
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-dimensional structural alignment based discovery and molecular basis of AtoB, catalyzing linear tetracyclic formation†
Ke
Ma
 a,
Jie
Liu
a,
Zequan
Huang
a,
Mengyue
Wu
a,
Dong
Liu
a,
Jinwei
Ren
b,
Aili
Fan
*a and
Wenhan
Lin
*ac
a,
Jie
Liu
a,
Zequan
Huang
a,
Mengyue
Wu
a,
Dong
Liu
a,
Jinwei
Ren
b,
Aili
Fan
*a and
Wenhan
Lin
*ac
aState Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China. E-mail: fanaili@bjmu.edu.cn; whlin@bjmu.edu.cn
bState Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China
cNingbo Institute of Marine Medicine, Peking University, Ningbo 315832, Zhejiang, China
First published on 11th October 2024
Abstract
Enzymes from the nuclear transport factor 2-like (NTF2-like) superfamily represent a rare group of biocatalysts with diverse catalytic functions facilitating intriguing skeleton formations. However, most proteins of this family remain enigmatic and await further elucidation. In this study, a combination of protein structural alignment with clustering analysis uncovers a new aldolase, AtoB, belonging to the NTF2-like superfamily. AtoB catalyzes the key intramolecular aldol reaction in linear tetracyclic meroterpenoid biosynthesis. The X-ray crystal structures of AtoB and AtoB-ligand complex are established at 1.9 Å and 1.6 Å resolution, respectively, revealing the rotation of the α4 helix and key residues in the active site for substrate binding. Molecular docking and site-directed mutagenesis demonstrate an acid–base pair involved in the AtoB-catalyzed aldol reaction, of which Arg59 is responsible for stereocontrol of hydroxylated C-10a during condensation. These findings provide valuable information for understanding the catalytic mechanisms of the AtoB-catalyzed aldol reaction. Additionally, a branching biosynthetic pathway of aspertetranones is elucidated during the exploration of the natural substrate of AtoB.
Introduction
Fungal natural products display complex structures and versatile biological activities, continuously providing opportunities to develop life-saving medicines, such as immunosuppressive cyclosporine and mycophenolic acid, antimicrobial agents griseofulvin and caspofungin.1–4 The biosynthesis of fungal natural products draws significant focus from researchers, particularly on the tailoring enzymes that greatly expand the chemical space of these products. However, previous studies mostly focused on the ultra-large protein superfamily such as cytochrome P450, flavin dependent oxidase, and prenyltransferases.5–7 Novel enzyme families have rarely been reported.Among these understudied enzyme families, the nuclear transport factor 2-like (NTF2-like) superfamily, featuring a natural small alpha–beta fold first observed in the structure of the rat NTF2 protein, showcases astonishing attractiveness.8 Recently, a few fungal NTF2-like enzymes with distinct catalytic functions have been characterized. For example, SdnG, BvnE, NsrQ and Trt14 catalyze the Diels–Alder reaction, semipinacol rearrangement, isomerization, and intramolecular methoxy rearrangement in the biosynthesis of sordarin, brevianamides, blennolides and terretonins, respectively (Fig. 1A).9–12 It is noteworthy that genes encoding NTF2-like proteins are widely distributed among fungal and bacterial genomes, but with low similarity at the amino acid level. These facts imply that the undiscovered NTF2-like enzymes may hold the potential to catalyze intricate reactions, necessitating the development of a universal strategy to uncover new members of the NTF2-like superfamily.
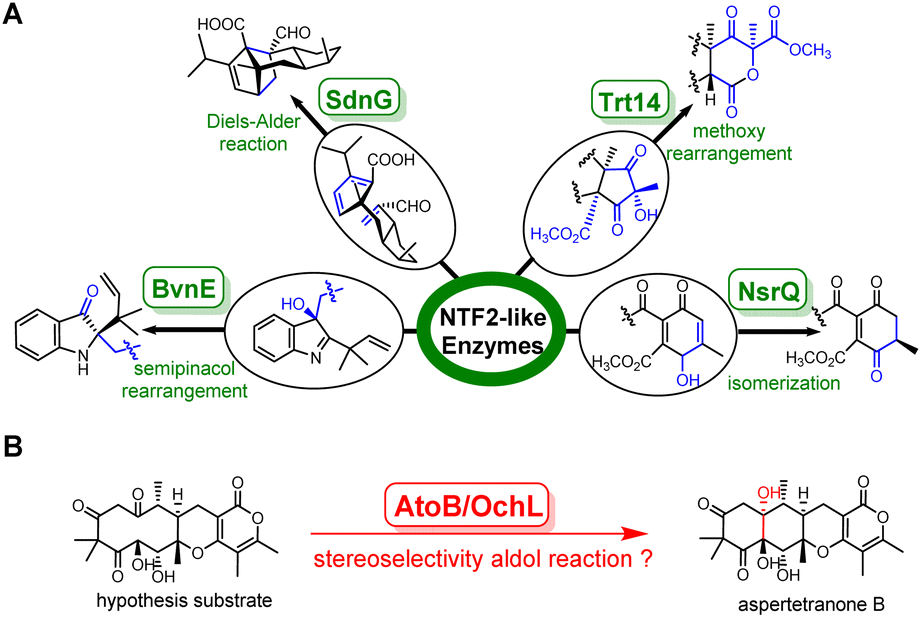 | ||
| Fig. 1 Representative fungal NTF2-like enzymes catalyzed distinct reactions. (A) SdnG, BvnE, NsrQ and Trt14 catalyzed Diels–Alder reaction, semipinacol reaction, isomerization, and intramolecular methoxy rearrangement, respectively. (B) Putative aldolase catalyzed 6/6/6/6 tetracyclic skeleton formation in the biosynthesis of fungal meroterpenoids. | ||
Herein, stemming from protein structural alignment and CLANS clustering method, we identified a novel NTF2-like aldolase, AtoB, involved in the biosynthesis of aspertetranone A from the marine-derived fungus Aspergillus ochraceus LZDX-32-15. Through target gene deletion in A. ochraceus and heterologous expression in A. nidulans, a branching biosynthetic pathway of aspertetranones was charted and AtoB was found to catalyze an intramolecular adol reaction to form the linear tetracycle of aspertetranones. Surprisingly, a recent online article reported an isozyme OchL,13 which is assumed to catalyze the same reaction to form ochraceopones based on the evidence from three-protein (OchGJL) bioconversion (Fig. 1B). However, the cryptic substrate and the mechanistic details of this new class of intramolecular NTF2-like aldolase remain unknown. To investigate the molecular basis for the AtoB-catalyzed reaction, we performed in vitro characterization of this enzyme using the isolated natural substrate and solved the X-ray crystal structures of AtoB and its complex with the substrate analogue. Structural analyses indicated that the rotatable α4 helix played a key role in substrate binding, and Arg59–Tyr114 based acid–base chemistry was employed for AtoB to catalyze the aldol reaction. Notably, Arg59 is also critical for exerting stereochemical control at C-10a via hydrogen bonds. Combining the structural analyses and mutational studies, a mechanism for this novel NTF2-like aldolase AtoB was proposed.
Results and discussion
Discovery of the NTF2-like enzyme AtoB and related gene cluster
To discover new NTF2-like enzymes capable of catalyzing novel reactions, we employed a recently developed tool, Foldseek, in association with CLANS and phylogenetic analysis.14,15 Foldseek is built up for identifying homologous proteins based on the three-dimensional backbone superposition and tertiary amino acid interactions, which could effectively address the challenge posed by the very low sequence similarities within the NTF2-like superfamily.Specifically, the superfamily members SdnG, BvnE, Trt14, and PrhC were used as probes to search for relevant NTF2-like enzymes against our in-house marine fungal genomic database. Ten putative protein sequences (with 10–40% amino acid identity) were obtained. However, the catalytic functions of these candidate proteins were hardly distinguished due to the diverse catalytic and non-catalytic roles and low peptide sequence similarities within the NTF2-like superfamily. Therefore, the structures of the 10 candidate proteins were predicted by RoseTTAFold2 for subsequent Foldseek searching.16 A total of 1432 deduplicated protein sequences were then obtained and processed for the sequence clustering by CLANS.14 The NTF2-like superfamily was segregated into nine main clusters, each containing more than 25 members (Fig. 2A). They presented diverse biological functions such as nuclear transport factors (cluster 1), catalytic enzymes (clusters 2–5), kinases (cluster 6), nutrient uptake related proteins (cluster 7), and cell shape related proteins (cluster 8). The known fungal NTF2-like enzymes (BvnE, NsrQ, and Trt14) and one of the candidate proteins (named AtoB) clearly constituted a distinct sequence cluster (cluster 2), suggesting that AtoB might have a potential catalytic function. The phylogenetic analysis of sequences in cluster 2 revealed that these NTF2-like members were classified into three clades, with Trt14, BvnE and PrhC in clade 1 and NsrQ and Dcr3 in clade 2 (Fig. 2B and S1†).
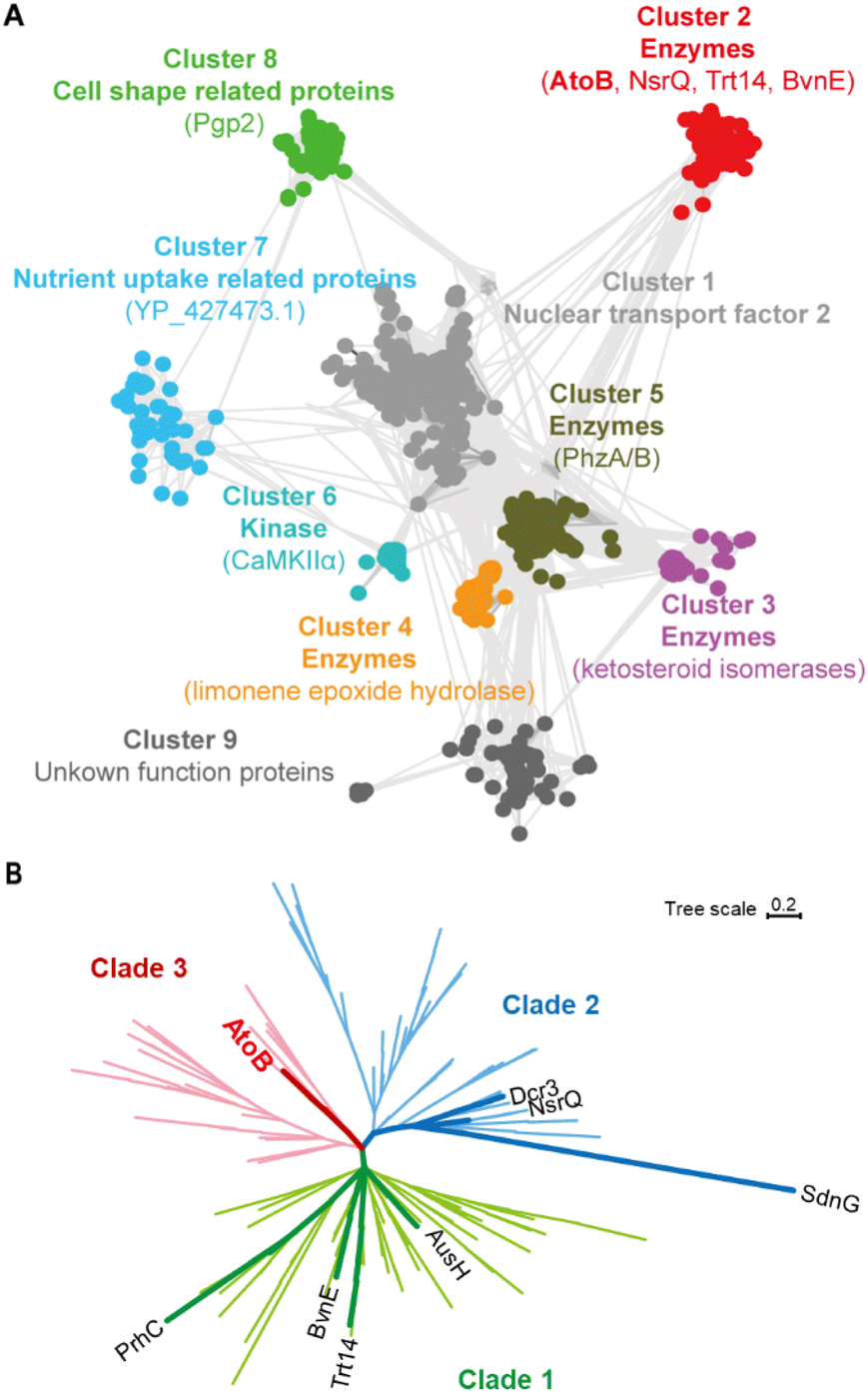 | ||
| Fig. 2 Sequence clustering and phylogenetic analysis of NTF2-like proteins. (A) NTF2-like superfamily was classified into nine main clusters by the sequence clustering method. (B) Phylogenetic analysis of NTF2-like protein in cluster 2. AtoB was in clade 3. | ||
Fascinatingly, AtoB significantly differed from the known fungal NTF2-like enzymes and was located in the distinct clade 3, further implying that AtoB could process a unique function.
Further analysis of the up-stream and down-stream genes of atoB in the genome of A. ochraceus LZDX-32-15 enabled postulation of a biosynthetic gene cluster (ato cluster, accession number PQ310357) containing a polyketide synthase (AtoC), a prenyltransferase (AtoE), a FAD-dependent monooxygenase (AtoH) and a terpene cyclase (AtoK) and eight more putative proteins (AtoA, AtoD, AtoF, AtoG, AtoI, AtoJ, AtoL, and AtoM; Table S1†). Significantly, the ato cluster showed low homology to the known fungal meroterpenoid BGCs (Fig. S2†).17–23
Characterization of AtoB involved in the biosynthetic pathway of aspertetranones
To investigate the function of AtoB, combining genetic deletions in A. ochraceus AoS1 strain with heterologous reconstitutions in Aspergillus nidulans LO8030 was conducted (Table S2†).24 The products of these mutants were analyzed by LC-MS and their structures were determined by NMR and MS spectroscopic data.To experimentally validate the products of ato cluster, the genes from atoA to atoM were co-expressed in A. nidulans LO8030, resulting in the mutant strain An-atocluster. As shown in Fig. 3, a product (1) was detected in the cultured An-atocluster and identified as aspertetranone A, while the A. ochraceus produced 1 and aspertetranone D (2). To verify the functions of atoC, atoE, atoH, and atoK as classical biosynthetic genes of fungal meroterpenoids, these four genes were co-expressed in A. nidulans to obtain the An-atoCEHK strain. Consistent with the hypothesis, An-atoCEHK generated a meroterpenoid precursor 3. These results suggested that ato genes might share comparable functions and similarity with och cluster, whose sequence is not released now (Fig. S3†).13 Compared to och cluster, AtoB might catalyze the same aldol reaction as the assumed isozyme OchL. However, the function and catalytic mechanism of OchL remained elusive due to the lack of a natural substrate.
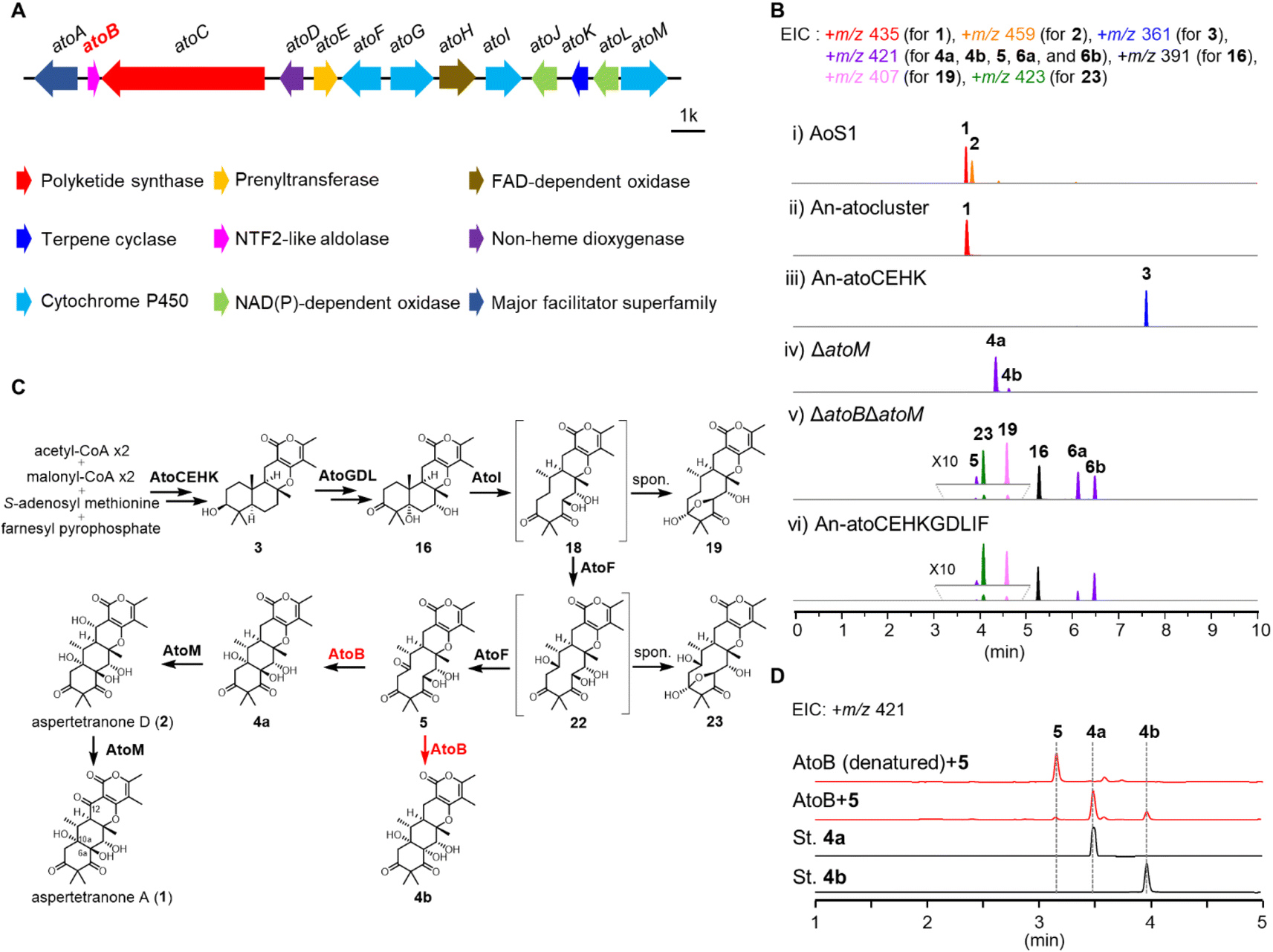 | ||
| Fig. 3 Biosynthetic pathway of aspertetranones and function of AtoB. (A) The ato cluster from A. ochraceus. (B) LC-MS analysis of the metabolites of A. nidulans transformants and A. ochraceus mutants: (i) A. ochraceus AoS1; (ii) heterologous expression of the whole ato cluster in A. nidulans; (iii) heterologous expression of atoCEHK genes in A. nidulans; (iv) atoM knock-out mutant in A. ochraceus AoS1; (v) atoM and atoB double-knockout mutant in A. ochraceus AoS1; (vi) heterologous expression of atoCEHKGDLIF genes in A. nidulans. (C) AtoB catalyzed the aldol reaction in the biosynthesis of aspertetranones. (D) In vitro enzymatic reaction of AtoB with its natural substrate 5. | ||
Therefore, thirteen single- or double-knockout strains were constructed to explore the natural substrate of AtoB including ΔatoD, ΔatoF, ΔatoG, ΔatoI, ΔatoJ, ΔatoL, ΔatoM, ΔatoGΔatoD, ΔatoGΔatoL, ΔatoDΔatoL, ΔatoIΔatoM, ΔatoFΔatoM, and ΔatoBΔatoM (Fig. 3, S3 and S4†). The metabolites of these mutant strains revealed a branching biosynthetic pathway of aspertetranones and five new derivatives (7, 8, 10, 11, and 14), based on the substrate promiscuity of the short-chain dehydrogenase AtoL and cytochrome P450 AtoM (Fig. S3†).
The mutant strain ΔatoM accumulated 4a in addition to an unreported minor 4b, indicating that AtoM catalyzed the hydroxylation and ketonization of the allylic position (C-12) of 4a (Fig. 3 and Table S7†). In the double-knockout strain ΔatoBΔatoM and heterologous expression mutant An-atoCEHKGDLIF, three main products were detected and characterized by LC-MS and NMR analysis, demonstrating that both mutants failed to produce the linear tetracyclic compound 4a or 4b. Instead, compound 16 and two shunt products, 6a and 6b, with a γ-lactone ring significantly accumulated as reported before (Fig. 3A and S4†).13 These results confirmed that AtoB could catalyze the same aldol reaction as the assumed isozyme OchL. Inspiringly, a trace compound 5 together with two metabolic intermediates 19 and 23 were detected, which could be the natural substrate of AtoB (Fig. 3A and S4†). After a scale-up fermentation of ΔatoBΔatoM strain, 5 was obtained with a molecular weight of 420, similar to 4a and 4b. Thus, we hypothesized that AtoB catalyzed the aldol reaction with 5 as a natural substrate to form the linear tetracyclic epimers 4a and 4b.
To verify the hypothesis, the His6-tagged AtoB was expressed in Escherichia coli BL21(DE3) strain and purified by Ni-NTA chromatography. The recombinant AtoB was incubated with the isolated 5, 19, and 23, respectively. The LC-MS analysis of reaction mixtures exhibited the conversion of 5 into epimers 4a and 4b, confirming that AtoB readily accepted 5 as the substrate (Fig. 3C). These results also illustrated that AtoB catalyzed the stereoselective aldol reaction of 5 to generate the 10aS-products with the chirality of C-6a retained (4a) or flipped (4b).
Overall structure of AtoB
To investigate the catalytic mechanism of AtoB, we solved its crystal structure presenting the apo form (PDB ID: 9JLM) and the complex with simulated substrate 19 (PDB ID: 8ZEC) at 1.9 Å and 1.6 Å resolution, respectively (Fig. 4, S5 and Table S5†). The asymmetric unit of apo-AtoB and AtoB–19 complex contained four and two protein molecules, respectively, which are arranged as symmetric homodimers. The monomeric AtoB adopts the distorted α + β barrel fold, which is mainly composed of four α-helices and six β-sheets enclosing a presumed active cavity, similar to the previously reported structures of NTF2-like enzymes.9–12,25 However, the overall shape of AtoB active pocket is not conserved among these enzymes (Fig. S6†). Different from other fungal NTF2-like enzymes, the cavity of AtoB was divided into two distinct parts with one side containing three basic amino acid residues and the other side two Glu residues and two Tyr residues. The dimer interface was held together by numerous electrostatic, hydrophilic, and hydrophobic interactions within the contact area (Fig. S7†).9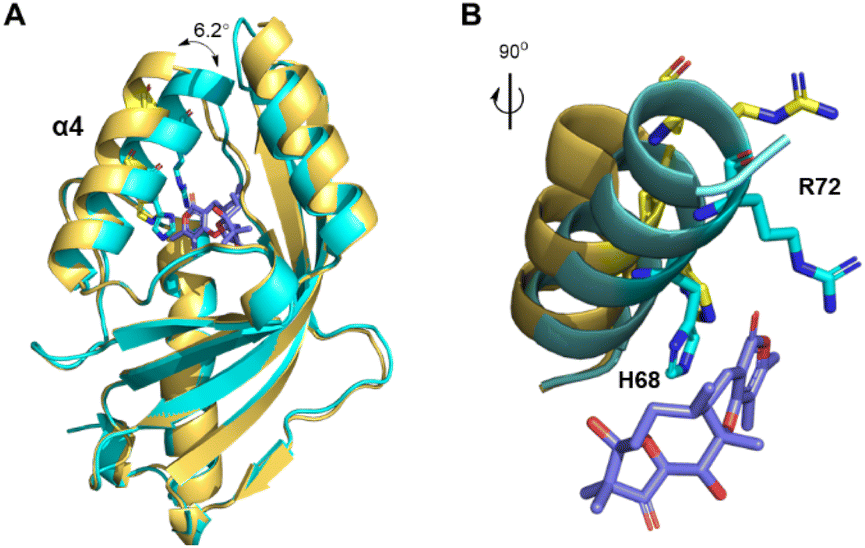 | ||
| Fig. 4 Comparison of the overall structures and the substrate binding sites of apo-AtoB and AtoB-ligand complex. (A) Main deviation between apo-AtoB (yellow) and AtoB–19 complex (protein in cyan and compound in slate). (B) Comparison of α4 helix, Arg72 and His68 in between apo-AtoB (protein and residues in yellow) and AtoB–19 complex (protein and residues in cyan and compound in slate). | ||
Notably, comparison of the structures of apo-AtoB and AtoB–19 complex structures showed that they entirely merged, except for the position of α4 helix (Fig. 4A). The α4 helix from the AtoB–19 complex rotated closer to the active cavity for an approximate 6.2°, featuring the side chains of Arg72 and His68 extending into the active cavity (Fig. 4B). This conformational change implied that the hydrogen bonding interactions between Arg72/His68 and the substrate might play important roles in the substrate binding process.
Active site architecture and catalytic mechanism of AtoB
To further understand the structural details of the AtoB-catalyzed aldol reaction, we investigated the binding mode of the native substrate 5 within the active cavity of AtoB (Fig. 5). It turned out that 5 was surrounded by the hydrophilic residues distributed on both sides of the cavity. The rotated residue Arg72 interacted with the carbonyl group of the pyrone ring in 5via a hydrogen bond, while the imidazole ring of the other rotated residue His68 was almost parallel to the pyrone ring, suggesting the presence of a potential π–π stacking interaction. These interactions revealed that the pyrone ring of 5 was fixed by the movement of the α4 helix to expose the flexible ten-membered ring of 5 at the entrance of the active pocket to interact with other binding and catalytic residues. In addition, the substrate was fixed in place by hydrogen bond interactions involving the C-9 carbonyl group and Glu143 as well as the C-6a hydroxyl/C-7 carbonyl group and Glu136 (Fig. 5A).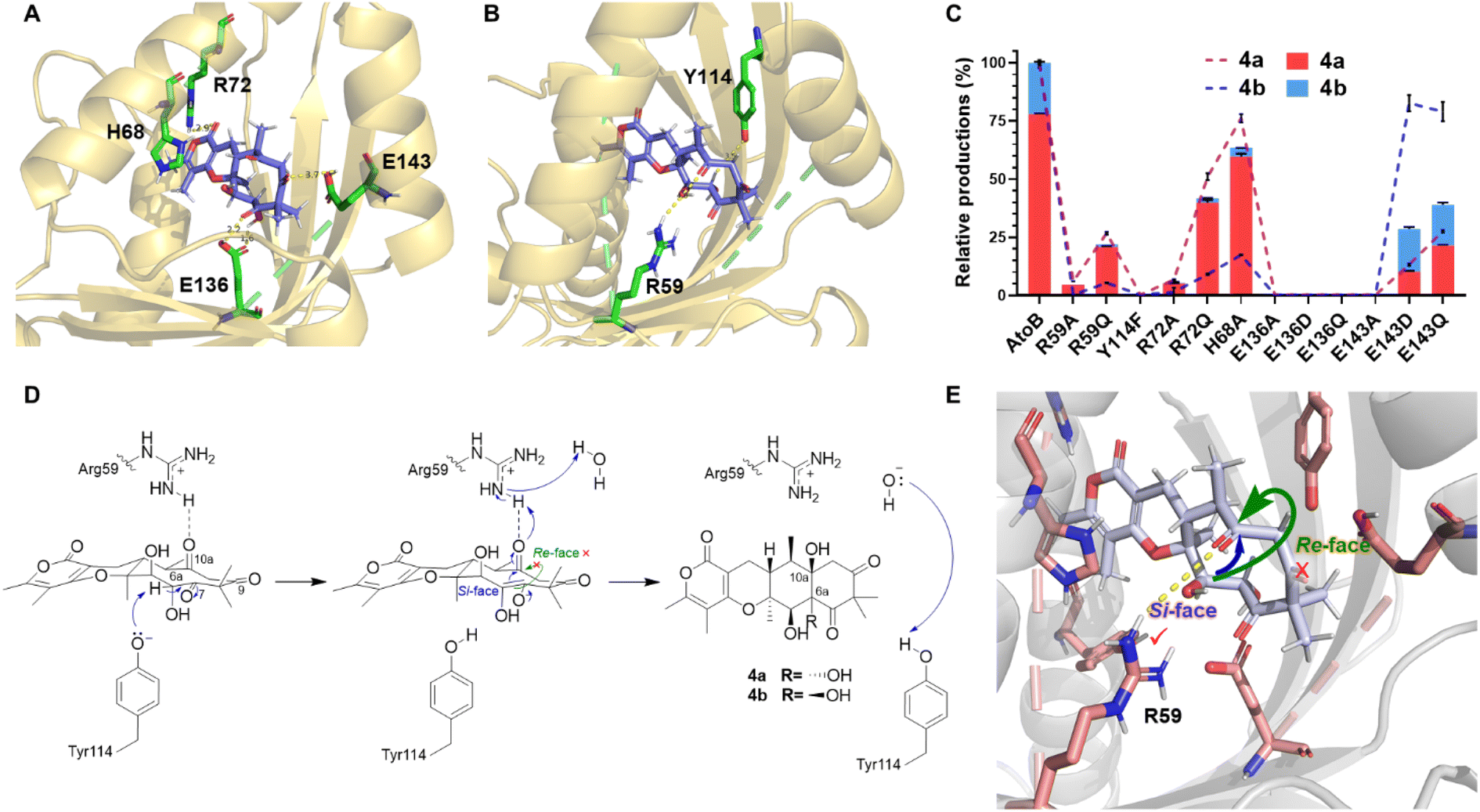 | ||
| Fig. 5 AtoB docking, site-directed mutagenesis activities and the proposed catalytic mechanism. (A) The docking model of AtoB–5 complex with the substrate binding residues, and hydrogen bonding distances shown in Å. (B) The key catalytic residues of AtoB. (C) In vitro activities of wild-type AtoB and its mutants. The relative production of 4a is shown as red bars and that of 4b as blue bars. The dashed line (4a in purple and 4b in blue) represents the percentage yield of products of mutants to wild-type AtoB. Triplicate assays were conducted for quantification. (D) Proposed mechanism for AtoB catalyzed aldol reaction. (E) The position of Arg59 determined the stereoselectivity at C-10a in 4a and 4b. | ||
For the aldol reaction catalyzed by AtoB, Tyr114 and Arg59 facilitated the condensation with Tyr114 positioned to interact with the Cα of the C-7 carbonyl group and Arg59 positioned to interact with the C-10a carbonyl group (Fig. 5B). The previously reported fungal NTF2-like enzymes usually share a similar acid–base pair to initiate the reaction, even though they catalyzed different reactions by the deprotonation of a hydroxyl group or Cα of a carbonyl group.10,11,26 Thus, Arg59–Tyr114 was considered as the catalytic acid–base pair to catalyze the aldol reaction and to form a 6/6/6/6 linear tetracyclic skeleton in aspertetranones.
To verify the binding mode of the substrate observed in the docking simulation, we systematically conducted the site-directed mutagenesis of AtoB and compared their activities to that of the wild type. The hydrophilic residues Arg59, Tyr114, Arg72, Glu136, Glu143, and His68 were substituted with Ala or other residues. Compared to the wild-type AtoB, the R72A mutant exhibited 6.65% yield of 4a, while R72Q maintained 51.20% yield of 4a (Fig. 5C and Table S6†). Similarly, the E136A, E136D, E136Q, E143A, E143D, and E143Q mutants showed the productivity of 4a with the range of 0% to 27.55%. These results indicated that Arg72, Glu136, and Glu143 played important roles in the substrate binding, especially the residue Glu136 which might be a key unit to fix the flexible ten-membered ring of 5. Additionally, the H68A mutant maintained 76.53% of 4a production, proving relatively weak π–π stacking interaction between the imidazole and the pyrone ring.
The activity of the Y114F variant was complete regression indicating that Tyr114 might trigger the aldol reaction as its ionized form to serve as a Lewis base to promote the deprotonation of Cα of the C-7 carbonyl group (Fig. 5D), following the spontaneous isomerization into the enol form with nucleophilicity. During this process, the hydrogen bond networks, especially the interactions between the carboxyl group of Glu136 and C-7 carbonyl/C-6a hydroxyl fix the conformation of the substrate to allow the nucleophilic enol attack at the C-10a carbonyl. Comparatively, Arg59 substituted R59A variant resulted in 5.91% yield of 4a. These in vitro assays revealed that Arg59 served as a proton donor for the C-10a carbonyl group, to finish the aldol reaction rather than as an initiator. Arg59 of AtoB is an unusual proton donor, as the calculated pKa value of its sidechain in solution is about 12.93.27 However, we noted that Arg329 of citrate synthase also acts as the proton donor with high-level QM/MM modelling evidence.28 Surprisingly, the R59Q variant maintained 26.74% yield of 4a. The side chain of glutamine might assist in proton transfer by interacting with a water molecule, which could also act as a proton donor. Furthermore, the interaction between Arg59 and C-10a carbonyl group was nearly perpendicular to the ten-member ring, setting an obstacle for conformational changes of the ten-membered ring and preventing the carbonyl group at C-10a from flipping to the opposite side of the ring plane (Fig. 5B and E). As a result, the stereocontrol of hydroxylated C-10a in the aldol product 4a and 4b was determined by the position of Arg59, suggesting that only a little distortion is satisfied when the nucleophile attacks from the si-face during the C–C bond formation step.
Interestingly, the enzyme reactions showed the ratios of 4a and 4b to be 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :22 for wild-type AtoB, while the ratios changed to 35:65 and 55:45 for E143D and E143Q, respectively (Fig. 5D and Table S6†). A plausible explanation is that variations of the hydrogen bond donor allowed a more stretched conformation of the ten-membered ring, resulting in reduction of the spatial hindrance of C-6a hydroxyl in 4b. Meanwhile, variants R72A, R72Q, and H68A produced a similar ratio of 4a and 4b (95:5). Accordingly, the loss of interactions between Arg72/His68 and the pyrone ring of 5 changed the location of the substrate and enhanced the hydrogen bonds between Glu136 and C-6a hydroxyl group, which prompted the retained chirality of C-6a. These observations indicated that the substrate was fixed in the active site through hydrogen bond networks, which governed the C-6a S or R configuration of 4a and 4b.
:22 for wild-type AtoB, while the ratios changed to 35:65 and 55:45 for E143D and E143Q, respectively (Fig. 5D and Table S6†). A plausible explanation is that variations of the hydrogen bond donor allowed a more stretched conformation of the ten-membered ring, resulting in reduction of the spatial hindrance of C-6a hydroxyl in 4b. Meanwhile, variants R72A, R72Q, and H68A produced a similar ratio of 4a and 4b (95:5). Accordingly, the loss of interactions between Arg72/His68 and the pyrone ring of 5 changed the location of the substrate and enhanced the hydrogen bonds between Glu136 and C-6a hydroxyl group, which prompted the retained chirality of C-6a. These observations indicated that the substrate was fixed in the active site through hydrogen bond networks, which governed the C-6a S or R configuration of 4a and 4b.
Conclusions
Since NTF2-like proteins possess low similarity of amino acid sequences with diverse biological functions, their catalytic potential has not been well explored. In this work, we established an effective strategy based on protein structural alignment in combination with clustering analysis to detect the catalytic functions of NTF2-like enzymes. This structure-based alignment approach enabled the discovery of uncharted enzymes, particularly when primary sequence alignment was restricted. As a result, a new NTF2-like aldolase AtoB was identified, and its catalytic functions were investigated by heterologous expression, genetic deletion, and in vitro assays. AtoB catalyzed the aldol condensation in the last step of formation of linear tetracyclic 4a and 4b. X-ray crystallographic characterization studies of AtoB and AtoB–19 complex along with the site-directed mutagenesis experiments revealed a catalytic acid–base pair, Arg59–Tyr114, and the rotated binding residues for the aldol reaction. Intriguingly, this study also indicated that Arg59 and E143 play a critical role in the stereoselectivity of aldol products. Overall, our investigations provide a structural and catalytic insight into the unique aldolase AtoB and open up new avenues for diversifying the catalytic repertoire of the NTF2-like superfamily.Data availability
The data supporting this article have been reported as part of the ESI.†Author contributions
W. L., A. F., and K. M. designed the research. K. M., J. L., and M. W. performed the bioinformatics analysis, the in vitro and in vivo assay, and the crystal structure determination. K. M., Z. H., D. L., and J. R. purified the compounds and elucidated the structures. All authors analyzed and discussed the results. K. M., A. F. and W. L. prepared the manuscript with input from all the authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Erwei Li (Institute of Microbiology, Chinese Academy of Sciences) for LC-MS data collection. This work was supported by the National Key Research & Development Program of China (2022YFC2804900), the National Natural Science Foundation of China (82104056, 81991525, 82173733), COMRA DY135-B-05, the Ningbo Key Science and Technology Development Program (2022Z144, 2021Z046), and the China Postdoctoral Science Foundation (2022M710267).References
- A. C. Allison and E. M. Eugui, Immunosuppressive and Other Effects of Mycophenolic-acid and an Ester Prodrug, Mycophenolate Mofetil, Immunol. Rev., 1993, 136, 5–28 CrossRef CAS.
- M. S. Butler, The role of natural product chemistry in drug discovery, J. Nat. Prod., 2004, 67, 2141–2153 CrossRef CAS.
- Y.-H. Chooi and Y. Tang, Navigating the Fungal Polyketide Chemical Space: From Genes to Molecules, J. Org. Chem., 2012, 77, 9933–9953 CrossRef CAS.
- B. Toenshoff, Immunosuppressive therapy post-transplantation in children: what the clinician needs to know, Expert Rev. Clin. Immunol., 2020, 16, 139–154 CrossRef CAS.
- N. Steffan, A. Grundmann, W. B. Yin, A. Kremer and S. M. Li, Indole Prenyltransferases from Fungi: A New Enzyme Group with High Potential for the Production of Prenylated Indole Derivatives, Curr. Med. Chem., 2009, 16, 218–231 CrossRef CAS.
- M.-C. Tang, Y. Zou, K. Watanabe, C. T. Walsh and Y. Tang, Oxidative Cyclization in Natural Product Biosynthesis, Chem. Rev., 2017, 117, 5226–5333 CrossRef CAS.
- X. Zhang, J. Guo, F. Cheng and S. Li, Cytochrome P450 enzymes in fungal natural product biosynthesis, Nat. Prod. Rep., 2021, 38, 1072–1099 RSC.
- T. L. Bullock, W. D. Clarkson, H. M. Kent and M. Stewart, The 1.6 angstrom resolution crystal structure of nuclear transport factor 2 (NTF2), J. Mol. Biol., 1996, 260, 422–431 CrossRef CAS.
- T. Mori, T. Iwabuchi, S. Hoshino, H. Wang, Y. Matsuda and I. Abe, Molecular basis for the unusual ring reconstruction in fungal meroterpenoid biogenesis, Nat. Chem. Biol., 2017, 13, 1066–1073 CrossRef CAS.
- Y. Ye, L. Du, X. Zhang, S. A. Newmister, M. McCauley, J. V. Alegre-Requena, W. Zhang, S. Mu, A. Minami, A. E. Fraley, M. L. Adrover-Castellano, N. A. Carney, V. V. Shende, F. Qi, H. Oikawa, H. Kato, S. Tsukamoto, R. S. Paton, R. M. Williams, D. H. Sherman and S. Li, Fungal-derived brevianamide assembly by a stereoselective semipinacolase, Nat. Catal., 2020, 3, 497–506 CrossRef CAS.
- J. Yang, T. Mori, X. Wei, Y. Matsuda and I. Abe, Structural Basis for Isomerization Reactions in Fungal Tetrahydroxanthone Biosynthesis and Diversification, Angew. Chem., Int. Ed., 2021, 60, 19458–19465 CrossRef CAS.
- S. H. Liu, J. L. Sun, Y. L. Hu, L. Zhang, X. Zhang, Z. Y. Yan, X. Guo, Z. K. Guo, R. H. Jiao, B. Zhang, R. X. Tan and H. M. Ge, Biosynthesis of Sordarin Revealing a Diels-Alderase for the Formation of the Norbornene Skeleton, Angew. Chem., Int. Ed., 2022, 61, e202205577 CrossRef CAS.
- Y. Li, M. Cong, W. Wang, X. Zhang, Y. Zhu, Y. Song, W. Zhang, H. Xiao, Y. Liu, C. Zhang, J. Wang and Y. Yan, An Enzymatic Carbon-Carbon Bond Cleavage and Aldol Reaction Cascade Converts an Angular Scaffold into the Linear Tetracyclic Core of Ochraceopones, Angew. Chem., Int. Ed., 2024, 63, e202403365 CrossRef CAS.
- T. Frickey and A. Lupas, CLANS: a Java application for visualizing protein families based on pairwise similarity, Bioinformatics, 2004, 20, 3702–3704 CrossRef CAS.
- M. van Kempen, S. S. Kim, C. Tumescheit, M. Mirdita, J. Lee, C. L. M. Gilchrist, J. Soeding and M. Steinegger, Fast and accurate protein structure search with Foldseek, Nat. Biotechnol., 2024, 42, 243–246 CrossRef CAS.
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch, R. D. Schaeffer, C. Millan, H. Park, C. Adams, C. R. Glassman, A. DeGiovanni, J. H. Pereira, A. V. Rodrigues, A. A. van Dijk, A. C. Ebrecht, D. J. Opperman, T. Sagmeister, C. Buhlheller, T. Pavkov-Keller, M. K. Rathinaswamy, U. Dalwadi, C. K. Yip, J. E. Burke, K. C. Garcia, N. V. Grishin, P. D. Adams, R. J. Read and D. Baker, Accurate prediction of protein structures and interactions using a three-track neural network, Science, 2021, 373, 871–876 CrossRef CAS.
- C.-J. Guo, B. P. Knox, Y.-M. Chiang, H.-C. Lo, J. F. Sanchez, K.-H. Lee, B. R. Oakley, K. S. Bruno and C. C. C. Wang, Molecular Genetic Characterization of a Cluster in A. terreu for Biosynthesis of the Meroterpenoid Terretonin, Org. Lett., 2012, 14, 5684–5687 CrossRef CAS.
- H.-C. Lo, R. Entwistle, C.-J. Guo, M. Ahuja, E. Szewczyk, J.-H. Hung, Y.-M. Chiang, B. R. Oakley and C. C. C. Wang, Two Separate Gene Clusters Encode the Biosynthetic Pathway for the Meroterpenoids Austinol and Dehydroaustinol in Aspergillus nidulans, J. Am. Chem. Soc., 2012, 134, 4709–4720 CrossRef CAS.
- Y. Matsuda, T. Awakawa and I. Abe, Reconstituted biosynthesis of fungal meroterpenoid andrastin A, Tetrahedron, 2013, 69, 8199–8204 CrossRef CAS.
- Y. Matsuda, T. Wakimoto, T. Mori, T. Awakawa and I. Abe, Complete Biosynthetic Pathway of Anditomin: Nature's Sophisticated Synthetic Route to a Complex Fungal Meroterpenoid, J. Am. Chem. Soc., 2014, 136, 15326–15336 CrossRef CAS.
- Y. Matsuda, T. Iwabuchi, T. Fujimoto, T. Awakawa, Y. Nakashima, T. Mori, H. Zhang, F. Hayash and I. Abe, Discovery of Key Dioxygenases that Diverged the Paraherquonin and Acetoxydehydroaustin Pathways in Penicillium brasilianum, J. Am. Chem. Soc., 2016, 138, 12671–12677 CrossRef CAS.
- Y. Matsuda, T. Bai, C. B. W. Phippen, C. S. Nodvig, I. Kjaerbolling, T. C. Vesth, M. R. Andersen, U. H. Mortensen, C. H. Gotfredsen, I. Abe and T. O. Larsen, Novofumigatonin biosynthesis involves a non-heme iron-dependent endoperoxide isomerase for orthoester formation, Nat. Commun., 2018, 9, 2587 CrossRef.
- B.-w. Qi, N. Li, B.-b. Zhang, Z.-k. Zhang, W.-j. Wang, X. Liu, J. Wang, T. Awakawa, P.-f. Tu, I. Abe, S.-p. Shi and J. Li, A Multifunctional Cytochrome P450 and a Meroterpenoid Cyclase in the Biosynthesis of Fungal Meroterpenoid Atlantinone B, Org. Lett., 2022, 24, 2526–2530 CrossRef CAS.
- Y.-M. Chiang, M. Ahuja, C. E. Oakley, R. Entwistle, A. Asokan, C. Zutz, C. C. C. Wang and B. R. Oakley, Development of Genetic Dereplication Strains in Aspergillus nidulans Results in the Discovery of Aspercryptin, Angew. Chem., Int. Ed., 2016, 55, 1662–1665 CrossRef CAS.
- N. Vuksanovic, X. Zhu, D. A. Serrano, V. Siitonen, M. Metsa-Ketela, C. E. Melancon III and N. R. Silvaggi, Structural characterization of three noncanonical NTF2-like superfamily proteins: implications for polyketide biosynthesis, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2020, 76, 372–383 CrossRef CAS.
- A. Sultana, P. Kallio, A. Jansson, J. S. Wang, J. Niemi, P. Mäntsälä and G. Schneider, Structure of the polyketide cyclase SnoaL reveals a novel mechanism for enzymatic aldol condensation, EMBO J., 2004, 23, 1911–1921 CrossRef CAS.
- M. H. M. Olsson, C. R. Sondergaard, M. Rostkowski and J. H. Jensen, PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS.
- M. W. van der Kamp, F. Perruccio and A. J. Mulholland, High-level QM/MM modelling predicts an arginine as the acid in the condensation reaction catalysed by citrate synthase, Chem. Commun., 2008, 16, 1874–1876 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05590j |
| This journal is © The Royal Society of Chemistry 2024 |