 < x< 1. When 1 − x exceeds 0.3, an irreversible structural transformation occurs. To relieve this irreversible structural transformation, Ti ions migrate from the TM layer into the Na layer. This was the first time that the Ti ions migrating into inter space was proposed as well. However, relative research studies on cation migration couldn't attract enough attention until Yoshida et al. utilized the Co substitution strategy for the NaFeO2 electrode to effectively suppress Fe ion migration into Na layers in 2013, which elaborated the structural stability and improved Na diffusivity.101 As for the O3-type NaFeO2 electrode, the direct visualization of the migration of Fe into the Na layer within NaFeO2 was achieved via high-resolution transmission electron microscopy (HRTEM) in 2016 by Li et al., offering a clear insight into the migration process.102 In the same O3-type NaFeO2 electrode, the migration of Fe ions from the TM layers into the Na layers was observed at the atomic level. Along a–b plane, the distribution of Fe and Na ions was detected via high angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) and annular bright field-scanning transmission electron microscopy (ABF-STEM) in 2018 by Li et al.103 In 2018, Guo et al. found out that at discharged states the O3-type Na0.8Ni0.3Co0.1Ti0.5O2 electrode showed TM ions in Na layers, as demonstrated by HAADF-STEM images and the corresponding line profiles.104 In 2019, Zhang et al. presented the reversible Co atoms migrating into Na layers based on HAADF-STEM.105 In 2020, the occurrence of migrated TM ions within the interlayer spacing of the Na0.2Ni0.45Zn0.05Mn0.4Ti0.1O2 electrode was detected by Mariyappan et al.106 In 2021, the roles of 3d cation migration and anion redox in controlling voltage fade were reported by Wang et al. in the O3-type material NaLi1/3Mn2/3O2 for anionic redox electrodes.107 In 2022, the details about Fe and Ni migrating to the Na layer, which resulted in the deterioration of both structure and performance, were acquired by Chen et al. via Energy Dispersive Spectroscopy (EDS) elemental mapping and its line profiles.108 In 2024, the migration of Ni and Fe ions was further discovered by Li et al. through Fast Fourier Transform (FFT) patterns and HAADF-STEM images, and the migration pathways of Ni and Fe ions were elucidated.98 Furthermore, they proposed mechanisms to suppress cation migration. As mentioned above in the related research, it could be clearly seen that since 2018, more and more researchers have been engaged in issues related to cation migration (Fig. 3), benefiting from the development of characterization techniques for microstructure testing. Moreover, the observation and study of cation migration in Na layers will contribute to the understanding of the behavior occurring in the internal microstructure of materials and inspire us to comprehend relevant reactions at the microscale. And the investigation of mechanisms of cation migration in sodium cathodes will provide guidance for designing cathode materials and create value for the development of advanced electrode materials.
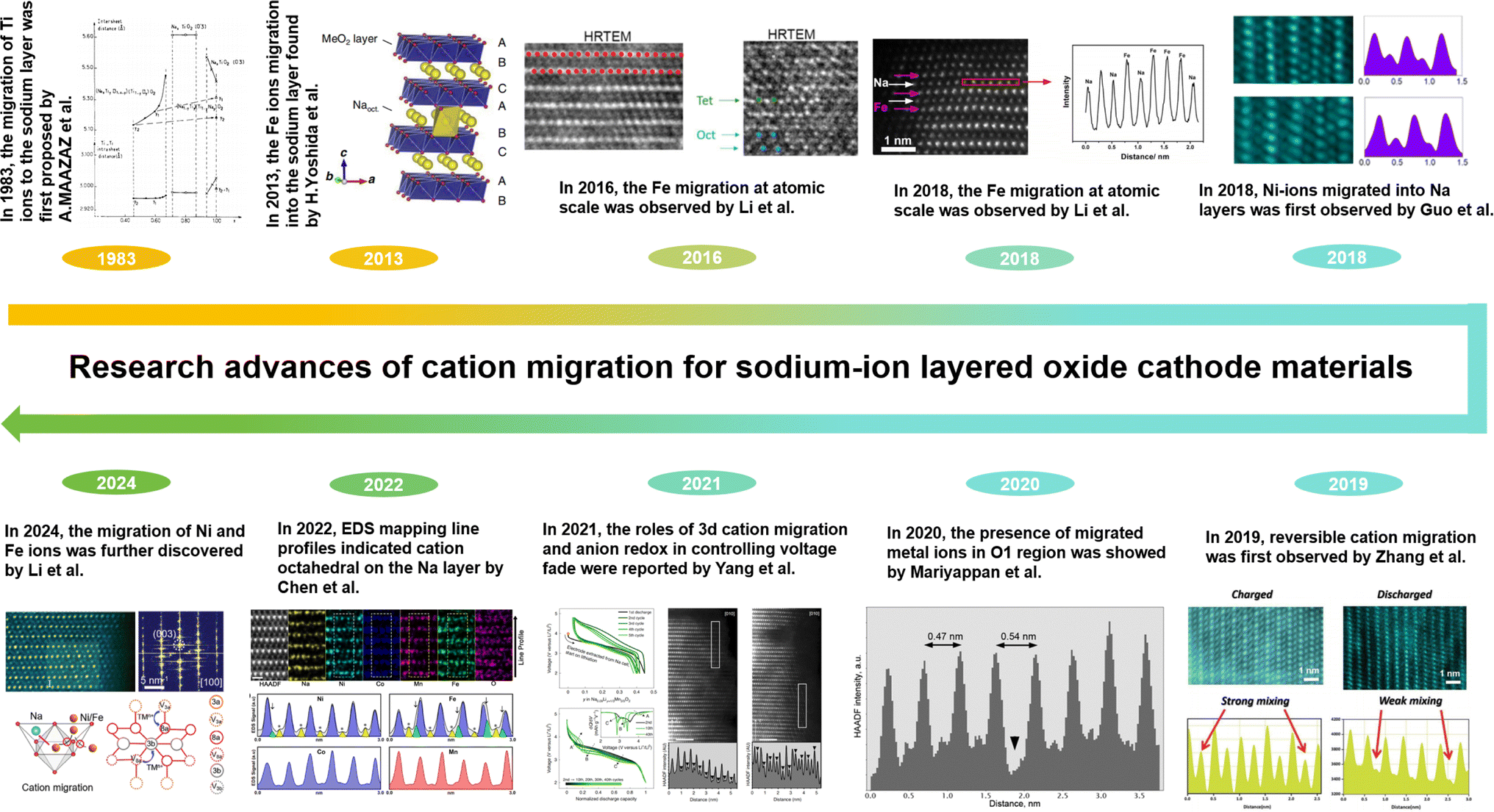 | ||
| Fig. 2 The cation migration research history of layered oxide cathode materials for SIBs. | ||
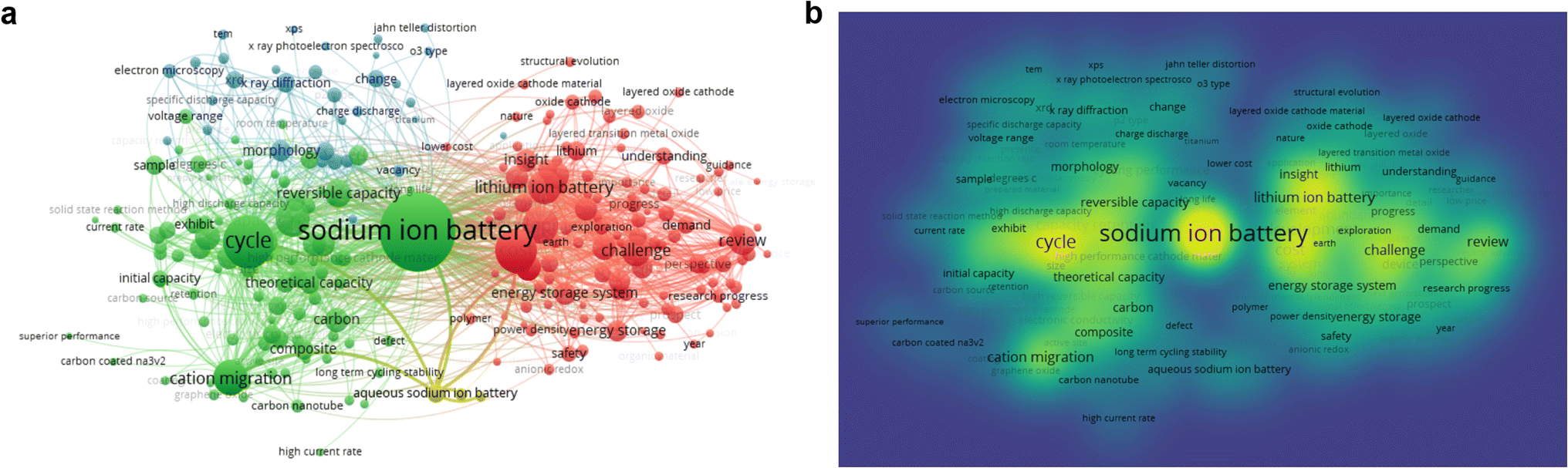 | ||
| Fig. 3 (a and b) Keyword data analysis of sodium-ion battery research hotspots. (The data were collected via Web of Science, September 2024). | ||
3. Detailed discussion of cation migration
3.1 TM migration and structure evolution
Although the concept of cation migration in layered oxide materials for SIBs was first proposed in 1983, it was evidenced by records of HRTEM and Selected Area Electron Diffraction (SAED) of NaFeO2 after 1 cycle, where the migration of Fe cations towards the sodium layer in NaFeO2 can be directly observed in Fig. 4a and b.100,102,103 Another research study on the NaFeO2 electrode at the fully charged state has obtained conclusive HAADF images, and the line profiles verified that the Fe ions migrate into Na layers as shown in Fig. 4c. As shown in Fig. 4d and e, the HAADF images and the contour map of electron energy loss spectroscopy (EELS) along the arrows in HAADF images have been observed, wherein the purple color means that Fe ions migrate into the sodium layer, and L3 and L2 edge peaks at around 708.7 and 721.7 eV are consistent with that of the Fe3+ ions in fresh NaFeO2. To further verify the complicated structural changes in NaFeO2, in situ XRD measurement results are shown in Fig. 4f, which proves that reverse migration of Fe ions and NaFeO2 is no longer the pristine state compared with discharge. Rietveld refinement reveals that approximately 29.0% of the Fe ions are situated within the sodium layer. The spin-polarized density functional theory (DFT) calculation results shown in Fig. 4g reveal the high activation energy for Fe migration in pristine NaFeO2 (3.68 eV), indicating that it is difficult for Fe ions to migrate from the Fe layer to the Na layer. However, it can be seen from Fig. 4h that when some of the Na ions are extracted, the energy barrier for the migration of the Fe ions at the octahedral site (OFe) is significantly reduced to 1.27 eV. The decreased energy barrier makes Fe migration feasible in the process of Na+ extraction. This research shows more systematic and all-round investigations of cation migration and structure evolution, enriching the understanding of structural transformation of Fe-containing oxides in the microstructure.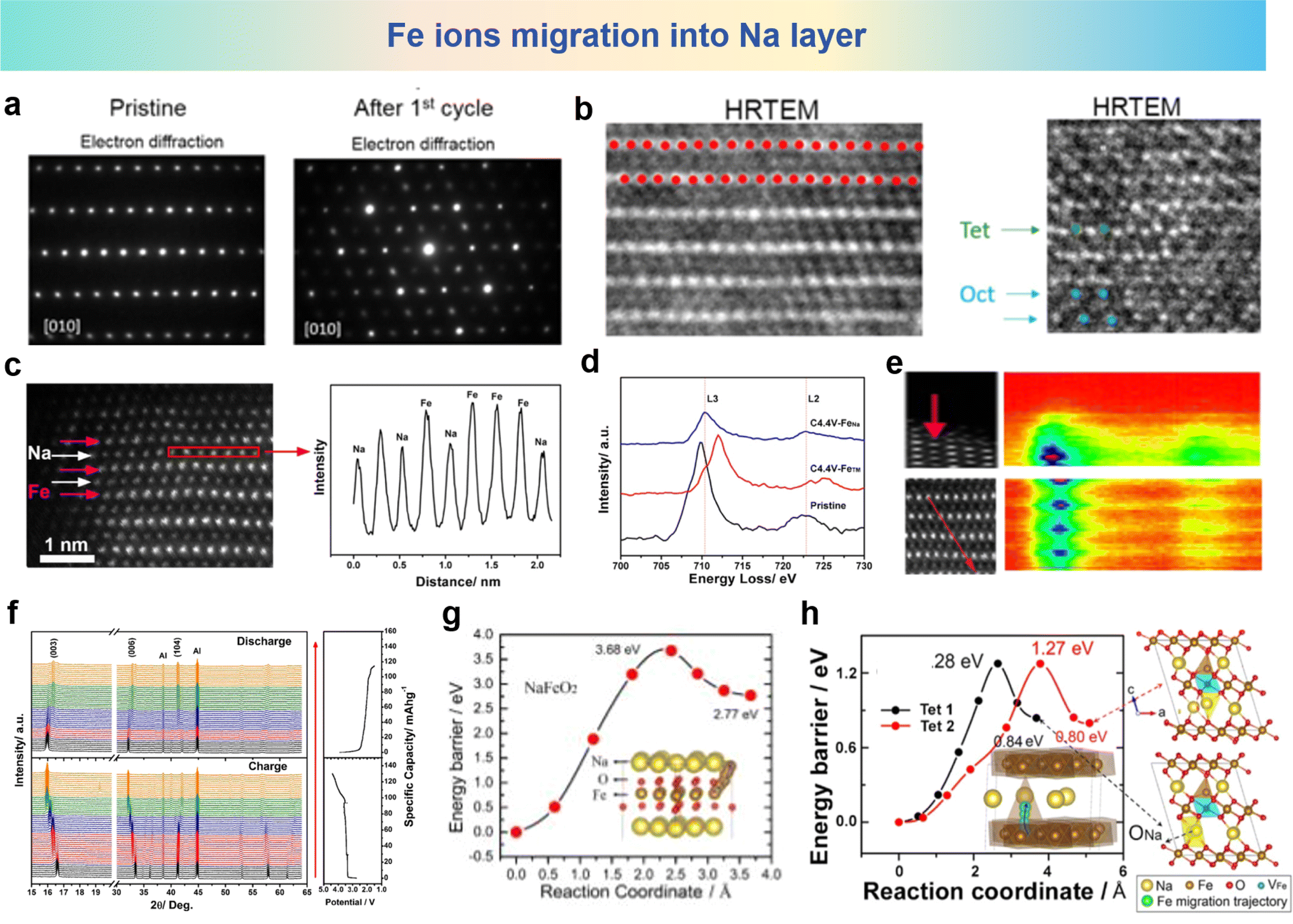 | ||
| Fig. 4 (a) SAED and (b) HRTEM images of NaFeO2 at the pristine stage (left) and after 1 cycle (right) between 2.0 and 4.5 V at C/10 rate. (a) and (b) are reprinted with permission from ref. 102. Copyright 2016 American Chemical Society. (c) HAADF images of the fully charged NaFeO2 electrode (left) and corresponding line profile (right) in the red rectangular region. (d) EELS spectra of Fe in pristine NaFeO2, in the Fe layers and in the Na layers of NaFeO2 charged to 4.4 V. (e) HAADF images (left) and EELS line-scanned contour map (right) along the Fe atoms (the red arrow). (f) In situ XRD patterns of NaFeO2 during the charge and discharge process. Pathways for direct Fe migration from the OFe to the TNa sites in (g) pristine NaFeO2 and (h) Na0.33FeO2. The yellow, brown, red, blue and green balls in the structural schematic are for Na, Fe, O, VFe and the Fe migration trajectory, respectively. (c)–(g) are reprinted with permission from ref. 103. Copyright 2018 Elsevier. | ||
As for the reversible rearrangement of the host structure between P-type and O-type stacking in layered sodium-containing oxides during charge and discharge could be suppressed by TM ion migration into the sodium layers.104 A O3 type Na0.8Ni0.3Co0.1Ti0.6O2 (NNCT) was synthesized by Zhou et al., Fig. 5a shows the structural changes during the charging process. Along with the migration of TM ions to the sodium layer, the O3 type stacking framework is transformed into P3 type. Then TM ions migrating into the sodium layer firmly grip the TM plate through strong TM–O bonds, thus inhibiting the P3–O3 conversion common in typical O3-type cathodes. It can be seen that although the initial charging phase changes to P3, the P3 stacking configuration is unexpectedly maintaining in the subsequent cycle. By incorporating the TM ions into the Na layer, the cation mixing nature in layered SIB cathodes can effectively stabilize the TM slabs through strong bonding between interlayered mixed TM and O, thus suppressing the host rearrangement and stabilize the P3-type stacking. STEM images and corresponding line profile results are shown in Fig. 5b, which show that TM ions irreversibly migrate into the Na layers during the initial charge process. To delineate the categories of TM ions that have undergone migration into the Na layers, Fig. 5c presents the annular dark field scanning transmission electron microscopy (ADF-STEM) images and scanning transmission electron microscopy-electron energy loss spectroscopy (EELS) line profiles after the initial charge. It can be observed that more Ni atoms are positioned within the sodium layers than within TM layers, while Ti and Co atoms mainly occupy the TM layers. Consequently, the TM ions migrating into the Na layers are mainly Ni atoms. The amount of Ni entering the Na layer is calculated by STEM image simulation. Fig. 5d and e show the simulated HAADF-STEM image of 53% Ni atom diffusion to the Na layer and the corresponding intensity distribution diagram, while Fig. 5f and g show the simulated HAADF-STEM image of 65% Ni atom diffusion to the Na layer and the corresponding intensity distribution diagram. The sodium layer is marked with an asterisk on the outline. Considering that 53% and 65% of the Ni atoms are diffused into the Na layer, the intensity ratio between the TM layer and the Na layer in the simulated image is equal to the minimum and maximum of the experimental values, respectively. Overall, it is estimated that about 53–65% of Ni atoms enter the Na layer irreversibly during the cycle. The possible TM migration paths in NNCT are shown in Fig. 5h, where the O3 stacking arrangement is reconfigured to the P3 arrangement in the charged state, and the TM migrate into the Na layer, which can suppress host rearrangement. This research highlights the significant impact of TM ion migration on the performance of SIB cathode materials and provides important clues for further addressing the challenges of layered oxides for SIBs.
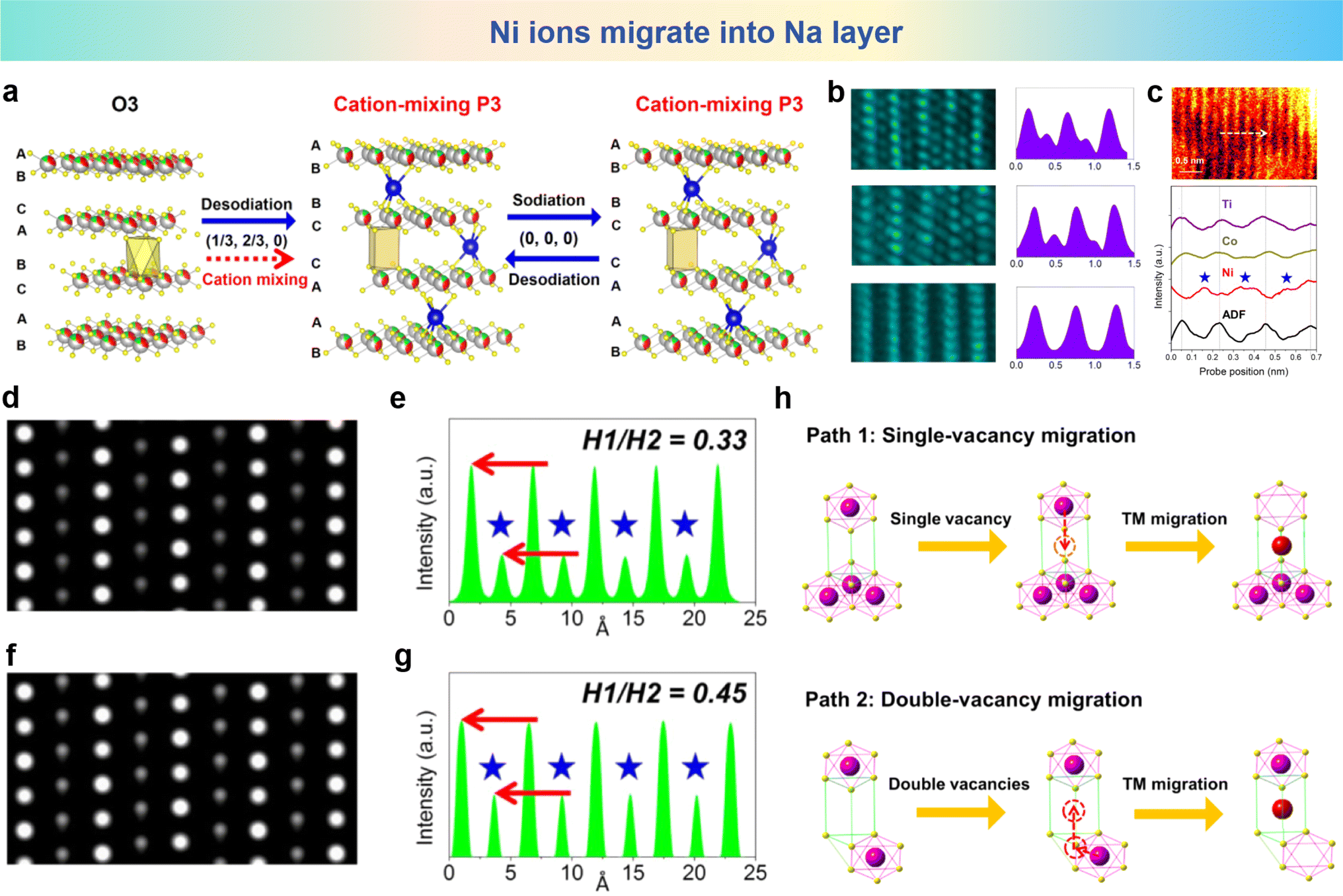 | ||
| Fig. 5 (a) Schematic illustration of structural evolution of NNT and NNCT electrodes in the charge/discharge process. (b) STEM images and the corresponding line profiles of pristine (bottom), fully charged (middle) and fully discharged (top) NNCT electrodes viewed along the [010] direction. (c) STEM-EELS line scan of the initially charged NNCT electrode. ABF-STEM image (top) viewed along the [010] direction indicates the typical TM layers (bright spots) and Na layers (dark spots) and STEM-EELS line profiles (down) describing the positions of TM layers are marked by dotted lines. (d) Simulated HAADF-STEM image considering that 53% Ni atoms diffuse into Na layers. (e) The corresponding intensity profile, wherein the intensity ratio of Na and TM layers is 0.33 and consistent with the minimum of experimental results. (f) Simulated HAADF-STEM image considering that 65% Ni atoms migrate into Na layers. (g) The corresponding intensity profile, wherein the intensity ratio of Na and TM layers is 0.45 and agreeable with the maximum of experimental results. (h) Two possible paths for TM ion migration from TM slabs into Na layers. One is single-vacancy migration (top) and the other is double-vacancy migration (bottom). (a)–(h) are reprinted with permission from ref. 108. Copyright 2018 Elsevier. | ||
Na0.8Co0.4Ti0.6O2 (NCT) was synthesized by Zhou et al., and the reversible migration of cations between TM layers and Na layers in the NCT electrode was also revealed by STEM images of the NCT electrode in the different stages (pristine, both in the fully charged and fully discharged stages) along with their respective line profiles shown in Fig. 6a and b.105 In the pristine states, the HAADF-STEM images show a well aligned structure. But in fully charged STEM images, the distinct spots emerge in adjacent TM slabs, and the presence of TM ions within the sodium layers serve as unequivocal evidence of their migration. As previously stated, the qualitative recognition of this migration process from TM layers to Na layers is well established. The roles of cation migration in structure evolution of the NCT electrode are investigated by in operando X-ray diffraction (XRD) and the corresponding patterns are depicted in Fig. 6c upon galvanostatic charging and discharging processes. At the pristine stage of the charging process, the O3 to P3 conversion leads to O3 and P3 biphasic coexistence. The O3 phase disappears as the charging progresses, and the P3 phase appears and becomes the pure phase until the end of the charge. Electron density maps portraying the distribution of electron density variations, arising from the application of Fourier transformation, which is applied to observe the TM migrating into Na inner layers compared with pristine states in Fig. 6d. It can be clearly visualized that after charging, the ball-like electron density for the TM ions appears in the sodium interlayer, which means that considerable amount of TM ions migrate to the sodium layers. As shown in Fig. 6e, the as-synthesized O3 type NCT electrode undergoes a phase transition from O3 to P3 during charging accompanied by a portion of cation migration occurring from the TM layers into the sodium layer. Following discharge, the electrode reverts to the O3 phase, demonstrating its reversible phase transition properties. During this period, a significant portion of the TM can be stated that the occupancy is within the Na layers. Reversible cation metal ion migration into Na layers is elaborated based on HAADF-STEM and the in operando XRD technique for the first time.
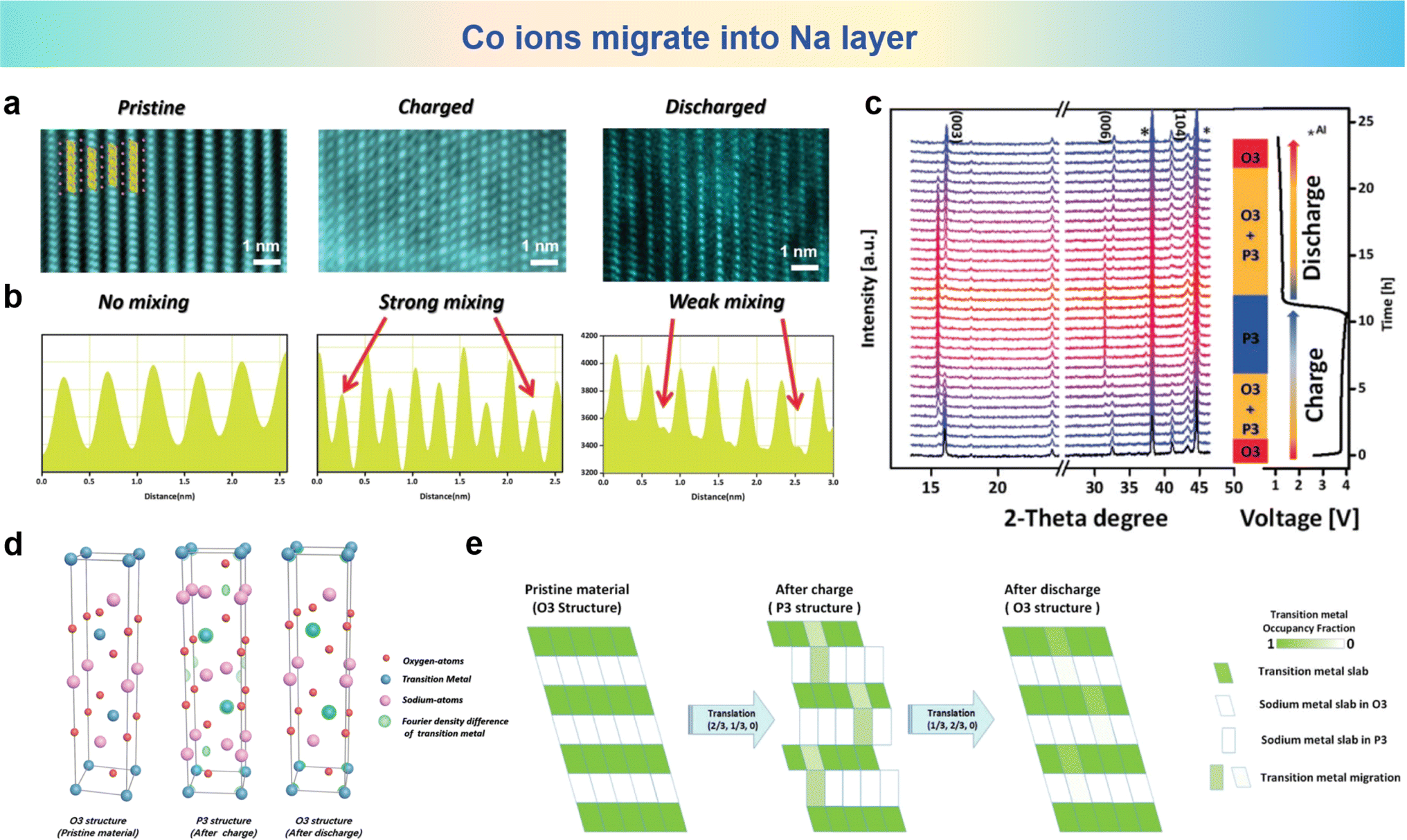 | ||
| Fig. 6 (a) The STEM images and (b) corresponding line profiles of the NCT electrode at pristine (left), fully charged (middle) and fully discharged (right) stages. (c) In operando XRD patterns measured at 10 mA g−1 and phase evolution during cycling. (d) Density difference electron density maps for different states produced by Fourier transforms. (e) Schematic plot of reversible TM migration upon cycling. (a)–(e) are reprinted with permission from ref. 105. Copyright 2019 Wiley. | ||
O3-type NaNi0.3Co0.12Mn0.18Fe0.4O2 (NCMF) was synthesized by Gao et al., who discovered successive migrations of TM ions to Na layers that accounted for structure and performance degradations.109 The TM migrations and structural transformations of a quaternary NCMF electrode are unveiled during charge/discharge processes. The XRD patterns of the pristine and cycled NCMF cathodes in Fig. 7a show that the O3 phase is well maintained after 50 cycles. Fig. 7b and c show the schematic of Ni ion and Fe ion migration into Na layers and the corresponding migration pathway in the microstructure. It can be seen that Fe and Ni ions migrate into the sodium layer from TMoct to Natet and then to Naoct. For further investigation of the structure changes of the NCMF electrode, atomic-resolution HAADF and geometric phase analysis (GPA) images of pristine and charged materials are shown in Fig. 7d and e. Apparently, the recycled material exhibits a fluctuating strain distribution along the c axis, compared with the pristine materials with relatively smaller and relatively uniform internal strain. The STEM-HAADF image of the pristine NCMF oriented along the [100] axis reveals that all TM cations occupy the TM layers, exhibiting a meticulously organized layered architecture. In contrast, the HAADF-STEM images of the charged NCMF cathode oriented along the [100] axis for pristine and the 4.0 V NCMF electrode charged to shown in Fig. 7f and g and corresponding line profiles taken along yellow lines in the HAADF-STEM image shown in Fig. 7h obviously show that TM ions have migrated into the sodium layer. In addition, the Line profiles for NCMF in pristine and charged states validate the migration of the TM into the Na layers as shown in Fig. 7i and j. The L3/L2 white-line ratios, utilizing the appropriate EELS spectra, were calculated to elucidate the alterations in the oxidation state of 3d TM ions during charging. The quantitative scanning transmission electron microscopy (QSTEM) image is shown in Fig. 7k, which is an enlarged HAADF-STEM image take from panel g and simulates the HAADF image with 40% TM ions migrating to the Na layer (inset). The cation migration is recorded by using line profiles of the HAADF-STEM image in Fig. 7l and m. The charged NCMF cathode further facilitates the direct observation of the dynamics of TM migration. Collectively, the irreversible alteration in the structure is associated with the process of migration from TM layers into Na layers in the cathode materials.
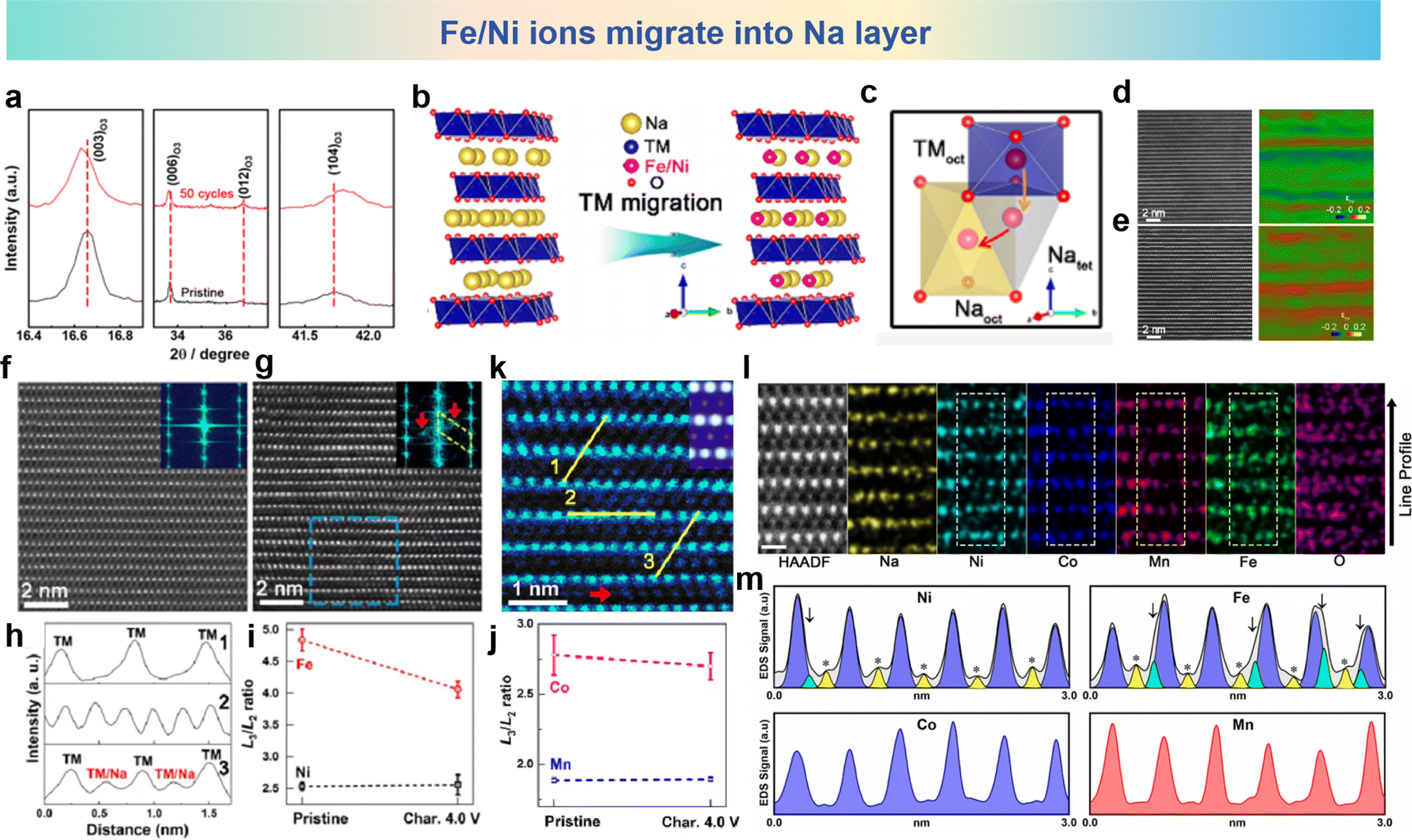 | ||
| Fig. 7 (a) Powder XRD patterns taken from pristine and cycled NCMF cathodes. (b) The schematic description of TM migration in subtle structure. (c) TM migration pathway in subtle structure. Atomic-resolution HAADF image and GPA image, respectively, for the (d) pristine and (e) cycled NCMF cathode. The STEM-HAADF image and its FFT patterns viewed along the [100] zone axis for (f) pristine and (g) charged to 4.0 V. (h) Line profiles taken along the STEM-HAADF image. White-line ratios (L3/L2) for (i) pristine and (j) charged to 4.0 V. (k) Enlarged STEM-HAADF image taken from panel g. (l) Atomic-resolution STEM-HAADF image and EDS elemental mappings of the NCMF cathode after 50 cycles along the [100] zone axis. (m) EDS mapping line profiles of Ni, Fe, Co, and Mn acquired from the white rectangles. Yellow peaks indicate cation octahedra on the Na layer (marked with asterisks), and green peaks indicate cation tetrahedra on the Na layer (marked with arrowheads). (a)–(m) are reprinted with permission from ref. 109. Copyright 2020 Chemical Society Japan. | ||
3.2 Failure mechanisms due to cation migration
Cation migration would unexpectedly happen between TM layers and Na layers in cathode materials during charging and discharging processes. More precisely, activated TM ions will migrate into the Na layer. These changes lead to the evolution of the microstructure, which causes structural collapse and surface deterioration, irreversible phase transitions and a rapid decrease in capacity/energy density, which in turn leads to the failure of the cathode material.96,110Li et al. found that when exposed to ambient air, commercial O3-type layered NaNi1/3Fe1/3Mn1/3O2 (O3-NNFM) materials undergoes the migration of cations from the TM layers towards the near-surface Na layers would occur.31 Additionally, air-exposed O3-NNFM materials exhibit increased Ni/Na disorder due to the lower Ni migration barrier, leading to harmful structural distortion and surface deterioration caused by cation mixing as shown in Fig. 8a. Bo et al. investigated the mechanism of irreversible phase transitions in NaxCrO2 layered materials for SIBs, as illustrated in the schematic diagram in Fig. 8b–d. The initial phase of structural transformation involves the process of sliding of layers and the extraction of Na+, converting the P′3-type configuration to an O3-type arrangement.111 In conjunction with this process, a disparity in the charge disproportion of Cr4+ ions exists, in which Cr6+ cations are generated and drop into the tetrahedral sites in the Na layer. The tetrahedrally coordinated Cr6+ cations may subsequently undergo mixed reactions with two adjacent Cr3+ cations, reorganizing the Cr4+ cations, escaping from the tetrahedral position, and ultimately entering the permanent octahedral position in the Na layer. The electrochemical reversibility of sodium extraction/insertion processes in Na1−xCrO2 at different upper cutoff voltages was investigated. As shown in Fig. 8e and f, the reversible storage capacity of these layered materials exhibits a marked decline when subjected to elevated cut-off voltages. The drastic reduction in capacity and energy density could be ascribed to the irreversible migration of TM from the oxide layers of TM to the intervening space between layers.112,113 As shown in Fig. 8g, Na0.85FeO2 (NF) exhibits severe TM migration in the deoxygenated state, leading to significant degradation in cycling performance.95
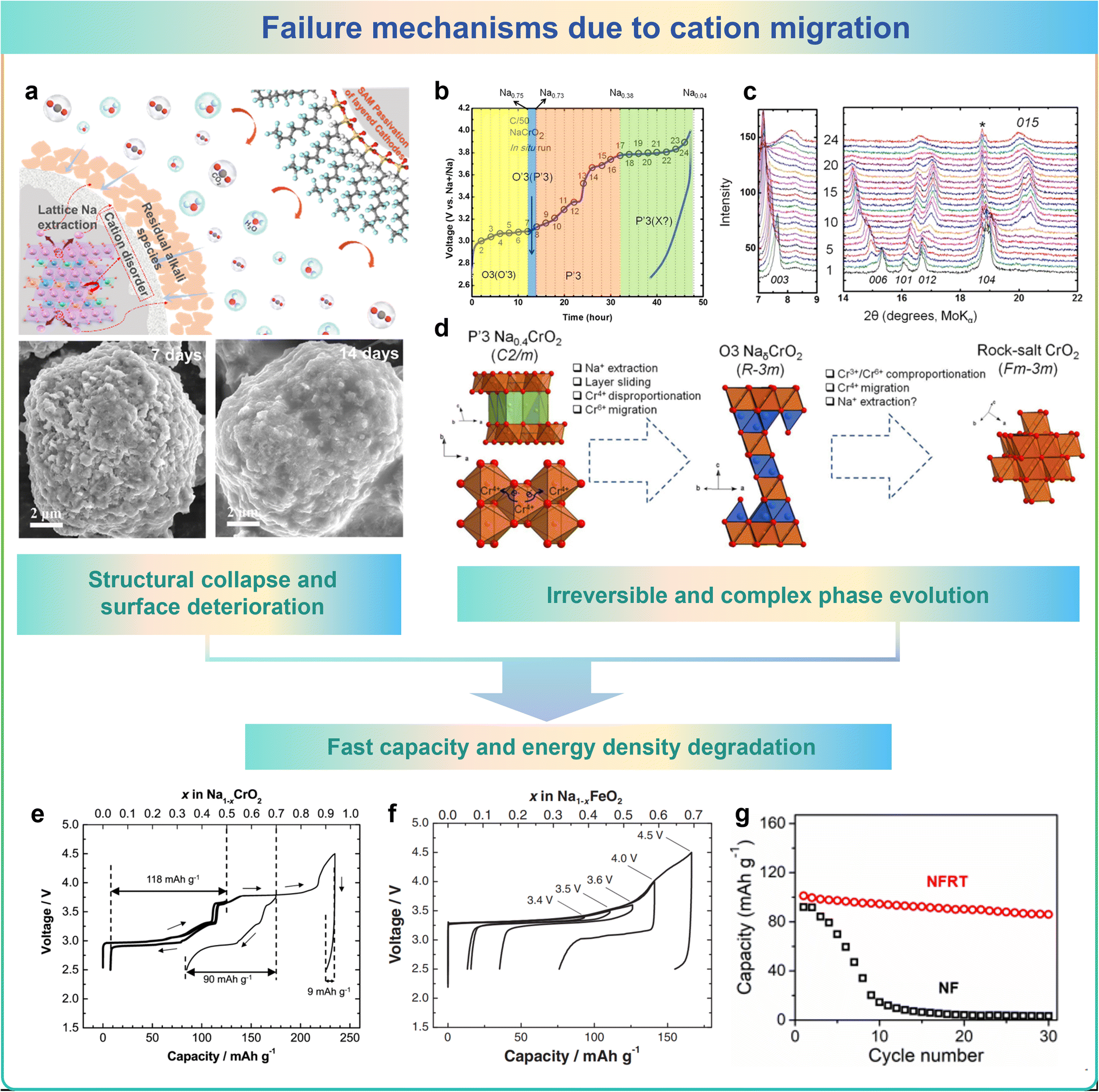 | ||
| Fig. 8 The failure mechanisms due to cation migration: (a) structural collapse and surface deterioration, (b–d) irreversible and complex phase evolution, and (e–g) fast capacity and energy density degradation. | ||
The above research clearly verified the small variations between TM layers and Na layers, which would entirely alter the cycling performance and intrinsic properties of layered oxide materials. The migration of TM into the Na layer in layered oxides has led to adverse structural reconstruction, slow Na+ diffusion, and subsequent performance degradation. Therefore, more efforts should be made to precisely investigate the mechanisms of microstructural changes and invent methods to suppress cathode material failure caused by cation migration.
4. Strategies for modulating cation migration
Herein, based on the influence of cation migration on the properties of cathode materials, effective approaches to suppress cation migration by element substitution and structure design have been investigated. More thorough details are shown in Fig. 9. Element doping introduces additional atoms or ions into the crystal structure, leading to lattice distortion, which can change the lattice parameters, crystal structure, and ion channels, thereby slowing down or hindering the migration of cations.114–116 In addition, element doping can also modify the electronic structure of the crystal, adjust the band structure and electron density of states, and affect the migration energy barrier of cations in the crystal, reducing the migration rate.117–119 Furthermore, multi-element doping can be used to design high-entropy structures, given that a complex crystal structure and thermodynamic behavior also limit cation migration.95,96 In material design and application, rational structural design can regulate the migration rate of cations, diffusion pathways, and ionic conductivity, thereby achieving precise control and optimization of cation migration.120–123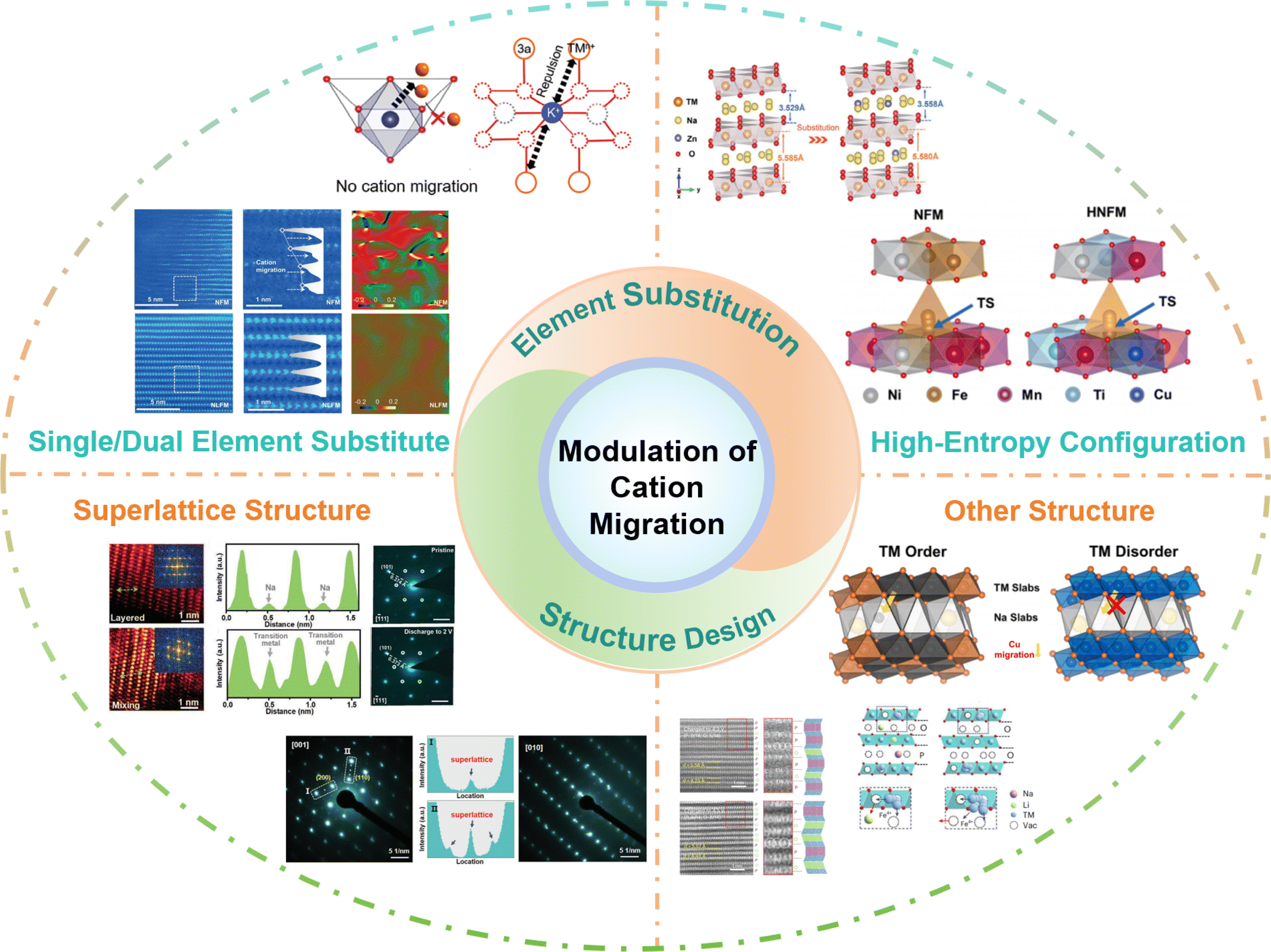 | ||
| Fig. 9 The practical strategies of modulating cation migration: single/dual element substitution, high-entropy configuration, superlattice structure and other structures. | ||
Element doping can have a significant impact on the cation migration performance by adjusting the ion size and charge state, introducing defects, and electronic structure modulation. The interactions and synergistic effects between different elements will affect the migration of cations.124–127 With the intention of changing the structure and characteristics, the high-entropy effect refers to the introduction of multiple different elements into a material to increase its entropy.128–131 The complexity and diversity resulting from doping of multiple elements may have unique effects on cation migration performance. However, the specific mechanisms of these effects still require further research and exploration. In addition, introducing some special structural features and defects can influence the migration of cations. For example, in a superlattice structure significant lattice distortions exist between the sub-lattices, which can restrict the diffusion and migration of cations.132–134 Additionally, the element distribution in the superlattice structure is highly ordered, which can suppress cation vacancy diffusion and hopping, thereby slowing down the cation migration rate. Moreover, the superlattice structure can also modulate the electronic structure of the material, altering the migration energy barrier for cations in the crystal, thus affecting the cation migration rate. In the case of an ordered structure or other intergrowth structures, distinct grain boundaries or interfaces exist between different crystal phases.135–138 These interfaces impede cation migration by introducing grain boundary energy barriers when cations traverse between different crystal phases, thereby increasing the energy cost of migration and slowing down the cation migration rate.
4.1 Element substitution strategy
Element substitution is a universally acknowledged strategy for elevating the structural stability and cycling performance, including single/dual element substitution and multiple element substitution, and the influence of element doping on the cation migration performance involves multiple aspects such as lattice distortion, defect regulation and electronic structure modulation.139–145 The cation migration is affected by the interaction and synergy between different elements. Meanwhile, researchers have increased the entropy of the material by doping more elements and designing high-entropy materials to inhibit the migration of cations.146–151 | ||
| Fig. 10 Atomic HAADF-STEM images of (a) NFO and (b) NFRO samples charged to 4.0 V. (c and d) Line-profile of the selected area by using a dashed line in (a) and (b). Nanobeam electron diffraction patterns of (e) NFO and (f) NFRO samples charged to 4.0 V. (g) HAADF-STEM images of (left) NFO and (right) NFRO samples charged to 4.0 V at low magnification. (h) GPA patterns of NaFeO2 (left) and Na4FeRuO6 (right) samples charged to 4.0 V. (i) In situ XRD patterns of the Na4FeRuO6 electrode at a current rate of 0.25C in the voltage range between 1.5 and 4.0 V. (a)–(i) are reprinted with permission from ref. 110. Copyright 2019 Wiley. | ||
The layered structure undergoes a series of phase transitions in which the fully desodiated O1 phase shows a huge reduction in cell volume together with cation migration, Mariyappan et al. solved both problems by Zn-doping.106 In the O3-NaNi0.5Mn0.4Ti0.1O2 (NMT) electrode, the Ni2+ cations are partially substituted by Zn2+, forming the O3-NaNi0.45Zn0.05Mn0.4Ti0.1O2 (ZNMT) electrode. By Zn-doping, the ZNMT electrode shows the reciprocal growth structure of P3–O1 during charge and discharge, in which the O1 phase exists only locally in the form of a nanoscale domain, which is conducive to suppressing the migration of TM ions into the interlayer space. The morphology of the charged ZNMT and NMT electrodes ais observed by TEM images in Fig. 11a and b. HAADF-STEM images from the [010] axis show that the ZNMT electrode contains a P3 and O1 coexisting structure. However, the O1 structure is found in large areas in the NMT electrode, which is attributed to the substantial migration of TM cations into the inner octahedral sites. Meanwhile, in Fig. 11c, the P3-type layers are easily discernible from the O1-type layers by the lateral shifts of the bright dots of the TM columns and a larger interlayer spacing of ∼5.4 Å compared to ∼4.7 Å in the O1 domains. In the O1 domain, obvious TM ion migration into the interlayer octahedral space occurs, while in the P3 domain, the migration of cations into the trigonal prismatic cavities is basically inhibited. The long-term cycling performance of 5% Zn-doped and no Zn-doped electrodes and their structural diagrams are shown in Fig. 11d. The phase structure of the 5% Zn-doped sample shows P3–O1 domains with better stability than the no Zn-doped electrode which presents only the O1 phase. For further investigation about the effect of Zn2+ doping on structural evolution in cathode materials, operando XRD analyses are shown in Fig. 11e and f. The differences of ZNMT and NMT electrodes appear at a voltage below 4.0 V. Compared with the NMT electrode, the ZNMT electrode has a shortened biphasic progression from O3 to O′3 to P3 and an increased solid-solution region of the P3 phase, while the pronounced difference appears above 4.0 V with a clear biphasic progression in the ZNMT electrode as opposed to a complex structural evolution in the NMT electrode. Therefore, the formation of P3–O1 in Zn-doped materials limits the structural degradation and the migration of Ni ions during the cycle, and improves the long-term cycle stability.
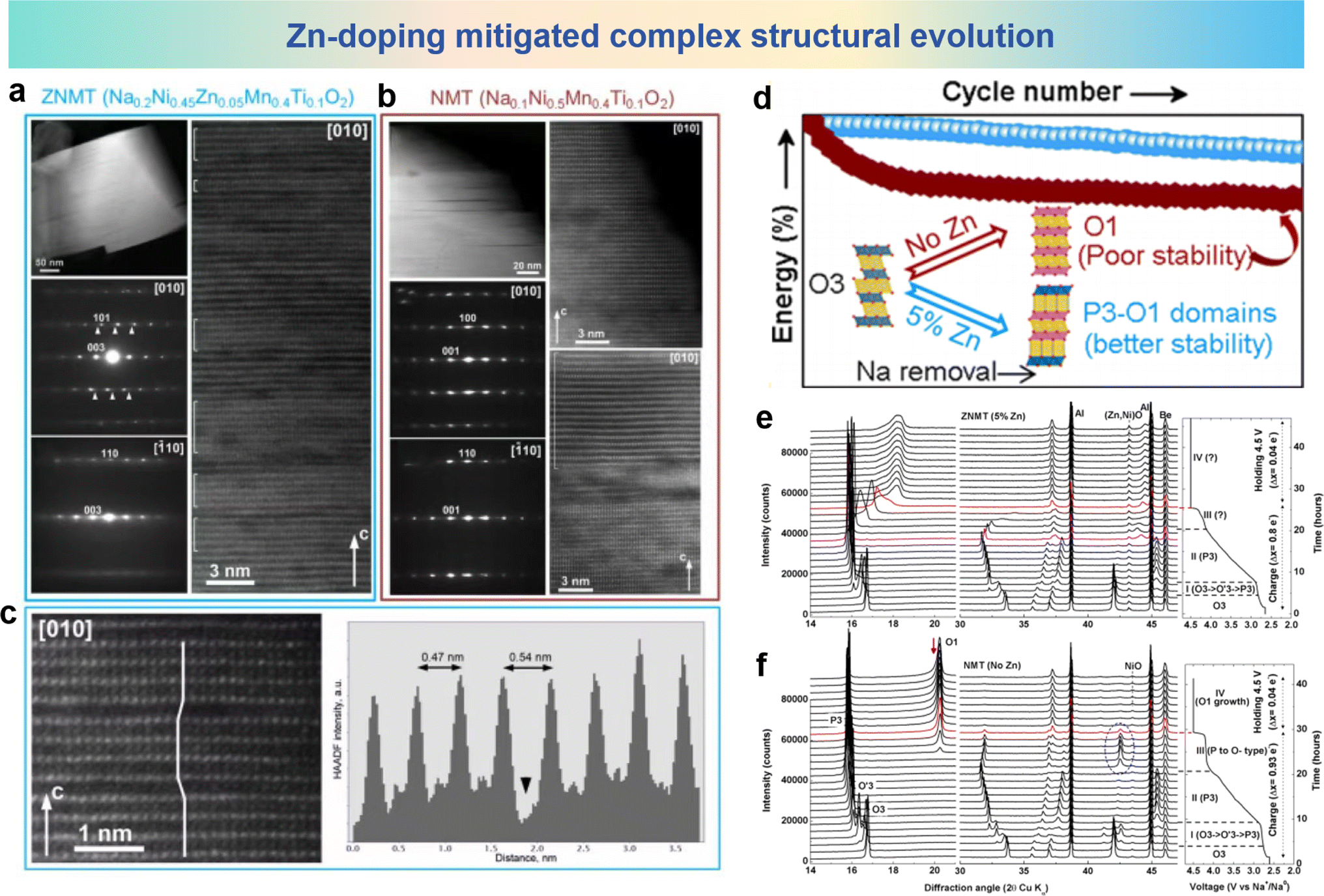 | ||
| Fig. 11 TEM analyses of (a) ZNMT and (b) NMT electrodes at the end of charge. (c) Zoomed in part of the HAADF-STEM image of the ZNMT material P3–O1 intergrowths (left) and the presence of migrated TM ions in the O1 region and P3 region. (d) A diagram about the structural changes of materials with/without Zn-doping and the corresponding cycling performance. Operando XRD analysis and corresponding structural changes during the first charge process of the (e) ZNMT and (f) NMT electrodes. (a)–(f) are reprinted with permission from ref. 106. Copyright 2020 American Chemical Society. | ||
Zhou et al. activated anion redox activity by Li-doping in Na2/3Fe2/3Mn1/3O2 (NFM) that inhibited Mn ion dissolution and TM ion migration.97 In Na0.8Li0.2Fe0.2Mn0.6O2 (NLFM), Li-substitution in TM slabs induces the possibility of anionic redox, and the anionic redox plays a dominant role in the structural stability of NLFM. As shown in Fig. 12a and b, the discrepancies in XRD patterns of NFM and NLFM with XRD refinement are presented. The peaks of NLFM can be assigned to the incorporated phases combining both P2 type and O3 type structures. The XRD patterns of the NFM and NLFM electrode soaked in water for 1 week and stored in air for 1 month are shown in Fig. 12c. NLFM presents unchanged patterns compared with the materials exposed to air for 1 month. In contrast, the NFM material shows an additional peak near the (002) peak, indicating that the structure of the material has changed. For better understanding the structural variation of NLFM materials, in situ XRD pattern for the first cycle is shown in Fig. 12d. The (002) peak shifting to a lower angle which demonstrates the slight expansion of the c-axis resulting from Na+ extraction. Then the (002) peak shifts to a higher angle at a high-charging voltage, which also indicates the redox of oxygen. During the discharge process, the (002) peak returns to its original undisturbed state, and the absence of phase transitions during this process indicates the stability of the structure. By comparing the volumetric variations in these two electrodes in initial cycle, Fig. 12e shows that the volume changes of NFM materials are 7.9% during charge and 10.4% during discharging. Enormous volume variation wrecks the structure stability and leads to capacity fading. It can be seen from Fig. 12f that, compared with NLFM, overexploitation of Mn in NFM leads to more serious Mn dissolution and weakens the structural stability. The phase evolution of NFM and NLFM materials in the microstructure was revealed using HAADF-STEM. As shown in Fig. 12g and h, the arrangement of TM is chaotic forming massive vacancies in NFM, which are caused by Fe migration and Mn dissolution. However, no TM vacancies and cation migration were observed even after 100 cycles of NLFM materials. GPA patterns corresponding to this analysis (Fig. 12i and j) of the two electrodes were used to investigate the lattice stress, showing apparent heterogeneity for NFM and uniformity for NLFM. The irregular lattice strain within NFM triggers a phase transition which leads to Fe ion migration and Mn ion dissolution during the charge and discharge progresses.
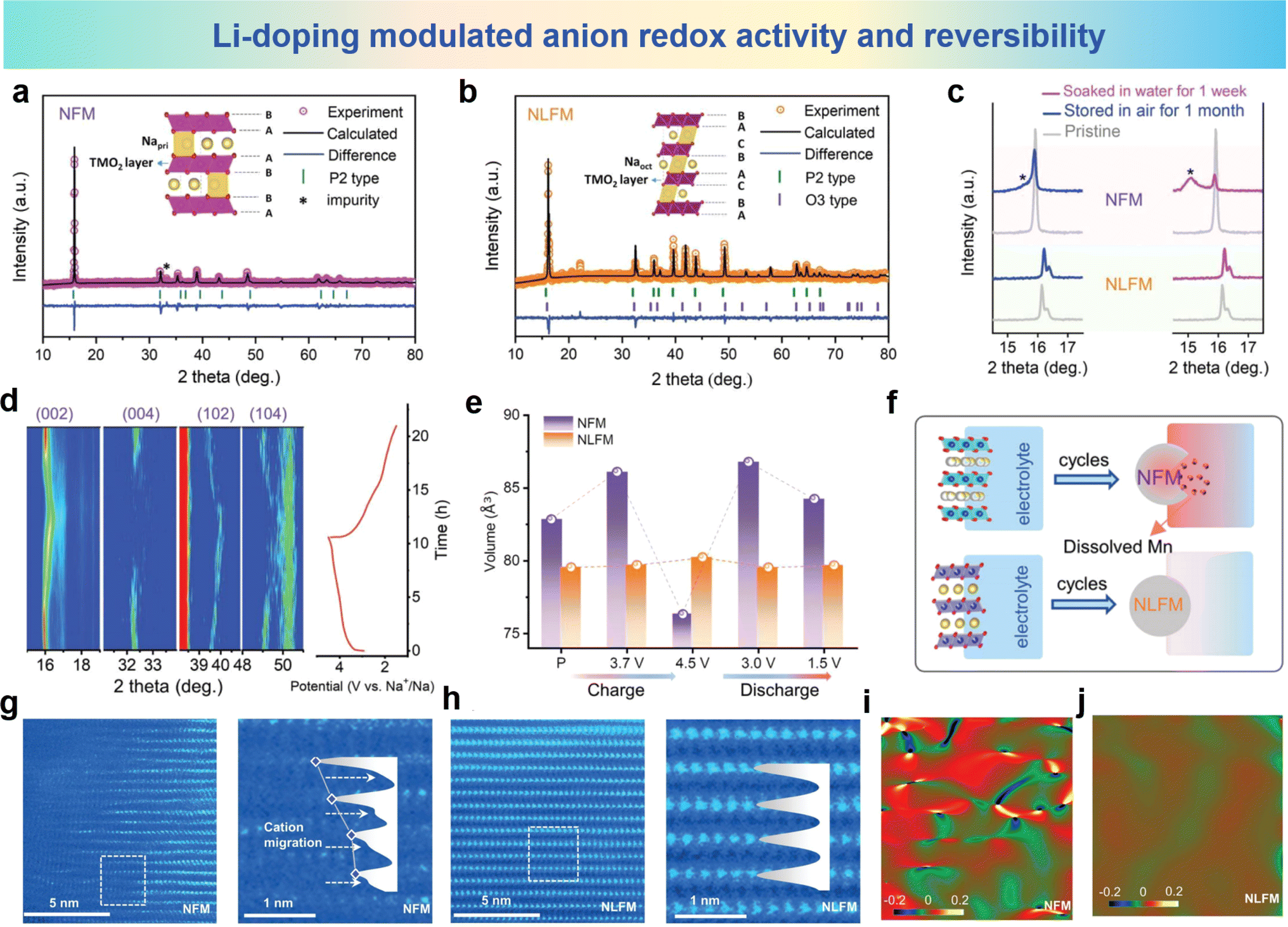 | ||
| Fig. 12 XRD refinement of (a) NFM and (b) NLFM electrodes. (c) Comparison of XRD patterns for NFM and NLFM materials exposed in air for one month and soaked in water for one week. (d) In situ XRD patterns of the NLFM electrode in the first cycle. (e) Evolution of cell volume for NFM and NLFM electrodes during the first cycle. (f) Schematic diagram of dissolved Mn with cycles. STEM images of (g) NFM and, (h) NLFM after 1000 cycles. GPA patterns of (i) NFM and (j) NLFM. (a)–(j) are reprinted with permission from ref. 97. Copyright 2022 Wiley. | ||
In order to solve the issues of irreversible cation migration and voltage decay at high potentials, Li et al. prepared O3–Na0.898K0.058Ni0.396Fe0.098Mn0.396Ti0.092O2 (KT-NFM) based on Na0.987Ni0.396Fe0.204Mn0.402O2 (NFM) using K and Ti co-doping.98 The HAADF-STEM images of the cycled sample are shown in Fig. 13a, in which the TM ions have migrated to the sodium layer. The crystal structure in Fig. 13b shows that K and Ti predominantly occupy the 3a and 3b sites in the Wyckoff notation, respectively. It can be seen from Fig. 13c that the doping of K and Ti significantly increases the overlap area and energy position of the Ni (3d-eg*)–O (2p) orbitals in KT-NFM, indicating an elevated Ni–O bonding energy. This strengthening of the TMO2 plane helps suppress undesired phase transitions. Fig. 13d illustrates the interlayer cation migration pathways in NFM and KT-NFM. In KT-NFM, the comparative energy values of Ni occupying the 8a and 3b sites are 1.24 eV and 3.93 eV, respectively, which are significantly higher compared to those in NFM (−3.02 and −2.10 eV as shown in Fig. 13e). Consistent patterns are discerned regarding the migration of Fe in both KT-NFM and NFM systems. These findings suggest the thermodynamic unfavourability of Ni and Fe ion migration within KT-NFM. This could be attributed to the contraction of the TMO2 plane induced by K occupying the Na site and the strong Coulomb repulsion generated, which suppresses the migration of Ni/Fe. Possible avenues for suppressing the interlayer migration of cations in KT-NFM are illustrated in Fig. 13f and g. In KT-NFM, TM ions are anchored by the large-sized K+ ions and the strong Coulomb repulsion between them and the NaO2 plane, which inhibits the migration of TM ions from neighboring 8a to 3b sites, thereby inhibiting the migration process of TM ions. Through structural modulation and inhibition of interlayer cation migration, KT-NFM exhibits improved stability and performance at high potential. These findings provide new insights and solutions for solving problems such as cation migration and Jahn–Teller distortion in O3-type materials under specific conditions.
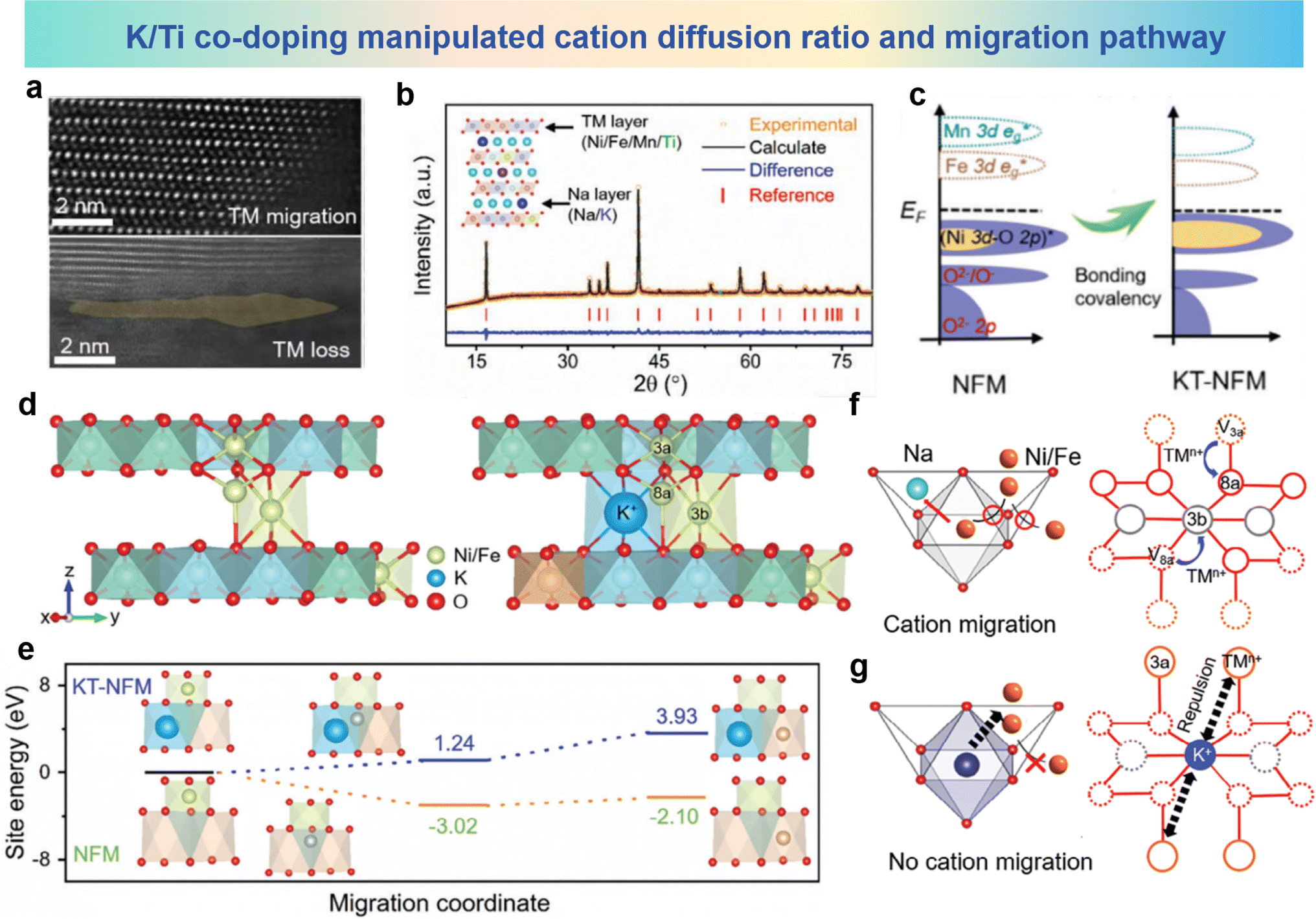 | ||
| Fig. 13 (a) HAADF-STEM image of cycled NFM. (b) XRD pattern and Rietveld refinement of KT-NFM and its inset crystallographic structure. (c) pDOS of NFM and KT-NFM. (d) TM migration paths from 3a–8a–3b sites in NFM (left) and KT-NFM (right). (e) Relative site energies of Ni migration in optimized structures of NFM and KT-NFM. (f and g) Proposed mechanism for inhibition of cation migration. (a)–(g) are reprinted with permission from ref. 98. Copyright 2024 Wiley. | ||
As a typical layered oxide cathode material, NaCrO2 undergoes TM migration at high charge states.95 In the charged Na layer, the emergence of a layer devoid of sodium, along with the occurrence of Cr has been identified by STEM and STEM-EDS analysis as shown in Fig. 14a. To address this issue, Guo et al. proposed an approach involving the joint introduction of Ru/Ti co-doping. NC is selected as the model material, and it can be seen from Fig. 14b that the TM migration phenomenon is due to the Na-free layer formed by a random desodiation reaction at high charge potential. The random desodiation can be attributed to Cr in the TM layer participating in charge compensation. During random desodiation, the homogeneous oxidation of Cr in two adjacent TM layers leads to random extraction of Na from the same Na layer, resulting in the formation of a Na-free layer. After Ru/Ti co-doping, hybridized Ru (4d)–O (2p) electrons are found within the first occupied band range, while no Cr (3d)–O (2p) electron density is observed. In the second occupied band range, significant hybridization between Cr 3d electrons and O 2p electrons is observed as shown in Fig. 14c. This suggests that the first contributing species to the charge compensation mechanism is through the Ru (4d)–O (2p) cationic oxidation–reduction reaction. Based on a specific understanding of the desodiation mechanism, it is believed that in Na0.85Cr0.85Ru0.10Ti0.05O2 (NCRT), a uniformly dispersed hybrid layer plays a key role in suppressing the formation of Na-free layers, thereby mitigating the likelihood of subsequent crystal structure degradation and TM ion migration. HAADF-STEM images of NaCrO2 (NC) and NCRT are shown in Fig. 14d, where significant fluctuations in the TM plate are observed in the NC image, with bright spots in the sodium layer attributed to Cr migration from the TM layer. This phenomenon could not be observed in NCRT, indicating the absence of Cr migration to the sodium layer. In situ XRD results in Fig. 14e show that the NC electrode has poor structure reversibility, whereas NCRT undergoes a reversible phase transition from O3 to P3 during the charge/discharge process. The structural and electrochemical irreversibility of the NC electrode is primarily attributed to the formation of sodium-free layers and subsequent Cr migration, which interrupts reversible sodium insertion, leading to subsequent structural irreversibility. The well-mixed layer formed by Ru/Ti co-doping has pivotal significance in hindering the emergence of sodium-free layers, mitigating the degradation of crystal architecture, and minimizing the migration of TM species. The co-doping strategy provides new insights and solutions for addressing the problem of cation migration in layered cathode materials for SIBs.
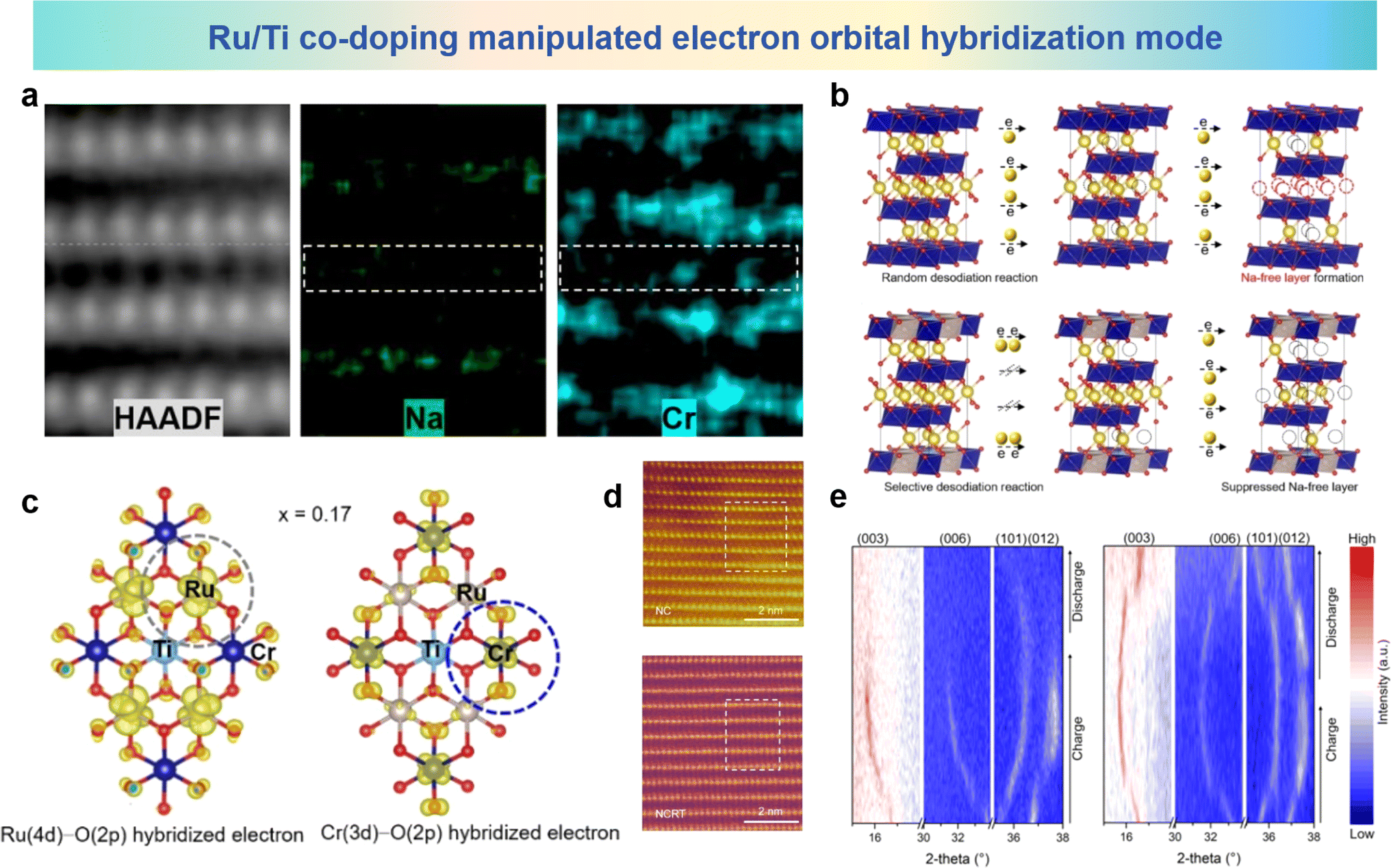 | ||
| Fig. 14 (a) Atomic HAADF-STEM image and corresponding atomic STEM-EDS mapping of charged NC. (b) Na-free layer formation derived from the random desodiation reaction and its suppression by the selective desodiation reaction in layered oxide cathodes. (c) Spatial electron distribution in the occupied bands. (d) HAADF-STEM images of fully charged NC and NCRT along the [100] zone axis. (e) In situ XRD patterns of NC and NCRT electrodes at 1.5–3.92 V. (a)–(e) are reprinted with permission from ref. 95. Copyright 2023 Wiley. | ||
Li et al. designed a high-entropy layered oxide cathode material Na0.85Li0.05Ni0.3Fe0.1Mn0.5Ti0.05O2 (LNFMT) with significantly reversible and high-rate capabilities based on the configurational entropy adjustment strategy.147 The distinctive configuration of LNFMT not only augmented the anionic reactivity but also mitigated the migration of TM under elevated voltage conditions. The high-entropy phenomenon enhances the migration of Li+ toward the Na layer, leading to the emergence of a quasi-tetrahedral configuration that hinders the migration of Fe3+ ions. The in situ XRD pattern of the original material Na0.85Ni0.3Fe0.2Mn0.5O2 (NFM) is shown in Fig. 15a, which indicates that a P2–O2 phase transition occurs during the first charge process in the voltage range of 2.7 V to 4.5 V. Furthermore, the P2–O2 phase transition is completely irreversible, indicating poor structural recovery and capacity loss. The principal factor contributing to the development of the O2 phase is the migration of TM, along with the slip of the TMO2 layer, leading to an increase in the electrostatic repulsion between adjacent oxygen atoms and consequently causing the degradation of the structure of NFM. Compared to NFM, the phase transition behavior of high-entropy LNFMT, as depicted in Fig. 15b, is relatively more straightforward. The entire progression of phase transition can be delineated as P2/O3–P2/P3–OP4/OP2–P2/P3. Throughout the entire process, the emergence of the O2 phase is not supported by any substantial evidence. During the charge–discharge cycling process, the predominant presence of the P2/P3 phase indicates that the adjustment of the configurational entropy effectively preserves the stability of the crystal structure, thereby preventing irreversible O2 phase transitions and consequently hindering the migration of TMs. Ex situ X-ray Absorption Spectroscopy (XAS) is shown in Fig. 15c and d, illustrating the investigation of the electronic and local environments of TM atoms in LNFMT. During the charge–discharge process, it was observed that the main absorption edge of Ni gradually shifted to higher energy regions, indicating the predominant presence of reversible Ni2+/Ni3+ and Ni3+/Ni4+ redox couples for the charge compensation mechanism. A similar reversible shift trend was also observed at the Fe K-edge, although the magnitude was not significant. Additionally, extended X-ray absorption fine structure (EXAFS) spectra of LNFMT are shown in Fig. 15e and f, where the distance between Ni–TM decreased during the charge process due to the oxidation of Ni ions. However, the length of the Fe–TM bonds remains almost unchanged during the charge–discharge process, reflecting that the local structure of FeO6 did not undergo significant changes. The maintenance of the unaltered atomic distance within the plane serves as an indirect testament to the effective suppression of Fe3+ migration. Fig. 15g presents first principles calculations of alterations observed during different Na+ extraction/insertion processes. Under the same conditions for LFNMT, the binding energy between Li and O is weak, causing Li+ to tend to migrate before Fe3+, forming a pseudo-tetrahedral configuration that confines the motility of Fe3+ ions. More specifically, the instability of Li+ in the sodium tetrahedral body prompts it to migrate to the TM layer in subsequent electrochemical cycles, and this movement effectively inhibits the migration of Fe3+. The binding energy of Ti–O also remains relatively unchanged in the high-voltage range, which helps to maintain the local stability of O–TM–O. Therefore, the high entropy effect formed by the interaction of multiple metal ions in LFNMT ensures the stability of the crystal structure.
 | ||
| Fig. 15 Contour map of operando XRD patterns during the initial charge/discharge process at 1.5–4.5 V of (a) NFM and (b) LNFMT. XANES (c) Ni K-edge and (d) Fe K-edge spectra of LNFMT during the first charge/discharge process. EXAFS (e) Ni K-edge and (f) Fe K-edge spectra of LNFMT during the first charge/discharge process. (g) First principles calculations of bonding energy changes after different degrees of Na+ de/intercalation. (a)–(g) are reprinted with permission from ref. 147. Copyright 2024 Elsevier. | ||
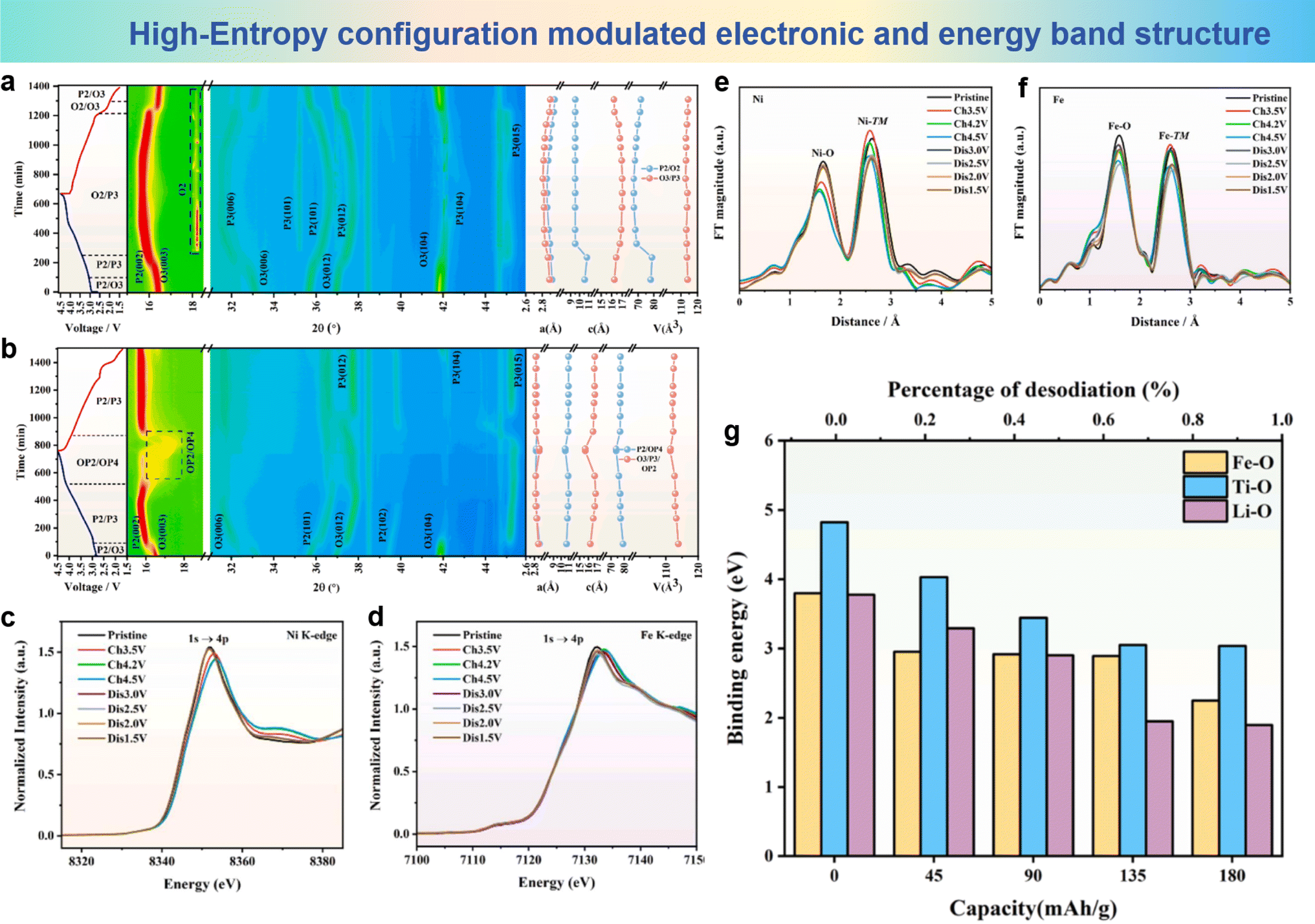
Another important advancement in the application of the high-entropy strategy in the cathode materials of SIBs is O3-NaNi1/3Fe1/3Mn1/3O2 (NFM), which is considered as a pioneer for commercialization.176–178 However, the working voltage of NFM is limited by the problem of iron migration above 4.2 V, which hinders further application. To address this problem, Zhang et al. proposed a high-entropy cathode material, NaMg0.08Cu0.12Ni0.2Fe0.2Mn0.2Ti0.2O2 (HNFM), which operates at a higher working voltage (4.3 V), and successfully suppressed the migration of iron ions.148 The Mössbauer spectra of Fe are show in Fig. 16a; as the Fe ions migrate into the tetrahedral positions within the Na layer, the O–Fe–O bond length in the FeO4 tetrahedron undergoes a substantial reduction in comparison to the O–vacancy–O bond length that exists in the tetrahedral site before the migration, thereby leading to a considerable decrement in the interlayer spacing of the Na layer. It is clearly observed that Fe ions migrate between layers at high voltage in NFM. As the sodium ion count diminishes progressively in the sodium layer, the energy level of the tetrahedral interstitial sites (characterized by a metastable, high-energy state) within the layer undergoes a decline, thereby elevating the likelihood of Fe ion migration towards these tetrahedral interstitial sites. Fig. 16b shows the plausible routes for Fe3+ ions to traverse from the octahedral sites within the TM layer to the tetrahedral sites in the sodium layer in both HNFM and NFM. The migration barriers for Fe3+ ions in both HNFM and NFM, calculated by density functional theory, are 0.75 eV and 0.47 eV, respectively (Fig. 16c). This indicates that Fe3+ ion migration is more difficult in HNFM compared to NFM. In situ XRD results show that NFM exhibits a phase transition mechanism encompassing the sequential transitions of O3–P-O′3–P-O3/P3 as illustrated in Fig. 16d, while HNFM undergoes a phase transition process of O3–O′3–P3 as illustrated in Fig. 16e, which indicates that HNFM can maintain structural stability at higher voltages and undergo reversible phase transitions. The findings indicate that the high-entropy approach significantly mitigates the mobility of Fe ions and enhances the structural stability of the material.
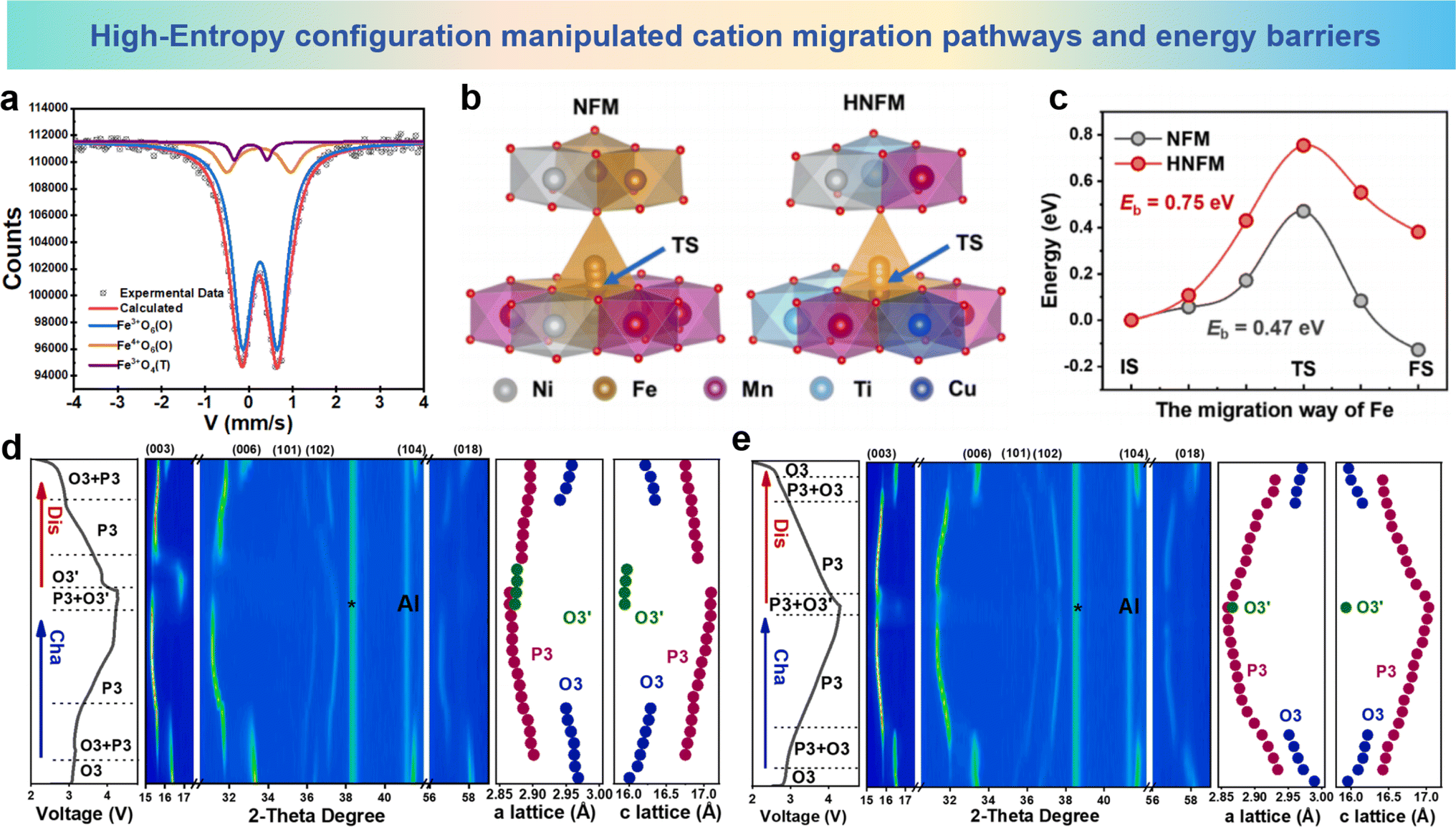 | ||
| Fig. 16 (a) Room-temperature Mössbauer spectrum of NFM treated with electrochemical sodium extraction. (b) The structural diagram of Fe migration from the octahedral site in the TM slabs to the tetrahedral site in the Na layer. (c) Fe migration energy barriers of HNFM and NFM (IS, TS, and FS respectively represent the initial state, transition state, and final state). 2D contour plots of in situ XRD during the structural evolution of (d) NFM and (e) HNFM cathodes. (a)–(e) are reprinted with permission from ref. 148. Copyright 2024 Elsevier. | ||
P2-type Na0.67Ni0.33Mn0.67O2 (NNMO) is considered as a promising cathode material for commercialization due to its open framework structure, high theoretical capacity, and ease of synthesis.179,180 When charged above 4.0 V, NNMO undergoes an anion redox reaction and delivers a capacity exceeding 200 mA h g−1. However, the redox reaction poses certain challenges, which leads to the formation of oxygen atoms with non-coordinated environments and non-bonding O 2p states within the lattice. These non-coordinated oxygen atoms and non-bonding states can cause detrimental cation migration and lattice oxygen distortion.181–183 To resolve these challenges, Liu et al. proposed the implementation of entropy adjustment and dual-site-substituted enhancement techniques to prepare Na0.67Zn0.05Ni0.22Cu0.06Mn0.66Ti0.01O2 (NZNCMTO) cathodes. The Zn formation energy results in Fig. 17a demonstrate that Zn ions prefer to reside within the Na layer, thereby reducing the formation of non-coordinated oxygen atoms and non-bonding states.184 The result of the Crystal Orbital Hamilton Population (COHP) values both prior to and following the process of Cu and Ti co-doping are shown in Fig. 17b. After doping, the COHP values of Mn and Ni are enhanced, indicating that doping strengthens the interaction between Mn/Ni and oxygen atoms. Furthermore, the incorporation of Cu and Ti into the system can disrupt the organized alignment of Ni and Mn in the TM layer, effectively reducing TM migration and improving material stability. The TM–O bond energies within NZNCMTO, as illustrated in Fig. 17c, reveal the augmentation of electrochemical cycling stability in the cathode materials, attributed to the incorporation of reinforced Cu–O bonds (1.8124 eV) and Ti–O bonds (2.1835 eV). To elucidate the impact of entropy modulation and dual-site substitution on the interaction mechanisms during charge/discharge of the NZNCMTO cathode material, in situ XRD tests were conducted as shown in Fig. 17d. The results show no new phases or transformations of OP4 or O2 phases occurring at the high voltage state (4.3 V), indicating a completely reversible evolution of the material during the process of charging and discharging. Moreover, the refined results demonstrate that NZNCMTO shows minimal variations in lattice parameters during the cycling process, with maximum variations of only 1.05%, 1.21%, and 0.78% (Fig. 17e). The near-zero strain alteration underscores the stabilizing influence of entropy modulation and dual-site substitution, which increases lattice stability and suppresses TM ion migration. In conclusion, the optimization of entropy and the employment of the dual-site doping methodologies hold significant influence in the realm of the NNMO material, reducing the formation of non-coordinated oxygen atoms and non-bonding states, suppressing detrimental cation migration, and enhancing lattice stability. The utilization of the high-entropy strategy offers a promising methodology for the design and fabrication of cathode materials with exceptional capacity and robust cycling stability for SIBs.
 | ||
| Fig. 17 (a) Formation energy and lattice diagram of Zn ions in the Na layer and TM layer. (b) Comparison of COHP values of Ni–O and Mn–O bonds in NZNCMTO and NNMO. (c) COHP results of entropy-tuned NZNCMTO. (d) In situ XRD patterns of entropy-tuned NZNCMTO. (e) Evolution of cell parameters and cell volume during the charge/discharge process for entropy-tuned NZNCMTO. (a)–(e) are reprinted with permission from ref. 184. Copyright 2024 Wiley. | ||
4.2 Structure design strategy
As is known to us all, the structural stability is a key factor for elaborating the performance and capability of electrodes during charge and discharge processes.185–188 By precisely tuning and designing the compositions and arrangements of TM layers in the electrode microstructure, the structural stability and cycle performance could be pronounced improved.189–192 The modulation of the cation migration rate, diffusion path and ionic conductivity can be realized by reasonable design of the material structure, which plays a crucial role in inhibiting the migration of cations.For the oxide cathodes of the same composition with different structures, the ordered honeycomb superlattice inhibits cation migration more effectively than the disordered structure. The Na3Ni2RuO6 cathode was prepared with ordered and disordered structures by different synthetic processes named ONNR (order) and DNNR (disorder).122 The structural characterization of the XRD Rietveld refinement results and the selected area electron diffraction (SAED) images of ONNR are shown in Fig. 18a, where the characteristic peak at 2θ ≈ 20° corresponding to the superlattice phase and SAED pattern of images projected along the [010] axis can be assigned to a standard O3-type crystal framework. In addition, the fine layered O3-type structure and the ordered structure of ONNR were confirmed by the HAADF images along the [310] and [312] directions in Fig. 18b. The in situ XRD patterns of the ONNR cathode are shown in Fig. 18c. When charged to 3.1 V, the O3-phase peaks decrease, while the intensity of the P3 phase peaks increase, resulting in the coexistence of two phases. Then the coexistence of the P3 and P′3-phases is maintained as the charging process continues. During the discharge process, ONNR exhibits a reversible transition of phases, specifically transitioning from P′3, P3 to O3, indicating the good cycle performance. The HAADF-STEM image taken after cycling and the corresponding FFT image for the both ordered and disordered cathodes are presented in Fig. 18d and e. Along the [310] axis, the TM atomic arrays exhibit a stratified configuration, organized in distinct layers, and the corresponding FFT image reveals the single crystalline property in the ONNR cathode. However, the DNNR cathode presenting a multi-crystalline structure and the degraded TM originating from the TM layers found in Na layers, demonstrate that some TMs migrated out of the metal-oxide layers after long-term cycling. The SAED patterns of the ONNR cathode in the pristine state charged to 3.8 V and discharged to 2.0 V are shown in Fig. 18f. In a pristine cathode material, an ordered superstructure and the superlattice of ONNR remain well-preserved after charge and discharge cycles. In addition, in the [111] orientation, the d-spacing of the (101) plane is 0.374 Å in the pristine state, increasing to 0.385 Å when charged to 3.8 V and decreasing to 0.373 Å when discharged to 2.0 V, pertaining to the augmentation of the crystalline structure during the extraction and intercalation of sodium. The depiction of the mechanism pertaining to both organized and disorganized cathodes is presented in Fig. 18g. The ordered arrangement of TM in layered oxides provides a more stable structure during long-term cycling compared to layered oxides. In ONNR cathode materials, TM ions exhibit an ordered honeycomb arrangement, providing a highly stable structure similar to the “ordered puzzle” connectivity, which can adjust the lattice by changing the crystal strain and suppress cation migration. In contrast, the arrangement of TM ions is irregular in DNNR cathode materials, which may lead to structural instability during Na-ion (de)intercalation into interlayers.
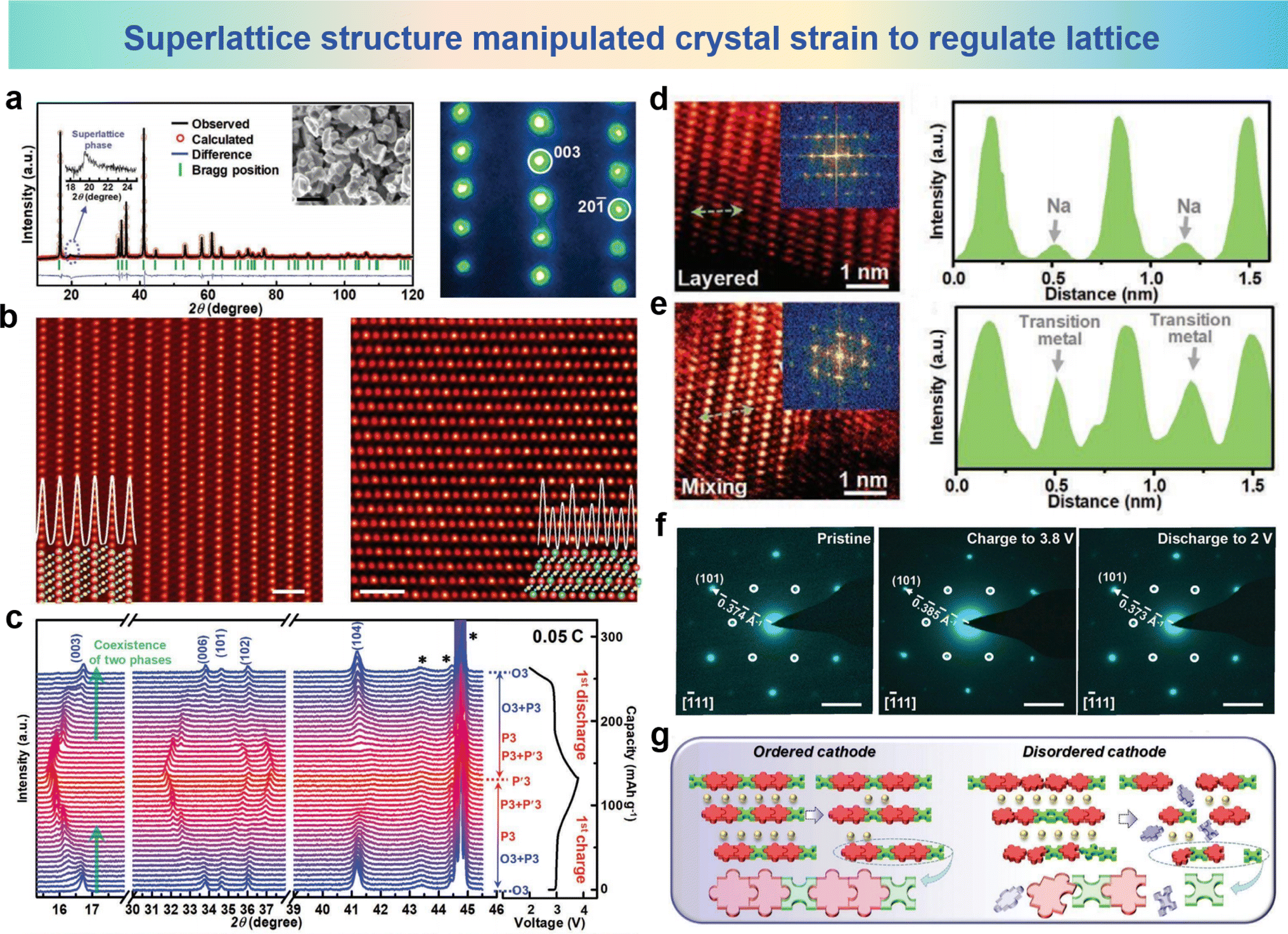 | ||
| Fig. 18 (a) XRD pattern and Rietveld plots (left) of NNR Na3Ni2RuO6 (magnified 2θ of 17.5°–25° and the SEM image was the inset) and SAED pattern (right) viewed along the [010] axis. (b) HAADF images of the NNR material oriented along the [010] (left) and [312] (right) axis and their corresponding atom models. (c) In situ XRD patterns of the NNR electrode in the 1st cycle at the voltage range of 2.0–3.8 V. HAADF-STEM image and FFT image of (d) ordered and (e) disordered cathodes (left) and the corresponding line profile (right) of the STEM image. (f) SAED patterns of the NNR cathode at pristine (left), charge to 3.8 V (middle) and discharge to 2.0 V (right) states. (g) Schematic diagram showing that the ordered cathode could perform better than the disordered cathode for long-term Na+ (de)intercalations. (a)–(g) are reprinted with permission from ref. 122. Copyright 2020 Wiley. | ||
A novel new queue-ordered superstructure is established in the O′3–NaMn0.6Al0.4O2 (NMA) cathode material.132 This superstructure enhances the layered structure, mitigates the impact of the Jahn–Teller effect, and inhibits the migration of TM during long-term cycles. The Rietveld XRD pattern of NMA is shown in Fig. 19a, and the purple star represents the ordered superlattices of metal ions in [Mn0.6Al0.4]O2. XRD patterns of NMA and NMO cathodes after 100 cycles are shown in Fig. 19b, suggesting that a significant portion of the novel NMA phase can revert to the O′3 phase configuration compared with the partial phase retentions of the cycled NMO cathode. As shown in Fig. 19c, the formation energy of NMA is more negative than that of NMO in the initial O′3 phase, which indicates the stable structure of the NMA electrode. Fig. 19d depicts further examination in the ordered configuration of the superlattice in NMA using SAED images, and the main diffraction points observed along the [001] axis present the O′3 phase, which exhibits a monoclinic crystal lattice configuration. Regular diffraction spots are clearly observed on the (200) and (110) planes, which belong to the superlattice structure. The corresponding line profiles of regions I and II exhibit superlattice positions. When selected SAED analysis is conducted along the [010] direction, these profiles become apparent with the spots of the superlattice being not obvious and the structure corresponding to the O′3 phase. Along the [001] and [010] axes the simulated crystal structure of the NMA electrode is shown in Fig. 19e, presents a “queue-ordered” arrangement of Mn3+ and Al3+ with a consistent proportion of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 in the TM layer of the NMA material. And it could also be observed that the Na layer and [Mn0.6Al0.4]O2 slabs present the conventional configuration of the O′3 phase. As shown in Fig. 19f, HRTEM images of NMA and NMO after 100 cycles display pronounced differences in the arrangement of TM slabs. Contrary to NMO, where some TM ions migrate from the TM layers, the NMA electrode inhibits the migration of TM ions due to the “queue-ordered” structure, which firmly maintains the slab structure. According to the above results, the “queue-ordered” arrangement in [Mn0.6Al0.4]O2 slabs could strengthen the TM layers and suppress cation migration of the NMA electrode and maintain good structural stability during cycling.
:2 in the TM layer of the NMA material. And it could also be observed that the Na layer and [Mn0.6Al0.4]O2 slabs present the conventional configuration of the O′3 phase. As shown in Fig. 19f, HRTEM images of NMA and NMO after 100 cycles display pronounced differences in the arrangement of TM slabs. Contrary to NMO, where some TM ions migrate from the TM layers, the NMA electrode inhibits the migration of TM ions due to the “queue-ordered” structure, which firmly maintains the slab structure. According to the above results, the “queue-ordered” arrangement in [Mn0.6Al0.4]O2 slabs could strengthen the TM layers and suppress cation migration of the NMA electrode and maintain good structural stability during cycling.
 | ||
| Fig. 19 (a) XRD profiles and Rietveld plots of NMA (inset SEM image of NMA). (b) XRD patterns of NMA and NMO cathodes after 100 cycles. (c) DFT-calculated formation energy of NMA and NMO. (d) SAED patterns of prepared NMA samples viewed along the [001] axis (left) and [010] axis (right). (Middle I and II are the corresponding line-profiles of the region in white rectangles in SAED patterns). (e) The simulated crystal structure of NMA along the [001] and [010] axes. (f) HRTEM images and amplified HRTEM image (inset) of NMA (left) and NMO (right) electrodes including line-profiles of relevant regions (middle I and II). (a)–(f) are reprinted with permission from ref. 132. Copyright 2021 Wiley. | ||
Kuang et al. introduced Cu into ordered/disordered O3-Nax(NiySb1−y)O2 to replace Ni, resulting in the ordered NaNi0.5Cu0.17Sb0.33O2 (ONNCSO0.17) and the disordered Na0.8Ni0.43Cu0.17Sb0.4O2 (D-NNCSO0.17).193 Kuang et al. discovered the irreversible migration of Cu ions from the TM layers to the Na layers in ordered ONNCSO0.17. The structural schematic and refined XRD results are shown in Fig. 20a and b. It can be observed that when the Ni/Sb molar ratio is 2/1, i.e. NaNi0.67Sb0.33O2 (O-NNSO), a common honeycomb-ordering structure is formed within the TM layers. In Na0.8Ni0.6Sb0.4O2 (D-NNSO) corresponding to a 3/2 ratio, the arrangement of Ni/Sb becomes completely disordered. HRTEM measurements were further conducted on the electrode cycled between 2.0 and 4.5 V, and from Fig. 20c and d, the distorted lattice and blurry FFT spots of O-NNCSO0.17 can be observed, whereas D-NNCSO0.17 does not exhibit these characteristics. The irreversible migration of Cu ions to the Na layer is the cause of this phenomenon, and the migration of Cu ions only occurs in the honeycomb ordered compound, not in the disordered compound. Observations along the [100] direction of the initially charged O-NNCSO0.17 and D-NNCSO0.17 (Fig. 20e and f) show clear TM layers (bright) and Na layers (dark). Elemental line scan analysis was performed along the white arrows. From Fig. 20e, it can be seen that in O-NNCSO0.17, some signal peaks of Cu appear in the interlayers, while Ni is mainly present in the TM layer. However, for D-NNCSO0.17, in Fig. 20f no signal peaks of Cu and Ni elements are observed within the TM layer. HRTEM images and the corresponding intensity-profiles after 50 cycles between 2.0 and 4.0 V are shown in Fig. 20g and h, and there are no heavy signals within the TM layer for both samples. It can be seen from Fig. 20i that a heavy signal within the TM layer in the HRTEM image and the corresponding intensity profile of O-NNCSO0.17 after 50 cycles between 2.0 and 4.5 V are attributed to the migration of Cu from the TM layer to the Na layer. In contrast, in Fig. 20j, the cycled D-NNCSO0.17 shows a complete TM plate with no visible signals. The above analysis indicates that the irreversible Cu migration occurs in the ordered TM phase and the voltage is over 4.0 V, rather than in the disordered phase (Fig. 20k).
 | ||
| Fig. 20 Crystal structure of honeycomb-ordered (left) and XRD Rietveld refinement patterns (right) for (a) O-NNCSO0.17 and (b) D-NNCSO0.17. HRTEM images and FFT patterns of (c) O-NNCSO0.17 and (d) D-NNCSO0.17 after 50 cycles between 2.0 and 4.5 V. HAADF-STEM images and corresponding STEM-EDS line profiles of Cu and Ni elements along the white arrow of (e) O-NNCSO0.17 and (f) D-NNCSO0.17 viewed along the [100] direction. HRTEM images along the [100]/[010] direction (left) and corresponding intensity-profiles (right) of (g) O-NNCSO0.17 and (h) D-NNCSO0.17 after 50 cycles between 2.0 and 4.0 V. HRTEM images along the [100] direction (left) and corresponding intensity-profiles (right) of (i) O-NNCSO0.17 and (j) D-NNCSO0.17 after 50 cycles between 2.0 and 4.5 V. (k) Schematic diagrams of crystal structures of TM ordered compounds and disordered compounds. (a)–(k) are reprinted with permission from ref. 193. Copyright 2024 Elsevier. | ||
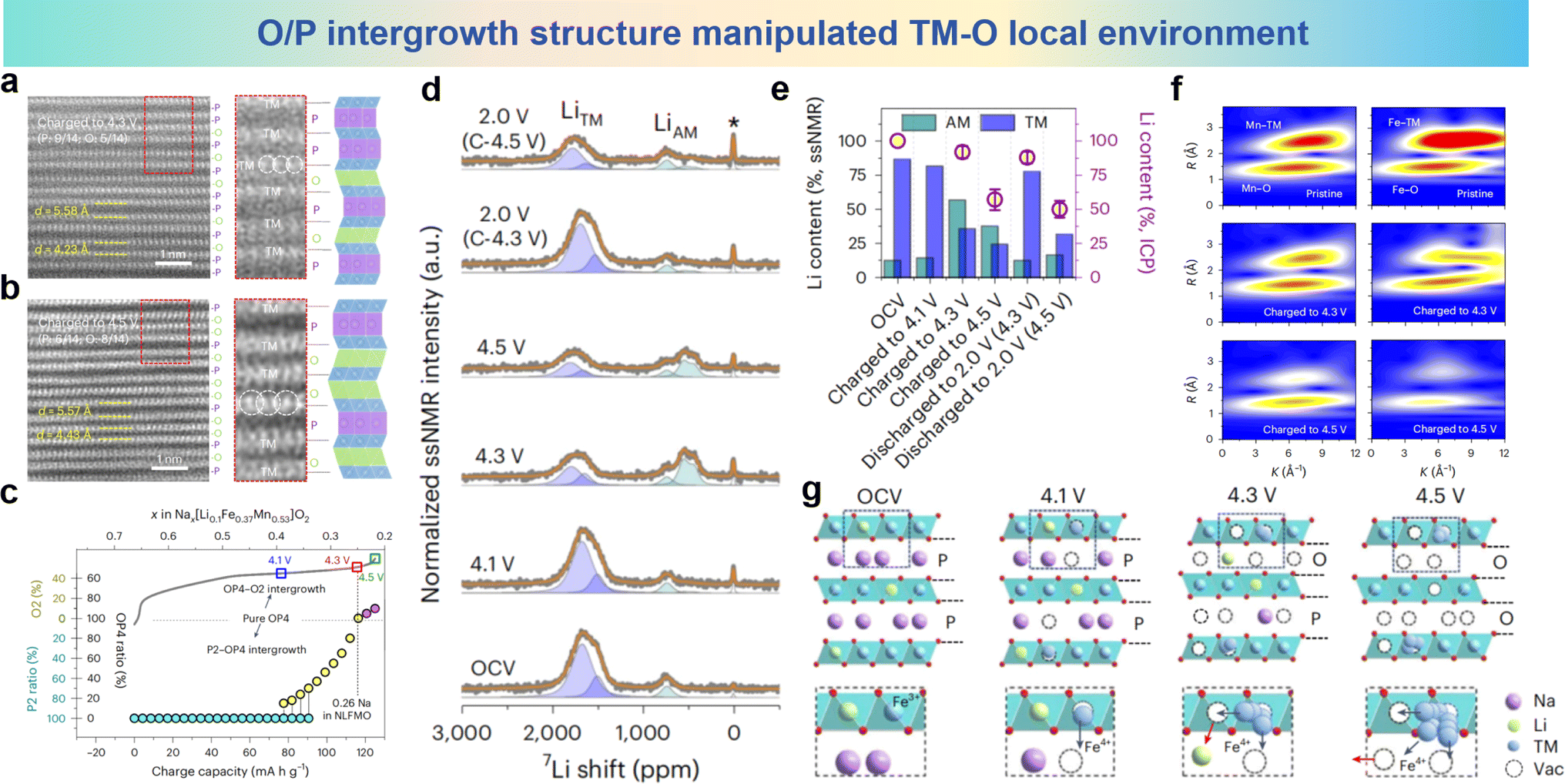 | ||
| Fig. 21 STEM images of different charged states at (a) 4.3 V and (b) 4.5 V, collected in HAADF mode. (c) Quantitative and qualitative assignment of the structural evolution of the P2–OP4–O2 intergrowth structures during the charging process (OCV to 4.5 V). (d) 7Li pj-MATPASS ssNMR spectra obtained in various charge/discharge states (pristine, 4.1 V charge, 4.3 V charge, 4.5 V charge, discharge to 2.0 V after 4.3 V charging and discharge to 2.0 V after 4.5 V charging) for the initial cycle. (e) The Li content (in the lattice of NLFMO) quantified by ssNMR (AM layer, green bars; TM layer, blue bars) and ICP (violet circles). (f) The WT-EXAFS spectra of NLFMO in different charge states. (g) Schematic diagrams of the structural evolution during desodiation. (a)–(g) are reprinted with permission from ref. 154. Copyright 2024 Nature Research. | ||
| Cathode material | Voltage range | Specific capacity | Capacity retention | Ref. |
|---|---|---|---|---|
| O3–Na4FeRuO6 | 2.0–4.0 V | 120 mA h g−1 @ 0.2C (1C = 100 mA g−1) | 80%/2C, 100 cycles | 110 |
| NaNi0.45Zn0.05Mn0.4Ti0.1O2 | 2.0–4.5 V | 187 mA h g−1 @ C/10 | 85%/C/10, 100 cycles | 106 |
| Na0.8Li0.2Fe0.2Mn0.6O2 | 1.5–4.5 V | 165 mA h g−1 @ 10 mA g−1 | 98.3%/200 mA g−1, 100 cycles | 97 |
| Na0.898K0.058Ni0.396Fe0.098Mn0.396Ti0.092O2 | 2.0–4.0 V | 138.6 mA h g−1 @ 5 mA g−1 | 92.8%/200 mA g−1, 500 cycles | 98 |
| Na0.85Cr0.85Ru0.10Ti0.05O2 | 1.5–3.92 V | 91.7 mA h g−1 @ 10 A g−1 | 86.2%/1 A g−1, 500 cycles | 95 |
| NaNi0.25Mg0.05Cu0.1Fe0.2Mn0.2Ti0.1Sn0.1O2 | 2.0–4.0 V | 130.8 mA h g−1 @ 0.1C (14 mA g−1) | 75%/1C, 500 cycles | 184 |
| NaMg0.08Cu0.12Ni0.2Fe0.2Mn0.2Ti0.2O2 | 2.0–4.3V | 131 mA h g−1 @ 0.2C (1C = 120 mA g−1) | 84%/1C, 200 cycles | 148 |
| Na0.85Li0.05Ni0.3Fe0.1Mn0.5Ti0.05O2 | 2.0–4.2V | 116 mA h g−1 @ 10C (1C = 150 mA g−1) | 94.23%/0.2C, 150 cycles | 147 |
| Na3Ni2RuO6 | 2.0–3.8V | 123 mA h g−1 @ 0.05C (1C = 100 mA g−1) | 81%/1C, 1000 cycles | 122 |
| NaMn0.6Al0.4O2 | 1.0–4.0 V | 160 mA h g−1 @ 20 mA g−1 | 81%/500 mA g−1, 500 cycles | 132 |
| Na0.67Li0.1Fe0.37Mn0.53O2 | 2.0–4.5V | 125.5 mA h g−1 @ 10 mA g−1 | 84.5%/100 mA g−1, 100 cycles | 154 |
5. Conclusion and prospects
In summary, this paper provides an overview of the cation migration phenomenon in layered oxide cathode materials and its impact on the performance of SIBs. Numerous research studies have shown that cation migration can lead to irreversible phase transitions, structural distortions, surface cracks and detrimental effects, resulting in a sharp decline in specific capacity and poor cycling stability.194–196 Extensive research and exploration have been conducted in previous studies. The historical background and current progress and strategies to suppress the structural instability caused by irreversible cation migration have been summarized. One strategy is doping effect to improve material performance, which can alter the electronic structure and ionic conductivity of the material, thereby suppressing cation migration.106 And the high-entropy effect can modify the crystal structure and chemical environment of the material, enhancing its stability and cycling lifespan.171 Another strategy is to design and synthesize novel layered oxide cathode materials with optimized structures that minimize cation migration.132 For example, by controlling factors such as the crystal structure, particle size, and interfaces, the rate of cation migration can be reduced, leading to improved cycling stability. This review provides guidance for the development of advanced materials with high specific capacity and stable cycling performance.Although great advances have been made in exploring and suppressing TM migration in SIBs, the current understanding of the underlying mechanism is still insufficient, and the methods to suppress cation migration are relatively limited. It is essential to deeply investigate the mechanisms and migration behavior of cations and to explore new methods of inhibiting cation migration, which will provide important guidance for the design and development of new positive electrode materials.
Firstly, further study on the migration pathways and mechanisms of cations in the layered oxide cathode materials during charging and discharging processes is helpful to design appropriate strategies to suppress harmful migration. A deeper understanding of the migration of TM ions is beneficial for the design of stable anion redox in layered oxide cathodes.
Secondly, methods such as element substitution and structural design can continue to be explored. Irreversible migration can be effectively suppressed by the introduction of specific elements to substitute some elements in the original structure, or by carefully designing the crystal structure and interfacial properties, thereby improving the structural stability and cycling performance of the material.197–199 Furthermore, the optimization of the ionic conductivity of the material is crucial. Manipulation of ion transport pathways and rates can slow the rate of cation migration, further enhancing the stability and cycling lifespan of the material. Local chemical modulation can be employed to regulate electron density and coordination environment near the TM ions. Additionally, for materials involved in anion redox reactions, cation migration is crucial for activating anion redox activity. It is expected that the combined application of these strategies will provide important guidance and foundations for the development of high-performance and stable cathode materials.
Thirdly, advanced techniques can be employed to understand the microscopic mechanism of cation migration as well as guide the design and synthesis of materials. As shown in Fig. 22, ABF-STEM and HAADF-STEM images and signal profiles can be used to observe the microstructure and atomic arrangement of materials, thereby revealing the positions and migration behavior in layered oxide cathode materials for SIBs, and providing some new insights for predicting and inhibiting cation migration. For cations that have migrated, the STEM-EDS mapping can provide relative abundance information of different elements, indicating the content ratio of elements in different regions. By comparing the elemental abundance in different regions, it is possible to understand the distribution differences and migration behavior of cations, providing some insights when designing new cathode materials. In terms of theoretical simulation, DFT calculations can be used to compute the energy barriers for cation migration within a lattice and be utilized to simulate the migration pathways of cations within materials, providing information about the ease and better understanding of cation migration, which is of great significance for the design and development of cathode materials. In addition, NMR (Nuclear Magnetic Resonance) technology can provide information about the chemical environment, migration rate, and position within the crystal structure of cations, which is crucial for understanding the migration behaviors of cations in materials and their impact on material performance. The combination of the above with ex/in situ XRD allows for a more in-depth analysis of the phase transitions and interaction mechanisms during charging and discharging of the material, including the individual cell parameters and volume changes. By combining these advanced techniques, researchers can obtain detailed information about cation migration pathways, interface reactions, and crystal structure changes, which will provide further guidance for material design and performance optimization, contributing to the development of more stable and high-performance cathode materials for SIBs.
 | ||
| Fig. 22 Advanced characterization techniques for detecting cation migration. | ||
Finally, as cation migration has an important effect on the cycling performance and stability of layered oxide cathodes, ongoing research and innovation is needed to develop high-performance and stable layered oxide cathode materials for SIBs in order to support their commercialization.153,200,201 Research into new synthesis methods, material structure design, and surface modification techniques which may positively regulate the ion migration in layered oxide cathodes will effectively increase the cycle life of cathode materials and reduce the capacity degradation problems associated with long term cycling of SIBs. Additionally, increasing energy density and capacity is an important goal for advancing the commercialization of SIBs, which can be achieved through the investigation of new materials, such as high-capacity or nanostructured materials for SIBs. Another challenge in layered oxide cathodes is cation migration, and the cycling performance and stability of layered oxide cathodes can be addressed by conducting in-depth research into migration mechanisms and implementing appropriate strategies to suppress harmful migration.202–207 As can be seen in Fig. 23, SIBs have the potential to be commercially applied in the field of energy storage through continuous research, innovation, and targeted solutions for technical challenges. They can provide reliable energy storage solutions for the widespread use of renewable energy and electric vehicles, contributing to the transition to clean energy and sustainable development. In summary, we believe that a thorough understanding of TM migration will gradually mitigate the negative effect and promote the development of SIB layered oxides to provide more efficient and stable solutions in the field of sustainable energy storage.
 | ||
| Fig. 23 Prospects of layered oxide cathodes for SIBs to suppress cation migration and further applications to achieve high energy density, cycling stability, reversible phase transitions and oxygen-redox. | ||
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Author contributions
Zhuang-Chun Jian: writing original draft, conceptualization and supervision; Jun-Xu Guo: writing original draft; Yi-Feng Liu: software; Yan-Fang Zhu: writing-review and editing; Jingqiang Wang: conceptualization and supervision; Yao Xiao: conceptualization, funding acquisition, supervision, and writing-review and editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52402301, 52472240, and 52202284), Natural Science Foundation of Zhejiang Province (LQ23E020002), Wenzhou Key Scientific and Technological Innovation Research Project (ZG2023053), and Wenzhou Natural Science Foundation (G20220019 and G20220021).References
- C. Zhao, Q. Wang, Z. Yao, J. Wang, B. Sánchez-Lengeling, F. Ding, X. Qi, Y. Lu, X. Bai, B. Li, H. Li, A. Aspuru-Guzik, X. Huang, C. Delmas, M. Wagemaker, L. Chen and Y.-S. Hu, Science, 2020, 370, 708–711 CrossRef CAS PubMed
.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC
- Z. Wu, G. Zeng, J. Yin, C.-L. Chiang, Q. Zhang, B. Zhang, J. Chen, Y. Yan, Y. Tang, H. Zhang, S. Zhou, Q. Wang, X. Kuai, Y.-G. Lin, L. Gu, Y. Qiao and S.-G. Sun, ACS Energy Lett., 2023, 8, 4806–4817 CrossRef CAS
- Q. Zhang, Y. Wang, Q. Deng, Y. Chu, P. Dong, C. Chen, Z. Wang, Z. Xia and C. Yang, Angew. Chem., Int. Ed., 2024, 63, e202401716 CrossRef CAS PubMed
- J. Yao, Y. Li, T. Xiong, Y. Fan, L. Zhao, X. Cheng, Y. Tian, L. Li, Y. Li, W. Zhang, P. Yu, P. Guo, Z. Yang, J. Peng, L. Xue, J. Wang, Z. Li, M. Xie, H. Liu and S. Dou, Angew. Chem., Int. Ed., 2024, 63, e202407898 CrossRef CAS PubMed
- Y. Liu, T. Yu, S. Xu, Y. Sun, J. Li, X. Xu, H. Li, M. Zhang, J. Tian, R. Hou, Y. Rao, H. Zhou and S. Guo, Angew. Chem., Int. Ed., 2024, 63, e202403617 CrossRef CAS PubMed
- Z. Zhuang, J. Li, H. Ji, Z. Piao, X. Wu, G. Ji, S. Liu, J. Ma, D. Tang, N. Zheng, J. Wang and G. Zhou, Adv. Mater., 2024, 36, 2313144 CrossRef CAS PubMed
- J. Wang, Y.-F. Zhu, Y. Su, J.-X. Guo, S. Chen, H.-K. Liu, S.-X. Dou, S.-L. Chou and Y. Xiao, Chem. Soc. Rev., 2024, 53, 4230–4301 RSC
- J.-J. Marie, R. A. House, G. J. Rees, A. W. Robertson, M. Jenkins, J. Chen, S. Agrestini, M. Garcia-Fernandez, K.-J. Zhou and P. G. Bruce, Nat. Mater., 2024, 23, 818–825 CrossRef CAS PubMed
- D. M. Davies, M. G. Verde, O. Mnyshenko, Y. R. Chen, R. Rajeev, Y. S. Meng and G. Elliott, Nat. Energy, 2018, 4, 42–50 CrossRef
- T. Cui, J. Xu, X. Wang, L. Liu, Y. Xiang, H. Zhu, X. Li and Y. Fu, Nat. Commun., 2024, 15, 4742 CrossRef CAS PubMed
- J. Zhang, J. Chou, X. X. Luo, Y. M. Yang, M. Y. Yan, D. Jia, C. H. Zhang, Y. H. Wang, W. P. Wang, S. J. Tan, J. C. Guo, Y. Zhao, F. Wang, S. Xin, L. J. Wan and Y. G. Guo, Angew. Chem., Int. Ed., 2023, 63, e202316087 CrossRef PubMed
- Y. Wu, Z. Zeng, M. Liu, C. Cai, S. Lei, H. Zhang, S. Cheng and J. Xie, Adv. Energy Mater., 2024, 14, 2401037 CrossRef CAS
- C. Shen, Y. Liu, L. Hu, W. Li, X. Liu, Y. Shi, Y. Jiang, B. Zhao and J. Zhang, Nano Energy, 2022, 101, 107555 CrossRef CAS
- X.-H. Meng, D. Xiao, Z.-Y. Zhou, W.-Z. Liu, J.-L. Shi, L.-J. Wan and Y.-G. Guo, J. Am. Chem. Soc., 2024, 146, 14889–14897 CrossRef CAS PubMed
- J. Tian, G. Wang, W. Zeng, J. Zhu, W. Tian, S. Zhang, Y. Zhang, J. Wang, Q. Li, H. Zhao, C. Li, X. Li, L. Chen and S. Mu, Energy Storage Mater., 2024, 68, 103344 CrossRef
- F. Zou, J.-B. Kim, J. Zhang, G.-H. Lee, L. Lyu, J.-H. Choi, T. Kankaanpää, Y. M. Lee and Y.-M. Kang, Energy Environ. Sci., 2024, 17, 4319–4326 RSC
- X. Xu, S. Chu, S. Xu, S. Guo and H. Zhou, Energy Environ. Sci., 2024, 17, 3052–3059 RSC
- M. Wang, Y. Yao, Y. Tian, Y. Yuan, L. Wang, F. Yang, J. Ren, X. Hu, F. Wu, S. Zhang, J. Wu and J. Lu, Adv. Mater., 2023, 35, 2210658 CrossRef CAS PubMed
- H.-H. Ryu, H.-W. Lim, S. G. Lee and Y.-K. Sun, Nat. Energy, 2023, 9, 47–56 CrossRef
- J. Ren, H. Zhu, Y. Fang, W. Li, S. Lan, S. Wei, Z. Yin, Y. Tang, Y. Ren and Q. Liu, Carbon Neutralization, 2023, 2, 339–377 CrossRef CAS
- G. T. Park, S. B. Kim, J. I. Yoon, N. Y. Park, M. C. Kim, S. M. Han, D. H. Kim, M. S. Kim and Y. K. Sun, Adv. Energy Mater., 2024, 14, 2400130 CrossRef CAS
- M. Juelsholt, J. Chen, M. A. Pérez-Osorio, G. J. Rees, S. De Sousa Coutinho, H. E. Maynard-Casely, J. Liu, M. Everett, S. Agrestini, M. Garcia-Fernandez, K.-J. Zhou, R. A. House and P. G. Bruce, Energy Environ. Sci., 2024, 17, 2530–2540 RSC
- Y. S. Jiang, Z. M. Liao, F. D. Yu, W. Ke, X. Y. Li, Y. Xia, G. J. Xu, G. Sun, Y. G. Xia, W. Yin, L. Deng, L. Zhao and Z. B. Wang, Adv. Mater., 2024, 36, 2405238 CrossRef PubMed
- Q. Gan, N. Qin, H. Guo, F. Zhang, H. Yuan, W. Luo, Z. Li, Y. Li, L. Lu, Z. Xu, L. Wang, J. Lu and Z. Lu, ACS Energy Lett., 2024, 9, 1562–1571 CrossRef CAS
- Z. Dai, Y. Liu, X. Lu, H. Zhao and Y. Bai, Adv. Mater., 2024, 36, 2313500 CrossRef CAS PubMed
- Y. Chu, Y. Mu, H. Gu, Y. Hu, X. Wei, L. Zou, C. Yu, X. Xu, S. Kang, K. Li, M. Han, Q. Zhang and L. Zeng, Adv. Mater., 2024, 36, e2405628 CrossRef PubMed
- X. Liang, J. Y. Hwang and Y. K. Sun, Adv. Energy Mater., 2023, 13, 2301975 CrossRef CAS
- S. Zhang, J. Wu, N. Jiang, H. Sun, H. Yang, L. Shen, M. Zhou, W. Liu, H. Zhou and H. Zhao, Adv. Energy Mater., 2024, 14, 2401123 CrossRef CAS
- X. Xiang, K. Zhang and J. Chen, Adv. Mater., 2015, 27, 5343–5364 CrossRef CAS PubMed
- Y. Huang, W. Zhang, Y. Zhou, Y. Wang, L. Li, H. Shao, X. Li, Z. Hong, H. Xia, Y. Shen and L. Chen, ACS Nano, 2024, 18, 13106–13116 CrossRef CAS PubMed
- P. Vanaphuti, Z. Yao, Y. Liu, Y. Lin, J. Wen, Z. Yang, Z. Feng, X. Ma, A. C. Zauha, Y. Wang and Y. Wang, Small, 2022, 18, 2201086 CrossRef CAS PubMed
- Y. Han, W. Xie, G. T. Hill, P. Smeets, X. Hu, G. Yan, S. Zou, J. Liu, R. Wu, F. Shi, H. Zhou, P. Canepa and C. Liu, Nat. Mater., 2024, 23, 951–959 CrossRef CAS PubMed
- Q. Xu, Z. Liu, Y. Jin, X. Yang, T. Sun, T. Zheng, N. Li, Y. Wang, T. Li, K. Wang and J. Jiang, Energy Environ. Sci., 2024, 17, 5451–5460 RSC
- F. Zhang, B. He, Y. Xin, T. Zhu, Y. Zhang, S. Wang, W. Li, Y. Yang and H. Tian, Chem. Rev., 2024, 124, 4778–4821 CrossRef CAS PubMed
- J. Wang, Q.-Q. Sun, J. Yu, J.-X. Guo, N.-K. Mo, H.-W. Li, Y. Su, S. Zhao, Y.-F. Zhu, H. Chu, S. Dou and Y. Xiao, Composites, Part B, 2024, 284, 111664 CrossRef CAS
- K. Kawai, X.-M. Shi, N. Takenaka, J. Jang, B. M. de Boisse, A. Tsuchimoto, D. Asakura, J. Kikkawa, M. Nakayama, M. Okubo and A. Yamada, Energy Environ. Sci., 2022, 15, 2591–2600 RSC
- X. H. Meng, X. D. Zhang, H. Sheng, M. Fan, T. Lin, D. Xiao, J. Tian, R. Wen, W. Z. Liu, J. L. Shi, L. J. Wan and Y. G. Guo, Angew. Chem., Int. Ed., 2023, 62, e202302170 CrossRef CAS PubMed
- H. Ren, Y. Li, Q. Ni, Y. Bai, H. Zhao and C. Wu, Adv. Mater., 2022, 34, 2106171 CrossRef CAS PubMed
- N. Hong, J. Li, H. Wang, X. Hu, B. Zhao, F. Hua, Y. Mei, J. Huang, B. Zhang, W. Jian, J. Gao, Y. Tian, X. Shi, W. Deng, G. Zou, H. Hou, Z. Hu, Z. Long and X. Ji, Adv. Funct. Mater., 2024, 34, 2402398 CrossRef CAS
- Y. Qiao, S. Guo, K. Zhu, P. Liu, X. Li, K. Jiang, C.-J. Sun, M. Chen and H. Zhou, Energy Environ. Sci., 2018, 11, 299–305 RSC
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed
- Y. Liu, S. Roy, S. Sarkar, J. Xu, Y. Zhao and J. Zhang, Carbon Energy, 2021, 3, 795–826 CrossRef CAS
- J. Peng, W. Zhang, Q. Liu, J. Wang, S. Chou, H. Liu and S. Dou, Adv. Mater., 2022, 34, 2108384 CrossRef CAS PubMed
- C. X. Zhao, J. N. Liu, B. Q. Li, D. Ren, X. Chen, J. Yu and Q. Zhang, Adv. Funct. Mater., 2020, 30, 2003619 CrossRef CAS
- Q. Liu, Z. Hu, M. Chen, C. Zou, H. Jin, S. Wang, S. L. Chou, Y. Liu and S. X. Dou, Adv. Funct. Mater., 2020, 30, 1909530 CrossRef CAS
- Z. Li, L. Qiu, P. Li, H. Liu, D. Wang, W. Hua, T. Chen, Y. Song, F. Wan, B. Zhong, Z. Wu and X. Guo, J. Mater. Chem. A, 2024, 12, 7777–7787 RSC
- W. Zuo, X. Liu, J. Qiu, D. Zhang, Z. Xiao, J. Xie, F. Ren, J. Wang, Y. Li, G. F. Ortiz, W. Wen, S. Wu, M.-S. Wang, R. Fu and Y. Yang, Nat. Commun., 2021, 12, 4903 CrossRef CAS PubMed
- W. Zhao, S. Lei, J. Li, F. Jiang, T. Wu, Y. Yang, W. Sun, X. Ji and P. Ge, Adv. Energy Mater., 2024, 14, 2304431 CrossRef CAS
- Y. Xiao, Y.-F. Zhu, L. Li, P.-F. Wang, W. Zhang, C. Li, S.-X. Dou and S.-L. Chou, Cell Rep. Phys. Sci., 2021, 2, 100547 CrossRef CAS
- Y. Xiao, J. Xiao, H. Zhao, J. Li, G. Zhang, D. Zhang, X. Guo, H. Gao, Y. Wang, J. Chen, G. Wang and H. Liu, Small, 2024, 20, 2401957 CrossRef CAS PubMed
- Q. Zhang, Z. Zheng, R. Gao, X. Xiao, M. Jiao, B. Wang, G. Zhou and H. M. Cheng, Adv. Mater., 2024, 36, 2401018 CrossRef CAS PubMed
- Z.-C. Jian, Y.-F. Liu, Y.-F. Zhu, J.-Y. Li, H.-Y. Hu, J. Wang, L.-Y. Kong, X.-B. Jia, H.-X. Liu, J.-X. Guo, M.-Y. Li, Y.-S. Xu, J.-F. Mao, S.-L. Zhang, Y. Su, S.-X. Dou, S.-L. Chou and Y. Xiao, Nano Energy, 2024, 125, 109528 CrossRef CAS
- X. Xia, T. Liu, C. Cheng, H. Li, T. Yan, H. Hu, Y. Shen, H. Ju, T. S. Chan, Z. Wu, Y. Su, Y. Zhao, D. Cao and L. Zhang, Adv. Mater., 2022, 35, 2209556 CrossRef PubMed
- J. Yu, R. Ran, Y. Zhong, W. Zhou, M. Ni and Z. Shao, Energy Environ. Mater., 2020, 3, 121–145 CrossRef CAS
- Y. Sun, S. Guo and H. Zhou, Energy Environ. Sci., 2019, 12, 825–840 RSC
- H. Wang, H. Chen, Y. Mei, J. Gao, L. Ni, N. Hong, B. Zhang, F. Zhu, J. Huang, K. Wang, W. Deng, D. S. Silvester, C. E. Banks, S. Yasar, B. Song, G. Zou, H. Hou and X. Ji, ACS Nano, 2024, 18, 13150–13163 CrossRef CAS PubMed
- H. Su, G. Guo, Y. Ren, X. Yu, X. Zhang, T. Ma, Y. Lu, Z. Zhang, H. Ma, M. Sui, H. Li, C. Sun, Z. Chen, G. Xu, R. Wang, K. Amine and H. Yu, Energy Environ. Sci., 2020, 13, 4371–4380 RSC
- L. Mu, X. Feng, R. Kou, Y. Zhang, H. Guo, C. Tian, C. J. Sun, X. W. Du, D. Nordlund, H. L. Xin and F. Lin, Adv. Energy Mater., 2018, 8, 1801975 CrossRef
- J. Li, H. Hu, J. Wang and Y. Xiao, Carbon Neutralization, 2022, 1, 96–116 CrossRef
- G. G. Eshetu, G. A. Elia, M. Armand, M. Forsyth, S. Komaba, T. Rojo and S. Passerini, Adv. Energy Mater., 2020, 10, 2000093 CrossRef CAS
- L. Zhang, C. Guan, J. Zheng, H. Li, S. Li, S. Li, Y. Lai and Z. Zhang, Sci. Bull., 2023, 68, 180–191 CrossRef CAS PubMed
- Q. Lin, W. Guan, J. Meng, W. Huang, X. Wei, Y. Zeng, J. Li and Z. Zhang, Nano Energy, 2018, 54, 313–321 CrossRef CAS
- X. Cao, Y. Qiao, M. Jia, P. He and H. Zhou, Adv. Energy Mater., 2021, 12, 2003972 CrossRef
- M. Bianchini, J. Wang, R. J. Clément, B. Ouyang, P. Xiao, D. Kitchaev, T. Shi, Y. Zhang, Y. Wang, H. Kim, M. Zhang, J. Bai, F. Wang, W. Sun and G. Ceder, Nat. Mater., 2020, 19, 1088–1095 CrossRef CAS PubMed
- S. Chen, L. Sun, Y. Huang, D. Yang, M. Zhou and D. Zheng, Carbon Neutralization, 2023, 2, 494–509 CrossRef CAS
- C. Sun, X. Liao, F. Xia, Y. Zhao, L. Zhang, S. Mu, S. Shi, Y. Li, H. Peng, G. Van Tendeloo, K. Zhao and J. Wu, ACS Nano, 2020, 14, 6181–6190 CrossRef CAS PubMed
- S. Liu, X. Feng, X. Wang, X. Shen, E. Hu, R. Xiao, R. Yu, H. Yang, N. Song, Z. Wang, X. Yang and L. Chen, Adv. Energy Mater., 2018, 8, 1703092 CrossRef
- Y. Xiao, Y. Zhu, T. Gao, B. Zhong and X. Guo, Ionics, 2016, 23, 27–34 CrossRef
- X. Song, R. Liu, J. Jin, X. Zhao, Y. Wang, Q. Shen, Z. Sun, X. Qu, L. Jiao and Y. Liu, Energy Storage Mater., 2024, 69, 103377 CrossRef
- M. H. Han, E. Gonzalo, G. Singh and T. Rojo, Energy Environ. Sci., 2015, 8, 81–102 RSC
- M. Zhang, L. Qiu, W. Hua, Y. Song, Y. Deng, Z. Wu, Y. Zhu, B. Zhong, S. Chou, S. Dou, Y. Xiao and X. Guo, Adv. Mater., 2024, 36, 2311814 CrossRef CAS PubMed
- Y. Wang, J. Jin, X. Zhao, Q. Shen, X. Qu, L. Jiao and Y. Liu, Angew. Chem., Int. Ed., 2024, 63, e202409152 CrossRef CAS PubMed
- X. L. Li, J. Bao, Y. F. Li, D. Chen, C. Ma, Q. Q. Qiu, X. Y. Yue, Q. C. Wang and Y. N. Zhou, Adv. Sci., 2021, 8, 2004448 CrossRef CAS PubMed
- N. Li, W. Yin, B. Wang, F. Wang, X. Xiao, J. Zhao and E. Zhao, Energy Environ. Mater., 2023, 7, e12671 CrossRef
- S. Chu, C. Zhang, H. Xu, S. Guo, P. Wang and H. Zhou, Angew. Chem., Int. Ed., 2021, 60, 13366–13371 CrossRef CAS PubMed
- Z. Chen, Y. Deng, J. Kong, W. Fu, C. Liu, T. Jin and L. Jiao, Adv. Mater., 2024, 36, 2402008 CrossRef CAS PubMed
- X. Xu, S. Hu, Q. Pan, Y. Huang, J. Zhang, Y. Chen, H. Wang, F. Zheng and Q. Li, Small, 2023, 20, 2307377 CrossRef PubMed
- H. Ji, W. Ji, H. Xue, G. Chen, R. Qi, Z. Huang, H. Fang, M. Chu, L. Liu, Z. Ma, S. Xu, J. Zhai, W. Zeng, C. Schulz, D. Wong, H. Chen, J. Xu, W. Yin, F. Pan and Y. Xiao, Sci. Bull., 2023, 68, 65–76 CrossRef CAS PubMed
- D. Wu, X. Yang, S. Feng, Y. Zhu and M. Gu, Nano Lett., 2021, 21, 9619–9624 CrossRef CAS PubMed
- L. Zhang, J. Wang, J. Li, G. Schuck, M. Winter, G. Schumacher and J. Li, Nano Energy, 2020, 70, 104535 CrossRef CAS
- C. Zhao, C. Li, Q. Yang, Q. Qiu, W. Tong, S. Zheng, J. Ma, M. Shen and B. Hu, Energy Storage Mater., 2021, 39, 60–69 CrossRef
- C.-Y. Yu, J.-S. Park, H.-G. Jung, K.-Y. Chung, D. Aurbach, Y.-K. Sun and S.-T. Myung, Energy Environ. Sci., 2015, 8, 2019–2026 RSC
- Y. F. Zhu, Y. Xiao, W. B. Hua, S. Indris, S. X. Dou, Y. G. Guo and S. L. Chou, Angew. Chem., Int. Ed., 2020, 59, 9299–9304 CrossRef CAS PubMed
- A. Joshi, S. Chakrabarty, S. H. Akella, A. Saha, A. Mukherjee, B. Schmerling, M. Ejgenberg, R. Sharma and M. Noked, Adv. Mater., 2023, 35, 2304440 CrossRef CAS PubMed
- C. Ma, A. Abulikemu, J. Bao, T. Uchiyama, Y. Y. Xia, X. L. Li, Y. Uchimoto and Y. N. Zhou, Small, 2023, 19, 2302332 CrossRef CAS PubMed
- Y. Yang, J. Cai, Y. Zuo, K. Zhang, C. Gao, L. Zhou, Z. Chen, W. Chu and D. Xia, Energy Storage Mater., 2024, 71, 103587 CrossRef
- L. Yang, J. M. L. del Amo, Z. Shadike, S. M. Bak, F. Bonilla, M. Galceran, P. K. Nayak, J. R. Buchheim, X. Q. Yang, T. Rojo and P. Adelhelm, Adv. Funct. Mater., 2020, 30, 2003364 CrossRef CAS
- Y. Wang, L. Wang, H. Zhu, J. Chu, Y. Fang, L. Wu, L. Huang, Y. Ren, C. J. Sun, Q. Liu, X. Ai, H. Yang and Y. Cao, Adv. Funct. Mater., 2020, 30, 1910327 CrossRef CAS
- B. Peng, G. Wan, N. Ahmad, L. Yu, X. Ma and G. Zhang, Adv. Energy Mater., 2023, 13, 2300334 CrossRef CAS
- C. Hu, X. Lou, X. Wu, J. Li, Z. Su, N. Zhang, J. Li, B. Hu and C. Li, ACS Nano, 2024, 18, 13397–13405 CrossRef CAS PubMed
- L. Gan, X. G. Yuan, J. J. Han, X. Yang, L. Zheng, Z. Huang and H. R. Yao, Adv. Funct. Mater., 2022, 33, 2209026 CrossRef
- X.-Y. Zhang, H.-Y. Hu, X.-Y. Liu, J. Wang, Y.-F. Liu, Y.-F. Zhu, L.-Y. Kong, Z.-C. Jian, S.-L. Chou and Y. Xiao, Nano Energy, 2024, 128, 109905 CrossRef CAS
- P. Zou, L. Yao, C. Wang, S. J. Lee, T. Li and H. L. Xin, Angew. Chem., Int. Ed., 2023, 62, e20230462 Search PubMed
- S. Chu, D. Kim, G. Choi, C. Zhang, H. Li, W. K. Pang, Y. Fan, A. M. D'Angelo, S. Guo and H. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202216174 CrossRef CAS PubMed
- S. Chu and S. Guo, Adv. Funct. Mater., 2023, 34, 2313234 CrossRef
- X. Li, J. Xu, H. Li, H. Zhu, S. Guo and H. Zhou, Adv. Sci., 2022, 9, 2105280 CrossRef CAS PubMed
- S. Gao, Z. Zhu, H. Fang, K. Feng, J. Zhong, M. Hou, Y. Guo, F. Li, W. Zhang, Z. Ma and F. Li, Adv. Mater., 2024, 36, 2311523 CrossRef CAS PubMed
- Y. Li, K. A. Mazzio, N. Yaqoob, Y. Sun, A. I. Freytag, D. Wong, C. Schulz, V. Baran, A. S. J. Mendez, G. Schuck, M. Zając, P. Kaghazchi and P. Adelhelm, Adv. Mater., 2024, 36, 2309842 CrossRef CAS PubMed
- C. D. a. P. H. A. MAAZAZ and J. lncl, Phenom, 1983, 1, 45–51 Search PubMed
- H. Yoshida, N. Yabuuchi and S. Komaba, Electrochem. Commun., 2013, 34, 60–63 CrossRef CAS
- X. Li, Y. Wang, D. Wu, L. Liu, S.-H. Bo and G. Ceder, Chem. Mater., 2016, 28, 6575–6583 CrossRef CAS
- Y. Li, Y. Gao, X. Wang, X. Shen, Q. Kong, R. Yu, G. Lu, Z. Wang and L. Chen, Nano Energy, 2018, 47, 519–526 CrossRef CAS
- S. Guo, Y. Sun, P. Liu, J. Yi, P. He, X. Zhang, Y. Zhu, R. Senga, K. Suenaga, M. Chen and H. Zhou, Sci. Bull., 2018, 63, 376–384 CrossRef CAS PubMed
- X. Zhang, S. Guo, P. Liu, Q. Li, S. Xu, Y. Liu, K. Jiang, P. He, M. Chen, P. Wang and H. Zhou, Adv. Energy Mater., 2019, 9, 1900189 CrossRef
- S. Mariyappan, T. Marchandier, F. Rabuel, A. Iadecola, G. Rousse, A. V. Morozov, A. M. Abakumov and J.-M. Tarascon, Chem. Mater., 2020, 32, 1657–1666 CrossRef CAS
- Q. Wang, S. Mariyappan, G. Rousse, A. V. Morozov, B. Porcheron, R. Dedryvère, J. Wu, W. Yang, L. Zhang, M. Chakir, M. Avdeev, M. Deschamps, Y.-S. Yu, J. Cabana, M.-L. Doublet, A. M. Abakumov and J.-M. Tarascon, Nat. Mater., 2021, 20, 353–361 CrossRef CAS PubMed
- M. Jiang, G. Qian, X.-Z. Liao, Z. Ren, Q. Dong, D. Meng, G. Cui, S. Yuan, S.-J. Lee, T. Qin, X. Liu, Y. Shen, Y.-S. He, L. Chen, Y. Liu, L. Li and Z.-F. Ma, J. Energy Chem., 2022, 69, 16–25 CrossRef CAS
- C. Chen, Z. Ding, Z. Han, C. Liang, X. Lan, P. Wang, P. Gao and W. Wei, J. Phys. Chem. Lett., 2020, 11, 5464–5470 CrossRef CAS PubMed
- J. Xu, Z. Han, K. Jiang, P. Bai, Y. Liang, X. Zhang, P. Wang, S. Guo and H. Zhou, Small, 2019, 16, 1904388 CrossRef PubMed
- S.-H. Bo, X. Li, A. J. Toumar and G. Ceder, Chem. Mater., 2016, 28, 1419–1429 CrossRef CAS
- N. Yabuuchi, H. Yoshida and S. Komaba, Electrochem, 2012, 80, 716–719 CrossRef CAS
- K. Kubota, I. Ikeuchi, T. Nakayama, C. Takei, N. Yabuuchi, H. Shiiba, M. Nakayama and S. Komaba, J. Phys. Chem. C, 2014, 119, 166–175 CrossRef
- Z. Cheng, B. Zhao, Y. J. Guo, L. Yu, B. Yuan, W. Hua, Y. X. Yin, S. Xu, B. Xiao, X. Han, P. F. Wang and Y. G. Guo, Adv. Energy Mater., 2022, 12, 2103461 CrossRef CAS
- T. Cui, L. Liu, Y. Xiang, C. Sheng, X. Li and Y. Fu, J. Am. Chem. Soc., 2024, 146, 13924–13933 CrossRef CAS PubMed
- C. Fang, Y. Huang, W. Zhang, J. Han, Z. Deng, Y. Cao and H. Yang, Adv. Energy Mater., 2015, 6, 1501727 CrossRef
- S. Feng, Y. Lu, X. Lu, H. Chen, X. Wu, M. Wu, F. Xu and Z. Wen, Adv. Energy Mater., 2024, 14, 2303773 CrossRef CAS
- E. Gabriel, Z. Wang, V. V. Singh, K. Graff, J. Liu, C. Koroni, D. Hou, D. Schwartz, C. Li, J. Liu, X. Guo, N. C. Osti, S. P. Ong and H. Xiong, J. Am. Chem. Soc., 2024, 146, 15108–15118 CrossRef CAS PubMed
- K. Wang, H. Wan, P. Yan, X. Chen, J. Fu, Z. Liu, H. Deng, F. Gao and M. Sui, Adv. Mater., 2019, 31, 1904816 CrossRef CAS PubMed
- X. Liang and Y. K. Sun, Adv. Funct. Mater., 2022, 32, 2206154 CrossRef CAS
- D. Eum, B. Kim, J.-H. Song, H. Park, H.-Y. Jang, S. J. Kim, S.-P. Cho, M. H. Lee, J. H. Heo, J. Park, Y. Ko, S. K. Park, J. Kim, K. Oh, D.-H. Kim, S. J. Kang and K. Kang, Nat. Mater., 2022, 21, 664–672 CrossRef CAS PubMed
- Q. Li, S. Xu, S. Guo, K. Jiang, X. Li, M. Jia, P. Wang and H. Zhou, Adv. Mater., 2020, 32, 1907936 CrossRef CAS PubMed
- X. Li, A. Gao, Q. Zhang, H. Yu, P. Ji, D. Xiao, X. Wang, D. Su, X. Rong, X. Yu, H. Li, Y. S. Hu and L. Gu, Carbon Energy, 2023, 6, e389 CrossRef
- Y. Lai, H. Xie, P. Li, B. Li, A. Zhao, L. Luo, Z. Jiang, Y. Fang, S. Chen, X. Ai, D. Xia and Y. Cao, Adv. Mater., 2022, 34, 2206039 CrossRef CAS PubMed
- J.-C. Li, J. Tang, J. Tian, C. Cheng, Y. Liao, B. Hu, T. Yu, H. Li, Z. Liu, Y. Rao, Y. Deng, L. Zhang, X. Zhang, S. Guo and H. Zhou, J. Am. Chem. Soc., 2024, 146, 7274–7287 CrossRef CAS PubMed
- H. Liu, C. Zhao, X. Wu, C. Hu, F. Geng, M. Shen, B. Hu, B. Hu and C. Li, Energy Environ. Sci., 2024, 17, 668–679 RSC
- O. Y. Gorobtsov, H. Hirsh, M. Zhang, D. Sheyfer, L. H. B. Nguyen, S. D. Matson, D. Weinstock, R. Bouck, Z. Wang, W. Cha, J. Maser, R. Harder, Y. S. Meng and A. Singer, Adv. Energy Mater., 2023, 13, 2203654 CrossRef CAS
- L. Zhu, M. Wang, S. Xiang, D. Sun, Y. Tang and H. Wang, Adv. Energy Mater., 2023, 13, 2302046 CrossRef CAS
- M. T. Ahsan, D. Qiu, Z. Ali, Z. Fang, W. Zhao, T. Shen and Y. Hou, Adv. Energy Mater., 2023, 14, 2302733 CrossRef
- N. G. Garcia, J. M. Gonçalves, C. Real, B. Freitas, J. G. Ruiz-Montoya and H. Zanin, Energy Storage Mater., 2024, 67, 103213 CrossRef
- Y. Xiao, H. R. Wang, H. Y. Hu, Y. F. Zhu, S. Li, J. Y. Li, X. W. Wu and S. L. Chou, Adv. Mater., 2022, 34, 2202695 CrossRef CAS PubMed
- Z. Ma, Z. Zhao, H. Xu, J. Sun, X. He, Z. Lei, Z. h. Liu, R. Jiang and Q. Li, Small, 2021, 17, 2006259 CrossRef CAS PubMed
- H.-Y. Hu, J.-Y. Li, Y.-F. Liu, Y.-F. Zhu, H.-W. Li, X.-B. Jia, Z.-C. Jian, H.-X. Liu, L.-Y. Kong, Z.-Q. Li, H.-H. Dong, M.-K. Zhang, L. Qiu, J.-Q. Wang, S.-Q. Chen, X.-W. Wu, X.-D. Guo and Y. Xiao, Chem. Sci., 2024, 15, 5192–5200 RSC
- D. Eum, B. Kim, S. J. Kim, H. Park, J. Wu, S.-P. Cho, G. Yoon, M. H. Lee, S.-K. Jung, W. Yang, W. M. Seong, K. Ku, O. Tamwattana, S. K. Park, I. Hwang and K. Kang, Nat. Mater., 2020, 19, 419–427 CrossRef CAS PubMed
- M. Liu, Z. Cheng, X. Zhu, H. Dong, T. Yan, L. Zhang, L. Zheng, H. R. Yao, X. Z. Wang, L. Yu, B. Xiao, Y. Xiao and P. F. Wang, Carbon Energy, 2024, 6, e565 CrossRef CAS
- X. Liang, T.-Y. Yu, H.-H. Ryu and Y.-K. Sun, Energy Storage Mater., 2022, 47, 515–525 CrossRef
- G. Zhang, J. Li, Y. Fan, Y. Liu, P. Zhang, X. Shi, J. Ma, R. Zhang and Y. Huang, Energy Storage Mater., 2022, 51, 559–567 CrossRef
- R. Liu, W. Huang, J. Liu, Y. Li, J. Wang, Q. Liu, L. Ma, G. Kwon, S. N. Ehrlich, Y. Wu, T. Liu, K. Amine and H. Li, Adv. Mater., 2024, 36, 2401048 CrossRef CAS PubMed
- S. He, X. Shen, M. Han, Y. Liao, L. Xu, N. Yang, Y. Guo, B. Li, J. Shen, C. Zha, Y. Li, M. Wang, L. Wang, Y. Su and F. Wu, ACS Nano, 2024, 18, 11375–11388 CrossRef CAS PubMed
- C. Hu, Y. Li, D. Wang, C. Wu, F. Chen, L. Zhang, F. Wan, W. Hua, Y. Sun, B. Zhong, Z. Wu and X. Guo, Angew. Chem., Int. Ed., 2023, 62, e202312310 CrossRef CAS PubMed
- Y. Liu, Y.-H. Zhang, J. Ma, J. Zhao, X. Li and G. Cui, Chem. Mater., 2023, 36, 54–73 CrossRef
- Y. Tian, Y. Cai, Y. Chen, M. Jia, H. Hu, W. Xie, D. Li, H. Song, S. Guo and X. Zhang, Adv. Funct. Mater., 2024, 34, 2316342 Search PubMed
- Q. Wang, D. Zhou, C. Zhao, J. Wang, H. Guo, L. Wang, Z. Yao, D. Wong, G. Schuck, X. Bai, J. Lu and M. Wagemaker, Nat. Sustain., 2024, 7, 338–347 Search PubMed
- Y. Yang, Y. Feng, Z. Chen, Y. Feng, Q. Huang, C. Ma, Q. Xia, C. Liang, L. Zhou, M. S. Islam, P. Wang, L. Zhou, L. Mai and W. Wei, Nano Energy, 2020, 76, 105061 CrossRef CAS
- J. Ahn, H. Park, W. Ko, Y. Lee, J. Kang, S. Lee, S. Lee, E. Sim, K. Ihm, J. Hong, J.-K. Yoo, K. Ku and J. Kim, J. Energy Chem., 2023, 84, 153–161 CrossRef CAS
- F. Ding, C. Zhao, D. Xiao, X. Rong, H. Wang, Y. Li, Y. Yang, Y. Lu and Y.-S. Hu, J. Am. Chem. Soc., 2022, 144, 8286–8295 CrossRef CAS PubMed
- D. Hao, G. Zhang, D. Ning, D. Zhou, Y. Chai, J. Xu, X. Yin, R. Du, G. Schuck, J. Wang and Y. Li, Nano Energy, 2024, 125, 109562 CrossRef CAS
- X. Liu, Y. Wan, M. Jia, H. Zhang, W. Xie, H. Hu, X. Yan and X. Zhang, Energy Storage Mater., 2024, 67, 103313 CrossRef
- K. Walczak, A. Plewa, C. Ghica, W. Zając, A. Trenczek-Zając, M. Zając, J. Toboła and J. Molenda, Energy Storage Mater., 2022, 47, 500–514 CrossRef
- Y. Xiao, N. M. Abbasi, Y. F. Zhu, S. Li, S. J. Tan, W. Ling, L. Peng, T. Yang, L. Wang, X. D. Guo, Y. X. Yin, H. Zhang and Y. G. Guo, Adv. Funct. Mater., 2020, 30, 2001334 CrossRef CAS
- S. Liu, J. Wan, M. Ou, W. Zhang, M. Chang, F. Cheng, Y. Xu, S. Sun, C. Luo, K. Yang, C. Fang and J. Han, Adv. Energy Mater., 2023, 13, 2203521 CrossRef CAS
- T. Song, C. Wang and C. S. Lee, Carbon Neutralization, 2022, 1, 68–92 CrossRef
- Y. Xiao, Y. F. Liu, H. W. Li, J. Y. Li, J. Q. Wang, H. Y. Hu, Y. Su, Z. C. Jian, H. R. Yao, S. Q. Chen, X. X. Zeng, X. W. Wu, J. Z. Wang, Y. F. Zhu, S. X. Dou and S. L. Chou, InfoMat, 2023, 5, e12475 CrossRef CAS
- X. Wang, Q. Zhang, C. Zhao, H. Li, B. Zhang, G. Zeng, Y. Tang, Z. Huang, I. Hwang, H. Zhang, S. Zhou, Y. Qiu, Y. Xiao, J. Cabana, C.-J. Sun, K. Amine, Y. Sun, Q. Wang, G.-L. Xu, L. Gu, Y. Qiao and S.-G. Sun, Nat. Energy, 2024, 9, 184–196 CrossRef CAS
- Y. Wang, X. Zhao, J. Jin, Q. Shen, Y. Hu, X. Song, H. Li, X. Qu, L. Jiao and Y. Liu, J. Am. Chem. Soc., 2023, 145, 22708–22719 CrossRef CAS PubMed
- L.-Y. Kong, H.-X. Liu, Y.-F. Zhu, J.-Y. Li, Y. Su, H.-W. Li, H.-Y. Hu, Y.-F. Liu, M.-J. Yang, Z.-C. Jian, X.-B. Jia, S.-L. Chou and Y. Xiao, Sci. China: Chem., 2023, 67, 191–213 CrossRef
- L. Sun, Z. Wu, M. Hou, Y. Ni, H. Sun, P. Jiao, H. Li, W. Zhang, L. Zhang, K. Zhang, F. Cheng and J. Chen, Energy Environ. Sci., 2024, 17, 210–218 RSC
- J. Jin, Y. Liu, Q. Shen, X. Zhao, J. Zhang, Y. Song, T. Li, X. Xing and J. Chen, Adv. Funct. Mater., 2022, 32, 2203424 CrossRef CAS
- Y. You, S. Xin, H. Y. Asl, W. Li, P.-F. Wang, Y.-G. Guo and A. Manthiram, Chem, 2018, 4, 2124–2139 CAS
- P. F. Wang, H. Xin, T. T. Zuo, Q. Li, X. Yang, Y. X. Yin, X. Gao, X. Yu and Y. G. Guo, Angew. Chem., Int. Ed., 2018, 57, 8178–8183 CrossRef CAS PubMed
- J. Feng, Y. Liu, D. Fang and J. Li, Nano Energy, 2023, 118, 109030 CrossRef CAS
- C. Zhao, F. Ding, Y. Lu, L. Chen and Y. S. Hu, Angew. Chem., Int. Ed., 2019, 59, 264–269 CrossRef PubMed
- B. Wang, J. Ma, K. Wang, D. Wang, G. Xu, X. Wang, Z. Hu, C. W. Pao, J. L. Chen, L. Du, X. Du and G. Cui, Adv. Energy Mater., 2024, 14, 2401090 CrossRef CAS
- Z. Liu, R. Liu, S. Xu, J. Tian, J. Li, H. Li, T. Yu, S. Chu, A. M. D'Angelo, W. K. Pang, L. Zhang, S. Guo and H. Zhou, Angew. Chem., Int. Ed., 2024, 63, e202405620 CrossRef CAS PubMed
- H. Wang, X. Gao, S. Zhang, Y. Mei, L. Ni, J. Gao, H. Liu, N. Hong, B. Zhang, F. Zhu, W. Deng, G. Zou, H. Hou, X.-Y. Cao, H. Chen and X. Ji, ACS Nano, 2023, 17, 12530–12543 CrossRef CAS PubMed
- E. Gonzalo, M. Zarrabeitia, N. E. Drewett, J. M. López del Amo and T. Rojo, Energy Storage Mater., 2021, 34, 682–707 Search PubMed
- C.-C. Lin, H.-Y. Liu, J.-W. Kang, C.-C. Yang, C.-H. Li, H.-Y. T. Chen, S.-C. Huang, C.-S. Ni, Y.-C. Chuang, B.-H. Chen, C.-K. Chang and H.-Y. Chen, Energy Storage Mater., 2022, 51, 159–171 CrossRef
- P. Zhou, Z. Che, J. Liu, J. Zhou, X. Wu, J. Weng, J. Zhao, H. Cao, J. Zhou and F. Cheng, Energy Storage Mater., 2023, 57, 618–627 CrossRef
- W. Zheng, G. Liang, Q. Liu, J. Li, J. A. Yuwono, S. Zhang, V. K. Peterson and Z. Guo, Joule, 2023, 7, 2732–2748 CrossRef CAS
- Z. Zeng, A. Abulikemu, J. Zhang, Z. Peng, Y. Zhang, Y. Uchimoto, J. Han and Q.-C. Wang, Nano Energy, 2024, 128, 109813 CrossRef CAS
- X. Gao, X. Zhang, X. Liu, Y. Tian, Q. Cai, M. Jia and X. Yan, Small Methods, 2023, 7, 2300152 CrossRef CAS PubMed
- R. Li, X. Qin, X. Li, J. Zhu, L. R. Zheng, Z. Li and W. Zhou, Adv. Energy Mater., 2024, 14, 2400127 CrossRef CAS
- L. Yao, P. Zou, C. Wang, J. Jiang, L. Ma, S. Tan, K. A. Beyer, F. Xu, E. Hu and H. L. Xin, Adv. Energy Mater., 2022, 12, 2201989 CrossRef CAS
- Z. Y. Gu, J. Z. Guo, J. M. Cao, X. T. Wang, X. X. Zhao, X. Y. Zheng, W. H. Li, Z. H. Sun, H. J. Liang and X. L. Wu, Adv. Mater., 2022, 34, 2110108 CrossRef CAS PubMed
- X. Liu, G. Zhong, Z. Xiao, B. Zheng, W. Zuo, K. Zhou, H. Liu, Z. Liang, Y. Xiang, Z. Chen, G. F. Ortiz, R. Fu and Y. Yang, Nano Energy, 2020, 76, 104997 CrossRef CAS
- H. Yang, Q. Zhang, M. Chen, Y. Yang and J. Zhao, Adv. Funct. Mater., 2023, 34, 2308257 CrossRef
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., Int. Ed., 2017, 57, 102–120 CrossRef PubMed
- L. Sun, Y. Xie, X. Z. Liao, H. Wang, G. Tan, Z. Chen, Y. Ren, J. Gim, W. Tang, Y. S. He, K. Amine and Z. F. Ma, Small, 2018, 14, 1704523 CrossRef PubMed
- H. Li, J. Wang, S. Xu, A. Chen, H. Lu, Y. Jin, S. Guo and J. Zhu, Adv. Mater., 2024, 36, 2403073 CrossRef CAS PubMed
- X. Z. Wang, Y. Zuo, Y. Qin, X. Zhu, S. W. Xu, Y. J. Guo, T. Yan, L. Zhang, Z. Gao, L. Yu, M. Liu, Y. X. Yin, Y. Cheng, P. F. Wang and Y. G. Guo, Adv. Mater., 2024, 36, 2312300 CrossRef CAS PubMed
- X. Jian, Q. Shen, X. Zhao, J. Jin, Y. Wang, S. Li, X. Qu, L. Jiao and Y. Liu, Adv. Funct. Mater., 2023, 33, 2302200 CrossRef CAS
- W. Wang, Y. Shi, P. Li, R. Wang, F. Ye, X. Zhang, W. Li, Z. Wang, C. Xu, D. Xu, Q. Xu and X. Cui, Energy Environ. Sci., 2024, 17, 4283–4294 RSC
- H. Yu, A. Gao, X. Rong, S. Shen, X. Zheng, L. Yan, H. Wang, D. Su, Z. Hu, W. H. Kan, H. Chen, W. Yin, Y. Lu, Q. Zhang, L. Gu, C. Delmas, L. Chen, S. Wang and Y.-S. Hu, Energy Environ. Sci., 2024, 17, 3855–3867 RSC
- J. Liu, W. Huang, R. Liu, J. Lang, Y. Li, T. Liu, K. Amine and H. Li, Adv. Funct. Mater., 2024, 34, 2315437 CrossRef CAS
- S. Li, Z. Liu, L. Yang, X. Shen, Q. Liu, Z. Hu, Q. Kong, J. Ma, J. Li, H.-J. Lin, C.-T. Chen, X. Wang, R. Yu, Z. Wang and L. Chen, Nano Energy, 2022, 98, 107335 CrossRef CAS
- X. G. Yuan, Y. J. Guo, L. Gan, X. A. Yang, W. H. He, X. S. Zhang, Y. X. Yin, S. Xin, H. R. Yao, Z. Huang and Y. G. Guo, Adv. Funct. Mater., 2022, 32, 2111466 CrossRef CAS
- R.-M. Gao, Z.-J. Zheng, P.-F. Wang, C.-Y. Wang, H. Ye and F.-F. Cao, Energy Storage Mater., 2020, 30, 9–26 CrossRef
- T. Yuan, S. Li, Y. Sun, J.-H. Wang, A.-J. Chen, Q. Zheng, Y. Zhang, L. Chen, G. Nam, H. Che, J. Yang, S. Zheng, Z.-F. Ma and M. Liu, ACS Nano, 2022, 16, 18058–18070 CrossRef CAS PubMed
- J. Vergnet, M. Saubanère, M.-L. Doublet and J.-M. Tarascon, Joule, 2020, 4, 420–434 CrossRef CAS
- K. Xi, S. Chu, X. Zhang, X. Zhang, H. Zhang, H. Xu, J. Bian, T. Fang, S. Guo, P. Liu, M. Chen and H. Zhou, Nano Energy, 2020, 67, 104215 CrossRef CAS
- K. Zhang, D. Kim, Z. Hu, M. Park, G. Noh, Y. Yang, J. Zhang, V. W.-h. Lau, S.-L. Chou, M. Cho, S.-Y. Choi and Y.-M. Kang, Nat. Commun., 2019, 10, 5203 CrossRef CAS PubMed
- S.-J. Park, J. Lee, G.-H. Yoon, C. Koo, S.-H. Lee, S. Koo, D. Kwon, S. H. Song, T.-Y. Jeon, H. Baik, H. Kim, D. Kim and S.-H. Yu, Energy Storage Mater., 2022, 53, 340–351 CrossRef
- Y. Jin, Y. Peng, Y. Li, H. Zhou, J. Feng, Q. Fan, Q. Kuang, Y. Dong, X. Yang and Y. Zhao, Chem. Eng. J., 2024, 495, 152788 CrossRef CAS
- Z. Chen, M. Yang, G. Chen, G. Tang, Z. Huang, M. Chu, R. Qi, S. Li, R. Wang, C. Wang, T. Zhang, J. Zhai, W. Zhao, J. Zhang, J. Chen, L. He, J. Xu, W. Yin, J. Wang and Y. Xiao, Nano Energy, 2022, 94, 106958 CrossRef CAS
- H. Xu, C. Cheng, S. Chu, X. Zhang, J. Wu, L. Zhang, S. Guo and H. Zhou, Adv. Funct. Mater., 2020, 30, 2005164 CrossRef CAS
- Z. Liu, J. Wu, J. Zeng, F. Li, C. Peng, D. Xue, M. Zhu and J. Liu, Adv. Energy Mater., 2023, 13, 2301471 CrossRef CAS
- Y. Wang, X. Zhao, J. Jin, Q. Shen, N. Zhang, X. Qu, Y. Liu and L. Jiao, Energy Storage Mater., 2022, 47, 44–50 CrossRef
- S. Wu, B. Su, K. Ni, F. Pan, C. Wang, K. Zhang, D. Y. W. Yu, Y. Zhu and W. Zhang, Adv. Energy Mater., 2020, 11, 2002737 CrossRef
- X. Cao, H. Li, Y. Qiao, X. Li, M. Jia, J. Cabana and H. Zhou, Adv. Energy Mater., 2020, 10, 1903785 CrossRef CAS
- H. Liu, L. Kong, H. Wang, J. Li, J. Wang, Y. Zhu, H. Li, Z. Jian, X. Jia, Y. Su, S. Zhang, J. Mao, S. Chen, Y. Liu, S. Chou and Y. Xiao, Adv. Mater., 2024, 36, 2407994 CrossRef CAS PubMed
- Y.-F. Zhu, Y. Xiao, S.-X. Dou, Y.-M. Kang and S.-L. Chou, eScience, 2021, 1, 13–27 CrossRef
- X. Wang, M. Yu and X. Feng, eScience, 2023, 3, 100141 CrossRef
- R. Liu, L. Yu, X. He, H. Liu, X. Ma, Z. Tao, G. Wan, N. Ahmad, B. Peng, L. Shi and G. Zhang, eScience, 2023, 3, 100138 CrossRef
- Y. Su, X.-Y. Liu, R. Zhang, S. Zhang, J. Wang, Y.-D. Qian, Z.-C. Jian, Y.-F. Zhu, J.-F. Mao, S. Xu, S. Dou and Y. Xiao, Energy Storage Mater., 2024, 71, 103638 CrossRef
- Y. He, W. Shang and P. Tan, Carbon Neutralization, 2024, 3, 773–780 CrossRef
- T. Bai, J. Wang, H. Zhang, F. Ji, W. Song, S. Xiao, D. Gao, J. Lu, L. Ci and D. Li, eScience, 2024, 100310 CrossRef
- J. Wang, Y. Su, Y.-J. Li, H.-W. Li, J.-X. Guo, Q.-Q. Sun, H.-Y. Hu, Y.-F. Liu, X.-B. Jia, Z.-C. Jian, L.-Y. Kong, H.-X. Liu, J.-Y. Li, H. Chu, S.-X. Dou and Y. Xiao, ACS Appl. Mater. Interfaces, 2024, 16, 2330–2340 CrossRef CAS PubMed
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |