
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
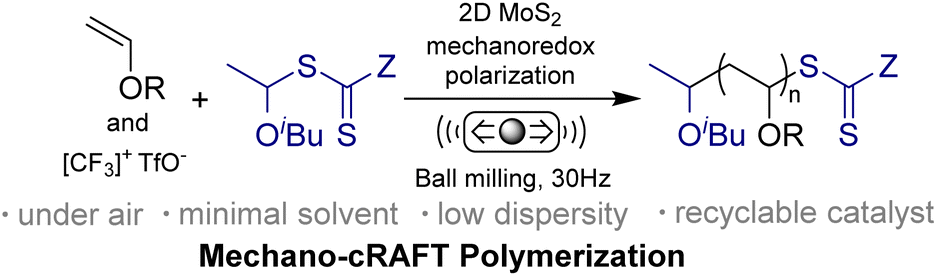
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanically induced cationic reversible addition–fragmentation chain transfer polymerization of vinyl ethers†
Longfei
Zhang
a,
Xiuyang
Zou
b,
Chengqiang
Ding
a and
Zhao
Wang
 *a
*a
aState and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, Jiangsu Key Laboratory of Advanced Functional Polymer Design and Application, Suzhou Key Laboratory of Macromolecular Design and Precision Synthesis, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: wangzhao@suda.edu.cn
bJiangsu Province Engineering Research Center of Environment Functional Materials, School of Chemistry and Chemical Engineering, Huaiyin Normal University, Huaian 223300, China
First published on 22nd October 2024
Abstract
Mechanoredox catalysis has emerged as a sustainable approach for organic transformations. Mechanically controlled polymerization that uses mechanoredox catalysts enables synthesis of complex polymers and mechanoresponsive materials with diverse applications. Despite its potential, the focus has predominantly been on free radical polymerization and acrylate monomers. The mechanochemical synthesis of poly(vinyl ether)s (PVEs) poses a significant challenge in the field. Herein, we report an efficient mechanically induced cationic reversible addition–fragmentation chain transfer (mechano-cRAFT) polymerization using 2D MoS2 as a mechanoredox catalyst, where free radical intermediates can be further oxidized to cations to promote cationic polymerization of vinyl ethers. This mechano-cRAFT polymerization can be conducted in air and with minimal organic solvent, resulting in quantitative monomer conversion. This strategy is applicable to a range of vinyl ether monomers, yielding polymers with controlled molecular weight and narrow dispersity. We also performed trapping experiments to investigate the piezoelectrically mediated redox process, and further validated the mechanism through density functional theory (DFT) calculations.
Introduction
Living cationic polymerization (LCP) is a well-developed polymerization technology that provides a valuable strategy for the preparation of well-controlled macromolecules.1,2 Vinyl ethers (VEs), a class of prevalent monomers in LCP, enable the synthesis of poly(vinyl ether)s (PVEs) with diverse applications, for instance as adhesives, lubricants, and anticorrosive agents.3 Since the development of LCP of vinyl ether in the 1980s, the polymerization often relies on Lewis acid catalysts to activate dormant species C–X (X: halogen or OC(O)R).4 However, maintaining low concentrations of cationic active species often requires extremely low temperatures and involves metal residues. Reversible addition–fragmentation chain transfer (RAFT) polymerization has emerged as a highly promising technique due to its broad monomer applicability, mild reaction conditions, and functional group tolerance.5 In 2015, Kamigaito and Sugihara's groups independently disclosed cationic RAFT polymerization using strong acids, but it still needs to be carried out below 0 °C.6,7 Therefore, there is a need to develop new chemistry that allows LCP under mild conditions.In recent years, extensive attention has been given to polymerization reactions controlled by an external field, such as light, electrical field and mechanical force.8 Light-induced cationic polymerization, in particular, has gained increasing attention due to its mild reaction conditions and non-invasive nature.9–12 Mechanochemistry, on the other hand, utilizing mechanical force to promote chemical transformations has been widely applied in polymer science, as well as organic and inorganic synthesis due to its high reactivity and sustainability.13–16 Mechanochemical reaction using ball milling exhibits significant advantages such as solvent independence, reduced side reactions, and large-scale production compared to traditional solution-phase chemistry.17 In free radical polymerization, mechanical force was used to activate solvents, vinyl monomers or mechanophores through homolytic cleavage of chemical bonds.18–22 However, the application of mechanical force to promote cationic polymerization remains a challenge.23
Recently, a burgeoning field of utilizing mechanoredox catalysts to promote organic transformations has emerged.24–26 This mechanoredox approach has been reported to induce a series of redox events through a single electron transfer (SET) process from piezoelectric nanoparticles to reactants.27–30 Recently, we and others have demonstrated the mechanically controlled free radical polymerization (FRP),31–33 reversible-deactivation radical polymerization (RDRP)34–39 and click polymerization.40,41 Most of these polymerizations are initiated by piezoelectric reduction of the initiator through SET mechanisms. An additional sacrificial electron donor was added to suppress electron–hole recombination.36,38 The oxidizing capability of mechanoredox catalysts for polymerization has not been thoroughly explored in previous research.
Herein, we propose a mechanically induced cationic RAFT polymerization (mechano-cRAFT) by 2D MoS2 and ball milling. It has been reported that odd-layered MoS2 exhibits excellent piezoelectric properties compared to traditional bulk piezoelectric materials.42 The spontaneous polarization of 2D MoS2 under an external force can promote the rapid separation of electrons and holes to trigger redox reactions. The Umemoto reagent was employed as the initiator, due to its suitable reduction potential (Ered = −0.26 V vs. SCE).43 We hypothesize that ball milling can efficiently polarize 2D MoS2 and generate CF3 radicals through piezoelectric reduction of the Umemoto reagent.42 The CF3 radicals then react with vinyl ether to produce free radical intermediates, which are subsequently oxidized to cations by polarized MoS2, thus initiating the cationic RAFT equilibrium (Scheme 1). The mechano-cRAFT can be carried out in a highly controlled fashion under solvent-free or solvent-less conditions, resulting in near-complete monomer conversion. The produced PVEs can be utilized for synthesizing block copolymers through in situ chain extension. This mechanochemical approach provides an alternative to solution-based chemistry for the synthesis of PVEs.
 | ||
| Scheme 1 Mechano-cRAFT polymerization in ball milling. | ||
Results and discussion
2D MoS2 was prepared by the hydrothermal method.42 The X-ray diffraction (XRD) pattern of the as-synthesized MoS2 showed a stable hexagonal (2H) phase (Fig. S1†). The (002) peak shifted to a lower 2θ value by 0.40°, indicating a d spacing of 0.63 nm, slightly larger than the reported value of 0.62 nm for MoS2 (JCPDS card no. 37-1492). The chemical states of Mo and S in MoS2 were characterized by X-ray photoelectron spectroscopy (XPS). Fig. S2a† shows two peaks at 233 eV and 229.8 eV, which are attributed to Mo 3d3/2 and Mo 3d5/2, respectively. In addition, the peaks positioned at 163.7 eV and 162.6 eV were assigned to S 2p1/2 and S 2p3/2, respectively (Fig. S2b†). Scanning electron microscopy (SEM) revealed a flower-like morphology of the 2D MoS2 sample (Fig. S3a†). Transmission electron microscopy (TEM) images further validated its layered structure with an interlayer spacing of 0.63 nm, consistent with the XRD result (Fig. S3b and c†). High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM, Fig. S3d†) and corresponding energy-dispersive spectroscopy (EDS, Fig. S3e and f†) mapping were used to further demonstrate the composition of 2D MoS2.First, we conducted mechanically induced cationic polymerization of isobutyl vinyl ether (IBVE) using the Umemoto reagent as an initiator and 2D MoS2 as a mechanoredox catalyst under ball milling conditions (30 Hz). It was reported that trifluoromethylation reactions can be conducted under air.43 Therefore, we examined the polymerization without inert gas protection. Bulk polymerization was carried out at various feed ratios ([IBVE]/[Umemoto] = 100, 200 and 1000) with 3 wt% MoS2 (Table S1†). The polymerization process was characterized by measuring monomer conversion by 1H NMR, analyzing the number average molecular weight (Mn) and dispersity (Đ) using gel permeation chromatography (GPC). Although a relatively high conversion (75%) was achieved within 90 min, the resulting polymers had high molecular weight (Mn = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 Da) and broad dispersity (Đ = 1.59) (Table S1, entry 1†). Changing the ratio of monomer and initiator leads to a variation in Mn (Table S1, entries 2 and 3†). Kinetic analysis of polymerization also confirmed a fast chain growth mechanism but in an uncontrolled manner (Fig. S6†).
600 Da) and broad dispersity (Đ = 1.59) (Table S1, entry 1†). Changing the ratio of monomer and initiator leads to a variation in Mn (Table S1, entries 2 and 3†). Kinetic analysis of polymerization also confirmed a fast chain growth mechanism but in an uncontrolled manner (Fig. S6†).
To achieve controlled cationic polymerization, we employed dithiocarbamate (DTCB) as the cationic RAFT agent due to its high chain transfer constant (Ctr) value in cationic RAFT polymerization.44 We began our investigation by combining IBVE and MoS2 with varying amounts of initiator and DTCB (Table 1). Bulk polymerization with a target degree of polymerization (DP) of 100 was carried out under solvent-free conditions, and a relatively high monomer conversion (86%) was observed after 80 min (Table 1, entry 1). Thermography was used to measure the temperature in the milling jar before and after polymerization, which increased from 27.3 °C to 31.4 °C (Fig. S7†). GPC analysis revealed the Mn value of poly(isobutyl vinyl ether) (PIBVE) was 8800 Da, which matched the theoretical value, with narrow dispersity (Đ = 1.06) (Fig. S9†). Subsequently, kinetic analysis was implemented to confirm the living characteristics of the bulk polymerization (Fig. 1a–c). 1H NMR results showed that the resonance signals corresponding to the vinyl group of IBVE (6.5 ppm) diminished with increasing reaction time, while the assigned peaks from the resulted PIBVE (0.9 ppm) was enhanced (Fig. S10†). A first-order relationship was observed between ln([IBVE]0/[IBVE]) and reaction time (Fig. 1b). The Mn increased linearly with the conversion and dispersity remained narrow, as expected (Fig. 1c). However, the solvent-free conditions did not allow for complete conversion (Fig. 1a).
|
|
|||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| Entrya | Target DP | CF3 reagent | Time (min) | Conv.b [%] | M n,th [Da] | M n,GPC [Da] | Đ |
| a [DTCB]/[CF3 reagent] = 10/1. IBVE (3.1 mmol), 2.4 wt% MoS2 (0.06 mmol). Ball mill (1.5 mL stainless-steel jar, 5 mm stainless-steel milling ball), LAG (η = 0.2 μL mg−1) with DCM. Room temperature, under air. b Determined by 1H NMR in CDCl3. c M n,th = [IBVE]0/[DTCB]0 × conversion × MIBVE + MDTCB. d Determined by GPC using polystyrene as a calibration standard in THF. e Solvent-free conditions. f [DTCB]/[Umemoto] = 2/1. g 0.5 wt% MoS2. h Scale-up experiment using 15.4 mmol of IBVE. | |||||||
| 1e | 100 | a | 80 | 86 | 8900 | 8800 | 1.06 |
| 2 | 100 | a | 70 | 95 | 9800 | 9800 | 1.06 |
| 3 | 25 | a | 75 | 96 | 2700 | 2600 | 1.06 |
| 4 | 50 | a | 75 | 94 | 5000 | 4900 | 1.05 |
| 5 | 200 | a | 90 | 98 | 19900 |
15200 |
1.10 |
| 6 | 300 | a | 90 | 98 | 29800 |
19200 |
1.19 |
| 7 | 400 | a | 90 | 99 | 39800 |
23200 |
1.19 |
| 8 | 600 | a | 90 | 98 | 59400 |
26400 |
1.26 |
| 9f | 100 | a | 70 | 96 | 9900 | 9900 | 1.08 |
| 10g | 100 | a | 120 | 97 | 10000 |
10000 |
1.05 |
| 11 | 100 | b | 70 | 96 | 9900 | 10000 |
1.06 |
| 12 | 100 | c | 70 | 96 | 9900 | 9900 | 1.08 |
| 13 | 100 | d | 70 | 95 | 9800 | 9800 | 1.05 |
| 14h | 100 | a | 90 | 96 | 9900 | 9600 | 1.09 |
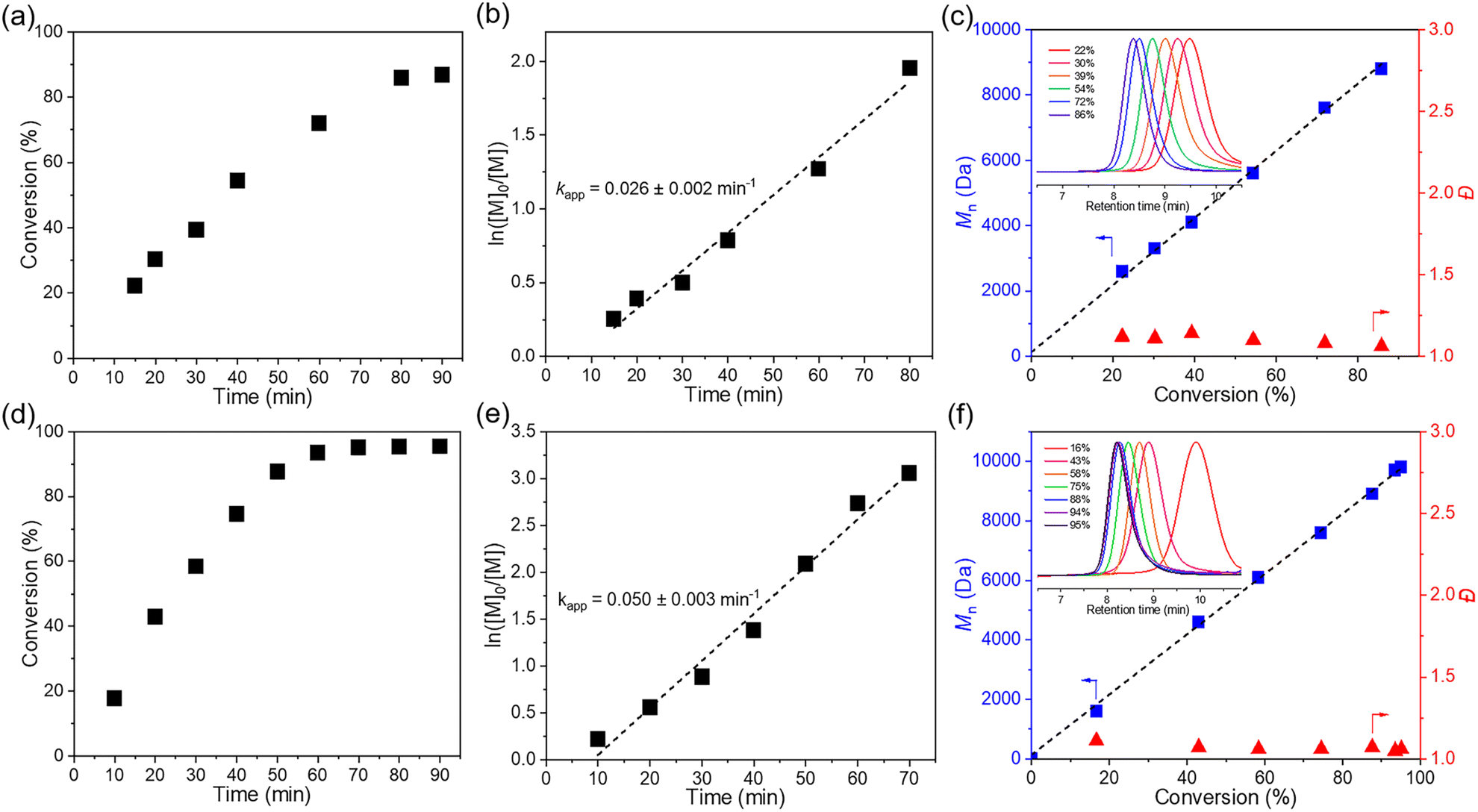 | ||
| Fig. 1 Kinetic data of mechano-cRAFT polymerization of IBVE (target DP = 100) in a ball mill without solvents. (a) Conversion vs. time. (b) ln([IBVE]0/[IBVE]) vs. time. (c) Mn and Đ vs. conversion. Kinetic data of mechano-cRAFT polymerization of IBVE (target DP = 100) in a ball mill under LAG conditions. (d) Conversion vs. time. (e) ln([IBVE]0/[IBVE]) vs. time. (f) Mn and Đ vs. conversion. Inset: overlay of GPC traces of PIBVE at different conversions. | ||
To ensure the full conversion of vinyl ethers, we employed the liquid-assisted grinding (LAG) technique to facilitate the polymerization process. The LAG technology involves the use of a small amount of solvent during mechanical grinding as lubricants to improve mixing efficiency and reaction kinetics.45 It was found that the addition of a small amount of DCM (η = 0.2 μL mg−1) enabled the quantitative monomer conversion (≥95%). GPC analysis of the resulting polymers showed Mn of 9800 Da and narrow dispersity (Đ = 1.06) (Table 1, entry 2). Subsequently, other organic solvents, including toluene, acetone, tetrahydrofuran (THF), acetonitrile (MeCN) and N,N-dimethylacetamide (DMAc), were introduced to investigate the effect of LAG on polymerization (Table S2 and Fig. S11†). The addition of acetone and MeCN resulted in the quantitative monomer conversions. In contrast, the conversion under THF and toluene conditions was low (60% within 90 min), and no polymerization observed after the addition of DMAc. In addition, the effects of varying LAG values were also investigated. The results indicated that increasing the LAG values did not significantly affect the polymerization reaction (Fig. S12†).
Kinetic analysis of IBVE polymerization (target DP = 100) under the LAG reaction conditions was carried out (Fig. 1d–f). Polymerization under LAG conditions demonstrated a faster reaction kinetics compared to bulk polymerization (0.05 vs. 0.025 min−1), and the semilogarithmic kinetic plot followed a first-order relationship (Fig. 1d and e). The molecular weight was also in line with the expected molecular weight, while the dispersity remains low (Đ < 1.1) (Fig. 1f). Subsequently, varying the molar ratio between IBVE and DTCB from 25 to 600 enabled the synthesis of PIBVE with different molecular weights and narrow distributions (Đ < 1.3) (Table 1 and Fig. S14†). With the increase of the target degree of polymerization (DP), the 1H NMR analysis suggested the formation of acetal terminated “dead chains”, likely attributable to the impact of residual water under an air atmosphere (Fig. S15†).
In order to analyze the chemical structure of PIBVE synthesized by mechano-cRAFT polymerization, we used sodium dithiocarbamate as a quencher to terminate the polymerization and characterized the purified polymers by 1H NMR, 13C NMR and matrix-assisted laser desorption time-of-flight mass spectrometry (MALDI-TOF MS).46 The 1H NMR spectrum of PIBVE displayed distinct peaks corresponding to the main and side chains of each repeating unit. In addition, the characteristic peak around 6 ppm can be attributed to the protons at the chain end connected to the dithiocarbamate. The characteristic peak around 1.2 ppm can be assigned to the chain-end methyl group (Fig. S16†). Subsequently, we have also prepared a low molecular weight polymer (Mn,GPC = 2600 Da) under air for MALDI-TOF MS analysis. The main peak series corresponded to PIBVE with a dithiocarbamate chain end. The secondary peak series can be attributed to acetal “dead chains” resulting from residual water in air (Fig. S17†). In contrast, we also prepared PIBVE with higher molecular weight (Mn,GPC = 4900 Da) under Ar. As shown in Fig. S18,† the peaks corresponded to PIBVE with a hydrogen-terminated chain end without acetal “dead chains”. This observation was attributed to the decomposition of the RAFT chain end during the MALDI-TOF MS analysis.47 In addition, 13C NMR analysis was used to calculate the meso diad content (% m). The meso-rich diad tacticity of 57.6% indicates that the polymerization proceeded by a cationic mechanism (Fig. S19†).48
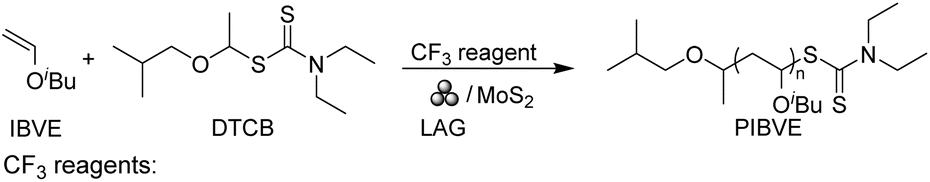
To further investigate the contributions of the Umemoto reagent and 2D MoS2, we changed the amount of Umemoto reagent (0.5 equiv.) and 2D MoS2 (0.5 wt%), respectively. The increase of the Umemoto reagent had little effect on the polymerization (Table 1, entry 9), while reducing the MoS2 loading resulted in a longer reaction time to achieve high monomer conversion (Table 1, entry 10). We also conducted polymerization under an Ar atmosphere. It is noteworthy that the monomer conversion reached 98% only after 12 min of ball milling. This polymerization exhibited a faster kinetics and was capable of obtaining a predictable molecular weight and narrow molecular weight distribution (Đ < 1.06) (Fig. S20†). While oxygen did not inhibit the polymerization, the results suggested that it could slow down the polymerization by reacting with free radical intermediates. The mechanism will be explored further in subsequent sections. In addition, the 2D MoS2 used in mechano-cRAFT polymerization in ball milling was easily recovered by centrifugation and recycled. After five cycles, no significant decrease in monomer conversion was observed, suggesting the high catalytic activity of 2D MoS2 remained after repeated usage (Fig. S21†).
Subsequently, we explored other commercially accessible electrophilic trifluoromethylation reagents as potential initiators. All of these reactions achieved quantitative monomer conversions (≥95%) under standard polymerization conditions (Table 1, entries 11–13). GPC characterization of the resulting polymer showed expected Mn and narrow dispersity (Đ < 1.08) (Fig. S22†). To demonstrate the practical utility of the mechano-cRAFT approach, we conducted scale-up experiments using a 5 mL stainless steel grinding jar with 15.4 mmol of IBVE (Table 1, entry 14). The results demonstrated a high monomer conversion of 96%, leading to the production of PIBVE with an Mn value of 9600 Da and Đ of 1.09 (Fig. S23†).
To demonstrate the versatility of this method, we investigated the mechano-cRAFT polymerization of various vinyl ether monomers under the LAG reaction conditions, including n-butyl vinyl ether (NBVE), n-propyl vinyl ether (NPVE), ethyl vinyl ether (EVE), cyclohexyl vinyl ether (CyVE), 2,3-dihydrofuran (DHF) and 2-chloroethyl vinyl ether (Cl-EVE). Almost quantitative monomer conversion was achieved for electron-rich vinyl ethers at various target DP values, with good agreement between experimental and theoretical Mn and low dispersity (Đ = 1.05–1.08) (Table 2, entries 1–6 and Fig. S24–S29†). Due to the influence of bulky groups, a broader dispersity (Đ = 1.38) was detected for cyclohexyl vinyl ether (CyVE) (Table 2, entry 7; Fig. S30 and S31†). For the polymerization of 2,3-dihydrofuran (DHF), a monomer conversion of 98% was achieved after 30 min, but the experimental Mn of the obtained poly(2,3-dihydrofuran) (PDHF) was slightly lower than the theoretical value (Table 2, entry 8; Fig. S32 and S33†). Moreover, the polymerization of 2-chloroethyl vinyl ether (Cl-EVE) was relatively slow due to the electron-withdrawing effect of the chlorine. But the expected Mn and narrow dispersity (Đ = 1.09) were still obtained after extended reaction time (Table 2, entry 9; Fig. S34 and S35†).
|
|
|||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| Entrya | M | Target DP | Time (min) | Conv.b [%] | M n,th [Da] | M n,GPC [Da] | Đ |
| a M = monomer, [DTCB]/[CF3 reagent] = 10/1, 2.4 wt% MoS2 (0.06 mmol). Ball mill (1.5 mL stainless-steel jar, 5 mm stainless-steel milling ball), LAG (η = 0.2 μL mg−1) with DCM. Room temperature, under air. b Determined by 1H NMR in CDCl3. c M n,th = [Monomer]0/[DTCB]0 × conversion × MMonomer + MDTCB. d Determined by GPC using polystyrene as a calibration standard in THF. | |||||||
| 1 | NBVE | 100 | 80 | 96 | 9900 | 9700 | 1.08 |
| 2 | NBVE | 50 | 80 | 97 | 5100 | 5100 | 1.05 |
| 3 | NPVE | 100 | 80 | 96 | 8500 | 8500 | 1.05 |
| 4 | NPVE | 50 | 80 | 98 | 4500 | 4600 | 1.05 |
| 5 | EVE | 100 | 30 | 98 | 7300 | 7300 | 1.05 |
| 6 | EVE | 50 | 45 | 98 | 3800 | 3800 | 1.05 |
| 7 | CyVE | 100 | 30 | 94 | 12100 |
9400 | 1.38 |
| 8 | DHF | 100 | 20 | 98 | 7100 | 5600 | 1.21 |
| 9 | Cl-EVE | 100 | 120 | 86 | 9400 | 9100 | 1.10 |

We investigated additional monomers suitable for cationic polymerization, including p-methoxystyrene (p-MOS), ethyl propenyl ether (EPE) and N-vinyl carbazole (NVC) (Table S5†). At a target DP of 100, the conversion of p-MOS reached 72% after 90 min under an Ar atmosphere. The resulting polymer exhibited an Mn of 8600 Da and dispersity of 1.14. While quantitative conversion of EPE was achieved within 90 min, the resulting polymer displayed a relatively low Mn and slightly broader dispersity (Fig. S36†). In addition, we found that NVC was not suitable for this polymerization.
To further clarify the living character of mechano-cRAFT polymerization, a chain extension experiment was conducted under the LAG reaction conditions (Fig. 2a). Initially, a poly(ethyl vinyl ether) (PEVE) with a target DP of 50 was synthesized (Mn = 3800 Da, Đ = 1.05) and was used for chain extension studies without isolation. IBVE (100 equiv.) was then added into the grinding jar along with Umemoto reagent and DCM to initiate the chain extension reaction. GPC analysis showed that PEVE-b-PIBVE was successfully synthesized with an Mn value of 10100 Da and a narrow dispersity (Đ = 1.08) (Fig. 2b). The resulting PEVE-b-PIBVE block copolymer was characterized by diffusion ordered spectroscopy (1H DOSY, Fig. 2c), with a single diffusion coefficient of 1.4 × 10−10 m2 s−1, confirming the continued growth of IBVE at the end of the PEVE chain.
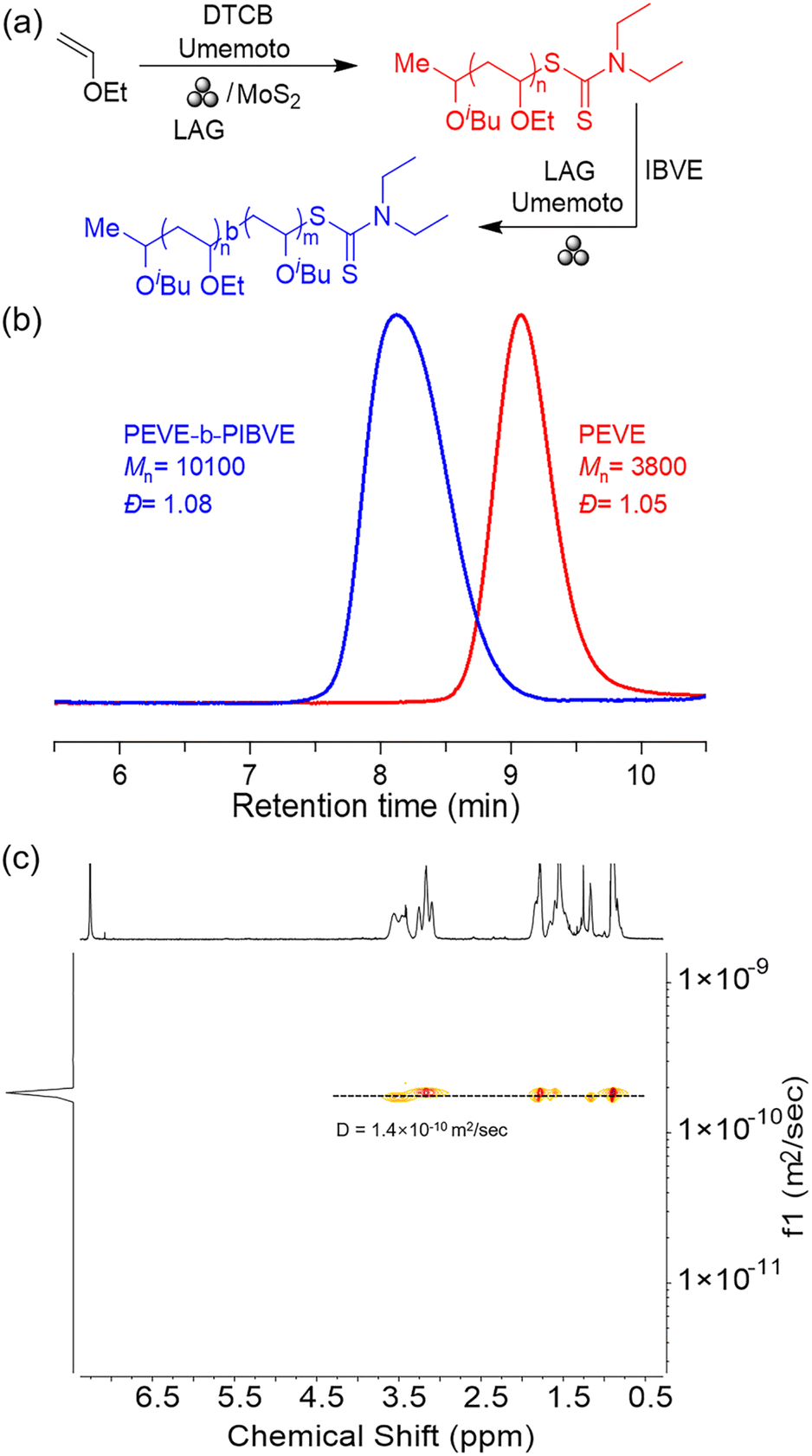 | ||
| Fig. 2 (a) Synthesis of the PEVE-b-PIBVE block copolymer via mechano-cRAFT under ball milling. (b) GPC traces for the PEVE-b-PIBVE block copolymer. (c) 1H DOSY for the PEVE-b-PIBVE block copolymer. | ||
To reveal the polymerization mechanism, various control experiments were conducted (Table S3†). No monomer conversion was detected when the polymerization was carried out in the absence of MoS2, BM, or the Umemoto reagent, respectively (Table S3, entries 1–3†). This demonstrated the essential role of these three components and further validated the mechanoredox process for promoting polymerization. In addition, no polymerization was found when the Umemoto reagent was replaced with sodium trifluoromethanesulfonate (NaOTf) (Table S3, entry 4†). This excluded the impact of the trifluoromethane sulfonate ion (TfO−) present in the Umemoto reagent on initiating polymerization. A control experiment was conducted at 35 °C using stirring instead of ball milling, and no polymerization occurred. This indicates that the polymerization was attributed to the mechanoredox mechanism (Table S3, entry 5†). Additionally, control experiments were performed using alternative piezoelectric BaTiO3 nanoparticles (Table S3, entry 6†). The monomer conversion reached 84% after 120 min (Fig. S42†). GPC analysis showed a single peak distribution with a shift to the higher molecular weight region over time (Fig. S43†). However, an induction period of 90 min was observed. This may be due to the slow activation of bulk piezoelectric materials and their low catalytic efficiency. Subsequently, non-piezoelectric TiO2 was selected to rule out the tribochemical influence of nanoparticles (Table S3, entry 7†).49 No polymerization was observed, confirming the importance of the mechanoredox process for initiating the polymerization.
In order to confirm the role of free radicals in the initiation step, 2,2,6,6-tetramethyl-1-piperinedinyloxy (TEMPO) was added as a radical scavenger. It was found that no monomer conversion was observed after 120 min of polymerization (Table S3, entry 8†). This suggested that the reaction may have occurred initially in the presence of free radicals. To verify the radical intermediates, the TEMPO trapping experiment was performed in the absence of IBVE. The TEMPO-CF3 intermediate was identified by gas chromatography-mass spectrometry (GC-MS) (Fig. S44†). To further confirm that the chain propagation was carried out by the cationic mechanism, radical inhibitors, e.g., 1,4-benzoquinone (BQ), 4-methoxyphenol (MEHQ) and TEMPO, were added after 15 min of reaction (Fig. 3a). The chain propagation was not inhibited by the radical scavengers. However, the monomer conversion was suppressed, probably due to the inhibition of the initiation process. After 30 minutes of reaction, the addition of cationic scavenger MeOH/Et3N led to the termination of polymerization, further supporting the cationic polymerization mechanism (Fig. S48†).
 | ||
| Fig. 3 (a) The relationship between monomer conversion and reaction time after adding different free radical inhibitors (control: without inhibitor). (b) Monomer conversion after the addition of electron or hole scavengers (control: without scavenger), reaction time: 60 min. | ||
To further explore the mechanoredox process, electron or hole scavengers were introduced during the polymerization (Fig. 3b, Table S4†). Specifically, 0.1 equivalent of EDTA-2Na (vs. MoS2) was added as a hole scavenger. After 60 minutes of reaction, the monomer conversion reached 29%, significantly lower than that in the control experiment (70%). After 120 minutes, the monomer conversion increased to 60%. In contrast, the introduction of 1 equivalent of EDTA-2Na demonstrated negligible monomer conversion (<5%) after 120 minutes, indicating the crucial role of the hole-mediated oxidation reaction in promoting mechano-cRAFT polymerization (Fig. S49†). In addition, the addition of 0.1 equivalent of AgNO3 (vs. MoS2) as an electron scavenger resulted in low monomer conversion (<5%). This finding solidified our conjecture that piezoelectrical reduction of the trifluoromethyl reagent to the CF3 radical stood as the pivotal step in mechano-cRAFT polymerization.
To gain a deeper understanding of the mechano-cRAFT reaction, density functional theory (DFT) methods were conducted to simulate the initiation process (Fig. 4). The DFT calculation results indicated that the CF3 radical generated through the SET mechanism undergoes electrophilic addition to the vinyl ether double bond, forming a free radical intermediate (INT 1). Subsequently, INT 1 was oxidized by a hole of 2D MoS2 to a cationic intermediate (INT 2), which required an activation energy of 5.81 kcal mol−1. This result provided further evidence for the short induction period observed in the polymerization reaction. In addition, we assessed the free radical addition of INT 1 with the monomer by DFT calculation (Fig. S51†). The results revealed that an activation energy of 48.23 kcal mol−1 was necessary to reach the transition state (TS 2). This significant reaction energy barrier would prevent the polymerization via the free radical mechanism. Based on the above results, we propose a potential mechanism for this mechano-cRAFT polymerization: (1) highly polarized 2D MoS2 under ball milling can activate the Umemoto reagent to generate the CF3 radical by the single electron transfer (SET) mechanism; (2) the CF3 radical readily adds to the double bond of isobutyl vinyl ether, leading to the formation of a radical intermediate due to its electrophilic nature; (3) the radical intermediate is subsequently oxidized by the holes of 2D MoS2 to yield the reactive cationic species to promote the RAFT polymerization (Fig. 5).
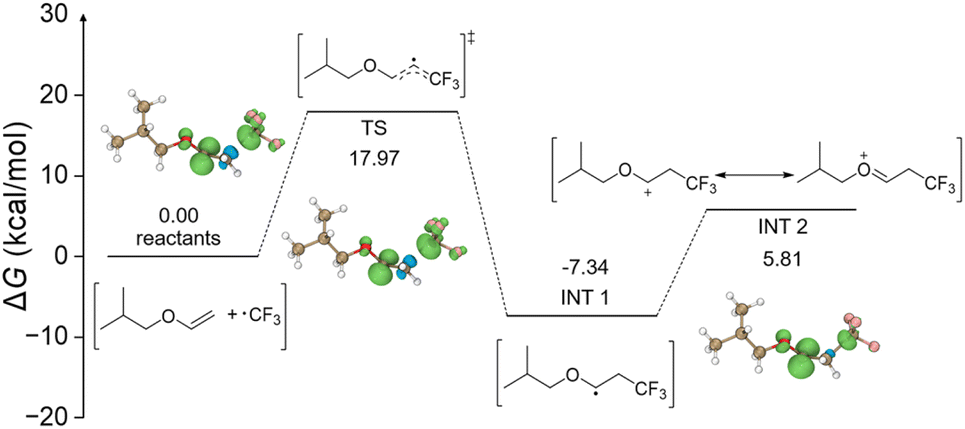 | ||
| Fig. 4 Energy profile for the initiation process of mechano-cRAFT polymerization. | ||
 | ||
| Fig. 5 Possible mechanism of mechano-cRAFT polymerization process. | ||
Conclusions
In summary, we have demonstrated a mechanically induced cationic RAFT polymerization of vinyl ethers under solvent-free or LAG conditions. The mechano-cRAFT polymerization can proceed in air and be initiated using commercially available electrophilic trifluoromethylation reagents. This method can be used with a variety of vinyl ether monomers, resulting in PVEs with predictable molecular weight and low dispersity. This method can also be used for one-pot synthesis of block copolymers. The mechanistic studies and DFT calculations supported the proposed piezoelectrically mediated redox mechanism. This methodology therefore expands the scope of mechanically controlled polymerization and allows numerous applications in the green synthesis of functional polymeric materials.Data availability
All experimental and characterization details are available in the ESI.†Author contributions
L. Z. performed the experiments, analysed all data, and drafted the manuscript. X. Z. and C. D. performed DFT calculations and checked the manuscript. Z. W. guided this work and corrected the manuscript. All authors read and confirmed the manuscript and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22471185 and 22101195), Natural Science Foundation of Jiangsu Province (BK20210732), Science and Technology Pro-gram of Suzhou (ZXL2022480), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Program of Innovative Research Team of Soochow University.References
- S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 109, 5245–5287 CrossRef CAS PubMed.
- Y. Chen, L. Zhang, Y. Jin, X. Lin and M. Chen, Macromol. Rapid Commun., 2021, 42, 2100148 CrossRef CAS.
- D. L. Langer, S. Oh and E. E. Stache, Chem. Sci., 2024, 15, 1840–1845 RSC.
- A. Kanazawa, S. Kanaoka and S. Aoshima, Chem. Lett., 2010, 39, 1232–1237 CrossRef CAS.
- M. R. Hill, R. N. Carmean and B. S. Sumerlin, Macromolecules, 2015, 48, 5459–5469 CrossRef CAS.
- M. Uchiyama, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2015, 54, 1924–1928 CrossRef CAS.
- S. Sugihara, N. Konegawa and Y. Maeda, Macromolecules, 2015, 48, 5120–5131 CrossRef CAS.
- Y. N. Zhou, J. J. Li, Y. Y. Wu and Z. H. Luo, Chem. Rev., 2020, 120, 2950–3048 CrossRef CAS PubMed.
- Q. Michaudel, V. Kottisch and B. P. Fors, Angew. Chem., Int. Ed., 2017, 56, 9670–9679 CrossRef CAS PubMed.
- V. Kottisch, Q. Michaudel and B. P. Fors, J. Am. Chem. Soc., 2016, 138, 15535–15538 CrossRef CAS PubMed.
- X. Zhang, Y. Jiang, Q. Ma, S. Hu and S. Liao, J. Am. Chem. Soc., 2021, 143, 6357–6362 CrossRef CAS.
- M. Ciftci, Y. Yoshikawa and Y. Yagci, Angew. Chem., Int. Ed., 2017, 56, 519–523 CrossRef CAS.
- A. Krusenbaum, S. Gratz, G. T. Tigineh, L. Borchardt and J. G. Kim, Chem. Soc. Rev., 2022, 51, 2873–2905 RSC.
- J. L. Do and T. Friscic, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- T. Friscic, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef CAS.
- E. Boldyreva, Chem. Soc. Rev., 2013, 42, 7719–7738 RSC.
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- T. G. McKenzie, E. Colombo, Q. Fu, M. Ashokkumar and G. G. Qiao, Angew. Chem., Int. Ed., 2017, 56, 12302–12306 CrossRef CAS PubMed.
- J. Collins, T. G. McKenzie, M. D. Nothling, S. Allison-Logan, M. Ashokkumar and G. G. Qiao, Macromolecules, 2018, 52, 185–195 CrossRef.
- Z. Wang, Z. Wang, X. Pan, L. Fu, S. Lathwal, M. Olszewski, J. Yan, A. E. Enciso, Z. Wang, H. Xia and K. Matyjaszewski, ACS Macro Lett., 2018, 7, 275–280 CrossRef CAS.
- S. L. Goodrich, M. E. Ross, J. B. Young and B. S. Sumerlin, Macromolecules, 2023, 56, 9350–9358 CrossRef CAS.
- J. Wang, I. Piskun and S. L. Craig, ACS Macro Lett., 2015, 4, 834–837 CrossRef CAS.
- H. Kinpara, Y. Hori, S. Shimada and H. Kashiwabara, Macromolecules, 1989, 22, 1277–1280 CrossRef.
- H. Xia and Z. Wang, Science, 2019, 366, 1451–1452 CrossRef CAS PubMed.
- J. Ayarza, Z. Wang, J. Wang, C. W. Huang and A. P. Esser-Kahn, ACS Macro Lett., 2020, 9, 1237–1248 CrossRef CAS PubMed.
- Z. Ren, Y. Peng, H. He, C. Ding, J. Wang, Z. Wang and Z. Zhang, Chin. J. Chem., 2022, 41, 111–128 CrossRef.
- K. Kubota, Y. Pang, A. Miura and H. Ito, Science, 2019, 366, 1500–1504 CrossRef CAS PubMed.
- T. Seo, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2023, 62, e202311531 CrossRef CAS PubMed.
- R. Qu, S. Wan, X. Zhang, X. Wang, L. Xue, Q. Wang, G. J. Cheng, L. Dai and Z. Lian, Angew. Chem., Int. Ed., 2024, 63, e202400645 CrossRef CAS.
- Z. Li, L. Zhang, R. Ding, J. Wang, D. Chen, Z. Ren, C. Ding, K. Chen, J. Wang and Z. Wang, Chem. Commun., 2024, 60, 6146–6149 RSC.
- M. D. Nothling, J. E. Daniels, Y. Vo, I. Johan and M. H. Stenzel, Angew. Chem., Int. Ed., 2023, 62, e202218955 CrossRef CAS PubMed.
- Z. Wang, J. Ayarza and A. P. Esser-Kahn, Angew. Chem., Int. Ed., 2019, 58, 12023–12026 CrossRef CAS PubMed.
- S. M. Zeitler, P. Chakma and M. R. Golder, Chem. Sci., 2022, 13, 4131–4138 RSC.
- H. Mohapatra, M. Kleiman and A. P. Esser-Kahn, Nat. Chem., 2016, 9, 135–139 CrossRef.
- J. Wang, L. Zhang, D. Chen, C. Wang, Z. Ren and Z. Wang, Macromolecules, 2024, 57, 6267–6274 CrossRef CAS.
- Z. Wang, X. Pan, L. Li, M. Fantin, J. Yan, Z. Wang, Z. Wang, H. Xia and K. Matyjaszewski, Macromolecules, 2017, 50, 7940–7948 CrossRef CAS.
- P. Chakma, S. M. Zeitler, F. Baum, J. Yu, W. Shindy, L. D. Pozzo and M. R. Golder, Angew. Chem., Int. Ed., 2023, 62, e202215733 CrossRef CAS.
- C. Ding, Y. Yan, Y. Peng, D. Wu, H. Shen, J. Zhang, Z. Wang and Z. Zhang, Macromolecules, 2022, 55, 4056–4063 CrossRef CAS.
- Z. Ren, C. Ding, R. Ding, J. Wang, Z. Li, R. Tan, X. Wang, Z. Wang and Z. Zhang, ACS Macro Lett., 2023, 12, 1159–1165 CrossRef CAS PubMed.
- Z. Wang, J. Wang, J. Ayarza, T. Steeves, Z. Hu, S. Manna and A. P. Esser-Kahn, Nat. Mater., 2021, 20, 869–874 CrossRef CAS.
- H. Mohapatra, J. Ayarza, E. C. Sanders, A. M. Scheuermann, P. J. Griffin and A. P. Esser-Kahn, Angew. Chem., Int. Ed., 2018, 57, 11208–11212 CrossRef CAS.
- J. M. Wu, W. E. Chang, Y. T. Chang and C. K. Chang, Adv. Mater., 2016, 28, 3718–3725 CrossRef CAS.
- Y. Pang, J. W. Lee, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2020, 59, 22570–22576 CrossRef CAS.
- M. Uchiyama, K. Satoh and M. Kamigaito, Prog. Polym. Sci., 2022, 124, 101485 CrossRef CAS.
- D. Tan and F. Garcia, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC.
- B. M. Peterson, V. Kottisch, M. J. Supej and B. P. Fors, ACS Cent. Sci., 2018, 4, 1228–1234 CrossRef CAS PubMed.
- J. Li, M. Chen, X. Lin, Q. Li, W. Zhang, G. Jin, X. Pan, J. Zhu and X. Zhu, ACS Macro Lett., 2020, 9, 1799–1805 CrossRef CAS PubMed.
- Y. Zhao, Y. Chen, H. Zhou, Y. Zhou, K. Chen, Y. Gu and M. Chen, Nat. Synth., 2023, 2, 653–662 CrossRef.
- C. Wang, R. Zhao, W. Fan, L. Li, H. Feng, Z. Li, C. Yan, X. Shao, K. Matyjaszewski and Z. Wang, Angew. Chem., Int. Ed., 2023, 62, e202309440 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05263c |
| This journal is © The Royal Society of Chemistry 2024 |