DOI:
10.1039/D4SC05524A
(Edge Article)
Chem. Sci., 2024,
15, 17873-17880
Arene extrusion as an approach to reductive elimination at boron: implication of carbene-ligated haloborylene as a transient reactive intermediate†
Received
16th August 2024
, Accepted 1st October 2024
First published on 3rd October 2024
Abstract
Herein, we report boron-centered arene extrusion reactions to afford putative cyclic(alkyl)(amino) carbene (CAAC)-ligated chloroborylene and bromoborylene intermediates. The borylene precursors, chloro-boranorbornadiene (ClB(C6Me6), 2Cl) and bromo-boranorbornadiene (BrB(C6Me6), 2Br) were synthesized through the reaction of the corresponding 1-halo-2,3,4,5-tetramethylborole dimer (XBC4Me4)2 (X = Cl, 1Cl; X = Br, 1Br) with 2-butyne. Treatment of 2Cl with CAACs resulted in the release of di-coordinate chloro-borylene (CAAC)BCl from hexamethylbenzene (C6Me6) at room temperature. In contrast, the reaction of 2Br with CAAC led to the formation of a boronium species [(CAAC)BC6Me6]+Br− (7) at room temperature. Heating 7 in toluene promoted the release of di-coordinate bromo-borylene (CAAC)BBr as a transient species. Surprisingly, heating 7 in dichloromethane resulted in the C–H activation of hexamethylbenzene. The conversion of a CAAC-stabilized bromo-borepin to a borylene, a boron-centered retro Büchner reaction, was also investigated.
1 Introduction
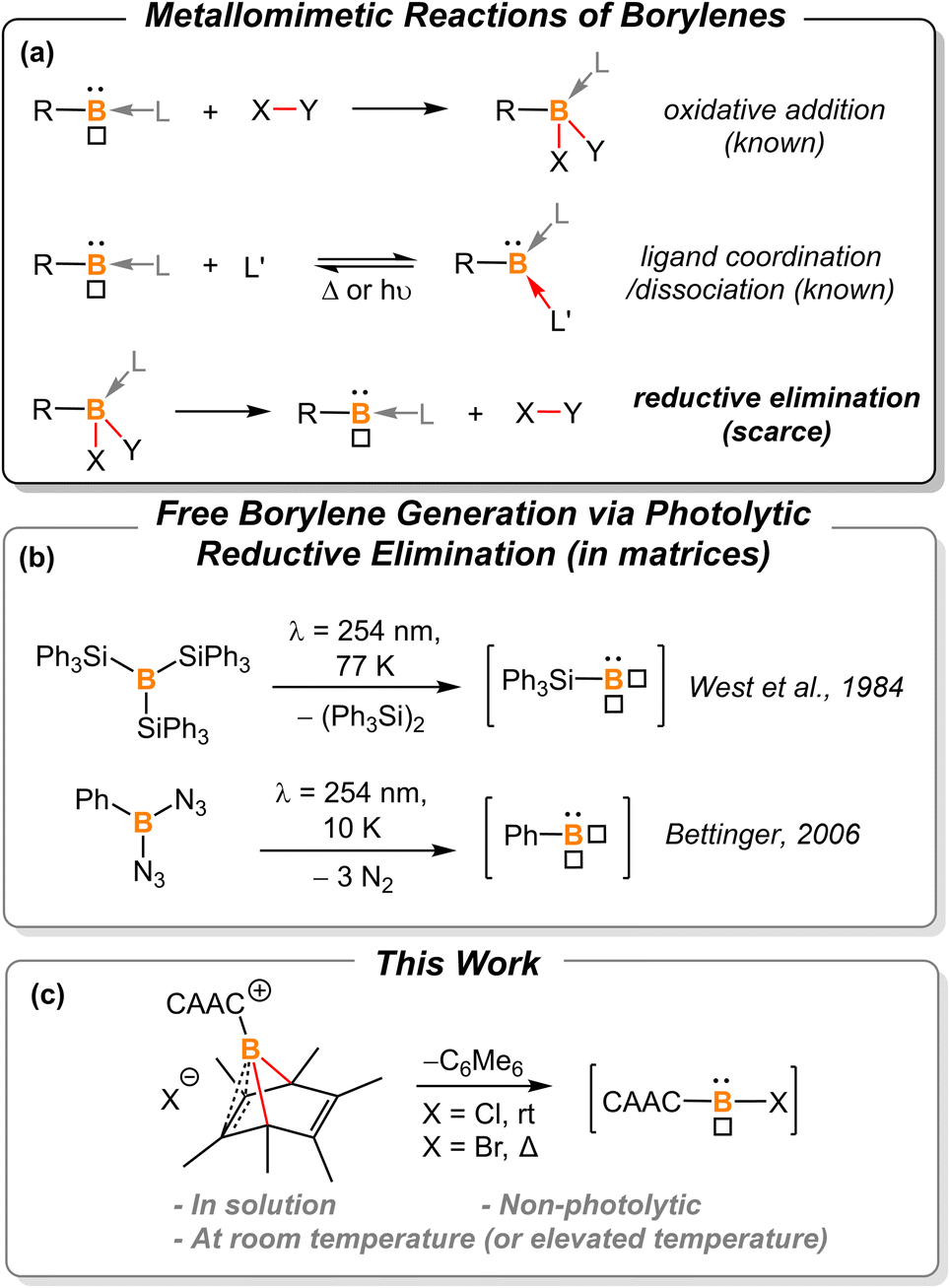
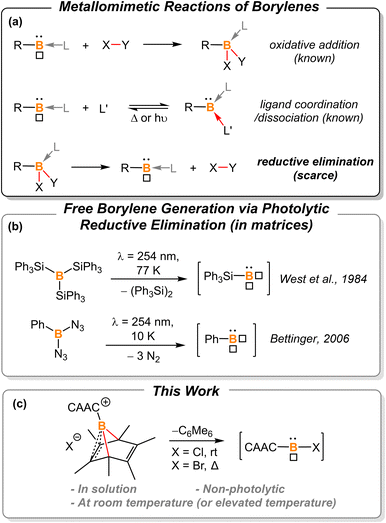
Borylenes are boron analogs of carbenes. Free borylenes (with the chemical formula R–B) are monocoordinate species that possess only four valence electrons, representing a class of hypovalent main-group species.1 While free monocoordinate borylenes only exist as highly reactive intermediates, Lewis-base stabilized borylenes are easier to handle, with some even being isolable at room temperature.2 Dicoordinate borylenes are conceptually expected to possess both a non-bonding electron pair and an empty p orbital, thus constituting a key class of metallomimetic boron species.3 Owing to the low-coordinate nature of borylenes, these B(I) species readily undergo oxidative addition reactions to form stable B(III)-centered molecules (Fig. 1a, top).2b,g,2j,4 The coordination of a ligand is also known to stabilize reactive borylene species. This ligand coordination/dissociation process mimics reaction steps that are common for transition metals (Fig. 1a, middle).2g,j,5 However, it is rationalized that B(III) species are less likely to undergo reductive elimination to afford corresponding borylenes due to the low electronegativity, small radius, and inherent electron-deficient nature of the boron atom (Fig. 1a, bottom). Indeed, to the best of our knowledge, there are only two examples of reductive elimination occurring at a single boron atom.
 |
| | Fig. 1 (a) Summarized reactions of borylenes, “□” represents the empty p-orbital of boron. (b) Previously reported photolytic reductive elimination to afford free borylenes; (c) borylene release from hexamethylboranorbornadiene. | |
In 1984, West et al. reported that photolysis of B(SiPh3)3 in a matrix resulted in the formation of triphenylsilylborylene (Ph3SiB) by elimination of equimolar Ph3Si–SiPh3 (Fig. 1b).1k In 2006, Bettinger described that the photolysis of bisazidophenylborane (PhB(N3)2) isolated in cryogenic matrices results in phenylborylene (PhB).1j These two examples demonstrate that reductive elimination can take place via photolysis of energetic B(III) compounds in matrices. However, a fundamental question still remains elusive: can boron behave like transition metals that undergo thermal reductive elimination?
In the last decade, the Cummins group developed a series of dibenzo-7-phosphanorbornadiene compounds, RPA (A = C14H10 or anthracene).6 Depending on the nature of the substituent, some of these compounds eliminate one equivalent of anthracene and release a reactive phosphinidene species into the solution or gas phase for further reactions or spectroscopic characterization. Such phosphorus-centered arene extrusion reactions are regarded as a particular type of reductive elimination, as the center atom becomes a corresponding subvalent species upon the extrusion of the arene molecule.6a,b,7 We postulate that aromatization could provide the extra driving force for boron-centered reductive elimination.
In 2013, Braunschweig et al. investigated the potential of the liberation of NHC-stabilized phenylborylene from (IMe)(Ph)B(C6Ph6).8 However, ring expansion to form NHC-borepin Lewis adducts was found to be the preferred reaction channel and no elimination of an NHC-stabilized borylene was detected. They attributed the observed behavior to molecular strain and steric factors. Therefore, we sought to find a platform with less molecular strain and steric factors for releasing the borylene fragment, and thus hexamethyl-boranorbornadiene was the targeted system (Fig. 1c). Herein, we demonstrate the first example of non-photolytic reductive elimination taking place at a single boron atom to afford putative cyclic(alkyl)(amino) carbene (CAAC)-stabilized haloborylenes.
2 Results and discussion
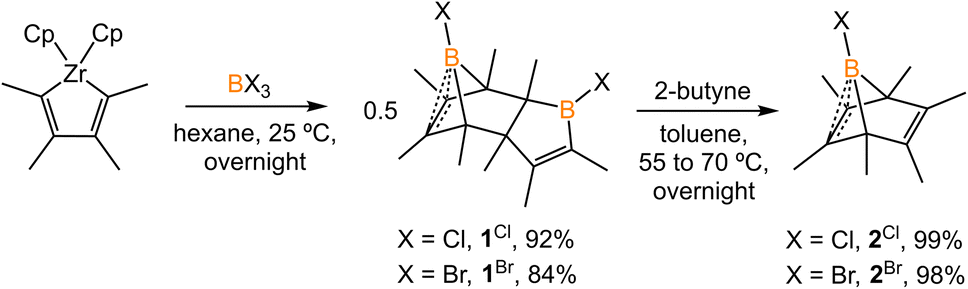
2.1 Synthesis of boranorbornadiene
The synthesis of 1-phenyl-2,3,4,5-tetramethylborole dimer using a zirconium reagent was developed by Fagan et al.9 The halogen-substituted borole dimers were obtained in a similar manner. The reaction of zirconium metallacycle Cp2Zr(C4Me4) with BCl3 (1.1 equiv.) led to the precipitation of Cp2ZrCl2 and production of 1-chloro-2,3,4,5-tetramethylborole dimer 1Cl (Scheme 1). The borole dimer 1Cl was separated from the zirconium salt by filtration, purified by recrystallization, and obtained in an excellent yield (92%). Similarly, the same procedure using BBr3 afforded 1-bromo-2,3,4,5-tetramethylborole dimer 1Br in 84% yield. The boron atoms of 1Cl and 1Br in vinylic positions are observed as broad singlets in the 11B{1H} NMR spectra at δB 66.2 and 67.6 ppm, respectively, in the range expected for tricoordinate boron centers. In contrast, the bridging boron atoms correspond to sharp singlet signals at δB 3.4 (1Cl) and −3.9 ppm (1Br), attributed to the non-classical interaction between the electron-deficient boron atoms and electron-rich C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds. Treatment of the borole dimers 1Cl and 1Br with 2-butyne (4 equiv.) at elevated temperatures afforded the corresponding Diels–Alder products 2Cl (99%) and 2Br (98%). Compounds 2Cl and 2Br are liquids at room temperature and solidify19 at −35 °C. In their 11B{1H} NMR spectra, the bridging boron atoms correspond to sharp singlets at δB −7.5 (2Cl), and −11.3 ppm (2Br). Fagan et al. reported the synthesis and NMR spectra of compound 2Ph, but its crystal structure remained elusive.9 In this work, we present the crystal structure of 2Ph in Fig. S52.†
C double bonds. Treatment of the borole dimers 1Cl and 1Br with 2-butyne (4 equiv.) at elevated temperatures afforded the corresponding Diels–Alder products 2Cl (99%) and 2Br (98%). Compounds 2Cl and 2Br are liquids at room temperature and solidify19 at −35 °C. In their 11B{1H} NMR spectra, the bridging boron atoms correspond to sharp singlets at δB −7.5 (2Cl), and −11.3 ppm (2Br). Fagan et al. reported the synthesis and NMR spectra of compound 2Ph, but its crystal structure remained elusive.9 In this work, we present the crystal structure of 2Ph in Fig. S52.†
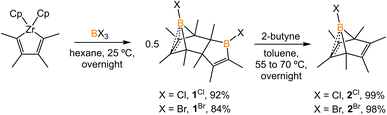 |
| | Scheme 1 Synthesis of borole dimers (1Cl and 1Br) and boranorbornadienes (2Cl and 2Br). | |
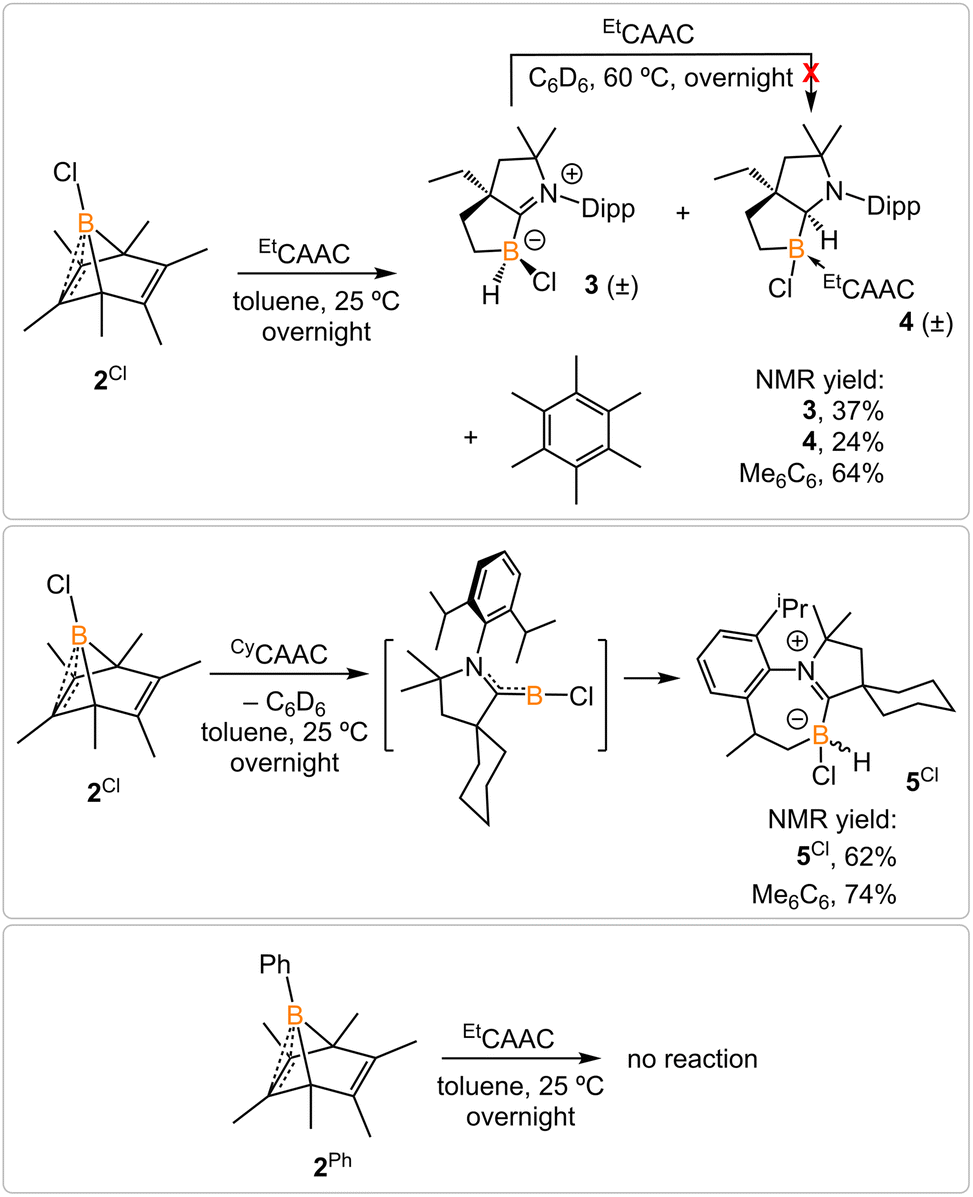
2.2 Chloroborylene release from boranorbornadiene
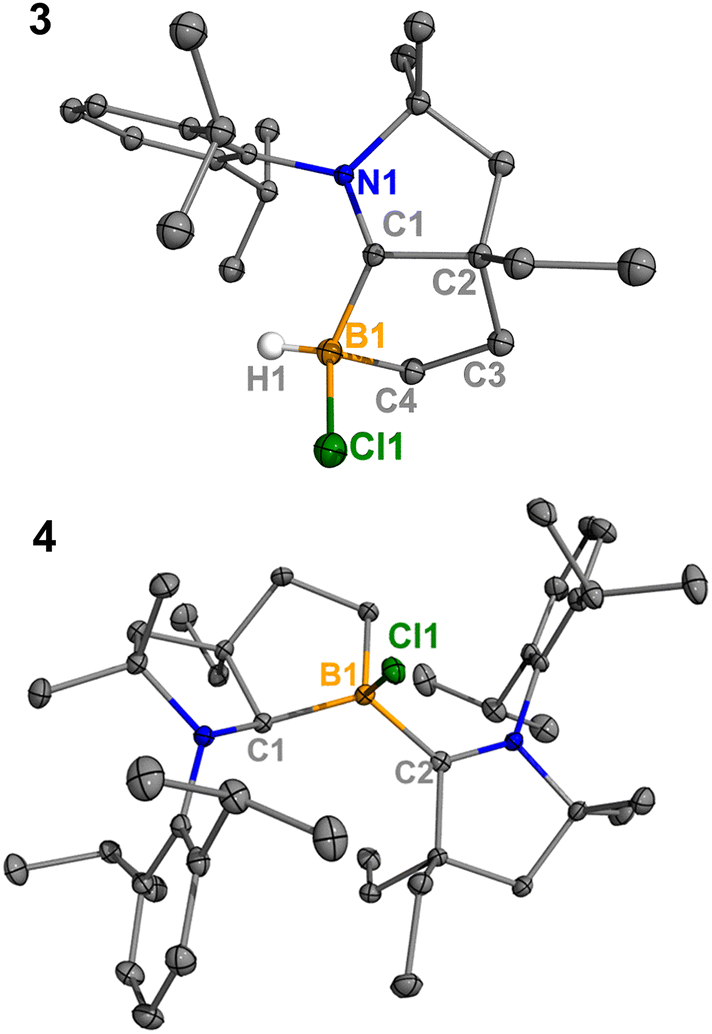
Treatment of 2Cl with cyclic(alkyl)(amino) carbene (EtCAAC) in toluene resulted in an immediate color change from colorless to orange (Scheme 2). The reaction led to the formation of several boron species based on 11B NMR spectroscopy. The 1H NMR spectrum revealed a significant amount of hexamethylbenzene (2.13 ppm in C6D6), suggesting the release of the “BCl” fragment from 2Cl. Two boron-containing species (3 and 4), along with hexamethylbenzene, crystallized out of the reaction mixture at −35 °C. Compound 3 displays a doublet at −10.9 ppm (1JH–B = 86.0 Hz) and 4 displays a singlet at 2.9 ppm in its 11B{1H} NMR spectrum. After washing with hexanes to remove hexamethylbenzene, the remaining solids were recrystallized from toluene at −35 °C and compound 3 precipitated out first. Its 11B{1H} NMR spectrum only displayed a singlet at −10.9 ppm, indicative of a boron hydride moiety. Its 13C DEPT-135 (Distortionless Enhancement by Polarization Transfer) NMR spectrum displayed an inverted broad signal at 20.23 ppm (FWHM = 115.6 Hz), suggesting a methylene unit adjacent to the boron atom. Therefore, compound 3 was assigned as a CH3 activation product. Storage of a concentrated toluene solution of 3 at −35 °C afforded single crystals suitable for single-crystal X-ray diffraction (XRD) analysis. The solid-state structure of 3 reveals an intramolecular C–H bond activation of the ethyl group in EtCAAC, a process that may occur via a transient dicoordinate chloroborylene (Fig. 2). Overall, the formation of 3 may be rationalized as a cascade reaction: the coordination of EtCAAC to the boron atom, borylene release from hexamethylbenzene, and intramolecular C–H activation. The crystal structure of 4 was obtained by XRD by selecting 4 from a crystalline mixture of 3 and 4. Initially we hypothesized that 4 was generated from 3, in which a hydride (H1) migration from B1 to the electrophilic carbon C1 occurred followed by coordination of a second EtCAAC to B1. However, heating compound 3 with EtCAAC in C6D6 for one day did not result in any observable reaction. At this stage, the pathway leading to compound 4 remains unclear.
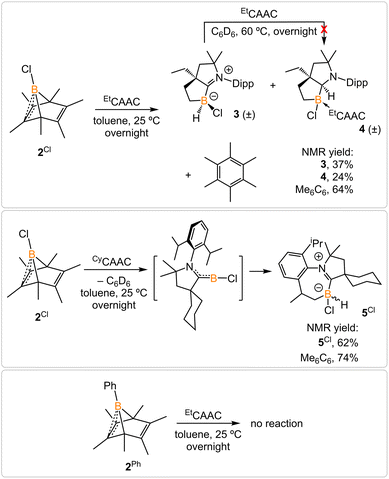 |
| | Scheme 2 Treatment of 2Cl with EtCAAC and CyCAAC and attempt to generate phenylborylene. | |
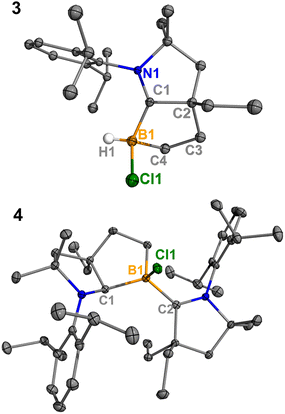 |
| | Fig. 2 Molecular structures of 3 and 4 in the solid-state. Hydrogen atoms, except for the one connected to B1, have been omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. | |
Treatment of 2Cl with CyCAAC led to the formation of hexamethylbenzene and the C–H activation product 5Cl. Unlike 3, compound 5Cl was obtained as two diastereomers (dr value = 67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :33) with different 11B NMR chemical shifts (δ = −4.7, −8.0 ppm) and coupling constants (1JB–H = 137.4 Hz, 67.6 Hz, respectively). The C–H activation products are quite similar to those generated from durylborylene (MeCAAC)(Dur)B: reported by Braunschweig et al.,1i further implicating the formation of the proposed borylene intermediate in this type of reaction. Interestingly, in contrast to the B-Dur species, 3 and 5Cl do not undergo a subsequent hydride migration from boron to carbon, an observation attributed to the stronger B–H bonds in 3 and 5Cl. Our attempt to generate a phenyl borylene using the same method was unsuccessful. The treatment of 2Ph with EtCAAC resulted in no reaction, likely a consequence of the steric hindrance of EtCAAC and the phenyl group.
:33) with different 11B NMR chemical shifts (δ = −4.7, −8.0 ppm) and coupling constants (1JB–H = 137.4 Hz, 67.6 Hz, respectively). The C–H activation products are quite similar to those generated from durylborylene (MeCAAC)(Dur)B: reported by Braunschweig et al.,1i further implicating the formation of the proposed borylene intermediate in this type of reaction. Interestingly, in contrast to the B-Dur species, 3 and 5Cl do not undergo a subsequent hydride migration from boron to carbon, an observation attributed to the stronger B–H bonds in 3 and 5Cl. Our attempt to generate a phenyl borylene using the same method was unsuccessful. The treatment of 2Ph with EtCAAC resulted in no reaction, likely a consequence of the steric hindrance of EtCAAC and the phenyl group.
2.3 Mechanistic studies
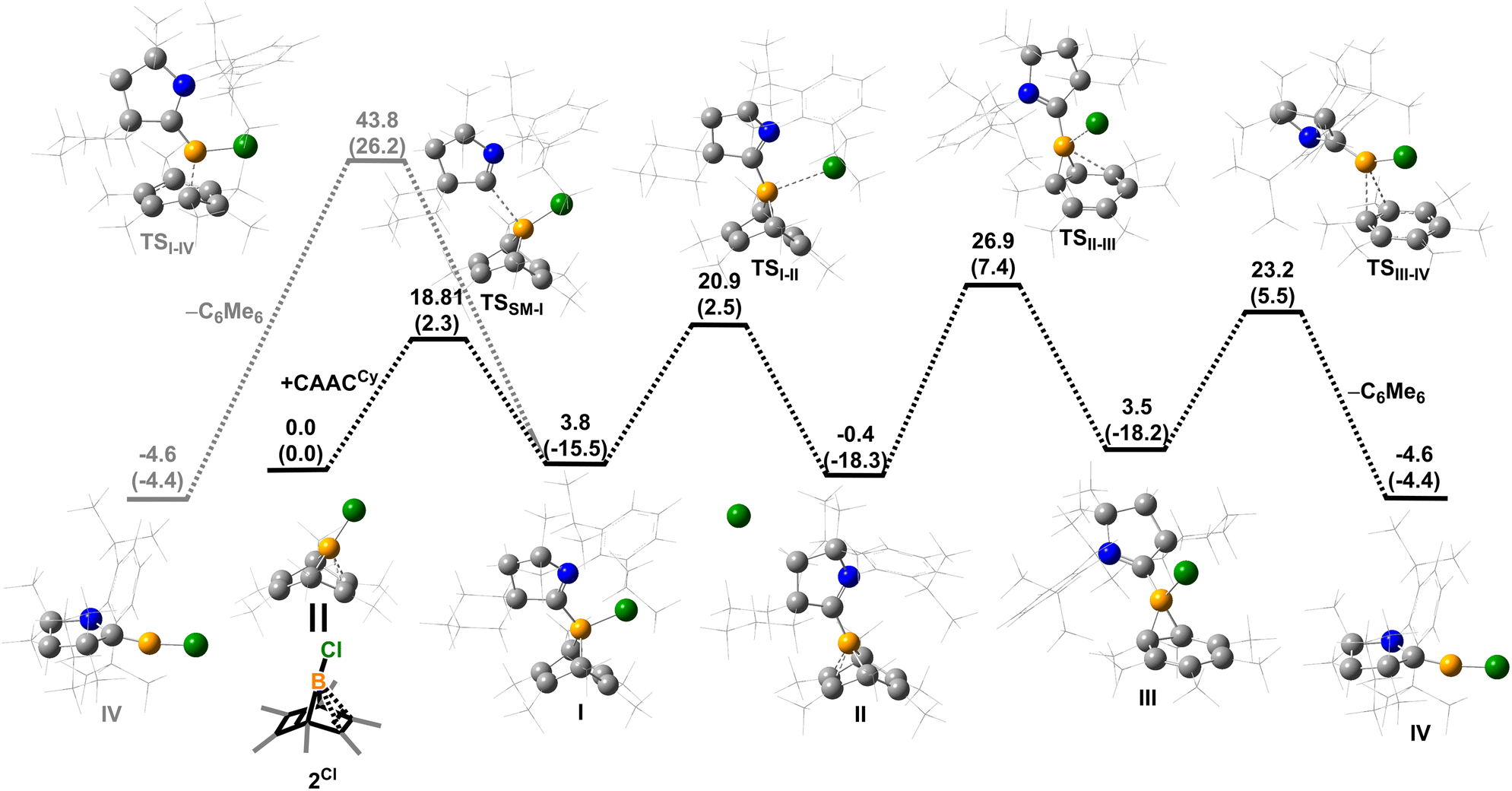
Density functional theory (DFT) calculations were performed to provide further insight into the borylene-releasing process. All calculations were performed at the ωB97xD/6-311G** level of theory together with single point energy corrections at the ωB97M-V/def2-QZVPP level. According to the computational results, 2Cl (SM) and CyCAAC first interact to form a Lewis acid–base adduct I (Fig. 3). While the chloroborylene (IV) could directly leave from hexamethylbenzene via transition state TSI–IV in an exergonic step (ΔG = −8.1 kcal mol−1), its high activation barrier (43.8 kcal mol−1) is inconsistent with the experimental fact that the hexamethylbenzene extrusion process is complete at 25 °C (room temperature) within several hours. Alternatively, the B–C bond in I could undergo a 1,3-suprafacial-sigma shift, transforming from boranorbornadiene (I) to boranorcaradiene (III), followed by a borylene-releasing process. The barrier of the rate-determining step in this alternative stepwise pathway is predicted to be 27.3 kcal mol−1, a value surmountable at room temperature. Interestingly, in contrast to the per-phenyl-boranorbornadiene NHC adduct (IMe)(Ph)B(C6Ph6),8,10 which converts to its boranorcaradiene structure linked only by one transition state, the conversion from I to III is connected by two transition states (TSI–II and TSII–III) and one intermediate (II), which is associated with a chloride dissociation and reassociation process. The intermediate II is considered to be a non-classical boronium species stabilized by a three-center two-electron bond arising from donation of the olefinic π bond to the Lewis-acidic boron center. Other possible reaction pathways from boranorcaradiene III to borylene IV are discussed in the ESI (Fig. S60†).
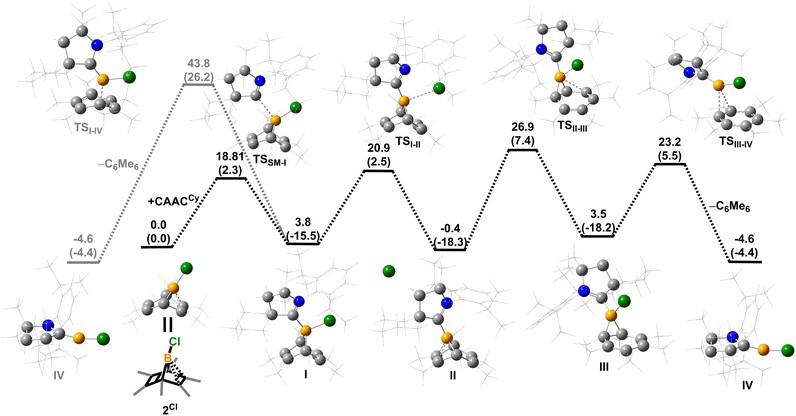 |
| | Fig. 3 Energy profiles calculated for the reaction from SM to IV and from I to IV. The relative Gibbs free energies (calculated at 298 K) and electronic energies (in parentheses) are given in kcal mol−1 (in scale). (ωB97xD/6-311G**//ωB97M-V/def2-QZVPP level of theory). | |
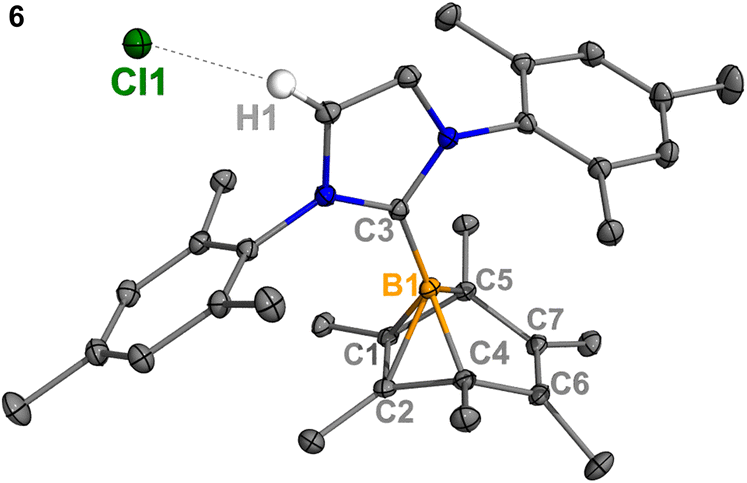
Experimentally, all attempts to isolate intermediates between I and IV were unsuccessful, presumably because of the spontaneity of the borylene-releasing process. We proposed that using an NHC ligand instead of a CAAC ligand would render the borylene IV less stable,2b,11 and the releasing process less favorable, thereby enabling us to isolate key intermediates. Leaving a 1:1 mixture of 2Cl and IMes (Mes = 2,4,6-trimethylphenyl) undisturbed overnight (Scheme 3) resulted in the precipitation of white crystalline solids. XRD analysis revealed a structure of the boranorbornadiene cation 6 (Fig. 4), which is analogous to the putative intermediate II. The B1–C1 (1.806(2) Å) and B1–C2 (1.798(2) Å) distances in 6 are shorter than those in 2Ph (1.816(2) Å and 1.811(2) Å, respectively, Fig. S52†), and the C1–C2 (1.393(2) Å) bond in 6 is longer than that in 2Ph (1.388(2) Å), indicating a stronger interaction between the boron atom and CC double bond. The center boron atom gave a single sharp resonance at −16.4 ppm in the 11B NMR spectrum. The formation of compound 6 corroborates our proposed mechanism and is consistent with the calculated energies [I (3.8 kcal mol−1), II (−0.4 kcal mol−1), III (3.5 kcal mol−1)], suggesting that II is the most stable isomer. However, 6 is even stable in boiling C6D6 for several hours, indicating that the borylene-releasing process in this case is extremely unfavorable.
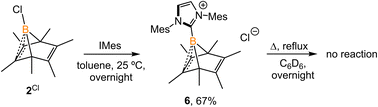 |
| | Scheme 3 Synthesis of 6. | |
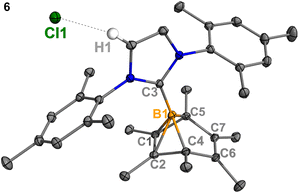 |
| | Fig. 4 Molecular structure of 6 in the solid-state. Hydrogen atoms, except for H1, have been omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. Selected bond lengths [Å] and angles [°]: B1–C1 1.798(2), B1–C2 1.807(2), B1–C3 1.575(3), B1–C4 1.630(2), B1–C5 1.623(2), C1–C2 1.393(3), C6–C7 1.336(2), H1–Cl1 2.512, C4–B1–C5 96.78(9), C1–B1–C2 45.47, C4–B1–C5–C1 82.23. | |
2.4 Bromoborylene release from boranorbornadiene
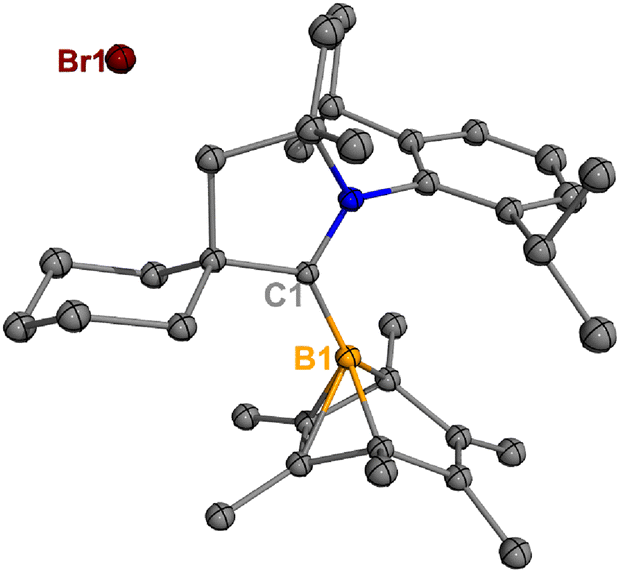
Treatment of 2Br with CyCAAC in toluene (Scheme 4) resulted in the precipitation of yellow crystalline solids (7). The 11B NMR spectrum of 7 features a resonance at δB −13.3 ppm. XRD analysis revealed a boronium structure analogous to that of 6 (Fig. 5). Compound 7 is nearly insoluble in toluene and does not release borylene at 25 °C. However, heating a suspension of 7 in toluene at 100 °C overnight afforded a mixture of hexamethylbenzene, an intramolecular C–H activation product 5Br, and a C–C activation product 8, indicating that the borylene-releasing process occurred at an elevated temperature. Interestingly, the C–C activation product was not produced in the reaction of 2Cl with CyCAAC (Scheme 2). Based on the studies conducted by Braunschweig11,12 and Lin et al.,12 the formation of 8 arises due to the enhanced Lewis acidity of CAAC-stabilized bromoborylene compared to chloroborylene. In the C(sp2)–C(sp3) activation process, bromoborylene may lower the barrier of the rate-determining π-coordination step, making the C(sp2)–C(sp3) bond activation more favorable. Treatment of 7 with bis(triphenylphosphine)iminium chloride ([PPN]Cl) in toluene at 25 °C also resulted in hexamethylbenzene extrusion and the formation of 5Cl (Scheme 5). Although we attempted to generate the CAAC-stabilized fluoroborylene in a similar manner, the reaction yielded intractable mixtures containing hexamethylbenzene and unidentified boron-containing species.
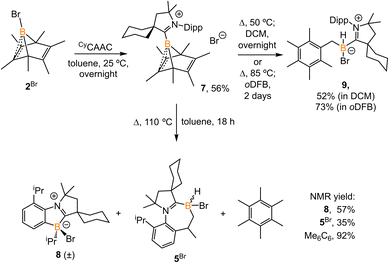 |
| | Scheme 4 Synthesis of 7 and its reaction at elevated temperature in different solvents. (oDFB = ortho-difluorobenzene). | |
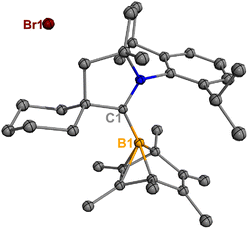 |
| | Fig. 5 Molecular structure of 7 in the solid state. Hydrogen atoms have been omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. | |
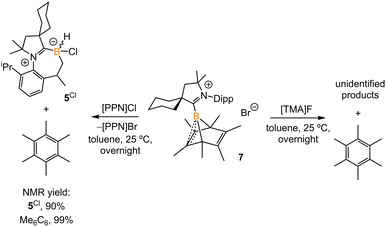 |
| | Scheme 5 Treatment of 7 with [PPN]Cl and [TMA]F. | |
Surprisingly, heating 7 in polar solvents like DCM or o-difluorobenzene (Scheme 4) led to the formation of a single boron species (9) displaying a broad singlet at δB −8.0 ppm, with no evidence of hexamethylbenzene formation via1H NMR spectroscopy. Single crystals suitable for XRD analysis were grown by slow diffusion of hexanes into a concentrated DCM solution of 7. The structure of 9 corresponds to the product of a process in which the B1 atom inserts into a C–H bond of the methyl group in hexamethylbenzene (Fig. 6). This reaction is relatively clean, and no other side products, including 5Br and 8, were observed in significant amounts, suggesting that the C–H activation process may not proceed through a borylene intermediate.
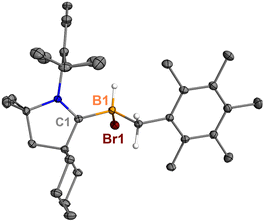 |
| | Fig. 6 Molecular structure of 9 in the solid state. Hydrogen atoms, except for the one connected to B1, have been omitted for clarity. Thermal ellipsoids are drawn at the 50% probability level. | |
To understand how solvents affected the reaction outcome, DFT calculations at the same level of theory (ωB97M-V/def2-QZVPP//ωB97xD/6-311G**) were performed (Fig. 7). While applying the dichloromethane solvation model (path in green), the barrier of the borylene-release process was elevated to 34.9 kcal mol−1, 7.6 kcal mol−1 higher than the chloroborylene-releasing process in toluene (Fig. 3). Alternatively, the reaction pathway leading to the formation of 9via transition state TS7–9Br has a lower energy barrier (32.4 kcal mol−1). The optimized structure of TS7–9Br features an agostic interaction between the boron center and the C–H bond. Therefore, the formation of 9 involves an intramolecular concerted process where (CAAC)B+ reductively eliminates from hexamethylbenzene and oxidatively inserts into the C–H bond. The pathway in black was calculated while applying the toluene solvation model. In toluene, the energy barrier from 7 to 9viaTS7–9Br increased to 35.4 kcal mol−1. In comparison, the energy barrier of the borylene-release process (from 7 to IVBr) decreased to 28.3 kcal mol−1 and thus became the more favorable process. Compared to the intermolecular C–H activation of hexamethylbenzene, the irreversible intramolecular C–H activation of the diisopropylphenyl (Dipp) group is more favorable, finally leading to the formation of compound 5Br. Overall, the computational results are consistent with our experimental observations.
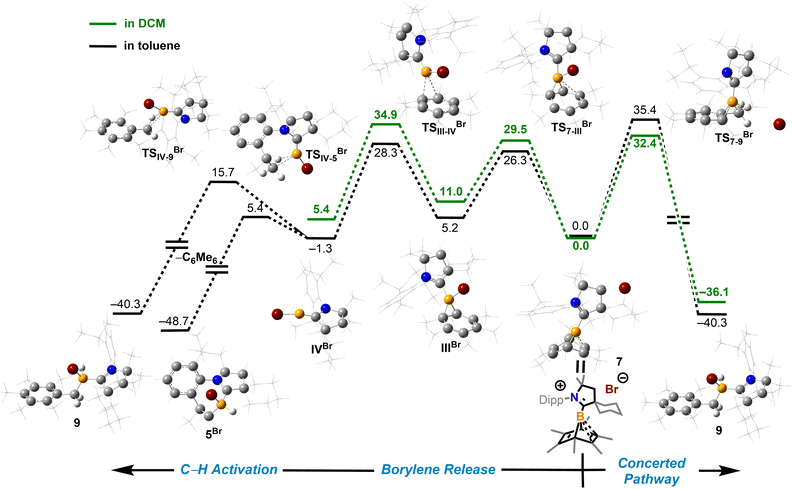 |
| | Fig. 7 Energy profiles calculated for the reaction from 7 to 9 and from 7 to VBr (or VIBr). The relative Gibbs free energies (calculated at 298 K) are given in kcal mol−1 (in scale). The calculation applied solvation models in toluene (path in black and gray) and DCM (path in green). (ωB97xD/6-311G**//ωB97M-V/def2-QZVPP level of theory). | |
2.5 Boron-centered Büchner reaction
The reaction of 1Br with an excess of 2-butyne (10 equiv.) at 25 °C for two days afforded a 1:1 mixture of bromoborepin compound 10Br and boranorbornadiene compound 2Br (Scheme 6). The boron atom in the borepin evinces a broad singlet signal at 57.9 ppm in its 11B NMR spectrum. Compound 10Br fully converts to 2Br at an elevated temperature and proved to be the key intermediate in the transformation of 1Br to 2Br.13 Compound 10Br was challenging to separate from 2Br. Treatment of the mixture of 10Br and 2Br with CyCAAC in toluene overnight resulted in the precipitation of yellow crystalline solids. All components in the reaction mixture displayed only one dominant signal at −13.3 ppm in the 11B NMR spectrum, attributed to compound 7. This assignment is supported by 1H NMR spectroscopy data. Therefore, the in situ formed Lewis acid–base adduct CyCAAC·10Br converts to 7 at 25 °C. Indeed, a similar borepin to boranorcaradiene transformation was observed by Taniguchi et al.14 In addition, compound 7 extrudes hexamethylbenzene upon heating in a non-polar solvent. Therefore, the transformation from CyCAAC·10Br to borylene IVBr is formally a boron-centered retro Büchner-ring-expansion reaction. It should be noted that other main-group element-centered Büchner reactions, including those for carbon,7b,15 silicon,7a,16 aluminum,17 and phosphorus,18 have been reported. The present work similarly demonstrates an example of a boron-centered retro Büchner reaction and its feasibility for releasing subvalent boron species.
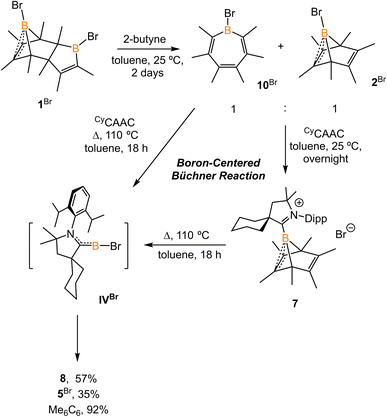 |
| | Scheme 6 Synthesis of 10Br and its conversion to 7. | |
3 Conclusions
In conclusion, we have synthesized chloro-boranorbornadiene and bromo-boranorbornadiene XB(C6Me6) (X = Cl and Br). The coordination of CAAC to XB(C6Me6) promotes hexamethylbenzene extrusion and the release of CAAC-ligated halogen–borylene (X = Cl, room temperature; X = Br, elevated temperature). The experimental and computational data suggest that the borylene-releasing process goes through a boranorbornadiene cation intermediate and a boranorcaradiene intermediate. Depending on the CAAC ligand and the substituents on boron, the subsequent transformations of di-coordinate borylene varied, producing different C–H and C–C activation products. CAAC ligands provide a better stabilization effect compared to NHC ligands, making the borylene-releasing process more kinetically and thermodynamically favorable. The release of borylene from hexamethylbenzene is the first example of a boron-centered arene extrusion reaction and the first example of thermal reductive elimination taking place at a single boron atom. These reactions have laid the groundwork for unusual borylene transfer chemistry and further studies remain ongoing in our laboratories.
Data availability
Experimental details, characterization, and computational details, including Fig. S1–S60 and Tables S1–S15,† X-ray crystallographic data for 1Cl, 2Ph, 3, 4, 5Cl, 5Br, 6, 7, 8, and 9 (CIF). CCDC identification codes 297451–2297460 are associated with the supplementary crystallographic data for this paper.
Author contributions
C. Z. conceptualized the project, synthesized and characterized the compounds, performed the computational work, and drafted the manuscript. R. J. G. and C. C. C. supervised the project, acquired financial support for the project, and revised the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. 2247252 (CCC) and No. 2046544 (RJG). RJG acknowledges additional laboratory support from the Arnold and Mabel Beckman Foundation. The authors acknowledge Research Computing at Massachusetts Institute of Technology (Engaging Cluster, Supercloud) for providing computational resources and technical support that have contributed to the results reported within this publication. The authors thank Dr Peter Müller for helping solve problems related to X-ray crystallography data.
Notes and references
-
(a) P. L. Timms, J. Am. Chem. Soc., 1968, 90, 4585–4589 CrossRef CAS;
(b) A. Meller, U. Seebold, W. Maringgele, M. Noltemeyer and G. M. Sheldrick, J. Am. Chem. Soc., 1989, 111, 8299–8300 CrossRef CAS;
(c) W. J. Grigsby and P. P. Power, J. Am. Chem. Soc., 1996, 118, 7981–7988 CrossRef CAS;
(d) K. Edel, M. Krieg, D. Grote and H. F. Bettinger, J. Am. Chem. Soc., 2017, 139, 15151–15159 CrossRef CAS PubMed;
(e) P. L. Timms, Acc. Chem. Res., 1973, 6, 118–123 CrossRef CAS;
(f) M. Ito, N. Tokitoh, T. Kawashima and R. Okazaki, Tetrahedron Lett., 1999, 40, 5557–5560 CrossRef CAS;
(g) M. Krasowska, M. Edelmann and H. F. Bettinger, J. Phys. Chem. A, 2016, 120, 6332–6341 CrossRef CAS PubMed;
(h) M. Krasowska and H. F. Bettinger, J. Am. Chem. Soc., 2012, 134, 17094–17103 CrossRef CAS PubMed;
(i) H. Braunschweig, I. Krummenacher, M. A. Légaré, A. Matler, K. Radacki and Q. Ye, J. Am. Chem. Soc., 2017, 139, 1802–1805 CrossRef CAS PubMed;
(j) H. F. Bettinger, J. Am. Chem. Soc., 2006, 128, 2534–2535 CrossRef CAS PubMed;
(k) B. Pachaly and R. West, Angew. Chem., Int. Ed., 1984, 23, 454–455 CrossRef;
(l) D. Mu and Q. Li, Phys. Chem. Chem. Phys., 2023, 25, 8074–8081 RSC.
-
(a) R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610–613 CrossRef CAS PubMed;
(b) F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159–193163 CrossRef CAS;
(c) A. D. Ledet and T. W. Hudnall, Dalton Trans., 2016, 45, 9820–9826 RSC;
(d) H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327–330 CrossRef CAS PubMed;
(e) M. A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed;
(f) M. Härterich, A. Matler, R. D. Dewhurst, A. Sachs, K. Oppel, A. Stoy and H. Braunschweig, Nat. Commun., 2023, 14, 2764 CrossRef PubMed;
(g) H. Wang, L. Wu, Z. Lin and Z. Xie, J. Am. Chem. Soc., 2017, 139, 13680–13683 CrossRef CAS PubMed;
(h) M. Heinz, M. Arrowsmith, J. I. Schweizer, I. Krummenacher, M. C. Holthausen and H. Braunschweig, J. Am. Chem. Soc., 2023, 145, 22685–22696 CrossRef CAS PubMed;
(i) X. Chen, Y. Yang, H. Wang and Z. Mo, J. Am. Chem. Soc., 2023, 145, 7011–7020 CrossRef CAS PubMed;
(j) H. Wang, L. Wu, Z. Lin and Z. Xie, Angew. Chem., Int. Ed., 2018, 57, 8708–8713 CrossRef CAS PubMed;
(k) W. Lv, Y. Dai, R. Guo, Y. Su, D. A. Ruiz, L. L. Liu, C. H. Tung and L. Kong, Angew. Chem., Int. Ed., 2023, 62, e202308467 CrossRef CAS PubMed;
(l) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed;
(m) M. Arrowsmith, D. Auerhammer, R. Bertermann, H. Braunschweig, G. Bringmann, M. A. Celik, R. D. Dewhurst, M. Finze, M. Grune, M. Hailmann, T. Hertle and I. Krummenacher, Angew. Chem., Int. Ed., 2016, 55, 14464–14468 CrossRef CAS PubMed.
-
(a) M. A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231–8261 CrossRef PubMed;
(b) H. Braunschweig, R. D. Dewhurst and V. H. Gessner, Chem. Soc. Rev., 2013, 42, 3197 RSC;
(c) H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924–3957 CrossRef CAS PubMed;
(d) M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10282–10292 CrossRef CAS PubMed;
(e) C. Zhang, C. C. Cummins and R. J. Gilliard, Science, 2024, 385, 327–331 CrossRef CAS PubMed;
(f) A. Jayaraman, B. Ritschel, M. Arrowsmith, C. Markl, M. Jürgensen, A. Halkić, Y. Konrad, A. Stoy, K. Radacki and H. Braunschweig, Angew. Chem., Int. Ed., 2024, e202412307 Search PubMed.
-
(a) P. Bissinger, H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kupfer, K. Radacki and K. Wagner, J. Am. Chem. Soc., 2011, 133, 19044–19047 CrossRef CAS PubMed;
(b) M. Arrowsmith, J. Böhnke, H. Braunschweig, H. Gao, M. A. Légaré, V. Paprocki and J. Seufert, Chemistry, 2017, 23, 12210–12217 CrossRef CAS PubMed;
(c) D. P. Curran, A. Boussonnière, S. J. Geib and E. Lacôte, Angew. Chem., Int. Ed., 2012, 51, 1602–1605 CrossRef CAS PubMed;
(d) P. Bissinger, H. Braunschweig, K. Kraft and T. Kupfer, Angew. Chem., Int. Ed., 2011, 50, 4704–4707 CrossRef CAS PubMed.
- C. Pranckevicius, M. Weber, I. Krummenacher, A. K. Phukan and H. Braunschweig, Chem. Sci., 2020, 11, 11055–11059 RSC.
-
(a) A. Velian and C. C. Cummins, J. Am. Chem. Soc., 2012, 134, 13978–19381 CrossRef CAS PubMed;
(b) W. J. Transue, A. Velian, M. Nava, C. García-Iriepa, M. Temprado and C. C. Cummins, J. Am. Chem. Soc., 2017, 139, 10822–10831 CrossRef CAS PubMed;
(c) T. Xin, M. B. Geeson, H. Zhu, Z. W. Qu, S. Grimme and C. C. Cummins, Chem. Sci., 2022, 13, 12696–12702 RSC.
-
(a) D. Wendel, A. Porzelt, F. A. D. Herz, D. Sarkar, C. Jandl, S. Inoue and B. Rieger, J. Am. Chem. Soc., 2017, 139, 8134–8137 CrossRef CAS PubMed;
(b) T. A. Perera, E. W. Reinheimer and T. W. Hudnall, J. Am. Chem. Soc., 2017, 139, 14807–14814 CrossRef CAS PubMed;
(c) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000–11003 CrossRef CAS PubMed;
(d) L. L. Liu, J. Zhou, R. Andrews and D. W. Stephan, J. Am. Chem. Soc., 2018, 140, 7466–7470 CrossRef CAS PubMed;
(e) L. L. Liu, J. Zhou, L. L. Cao, Y. Kim and D. W. Stephan, J. Am. Chem. Soc., 2019, 141, 8083–8087 CrossRef CAS PubMed.
- H. Braunschweig, J. Maier, K. Radacki and J. Wahler, Organometallics, 2013, 32, 6353–6359 CrossRef CAS.
- P. J. Fagan, E. G. Burns and J. C. Calabrese, J. Am. Chem. Soc., 1988, 110, 2979–2981 CrossRef CAS.
- F. Lindl, X. Guo, I. Krummenacher, F. Rauch, A. Rempel, V. Paprocki, T. Dellermann, T. E. Stennett, A. Lamprecht, T. Brückner, K. Radacki, G. Bélanger-Chabot, T. B. Marder, Z. Lin and H. Braunschweig, Chem.–Eur. J., 2021, 27, 11226–11233 CrossRef CAS PubMed.
- C. Pranckevicius, J. O. C. Jimenez-Halla, M. Kirsch, I. Krummenacher and H. Braunschweig, J. Am. Chem. Soc., 2018, 140, 10524–10529 CrossRef CAS PubMed.
- L. Wu, R. D. Dewhurst, H. Braunschweig and Z. Lin, Organometallics, 2021, 40, 766–775 CrossRef CAS.
- Z. Wang, Y. Zhou, J. X. Zhang, I. Krummenacher, H. Braunschweig and Z. Lin, Chem.–Eur. J., 2018, 24, 9612–9621 CrossRef CAS PubMed.
- M. Shimoi, I. Kevlishvili, T. Watanabe, K. Maeda, S. J. Geib, D. P. Curran, P. Liu and T. Taniguchi, Angew. Chem., Int. Ed., 2020, 59, 903–909 CrossRef CAS PubMed.
- B. Darses, P. Maldivi, C. Philouze, P. Dauban and J. F. Poisson, Org. Lett., 2021, 23, 300–304 CrossRef CAS.
-
(a) C. Xu, Z. Ye, L. Xiang, S. Yang, Q. Peng, X. Leng and Y. Chen, Angew. Chem., Int. Ed., 2021, 60, 3189–3195 CrossRef CAS PubMed;
(b) L. Zhu, J. Zhang and C. Cui, Inorg. Chem., 2019, 58, 12007–12010 CrossRef CAS PubMed.
-
(a) X. Zhang and L. L. Liu, Angew. Chem., Int. Ed., 2022, 61, e202116658 CrossRef CAS PubMed;
(b) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000–11003 CrossRef CAS PubMed.
- L. L. Liu, J. Zhou, L. L. Cao, R. Andrews, R. L. Falconer, C. A. Russell and D. W. Stephan, J. Am. Chem. Soc., 2018, 140, 147–150 CrossRef CAS PubMed.
-
(a) C. Fan, W. E. Piers, M. Parvez and R. McDonald, Organometallics, 2010, 29, 5132–5139 CrossRef CAS;
(b) J. J. Eisch, J. E. Galle, B. Shafii and A. L. Rheingold, Organometallics, 1990, 9, 2342–2349 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Robert J.
Gilliard
Jr
,
Robert J.
Gilliard
Jr