
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalysed Tsuji–Trost-type vinyl epoxide cross-coupling with umpolung hydrazones†
Evan F. W.
Chen
 and
Chao-Jun
Li
*
and
Chao-Jun
Li
*
Department of Chemistry, FRQNT Centre for Green Chemistry and Catalysis, McGill University, 801 Sherbrooke Street West, Montreal, Quebec H3A 0B8, Canada. E-mail: cj.li@mcgill.ca
First published on 16th October 2024
Abstract
Selective functionalisation of synthetically useful vinyl epoxides via carbon–carbon (C–C) bond formation has been a major challenge for many years due to its unique inherent chemical reactivity. Non-stabilised carbanions in the form of organometallic reagents have been shown to be robust and versatile reagents in C–C bond formation; however, they are employed in superstoichiometric quantities, require the protection of active functional groups, and generate copious amounts of metallic waste. Therefore, the development of mild carbanion sources as simple alternatives is highly desired. In this work, we report a highly chemo- and regioselective palladium-catalysed vinyl epoxide cross-coupling utilising hydrazones as organometallic equivalents (HOME). Hydrazones, generated from carbonyl-containing renewable feedstocks, enable a more sustainable reaction, and provide an alternative to highly reactive and sensitive unstabilized organometallic reagents. A broad substrate scope, with high functional group tolerance, is demonstrated along with the late-stage functionalisation of natural product derivatives.
Introduction
Characterised by a C–C double bond adjacent to an oxirane ring, vinyl epoxides have been recognised as a crucial building block in a diverse array of reactions.1 Vinyl epoxides exhibit unique reactivity, often acting as potent electrophiles2–4 and radical acceptors,5 and serve as substrates in sigmatropic rearrangements,6 showcasing their versatility in various synthetic transformations. With three different electrophilic sites, these substrates can be readily transformed into synthetically useful linear allylic or branched homoallylic alcohols via SN2′ (1,4-addition) and SN2-type (1,2-addition) ring openings. Over the past few decades, various studies have employed numerous unstabilized carbon nucleophiles for vinyl epoxide functionalisation.1,7–10 Despite these efforts, many alkylation methodologies frequently yielded mixtures of SN2 and SN2′ products,7,10,11 and site-selective alkylation of vinyl epoxides has proven to be difficult. Owing to the inherent steric interactions between the alkyl carbanion and the vinyl epoxide, SN2′ alkylation onto the least hindered alkenyl terminus is typically observed, while exclusive SN2 alkylation is uncommon.The development of transition metal catalysis provides a venue for the regioselective alkylation of vinyl epoxides. Since its discovery in 1973, the palladium-catalysed Tsuji–Trost allylation has been a cornerstone for allylic alkylations.12,13 Enhanced with the introduction of chiral ligands,14 this reaction has been demonstrated to be a powerful tool in crafting intricate, chiral molecules.15–17 With structural similarities shared between vinyl epoxides and allyl acetates, Trost and his colleagues broadened the scope to encompass the alkylation of vinyl epoxides in 1981.18 Non-stabilised alkyl carbanions, such as Grignard, organolithium, organozincate and organostannane reagents, have been shown to be robust and versatile nucleophiles in the generation of new C–C bonds with vinyl epoxides (Fig. 1A).19–22 Nevertheless, limitations are presented in these methodologies, as they employ highly reactive and air/moisture-sensitive reagents with low chemoselectivity, often requiring handling under inert conditions during preparation and use. Organometallic reagents also generate stoichiometric amounts of toxic metallic waste and show a high dependence on alkyl halide precursors from non-renewable petroleum feedstocks. The use of non-renewable carbon resources does not provide a route for sustainable chemistry, and therefore, the development of alternatives to organometallic reagents is highly desired.
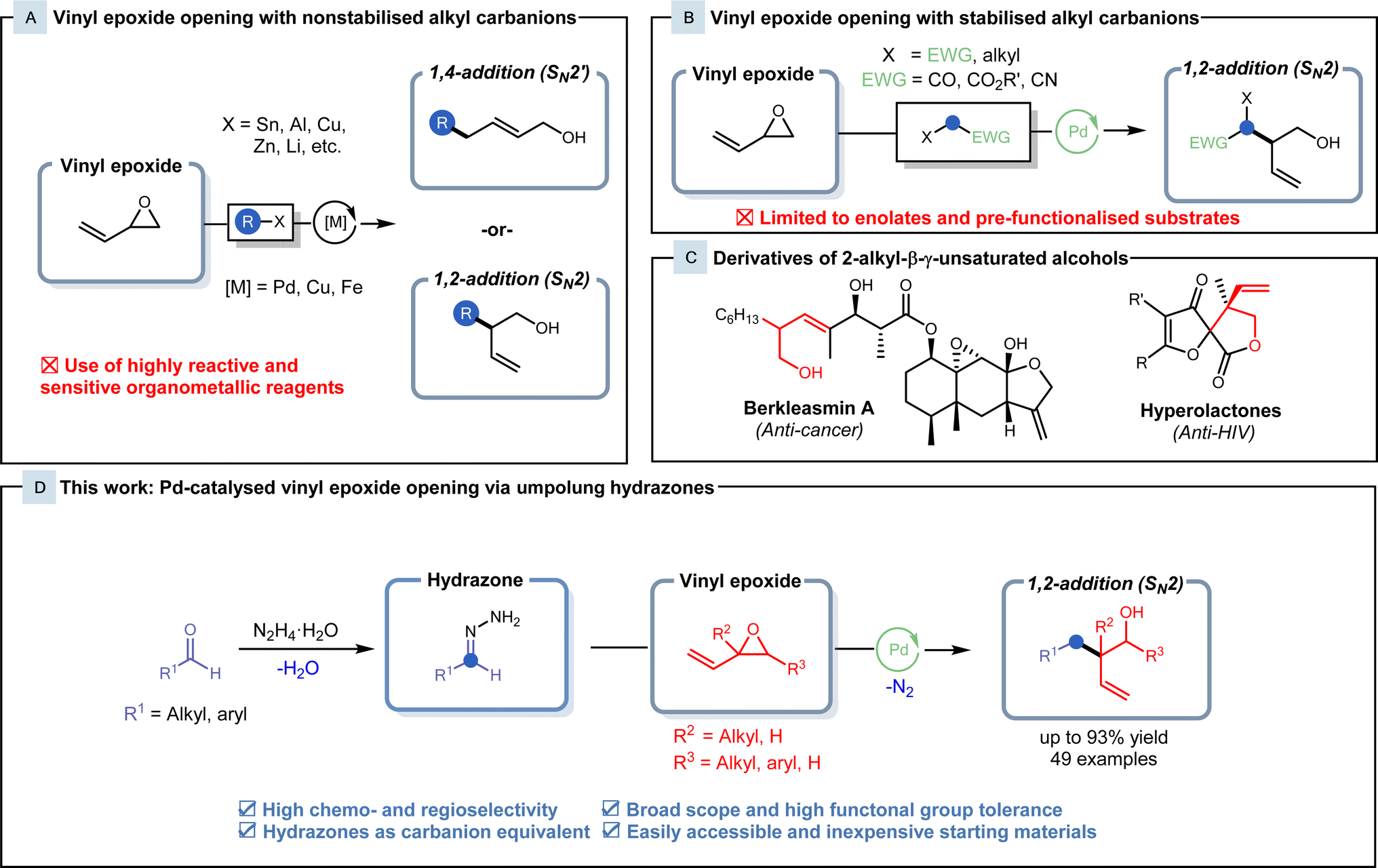 | ||
| Fig. 1 (A) Known methods in metal-catalysed vinyl epoxide opening with non-stabilised alkyl carbanions. (B) Recent reports on Pd-catalysed SN2-type vinyl epoxide opening with stabilised alkyl carbanions. (C) Bio-active natural products containing 2-alkyl-β-γ-unsaturated alcohol motifs. (D) This work: regioselective synthesis of 2-alkyl-β-γ-unsaturated alcohols via Pd-catalysed Tsuji–Trost type vinyl epoxide opening with HOME chemistry. | ||
Carbonyl-derived pronucleophiles in conjunction with palladium catalysis have recently been reported to display selectivity towards SN2-type vinyl epoxide openings (Fig. 1B). Conventionally, malonate substrates are utilised as stabilised nucleophiles,18 but recently, this field has expanded to include enolates derived from pre-functionalised esters.23,24 In 2021, Lundgren and his coworkers reported a Pd-catalysed benzylation of isoprene with 2,2,2-trifluoroethyl (hetero)arylacetate ester enolates to generate lactone intermediates that were subjected to subsequent hydrolysis and decarboxylation to form the branched homoallylic alcohol.25 These 2-alkyl-β-γ-unsaturated alcohol products also possess high synthetic value, as their derivatives appear as core motifs of various natural products and bio-active compounds, such as berkleasmins and hyperolactones (Fig. 1C).26,27 Although these methods provide highly efficient routes for the synthesis of these branched homoallylic alcohols, pre-functionalised and commercially unavailable starting materials hinder the applicability of these methods.
Hydrazones generated from inexpensive, naturally occurring carbonyls can serve as alkyl carbanion equivalents and undergo various reactions with carbon electrophiles.28,29 In our research group, we have been extensively developing this methodology and exploring the use of hydrazones as organometallic equivalents (HOME) in classical nucleophilic addition and cross-coupling reactions.28 This Umpolung strategy allows for efficient transformations under mild conditions comparable to classical organometallic reagents, with the benefit of generating nitrogen gas and water as inert byproducts. Hydrazones also exhibit lower basicity and reactivity along with air/moisture stability in comparison to organometallic reagents, allowing for safer handling. Herein, we report the palladium-catalysed cross-coupling of vinyl epoxides with hydrazones as alkyl organometallic reagent surrogates to generate branched homoallylic alcohols with high regio- and chemoselectivity (Fig. 1D).
Results and discussion
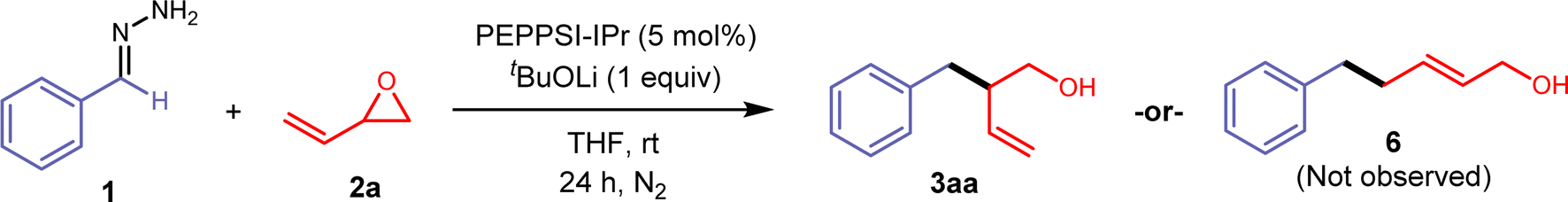
We began our investigation with the cross-coupling of benzaldehyde hydrazone 1aa (1.25 equiv) with vinyl epoxide (0.23 mmol) 2a using a Pd(0)/triphenylphosphine catalyst system (Table 1) with lithium tert-butoxide (2 equiv) as the base. Under these conditions, we primarily observed a C-alkylated product, and N-alkylated products were not detected. Only racemic branched homoallylic alcohol 3aa was observed in 29% yield and the linear allylic alcohol product 6 was not detected (entry 2). We hypothesised that only one equivalent of the base was required for the transformation, as the alkoxy-intermediate generated from the vinyl epoxide ring opening potentially acts as an equivalent of base to facilitate the deprotonation of the hydrazone (see ESI Fig. S1†). Vinyl epoxides are also known to undergo reactions and rearrangements in the presence of Lewis acids.6,30 While palladium is the catalyst for our system, it could also potentially act as a Lewis acid. This theory was validated through a control experiment with 1 equivalent of PEPPSI-IPr, where 3aa was not detected and we primarily observed decomposition of 2a. With these results, a lower Pd catalyst loading of 5% and 1 equivalent of base gave higher yields. Any further decrease of the Pd loading and base diminished the yield. N-heterocyclic carbene (NHC) ligands were previously reported to be an efficient ligand for the Pd-catalysed C-allylations of hydrazones.31,32 Electron-rich, bulky NHC ligands such as 1,3-bis(2,4,6-trimethylphenyl)imidazolium chloride (IMes HCl) and 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride (IPr HCl) were tested in combination with Pd2(dba)3 and afforded 3aa with yields of 19% (entry 3) and 36% (entry 4). A pre-ligated IPr (PEPPSI-IPr) was very efficient in this transformation and gave 3aa in a 90% yield (entry 1). Other tert-butoxide salts, organic bases (1,8-diazabicyclo(5.4.0)undec-7-ene (DBU), 1,5,7-triazabicyclo(4.4.0)dec-5-ene (TBD)), and lithium salt bases were screened but all gave reduced yields (entries 8 and 9, see ESI Table S2†). Cs2CO3 was the only other base that gave 3aa with a high yield of 82% (entry 7). Finally, various solvents such as 2-MeTHF, dioxane, toluene, and DMF were screened, but THF remained as the best solvent for this reaction (see ESI Table S3†). Control experiments show that no homoallylic alcohol 3aa was produced in the absence of a Pd catalyst and base.|
|
||
|---|---|---|
| Entry | Deviation from standard conditionsa | 3aa (yield%) |
| a Standard conditions: benzaldehyde hydrazone, 1a (0.28 mmol, 1.25 equiv), vinyl epoxide, 2a (0.23 mmol), PEPPSI-IPr (5 mol%), and tBuOLi (0.23 mmol, 1 equiv) in 1.5 mL THF for 24 h at room temperature under N2. Yields were determined by 1H NMR with 1,3,5-trimethoxybenzene as an internal standard. | ||
| 1 | None | 90 |
| 2 | Pd2(dba)3 (5 mol%), PPh3 (10 mol%), 2 equiv tBuOLi, 45 °C | 29 |
| 3 | Pd2(dba)3 (2.5 mol%), IMes HCl (5 mol%) | 19 |
| 4 | Pd2(dba)3 (2.5 mol%), IPr HCl (5 mol%) | 36 |
| 5 | [Pd(allyl)Cl]2 (2.5 mol%), IPr HCl (5 mol%) | 48 |
| 6 | SPhos Pd G4 (5 mol%) | 58 |
| 7 | Cs2CO3 instead of tBuOLi | 82 |
| 8 | K2CO3 instead of tBuOLi | 24 |
| 9 | DBU instead of tBuOLi | n.d. |
| 10 | 45 °C | 64 |
| 11 | Dioxane instead of THF | 84 |
| 12 | Toluene instead of THF | 78 |
| 13 | No base | n.d. |
| 14 | No catalyst | n.d. |
| 15 | Under air | 69 |
|
||
Substrate scope
With our optimised conditions for the cross-coupling of hydrazone 1aa and vinyl epoxide 2a, we proceeded to our substrate scope. Various substituted benzaldehyde hydrazones were explored (Fig. 2A). Initially, we observed complete conversion with benzaldehyde hydrazone 1aa and 2a within 24 hours, but incomplete conversion for para-substituted benzaldehyde hydrazones and polyaromatic hydrazones was observed. Lithium tert-butoxide was increased to 1.5 equivalents and a reaction time of 36 h was employed to ensure complete conversion and to improve reproducibility of the procedure (See ESI Table S4†). In some cases, the product was unable to be separated from the tert-butanol side-product, therefore cesium carbonate (1.25 equiv) was used instead of lithium tert-butoxide. In general, hydrazones with various substituents and substitution patterns gave moderate to excellent yields. Aryl hydrazones containing halogens, heteroatoms, and polyaromatic systems worked well under our conditions and this reaction exhibited excellent functional group tolerance. Para-substituted benzaldehyde hydrazones were first investigated. Hydrazones bearing electron-withdrawing groups, such as fluoro-, bromo-, chloro-, trifluoromethyl-, cyano- and phenyl-, at this position gave desired products 3ab–3ag at high yields of 71–93%. With electron-donating groups in the para-position, in the case of methyl-, isopropyl-, and benzyloxy-benzaldehyde hydrazones, the yield of the desired product 3ah–3aj had slightly diminished (59–79%). This is due to an electronic effect, where the electron-donating group destabilises the carbanion formed at the benzylic position. In the case of 3ak, 3al, and 3bc, the strong electron-donating group did not favour the formation of the carbanion, and we did not observe any desired product. Only the aryl hydrazone starting material was observed and recovered. Electron-donating and electron-withdrawing ortho-substituted benzaldehyde hydrazones also performed well in these conditions, giving desired products 3am–3ap in 68–78% yield. Regarding 3ap, the pre-coordination of the methoxy-group onto the Pd catalyst could direct the hydrazone coordination to the Pd catalyst after deprotonation, leading to an increased yield compared to the other cases with electron-donating groups in the ortho-position. Meta-substituted benzaldehyde hydrazones were also investigated and gave desired products 3aq–3as in moderately high yields of 69–82%. 1- and 2-Naphthaldehyde hydrazones proved to be effective substrates, giving the desired products 3au and 3at in high yields of 80% and 73%, respectively. Multiply-substituted benzaldehyde hydrazones worked well to yield alcohols 3av–3ay in moderate yields of 45–62%, where the steric hindrance and electron-donating properties of the ortho-substituents hinder the yield. Heteroaryl aldehyde hydrazones were also well tolerated (Fig. 2B), and moderately high yields of 60–71% were achieved for branched homoallylic alcohols 3az–3bd.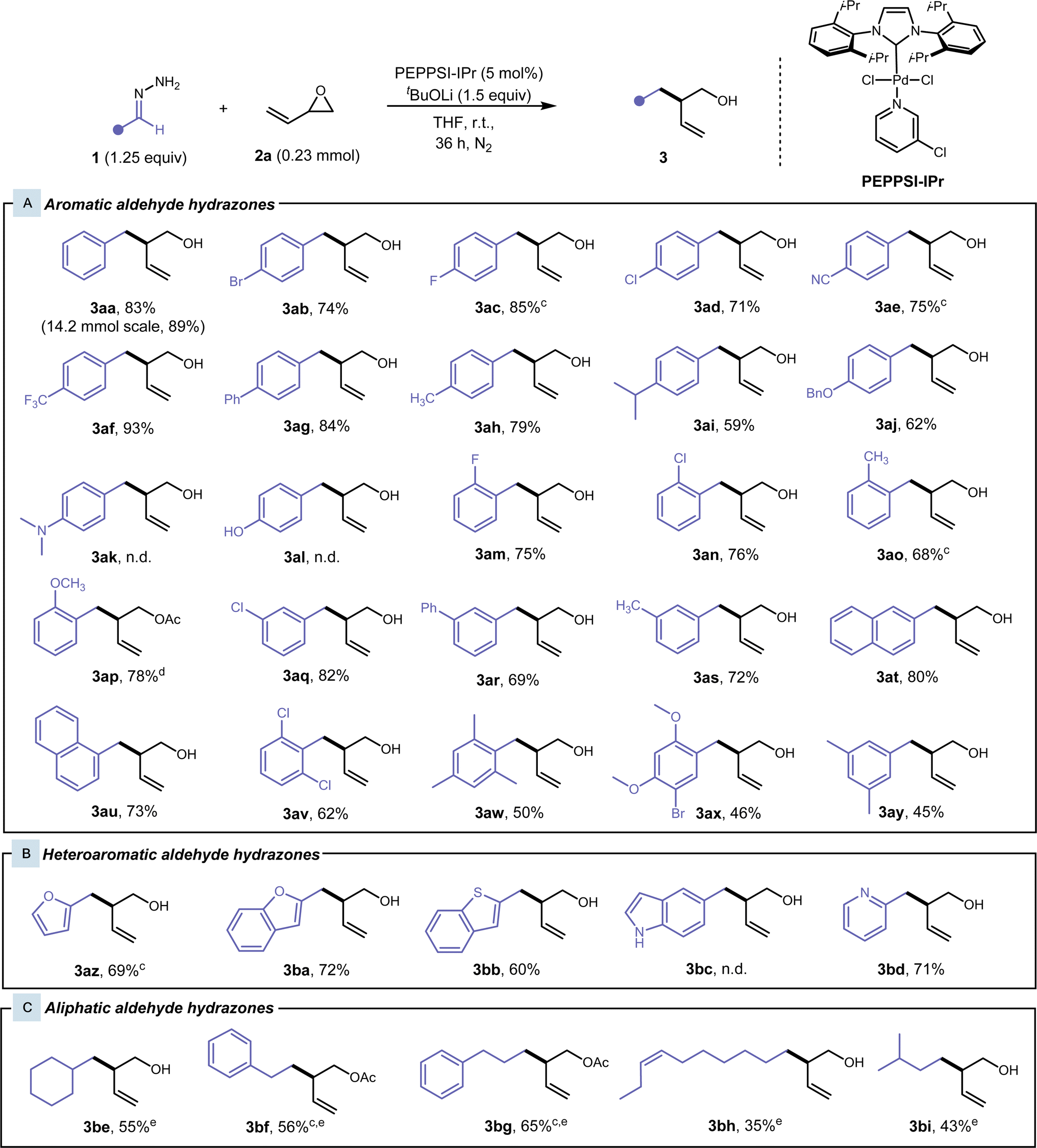 | ||
| Fig. 2 Aldehyde hydrazone substrate scope.a,b (a) Reaction conditions: Hydrazone 1 (0.28 mmol, 1.25 equiv), vinyl epoxide 2a (0.23 mmol), PEPPSI-IPr (5 mol%), and tBuOLi (0.34 mmol, 1.5 equiv) in 1.5 mL THF at room temperature for 36 h under N2. (b) Yields of isolated products. (c) Hydrazone 1 (0.28 mmol, 1.25 equiv), vinyl epoxide 2a (0.23 mmol), PEPPSI-IPr (5 mol%), and Cs2CO3 (0.28 mmol, 1.25 equiv) in 1.5 mL THF at room temperature for 36 h under N2. (d) Acetylation conditions: crude compound (purified with a silica plug), Ac2O (500 μL), and pyridine (500 μL) at room temperature overnight. (e) Hydrazone 1 (0.45 mmol, 2 equiv), vinyl epoxide 2a (0.23 mmol), PEPPSI-IPr (5 mol%), FeCl3 (10 mol%) and tBuOLi (0.34 mmol, 1.5 equiv), in 1.5 mL THF at 45 °C for 24 h under N2. | ||
We then shifted our focus to aliphatic aldehyde hydrazones (Fig. 2C), since generating carbanions from aliphatic aldehyde hydrazones presents a greater challenge. Without a neighbouring aromatic group, resonance stability of the carbanion is absent, and these hydrazones tend to react with themselves to form their inactive azine counterpart at an increased rate. Our initial optimised conditions showed low yields, and the reaction was reoptimized (See ESI Table S5† for more details). A Lewis acid catalyst (10 mol% FeCl3) was added to increase the reactivity of the vinyl epoxide, while the reaction was heated at 50 °C for 24 h to accelerate the reaction and to out-compete the azine side reaction. We were able to achieve the desired products 3be–3bi from cyclohexanecarboxaldehyde, 3-phenylpropionaldehyde, phenylacetaldehyde, cis-dec-7-enal, and isovaleraldehyde hydrazones in low to moderate yields of 35–65%, where the stability of the hydrazone mainly dictated the yield. Acetophenone-derived hydrazones were also subjected to the standard conditions and these modified conditions. However, only trace amounts of product were able to be detected by GCMS. This could be attributed to the added steric bulk about the benzylic position of the generated carbanion.
Subsequently, various substituted vinyl epoxides were then investigated (Fig. 3). Alkyl-substituted vinyl epoxides afforded the desired products 3bj–3bn in high yields of 61–88%. Next, benzaldehyde-derived vinyl epoxides present as a mixture of diastereomers were subjected to our protocol, and high yields of 76–89% were achieved for racemic alcohols 3bo–3bq. We also tested our conditions with a heteroaryl-substituted vinyl epoxide, which gave 3br in 53% yield.
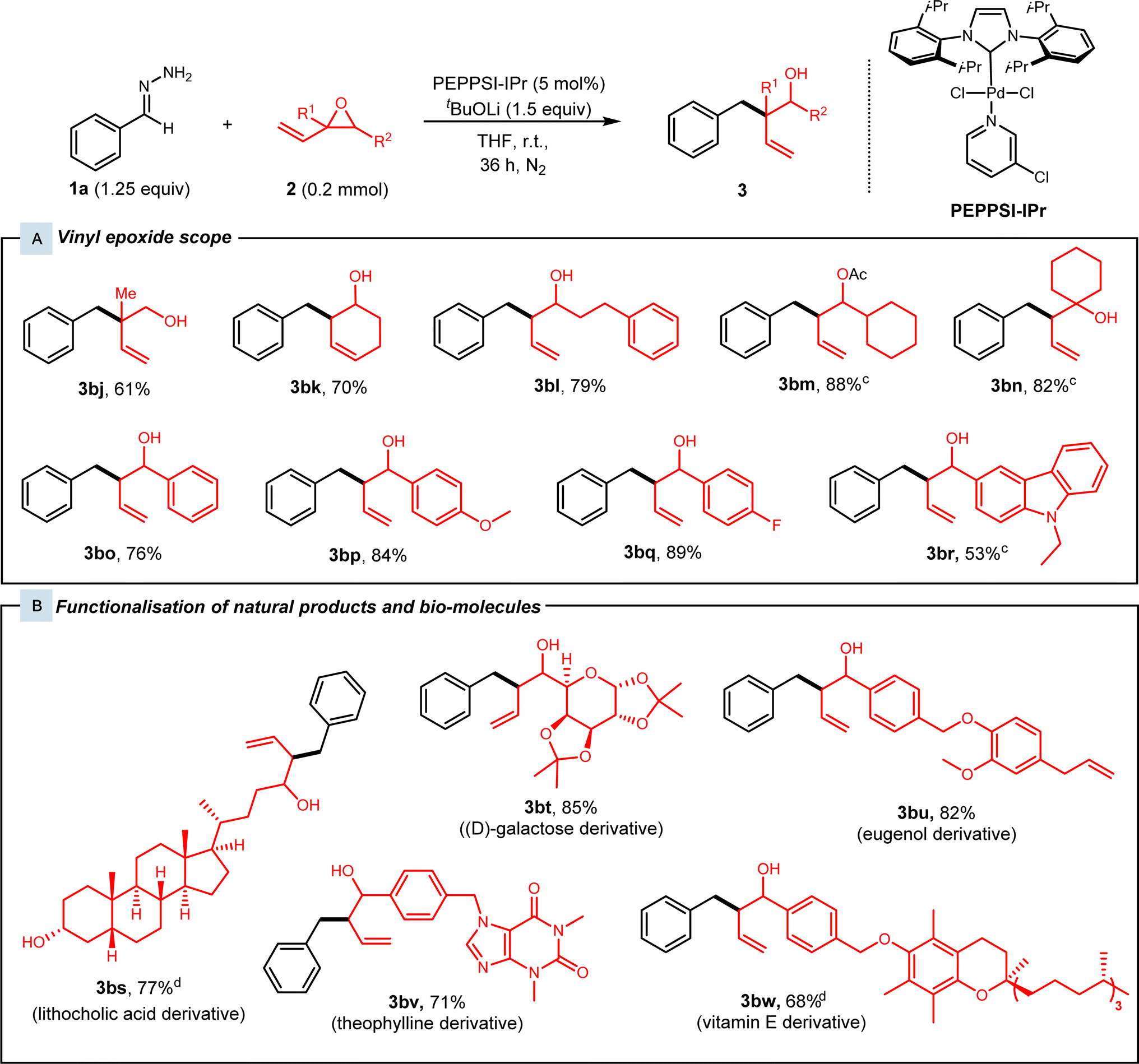 | ||
| Fig. 3 Vinyl epoxide substrate scope.a,b (a) Reaction conditions: Hydrazone 1 (0.25 mmol, 1.25 equiv), vinyl epoxide 2 (0.2 mmol), PEPPSI-IPr (5 mol%), and tBuOLi (0.38 mmol, 1.5 equiv) in 1.5 mL THF at room temperature for 36 h under N2. (b) Yields of isolated products. (c) Reaction conditions: Hydrazone 1 (0.20 mmol), vinyl epoxide 2 (0.25 mmol, 1.25 mmol), PEPPSI-IPr (5 mol%), and tBuOLi (0.38 mmol, 1.5 equiv) in 1.5 mL THF at room temperature for 36 h under N2. (d) 0.15 mmol vinyl epoxide. | ||
With the high functional group tolerance and efficiency of this protocol, we then turned our attention to the late-stage derivatisation of natural products (Fig. 3B). Vinyl epoxides synthesised from various natural product derivatives were subjected to standard conditions. Lithocholic acid and (D)-galactose-derived vinyl epoxides were superior substrates as they afforded the branched homoallylic alcohols 3bs and 3bt in yields of 77% and 85% respectively. Alkaloids such as theophylline-derived vinyl epoxide 2m were tested with our conditions and alcohol 3bv was achieved with 71% yield. Lastly, eugenol and vitamin E-derived vinyl epoxides 2n and 2o reacted smoothly under our catalytic system and gave alcohols 3bu and 3bw in 82% and 68% yield.
Mechanistic investigation
Several control experiments were performed to gain preliminary insight into the reaction mechanism (see the ESI† for more details). We first investigated the participation of the alkoxy-intermediate in our reaction. In previous literature, it was theorised that the alkoxy-intermediate deprotonates the nucleophile which directs the 1,2-addition onto the vinyl epoxide.33 In the absence of base, no reaction occurred between 1 and 2a (Fig. 4A). When 10 mol% base was employed, 3aa was only observed in 10% yield. These results suggest that the added base performs the initial deprotonation step to form a 2,3-diazaallyl anion. Otherwise, we would expect an adduct of the hydrazone and the vinyl epoxide if the alkoxy-intermediate performs the first deprotonation of the hydrazone. Subsequently, we performed deuterium isotope experiments. Deuterated benzaldehyde hydrazone 1 was subjected to our protocol and we observed deuteration of both the benzylic position (38% D) and the alcohol (60% D) of 3aa (Fig. 4B). Deuteration of the alcohol is evidence that the alkoxy-intermediate participates in one deprotonation step. We then subjected vinyl epoxide 2a with N-tosyl benzaldehyde hydrazone, a common carbene precursor. No reaction occurred, which deemed a carbene mechanism to be implausible and the reaction likely proceeds through a two-electron pathway (Fig. 4C). The high regioselectivity could be attributed to a 3,3′-elimination step analogous to diallylpalladium complexes in the work of Morken and coworkers (See ESI Fig. S2†).34,35 | ||
| Fig. 4 Mechanistic studies. (A) Base control studies. (B) Deuterium labelling studies. (C) Carbene mechanism probe experiment. | ||
Based on the previously described experimental observations, and preceding literature in similarly operating catalytic systems,31,34–36 we have proposed the following mechanism (Fig. 5). Initially, the Pd(II) precatalyst is activated with residual hydrazine in the reaction mixture leading to the IPr-Pd(0) active catalyst A. Hydrazine has been shown to reduce Pd(II) to Pd(0) efficiently.37A then undergoes oxidative addition with an activated vinyl epoxide 2 to form the π-allylpalladium complex B. Deprotonation of the hydrazone 1 with tBuOLi would allow the formation of a 2,3-diazaallyl anion, which then coordinates to B and forms C. A 3,3′-elimination would occur, leading to D in a regioselective manner. After ligand dissociation to reform active catalyst A, intermediate E undergoes intramolecular deprotonation and releases nitrogen with the assistance of a protonated base to form compound 3.
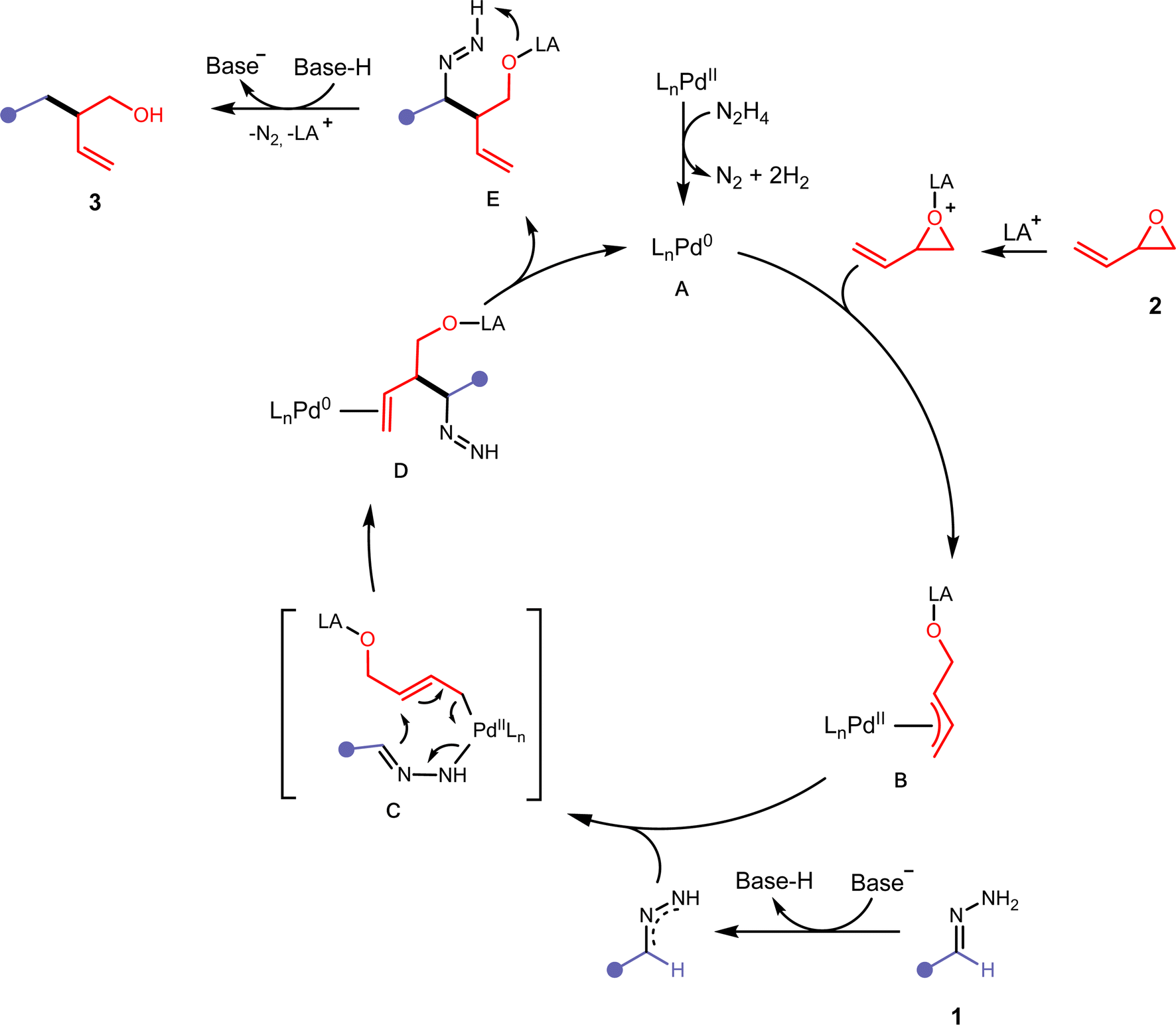 | ||
| Fig. 5 Proposed mechanism for the Pd-catalysed cross-coupling of hydrazones and vinyl epoxides. | ||
Conclusions
In summary, we have developed a regioselective and high-yielding Pd-catalysed method for the synthesis of 2-alkyl-β-γ-unsaturated alcohols utilising vinyl epoxides and Umpolung hydrazones. Highlights of this methodology include the efficient transformation of various substituted aryl/alkyl aldehyde-derived hydrazones and substituted vinyl epoxides. A broad substrate scope, high functional group tolerance, and late-stage modification of complex, diverse molecules demonstrate its synthetic utility. HOME chemistry provides a sustainable route for the functionalisation of vinyl epoxides with the deployment of carbonyl precursors as mild carbanion sources, and the absence of alkyl halides or external organometallic reagents. Our laboratory is currently conducting further research on the mechanism and exploring an asymmetric variant of this reaction.Data availability
All experimental procedures, mechanistic investigations, and characterisation data of all synthesised compounds are available in the ESI.†Author contributions
EC and CJL conceptualised the idea of this work. EC designed and conducted experiments for the optimisation and scope, and wrote the manuscript. CJL provided general guidance, project direction and manuscript revision.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by funding from the Canada Research Chair Foundation, the FRQNT Centre for Green Chemistry and Catalysis (CGCC), the National Science and Engineering Research Center (NSERC), the Canada Foundation for Innovation and McGill University. EC thanks NSERC for the Postgraduate Scholarship (PGS-D). The authors would like to thank Durbis J. Castillo-Pazos and Juan D. Lasso for proofreading this manuscript and Dr Alexander S. Wahba for running HRMS for all new compounds.References
- J. He, J. Ling and P. Chiu, Vinyl Epoxides in Organic Synthesis, Chem. Rev., 2014, 114(16), 8037–8128 CrossRef CAS PubMed.
- T. Murakami, T. Shimizu and K. Taguchi, Synthesis of Sphingadienine-type Glucocerebrosides, Tetrahedron, 2000, 56(4), 533–545 CrossRef CAS.
- B. M. Trost, G. Dong and J. A. Vance, Cyclic 1,2-Diketones as Core Building Blocks: A Strategy for the Total Synthesis of (−)-Terpestacin, Chem.–Eur. J., 2010, 16(21), 6265–6277 CrossRef CAS PubMed.
- I. Mangion, N. Strotman, M. Drahl, J. Imbriglio and E. Guidry, Dynamic Kinetic Asymmetric Allylation of Hydrazines and Hydroxylamines, Org. Lett., 2009, 11(15), 3258–3260 CrossRef CAS PubMed.
- N. Charrier, D. Gravestock and S. Z. Zard, Radical Additions of Xanthates to Vinyl Epoxides and Related Derivatives: A Powerful Tool for the Modular Creation of Quaternary Centers, Angew. Chem., Int. Ed., 2006, 45(39), 6520–6523 CrossRef CAS PubMed.
- X.-M. Deng, X.-L. Sun and Y. Tang, Highly Regioselective Rearrangement of 2-Substituted Vinylepoxides Catalyzed by Gallium(III) Triflate, J. Org. Chem., 2005, 70(16), 6537–6540 CrossRef CAS PubMed.
- R. W. Herr and C. R. Johnson, Comparison of the Reactions of Methylmagnesium, Methyllithium, and Methylcopper Reagents with 1,2-Epoxybutane and 3,4-Epoxy-1-butene, J. Am. Chem. Soc., 1970, 92(16), 4979–4981 CrossRef CAS.
- O. Equey, E. Vrancken and A. Alexakis, Regioselective SN2 Opening of Vinylic Epoxides with Trialkylzincates and Trialkylaluminates, Eur. J. Org. Chem., 2004, 2004(10), 2151–2159 CrossRef.
- A. B. Smith, S. M. Pitram, M. J. Gaunt and S. A. Kozmin, Dithiane Additions to Vinyl Epoxides: Steric Control over the SN2 and SN2‘ Manifolds, J. Am. Chem. Soc., 2002, 124(49), 14516–14517 CrossRef CAS PubMed.
- P. Restorp and P. Somfai, Regioselective and Divergent Opening of Vinyl Epoxides with Alkyne Nucleophiles, Eur. J. Org. Chem., 2005, 2005(18), 3946–3951 CrossRef.
- T. Hübscher and G. Helmchen, Enantioselective Formal Synthesis of Brefeldin A and Analogues via Anionic Cyclization of an Alkenyl Epoxide, Synlett, 2006, 2006(09), 1323–1326 CrossRef.
- J. Tsuji, H. Takahashi and M. Morikawa, Organic Syntheses by Means of Noble Metal Compounds XVII. Reaction of π-Allylpalladium Chloride with Nucleophiles, Tetrahedron Lett., 1965, 6(49), 4387–4388 CrossRef.
- B. M. Trost and T. J. Fullerton, New Synthetic Reactions. Allylic Alkylation, J. Am. Chem. Soc., 1973, 95(1), 292–294 CrossRef CAS.
- B. M. Trost, D. L. Van Vranken and C. Bingel, A Modular Approach for Ligand Design for Asymmetric Allylic Alkylations via Enantioselective Palladium-Catalyzed Ionizations, J. Am. Chem. Soc., 1992, 114(24), 9327–9343 CrossRef CAS.
- B. M. Trost and M. L. Crawley, Asymmetric Transition-Metal-Catalyzed Allylic Alkylations: Applications in Total Synthesis, Chem. Rev., 2003, 103(8), 2921–2944 CrossRef CAS PubMed.
- B. M. Trost and D. L. Van Vranken, Asymmetric Transition Metal-Catalyzed Allylic Alkylations, Chem. Rev., 1996, 96(1), 395–422 Search PubMed.
- O. Pàmies, J. Margalef, S. Cañellas, J. James, E. Judge, P. J. Guiry, C. Moberg, J.-E. Bäckvall, A. Pfaltz, M. A. Pericàs and M. Diéguez, Recent Advances in Enantioselective Pd-Catalyzed Allylic Substitution: From Design to Applications, Chem. Rev., 2021, 121(8), 4373–4505 CrossRef PubMed.
- B. M. Trost and G. A. Molander, Neutral Alkylations via Palladium(0) Catalysis, J. Am. Chem. Soc., 1981, 103(19), 5969–5972 Search PubMed.
- A. M. Echavarren, D. R. Tueting and J. K. Stille, Palladium-Catalyzed Coupling of Vinyl Epoxides with Organostannanes, J. Am. Chem. Soc., 1988, 110(12), 4039–4041 Search PubMed.
- M. Pineschi, F. Del Moro, P. Crotti, V. Di Bussolo and F. Macchia, Catalytic Regiodivergent Kinetic Resolution of Allylic Epoxides: A New Entry to Allylic and Homoallylic Alcohols with High Optical Purity, J. Org. Chem., 2004, 69(6), 2099–2105 CrossRef CAS PubMed.
- T. Hata, R. Bannai, M. Otsuki and H. Urabe, Iron-Catalyzed Regio- and Stereoselective Substitution of γ,δ-Epoxy-α,β-unsaturated Esters and Amides with Grignard Reagents, Org. Lett., 2010, 12(5), 1012–1014 Search PubMed.
- T. Mori, T. Nakamura and M. Kimura, Stereoselective Coupling Reaction of Dimethylzinc and Alkyne toward Nickelacycles, Org. Lett., 2011, 13(9), 2266–2269 Search PubMed.
- M. E. Fox, I. C. Lennona and V. Farina, Catalytic Asymmetric Synthesis of Ethyl (1R,2S)-Dehydrocoronamate, Tetrahedron Lett., 2007, 48, 945–948 CrossRef CAS.
- Y. Wu, C. Du, C. Hu, Y. Li and Z. Xie, Biomimetic Synthesis of Hyperolactones, J. Org. Chem., 2011, 76(10), 4075–4081 CrossRef CAS PubMed.
- M. G. J. Doyle, A. L. Gabbey, W. McNutt and R. J. Lundgren, Enantioselective Tertiary Electrophile (Hetero)Benzylation: Pd-Catalyzed Substitution of Isoprene Monoxide with Arylacetates, Angew. Chem., Int. Ed., 2021, 60(51), 26495–26499 CrossRef CAS PubMed.
- K. C. Nicolaou, T. R. Wu, D. Sarlah, D. M. Shaw, E. Rowcliffe and D. R. Burton, Total Synthesis, Revised Structure, and Biological Evaluation of Biyouyanagin A and Analogues Thereof, J. Am. Chem. Soc., 2008, 130(33), 11114–11121 CrossRef CAS PubMed.
- M. Isaka, U. Srisanoh, S. Veeranondha, W. Choowong and S. Lumyong, Cytotoxic Eremophilane Sesquiterpenoids From the Saprobic Fungus Berkleasmium Nigroapicale BCC 8220, Tetrahedron, 2009, 65(43), 8808–8815 CrossRef CAS.
- X.-J. Dai, C.-C. Li and C.-J. Li, Carbonyl Umpolung as an Organometallic Reagent Surrogate, Chem. Soc. Rev., 2021, 50(19), 10733–10742 RSC.
- C.-J. Li, HOME-Chemistry: Hydrazone as Organo-Metallic Equivalent, Pure Appl. Chem., 2023, 95(5), 465–474 CrossRef CAS.
- A. Alexakis, E. Vrancken and P. Mangeney, Effect of BF3·Et2O Reagent on the Base-Promoted Rearrangements of Epoxides Attached to Eight-Membered Rings, J. Chem. Soc., Perkin Trans., 2000,(20), 3354–3355 RSC.
- D. Zhu, L. Lv, C.-C. Li, S. Ung, J. Gao and C.-J. Li, Umpolung of Carbonyl Groups as Alkyl Organometallic Reagent Surrogates for Palladium-Catalyzed Allylic Alkylation, Angew. Chem., Int. Ed., 2018, 57(50), 16520–16524 CrossRef CAS PubMed.
- J. Kan, Z. Chen, Z. Qiu, L. Lv, C. Li and C.-J. Li, Umpolung Carbonyls Enable Direct Allylation and Olefination of Carbohydrates, Sci. Adv., 2022, 8(10), eabm6840 CrossRef CAS PubMed.
- B. M. Trost, R. C. Bunt, R. C. Lemoine and T. L. Calkins, Dynamic Kinetic Asymmetric Transformation of Diene Monoepoxides: A Practical Asymmetric Synthesis of Vinylglycinol, Vigabatrin, and Ethambutol, J. Am. Chem. Soc., 2000, 122(25), 5968–5976 Search PubMed.
- P. Zhang, L. A. Brozek and J. P. Morken, Pd-Catalyzed Enantioselective Allyl–Allyl Cross-Coupling, J. Am. Chem. Soc., 2010, 132(31), 10686–10688 Search PubMed.
- L. A. Brozek, M. J. Ardolino and J. P. Morken, Diastereocontrol in Asymmetric Allyl–Allyl Cross-Coupling: Stereocontrolled Reaction of Prochiral Allylboronates with Prochiral Allyl Chlorides, J. Am. Chem. Soc., 2011, 133(42), 16778–16781 CrossRef CAS PubMed.
- L. Lv and C.-J. Li, Palladium-Catalyzed Defluorinative Alkylation of gem-Difluorocyclopropanes: Switching Regioselectivity via Simple Hydrazones, Angew. Chem., Int. Ed., 2021, 60(23), 13098–13104 CrossRef CAS PubMed.
- Y. Wang, Y.-F. Shi, Y.-B. Chen and L.-M. Wu, Hydrazine Reduction of Metal Ions to Porous Submicro-Structures of Ag, Pd, Cu, Ni, and Bi, J. Solid State Chem., 2012, 191, 19–26 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05411c |
| This journal is © The Royal Society of Chemistry 2024 |