
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEmerging solid-state cycloaddition chemistry for molecular solar thermal energy storage
Cijil
Raju
 ,
Han P. Q.
Nguyen
and
Grace G. D.
Han
*
,
Han P. Q.
Nguyen
and
Grace G. D.
Han
*
Department of Chemistry, Brandeis University, 415 South Street, Waltham, MA 02453, USA. E-mail: gracehan@brandeis.edu
First published on 1st October 2024
Abstract
Recently discovered designs of solid-state molecular solar thermal energy storage systems are illustrated, including alkenes, imines, and anthracenes that undergo reversible [2 + 2] and [4 + 4] photocycloadditions for photon energy storage and release. The energy storage densities of various molecular designs, from 6 kJ mol−1 to 146 kJ mol−1 (or up to 318 J g−1), are compared and summarized, along with effective strategies for engineering their crystal packing structures that facilitate solid-state reactions. Many promising molecular scaffolds introduced here highlight the potential for achieving successful solid-state solar energy storage, guiding further discoveries and the development of new molecular systems for applications in solid-state solar thermal batteries.
 Cijil Raju | Cijil Raju was born in Kerala, India. He received his B.Sc. and M.Sc. degrees in Chemistry from Newman College, Thodupuzha (affiliated with M.G. University, Kottayam) in 2013 and 2015, respectively. In 2016, he worked as a research assistant in the Chemical Sciences & Technology Division at NIIST Thiruvananthapuram. He later joined the PhD program and obtained his PhD in Chemistry in 2023 from IISER Thiruvananthapuram under the supervision of Prof. Kana M. Sureshan. Currently, he is a postdoctoral research associate with Prof. Grace G. D. Han at Brandeis University. His research interests include chemical reactions and isomerization in confined media and their applications. His current research focuses on solid-state photoreactions for the development of molecular solar thermal energy storage materials and understanding their operating mechanisms. |
 Han P. Q. Nguyen | Han Nguyen was born in Ho Chi Minh City, Vietnam. She received her B.S. in Chemistry from Worcester State University and is currently pursuing a PhD in Chemistry at Brandeis University under the supervision of Prof. Grace G. D. Han. Her research focuses on developing advanced photon energy storage systems using intermolecular photocycloaddition reactions. Han's work involves exploring the mechanisms of photon energy storage and characterizing heat release from solar thermal materials at various scales. In the future, she aims to translate her research into practical applications, enhancing the scalability of these systems for commercial use and bridging the gap between laboratory research and real-world sustainable energy solutions. |
 Grace G. D. Han | Grace Han was born and raised in South Korea, where she attended POSTECH and received a B.S. in Chemistry. She spent a year at UC Berkeley and worked in the laboratory of Prof. Jeffrey Long to perform research on metal–organic frameworks. Grace received her PhD in 2015 from MIT, where she worked with Professor Timothy Swager on the development of organic materials for photovoltaics. She then joined the Department of Materials Science and Engineering at MIT as a postdoctoral associate with Professor Jeffrey Grossman. Grace joined the Department of Chemistry at Brandeis in July 2018 and was promoted to the rank of Associate Professor with tenure in 2024. Her group's research focuses on the discovery of molecular photoswitches and photochemistry for applications including solar energy conversion and storage, optically controlled catalyst recycling, and light-induced phase transitions. She will continue to study various light–molecule interactions, unique transformations, and the mechanisms of such phenomena. |
Introduction
Over the past couple of decades, there have been increasing interest and significant progress in the development of molecular solar thermal (MOST) energy storage systems.1–5 These molecular systems capture solar photon energy through photoinduced structural isomerization, storing it in the strained chemical bonds of metastable isomers (Fig. 1a). The reverse isomerization of these metastable states to their thermodynamically stable forms can be triggered by photochemical, thermal, electrochemical, or chemical means (such as catalysis), releasing the stored energy as heat.6,7 The primary metric for MOST systems is the amount of stored or released energy (ΔGstorage), which corresponds to the energy difference between the stable and metastable states. Other parameters including light absorption ranges, photostationary state (PSS) ratios, light penetration depths, and quantum yields of photoisomerization are also investigated to achieve efficient photon energy storage. In addition, the activation energy for thermal reverse isomerization (ΔG‡) is an important metric that determines the energy storage duration of MOST systems.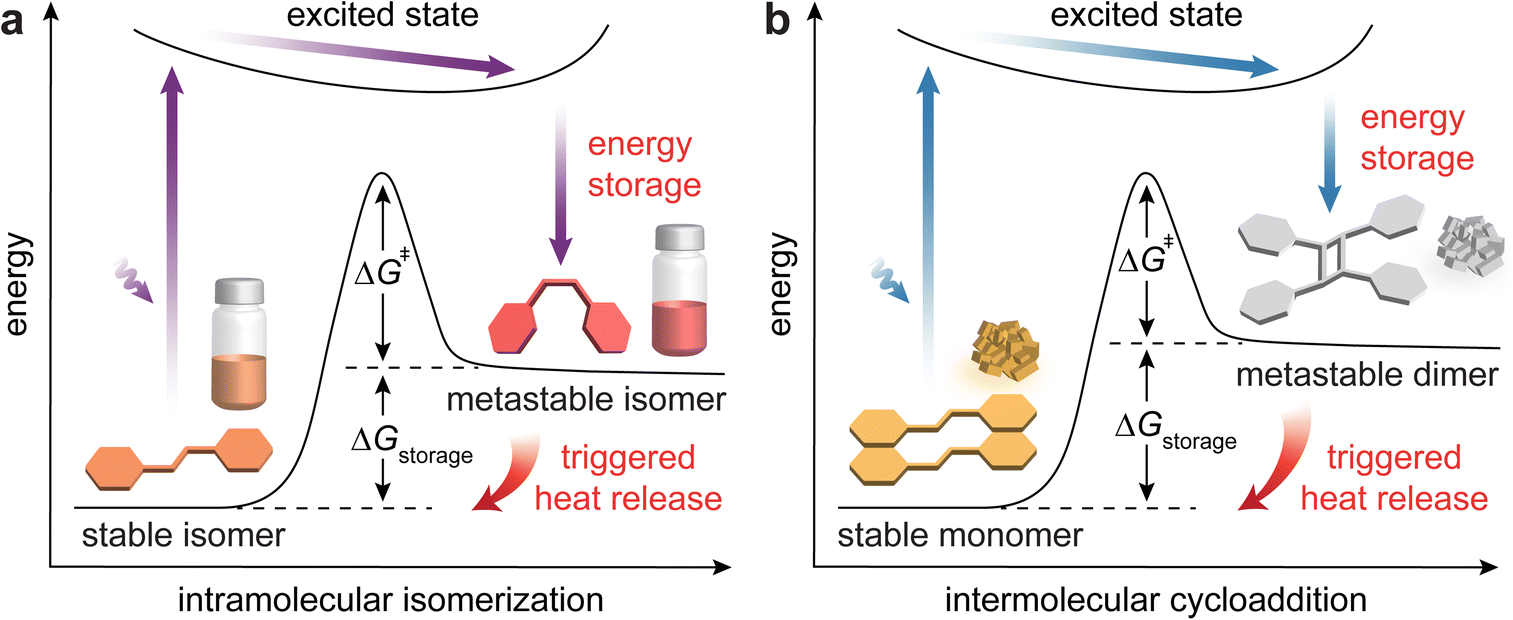 | ||
| Fig. 1 Energy diagrams illustrating solar photon energy storage and heat release through reversible (a) intramolecular isomerization and (b) intermolecular cycloaddition. | ||
These parameters have been fine-tuned and improved through new molecular designs and the optimization of known scaffolds.8,9 Various photoswitches, such as norbornadienes,10,11 azo(hetero)arenes,11–15 hydrazones,16 dihydroazulenes,17,18 and fulvalene dirutheniums,19,20 have been explored for their potential to store and release energy in the solution state. Recently, there have been developments in solvent-free MOST systems, including neat liquid-state photoswitches or solid-state compounds that transition to neat liquid upon isomerization, aimed at increasing the overall energy storage density.21–27 Nevertheless, these systems continue to present risks of organic substance leakage, which hinders their widespread implementation.
As a result, exclusively solid-state MOST compounds have been recently developed via efforts in enhancing the conformational freedom of photoswitches in solids. This led to several strategies such as integrating photoswitches in polymers or porous structures (frameworks or cages) or nano-carbon templates (e.g., graphene and graphene oxide), creating intrinsically amorphous solids, and engineering molecular stacking in crystals.28–43 Nevertheless, the number of examples is limited, as many conventional photoswitches undergo significant structural changes during E–Z isomerization or ring opening-closing transformations, which are hindered in a solid state.
This challenge has been recently addressed by developing topochemical photocycloaddition systems tailored for effective energy storage and release in crystalline solid states (Fig. 1b). Specifically, topochemical intermolecular [2 + 2] photo-cycloadditions between alkenes44 and [4 + 4] equivalents between fused aromatic compounds45 have been discovered to enable solar photon energy storage in their metastable cycloadducts, such as cyclobutanes and dianthracenes. Designing solid-state molecular systems for effective MOST energy storage is complex, requiring not only photochemical reactivity in the solid state but also meeting other essential criteria. This perspective will elaborate on the detailed molecular design criteria for achieving reversible solar energy storage and release, substantial energy densities, and high efficiencies via photoinduced cycloadditions in the solid state. We note that the fundamental design principles of MOST compounds and the general development of solvent-free MOST systems have been well documented in various reviews.11,12,26–29 Independently, many reviews on solid-state photoswitching materials46–49 and conventional topochemical reactions50–56 without energy storage considerations have been available. Distinctively, this perspective article will focus on the latest developments in topochemical reaction approaches for solid-state MOST energy storage.
Design principles of solid-state MOST compounds
Enabling photon energy storage via cycloaddition
For a successful topochemical photocycloaddition, the reactants are required to adopt a specific arrangement, akin to that of the transition state of reaction, in a crystalline state.51,52 Pioneering studies on topochemical cycloaddition reactions by Schmidt and co-workers established an empirical rule dictating the reactivity of molecules in crystals.57,58 Specifically, the rule predicts a high probability of topochemical cycloaddition of molecules that present a near parallel arrangement between the neighboring reactive units and their proximity within 4.2 Å. The principle has been widely accepted and demonstrated as a preliminary criterion to fulfill for crystalline–state reactions. Such desired molecular arrangements in crystals can be obtained by engineering non-covalent interactions among the reactants (e.g., H-bonding, halogen bonding, π interaction, and so on), which are often induced by the functional groups on the molecules (Fig. 2).59–62 Therefore, the type, position, and number of functional groups on a molecular scaffold have been the major design criteria to explore in the field of solid-state chemistry, as summarized in previous reviews.63–68 The molecular design strategies discovered in the fundamental investigations of solid-state photo-reactivity have served as important tools for achieving solid-state MOST compounds.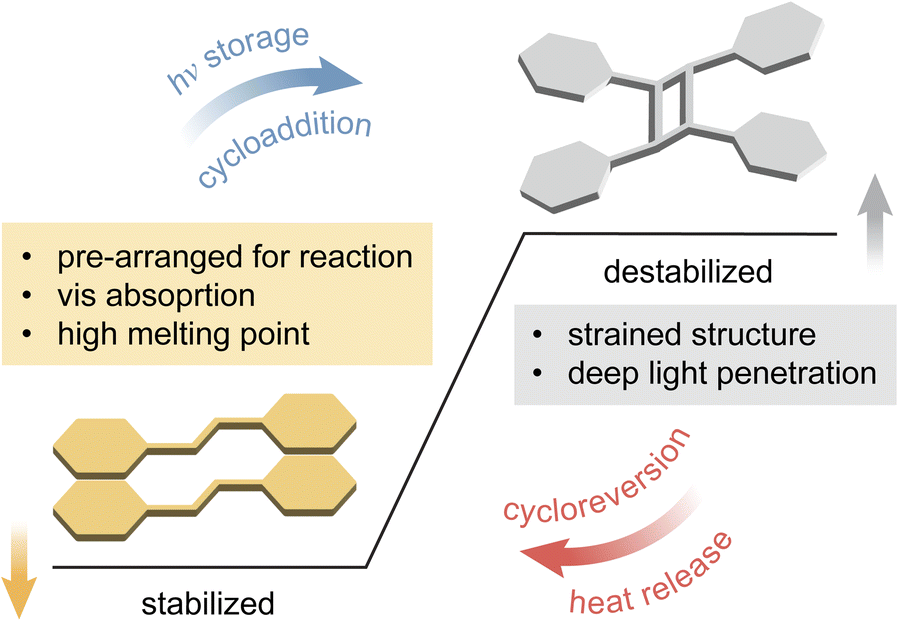 | ||
| Fig. 2 Molecular design principles for successful solid-state reactions that enable energy storage and release. | ||
In addition, for MOST applications, the light absorption profiles of reactants need to be carefully tailored to maximize the sunlight harvesting and the yield of solid-state cycloadditions on a large scale. To enable broad absorption of light in the visible range, the conjugation lengths across photochromic reactants are fine-tuned. Upon cycloaddition, the conjugation lengths are expected to significantly reduce in the metastable products, exhibiting negative photochromism, to limit any undesired photoinduced cycloreversion. Specifically, the functionalization of photochrome scaffolds with donor and acceptor groups, forming push–pull structures, has been incorporated to effectively red-shift their absorption in the visible range, which then turns colorless upon dimerization. Such molecular systems with large spectral separations between the reactants and products, particularly in the visible range, are able to achieve near quantitative PSS ratios, large light penetration depths, and complete energy storage. Lastly, the quantum yields of photocycloadditions are the fundamental characteristics of molecular systems, which should be carefully examined to maximize the efficiencies of solar energy conversion and storage in MOST systems.
Facilitating thermal energy release via cycloreversion
The cycloreversion of cycloadducts to the precursors or reactants releases the stored energy in solid-state MOST compounds, which is typically more challenging to achieve than the cycloaddition process. In principle, the cycloreversion of [2 + 2] and [4 + 4] cycloadducts can occur photochemically under the irradiation of short UV light. However, the photocycloreversion is often severely hindered due to the spectral overlap between the reactants and cycloadducts in the UV range, which lowers both the solid-state PSS ratios and light penetration depths. Moreover, the use of short UV can result in the photodegradation of molecules, and the large photon energy input for triggering the energy release from MOST systems is highly undesirable.Therefore, thermally induced cycloreversion of cycloadducts has emerged as an alternative triggering method that effectively releases the stored energy in the solid state. Despite the Woodward-Hoffman rules that intrinsically forbid the thermal cycloreversion of [2 + 2] and [4 + 4] cycloadducts, there have been observations of such reactions, particularly at high temperatures near 200 °C accompanying the degradation of compounds.69–78 Lowering the temperature of cycloreversion has been viable, however, through the careful molecular designs that allow for facile bond dissociations of cycloadducts. For example, functionalization at the reaction centers with sterically bulky or electron-withdrawing groups can weaken newly formed bonds of cycloadducts. The recent design strategies discovered to achieve mild thermal triggering conditions will be showcased in this perspective along with specific examples.
Another specific challenge associated with the energy release from solid-state MOST materials is the adverse reabsorption of the released heat by the melting of precursors that are generated during cycloreversion. Because of the endothermic melting transition, the net energy release from cycloreversion is significantly reduced. Thus, molecular designs to bypass the issue have to be considered, specifically to raise the melting point of the precursor molecule above the thermal cycloreversion temperature. Engineering strong directional non-covalent interactions (e.g., H-bonding, halogen bonding, π interaction, etc.) among the reactants in the solid state therefore plays a pivotal role in preserving the crystallinity of compounds and limiting their melting throughout the cycloreversion process. Functional groups that yield strong intermolecular interactions in crystals of reactants and products can thus facilitate cycloaddition/cycloreversion cycles preserving the crystallinity.
Achieving large energy storage densities (ΔGstorage)
In order to accomplish substantial energy storage densities in cycloaddition systems (40–100 kJ mol−1), comparable to those of conventional MOST photoswitches,6,7 one should fine-tune the relative energy levels of reactants and cycloadducts. In particular, either lowering the energy state of reactants or raising that of cycloadducts increases the energy gap between two states, resulting in larger energy storage in the MOST system. In recent reports, a strategy to dearomatize the reactant or break its conjugation through photocycloaddition has been emphasized, which enlarges the gap between the relative stabilization energy of reactants and cycloadducts.44,45 Another method for enhancing energy storage is to destabilize the cycloadduct by forming large ring strains through the functionalization of molecules with bulky groups. However, it should be noted that altering the structural features of molecules can affect other parameters associated with the MOST energy storage mechanism. For example, destabilizing cycloadduct structures may adversely facilitate their spontaneous cycloreversion, leading to unfavorably short energy storage periods. Modifying the reactant structure with bulky groups may significantly alter the molecular packing in crystals, resulting in the photostability of crystals. Hence, understanding and predicting the consequences of structural modifications will be crucial for finding the optimal and novel solid-state MOST compounds.Currently, the number of reported cycloadducts for MOST energy storage is limited to a few, and thus further molecular discoveries and fundamental studies remain vital tasks to accomplish solid-state MOST applications. Thus, we aim to highlight the state-of-the-art findings on the subject, which may elucidate important design principles and encourage the contribution from scholars in the diverse fields of photochemistry, solid-state chemistry, and beyond.
State-of-the-art solid-state MOST compounds
Intermolecular [2 + 2] and [4 + 4] photocycloadditions have been recently explored to store solar photon energy in cycloadducts including cyclobutanes, diazetidines, and dianthracenes. In the following examples, the energy storage densities have been measured by differential scanning calorimetry (DSC), and complete and reversible cycloaddition and reversion have been achieved. Also, the crystal packing of molecules has been analysed to shed light on the molecular design rules that lead to effective MOST energy storage and release.Styrylpyrylium/cyclobutane [2 + 2] cycloaddition
Topochemical [2 + 2] photocycloaddition reactions of alkenes to cyclobutanes have been vastly explored in solid-state chemistry, while the observation of complete and clean cycloreversion of cyclobutanes has been relatively less reported in the solid state.75–82 In 1985, Hesse and Hünig demonstrated the reversible cycloaddition of styrylpyrylium (STP) salts generating cyclobutane (CB)-bearing products,83 and in 1993, Novak and co-workers reported the observation of such reactions in single crystals.84 However, the potential of reversible STP/CB cycloaddition in MOST energy storage remained unexplored.Recently, STP derivatives with varied functional groups and counter anions (1–10) have been explored as candidates for MOST energy storage (Fig. 3a).44 All compounds exhibited an orange color with broad visible light absorption in the 400–600 nm range (Fig. 3b), attributed to the donor–acceptor design of STPs. An electron-rich phenyl ring, substituted with electron-donating groups (Me, OMe, and NMe2), and an electron-deficient pyrylium ring across the central alkene allow for the extensive delocalization of electron density in the STP structures. Such a donor–acceptor design also facilitates a favorable head-to-tail stacking of STPs in their crystals via strong complementary π interactions between electron-rich and electron-deficient aromatic rings on neighboring molecules.
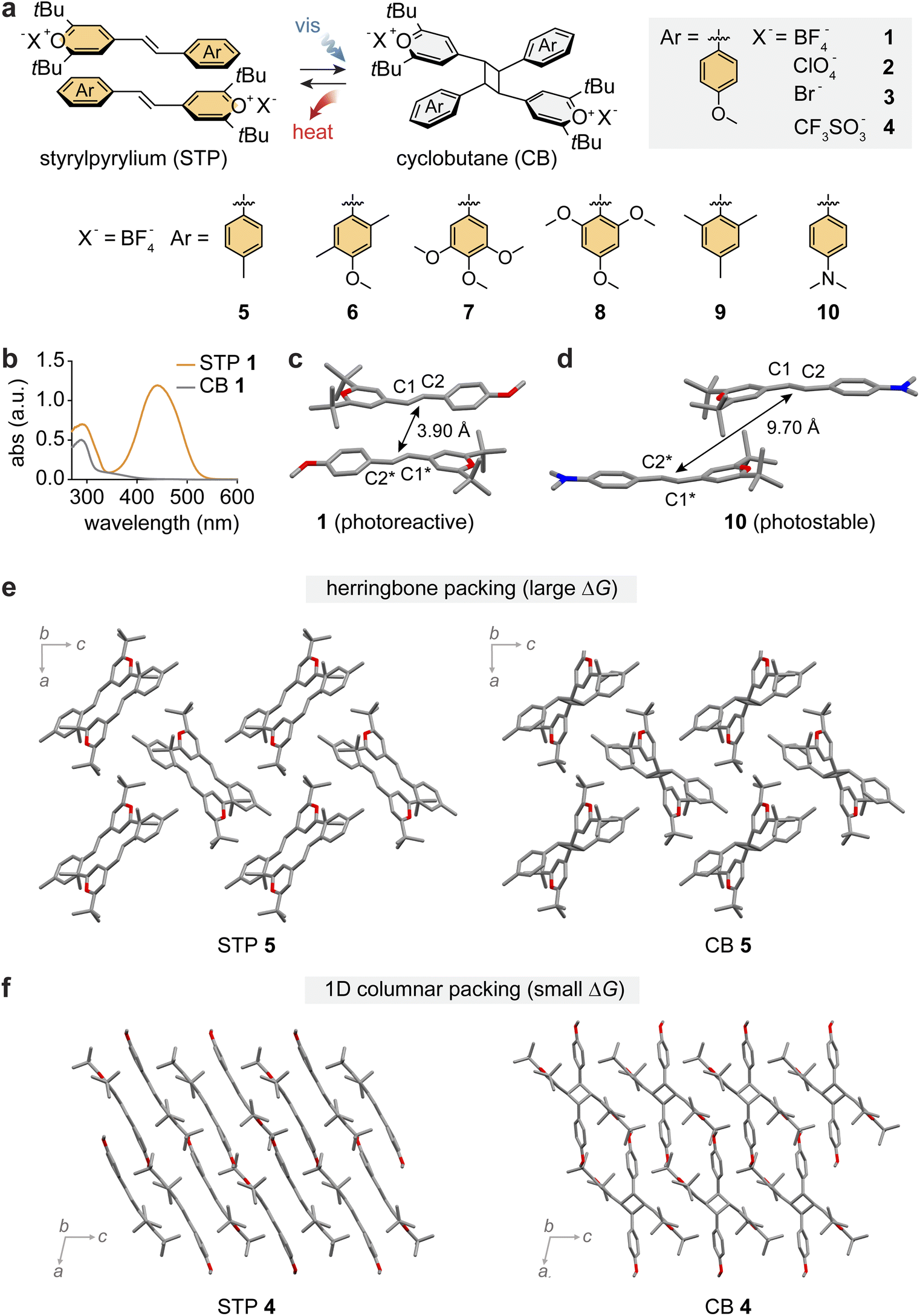 | ||
| Fig. 3 (a) Reversible photoinduced [2 + 2] cycloaddition and reversion between styrylpyrylium (STP) and cyclobutane (CB) with varied functional groups on the phenyl ring and varied counter-anions (X−). (b) UV-vis absorption spectra of STP 1 (orange) and CB 1 (gray). Crystal structures of (c) STP 1 showing proximal head-to-tail packing for [2 + 2] cycloaddition and (d) STP 10 showing offset packing unsuitable for cycloaddition. Crystal packing structures of (e) STP 5 and CB 5 and (f) STP 4 and CB 4 representing herringbone and 1D columnar packing, respectively. In panels (c)–(f), the hydrogen atoms are omitted in the crystal packing for clarity. This figure has been reproduced from ref. 44 with permission from Elsevier, copyright 2023. | ||
STPs 1–7 underwent solid-state [2 + 2] cycloaddition under visible light irradiation (470 or 530 nm) for 3–20 hours, or under the AM 1.5 standard solar spectrum for 15 hours, resulting in their products that store energy in the strained CB units. Due to the reduced conjugation in products, their absorption in the visible range is negligible, which can significantly increase the penetration depth of incident light through solid-state materials. In contrast, STPs 8–10 remained unreactive even after prolonged (48 hours) photoirradiation. Such different photo-reactivities of similar STP derivatives were rationalized by analysing their crystal structures. The representative crystal structures of STP 1 and 10 are shown in Fig. 3c and d. While all STPs adopted parallel stacking between paired molecules in crystals, the proximity between the reactive alkenes varied: shorter (1–7) or longer (8–10) than Schmidt's criterion, 4.2 Å. The different degrees of orbital overlap between paired STPs dictate their reactivity and capability to store photon energy by cycloaddition.
The cycloreversion from CB to STP releases the stored energy. Due to the overlapping absorption of STP and CB in the UV range (Fig. 3b), UV-induced cycloreversion was low-yielding even in thin films. On the other hand, thermally triggered cycloreversion was quantitative, effectively releasing heat, as monitored by DSC. The thermal triggering conditions for CBs ranged from 71 °C to 165 °C, reflecting the relative stability of CBs with varied substituents and counter anions. The reversibility of cycloaddition/cycloreversion of 1 was tested for 10 cycles, without any sign of degradation, and each cycloreversion process was completed in 10 minutes at 160 °C. It is indeed unusual to observe such facile thermal cycloreversion of cyclobutanes under mild heating conditions. It is hypothesized that the strong donor–acceptor substituents on CBs may stabilize the transition state energy of cycloreversion. The cycloreversion temperatures are well below the melting points of the resulting STPs (except for 5), enabling uncompromised energy release without any reabsorption of released heat. The high melting points of STPs (150–250 °C) are characteristic of ionic compounds with strong intermolecular coulombic interactions in crystals.
The energy storage densities measured for CBs 1, 2, 5 and 6 were markedly larger (31–42 kJ mol−1 or 39–51 J g−1) than those of CBs 4 and 7 (6–8 kJ mol−1 or 6–8 J g−1). Such drastic differences in the energy storage capabilities of molecules were strongly correlated with their crystal packing structures: STP/CB 1, 2, 5 and 6 adopt herringbone packing (Fig. 3e), while STP/CB 4 and 7 display 1D columnar packing (Fig. 3f). Detailed investigation on the crystal structures revealed different extents of coulombic interactions among cationic molecules and counter anions in each crystal packing, which enlarged the STP-CB energy gap for 1, 2, 5 and 6 and reduced it for 4 and 7. The STP/CB systems highlight the critical role of crystal packing in determining the efficacy of MOST energy storage via cycloaddition as well as the energy storage density.
Styryldipyrylium/cyclobutane [2 + 2] polymerization
The STP structure was further extended by the addition of another pyrylium group to the phenyl moiety, which formed a series of styryldipyryliums (11–14; Fig. 4a) with red-shifted absorption profiles.85 The extended compounds bearing two reactive alkenes were also expected to increase the molar and gravimetric energy storage densities. Although the molecular design of styryldipyryliums (SDPs) has been known,86 their crystal packing has been challenging to predict. Even with various substituents or counter anions, SDPs tend to form mostly photostable crystals rather than photoactive forms, which limits their application in MOST energy storage. Thus, various recrystallization methods or mechanical activation strategies have been explored to control the molecular arrangement in the solid state and reliably obtain photopolymerizable forms.85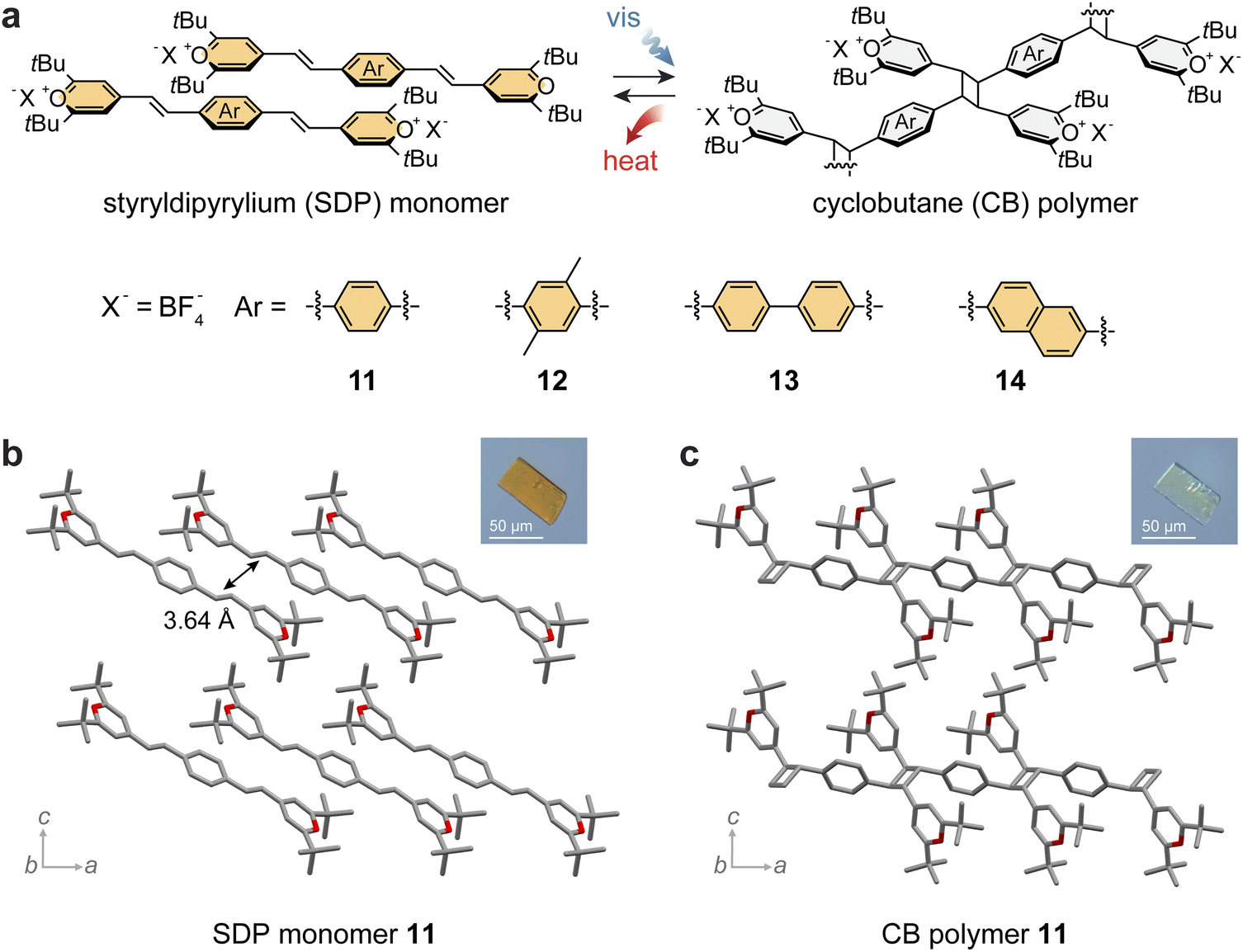 | ||
| Fig. 4 (a) Reversible photoinduced [2 + 2] cycloaddition and reversion between styryldipyrylium (SDP) monomers and cyclobutane (CB)-linked polymer derivatives with varied arene spacers. Crystal structures and microscopic optical crystal images of (b) SDP monomer 11 and (c) CB polymer 11. In panels (b) and (c), the hydrogen atoms are omitted in the crystal structure for clarity. This figure has been reproduced from ref. 85, an open access article of Springer Nature (Creative Commons CC BY license). | ||
For example, the series of SDPs 11–14 generally forms photostable crystals, where the distance between the nearest reactive alkene units exceeds 4.2 Å. For SDP 11, both photostable (major) and photoactive (minor) crystals were obtained, enabling the observation of light-induced topochemical polymerization in single crystals (Fig. 4b and c). Alternatively, the photoactive crystalline phase could be produced on a larger scale by the rapid reprecipitation of SDPs from solvent mixtures. The resulting orange crystalline powder demonstrated facile polymerization upon irradiation at 470 nm and thermally induced depolymerization at 263 °C, displaying a rather low energy storage and release (13 J g−1) compared to STPs.
The mechanical activation of photostable crystals by grinding effectively converted the initial crystalline phase to an amorphous phase that is photoactive and polymerizable. Light irradiation on the amorphized SDP powder yielded the generation of an amorphous CB polymer, similar to the crystalline polymer in Fig. 4c but with a large disorder. However, such an amorphous polymer displayed incomplete depolymerization, due to the presence of both all-trans (1r,2r,3r,4r) and cis–trans (1R,2R,3S,4S) CB isomers within the polymer backbone, in contrast to the crystalline polymer solely composed of the cis–trans CB isomer. DFT calculations suggest that an all-trans CB structure is essentially uncleavable under thermal triggering conditions, due to the stronger C–C bonds in the CB unit, limiting the energy release from the CB polymer.
Oxazole/diazetidine [2 + 2] cycloaddition
Photoinduced [2 + 2] cycloaddition of imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) groups yields a diazetidine, a four-membered heterocyclic ring containing two nitrogen atoms.87–89 Such a reaction remains less explored compared to the analogous photocycloaddition of alkenes (CC) yielding a large number of cyclobutane-bearing products. In the 1980s, Paillous and co-workers evaluated the photoreactivity of 2-phenylbenzoxazole (PB) 15; they observed the UV (300 nm)-induced solution-state [2 + 2] cycloaddition of the imine units of PBs forming 1,3-diazetidine cycloadducts.90,91 The strained diazetidine rings displayed exothermicity upon acid-mediated cycloreversion. Later, the photoreactivity of PB derivatives, functionalized with F, Cl, Br, or I, was tested, revealing the favorable cycloaddition of fluorinated (16) and chlorinated (17) derivatives in solution or at the hexane–water biphasic interface.92,93 However, the energy release from these cycloadducts was not examined for MOST energy storage applications.
N) groups yields a diazetidine, a four-membered heterocyclic ring containing two nitrogen atoms.87–89 Such a reaction remains less explored compared to the analogous photocycloaddition of alkenes (CC) yielding a large number of cyclobutane-bearing products. In the 1980s, Paillous and co-workers evaluated the photoreactivity of 2-phenylbenzoxazole (PB) 15; they observed the UV (300 nm)-induced solution-state [2 + 2] cycloaddition of the imine units of PBs forming 1,3-diazetidine cycloadducts.90,91 The strained diazetidine rings displayed exothermicity upon acid-mediated cycloreversion. Later, the photoreactivity of PB derivatives, functionalized with F, Cl, Br, or I, was tested, revealing the favorable cycloaddition of fluorinated (16) and chlorinated (17) derivatives in solution or at the hexane–water biphasic interface.92,93 However, the energy release from these cycloadducts was not examined for MOST energy storage applications.
Inspired by the potential to store large energy in their strained diazetidine rings, we recently investigated the MOST energy storage capabilities of PB derivatives 15–18 (Fig. 5a).94 The crystal structures of PBs display parallel arrangements between neighboring molecules that are either head-to-head (17 and 18) or head-to-tail (16) stacked with desired distances between reaction CN units (Fig. 5b and c). Despite having a favorable crystalline arrangement, PBs 16 and 17 failed to undergo photocycloaddition in the solid state, similar to other PB derivatives, displaying prominent fluorescence.95,96 Therefore, the hexane–water biphasic condition was used instead to facilitate their photocycloaddition at the interface under UV irradiation for 72 hours.
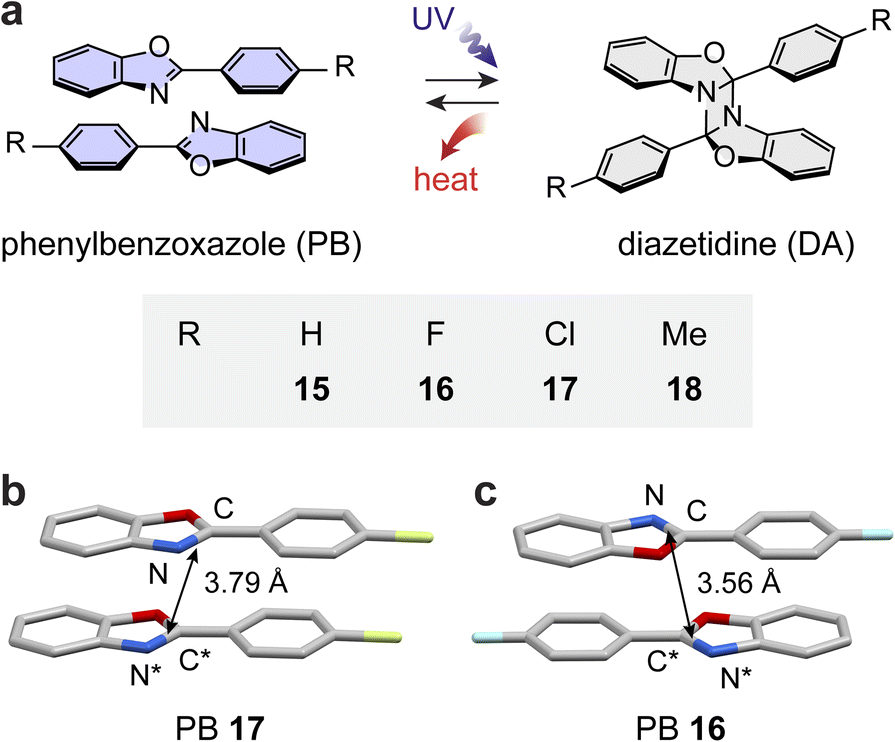 | ||
| Fig. 5 (a) Reversible photoinduced [2 + 2] cycloaddition and reversion between various phenylbenzoxazole (PB) and corresponding diazetidine (DA) derivatives. Proximal (b) head-to-head stacking of PB 17 and (c) head-to-tail PB 16 molecules in their crystals. In panels (b) and (c), the hydrogen atoms are omitted in the crystal structures for clarity. | ||
The thermal activation of all solid-state diazetidine (DA) derivatives successfully induced their complete cycloreversion at around 110 °C within 15 minutes. The energy release from DAs is remarkably large, ranging from 94 to 146 kJ mol−1 (224–318 J g−1), compared to all other solid-state MOST compounds reported to date. The exceptional energy storage capability of the PB/DA system is attributed to the photoinduced dearomatization of PBs and the formation of a highly strained four membered ring of DAs. However, most of the compounds displayed the unfavorable reabsorption of released heat due to the melting of regenerated PB, except for PB 17 that has a high melting point.
DAs can also be produced from the photocycloaddition of oxazolones, as reported by Wendlander and co-workers in 1984.97 The effective solid-state cycloaddition of oxazolones is promising for MOST energy storage, and the evaluation of their energy storage densities and release conditions is an ongoing effort, along with the optimization of molecular designs.
Anthracene/dianthracene [4 + 4] cycloaddition
The photoinduced [4 + 4] cycloaddition of anthracenes (A) has been reported to occur in solution, neat liquid, and solid state, creating new C–C bonds between the C9 and C10 positions of anthracenes.72–74,98–101 The dearomatized dianthracenes store energy and revert to anthracenes releasing energy in the range of 20–50 kJ mol−1 for most derivatives that were investigated.82,102–107 Exceptionally, in 1973, Donati and co-workers reported the solid-state cycloreversion of dianthracenes of 9-cyanoanthracene (19) and 9-cyano-10-acetoxyanthracene (22), releasing a substantial thermal energy of 74 and 82 kJ mol−1, respectively.108 Building upon this discovery, a comprehensive investigation on 9-cyanoanthracene derivatives 19–22 and their capability to store and release solar energy in the solid state has been recently performed (Fig. 6a).45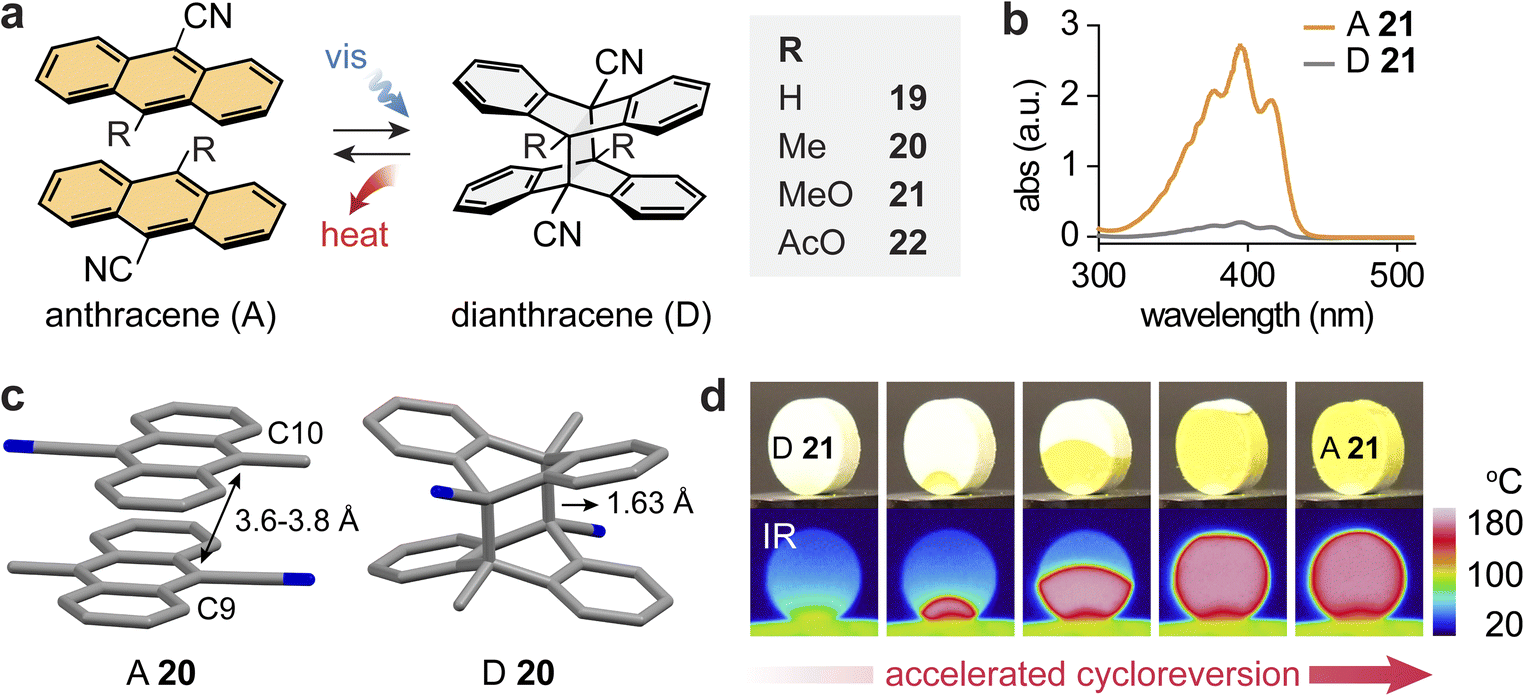 | ||
| Fig. 6 (a) Reversible photoinduced [4 + 4] cycloaddition and reversion between various anthracenes and corresponding dianthracenes. (b) UV-vis absorption spectra of A 21 (orange) and D 21 (gray). (c) Experimental crystal structure of A 20, showing the head-to-tail stacking of anthracenes and the simulated crystal structure of D 20, showing a shortened distance between reactive carbons. (d) Optical and IR images showing the color and temperature change of a pellet of D 21 during cycloreversion and heat release. In panel (c), the hydrogen atoms are omitted in the crystal structures for clarity. The figure was reproduced from ref. 45 with permission from Elsevier, copyright 2024. | ||
All anthracene derivatives exhibited a characteristic n–π* transition with an absorption band centred around 400 nm (Fig. 6b). Upon photoirradiation at 405 nm for 4–20 hours, they underwent solid-state cycloaddition to form respective dianthracenes, storing energy and turning colorless. Crystal structures show the favorable head-to-tail stacking between neighboring anthracenes that are separated by 3.5–4.2 Å between the reactive carbons (C9/C10), which enables a facile crystal-to-crystal transformation (Fig. 6c). Dianthracenes 20–22 underwent thermally induced cycloreversion to the corresponding anthracenes at reasonably low temperatures, exhibiting the onset of reversion at temperatures below 75 °C, while the onset of 19 was at 140 °C. However, because all anthracenes displayed high melting points in the range of 170–210 °C, the complete heat release from cycloreversion could be accurately measured: 82–102 kJ mol−1 or around 200 J g−1. Such large energy release is attributed to the rearomatization and stabilization of the resulting anthracenes relative to the dearomatized dianthracenes. The cyclability of the compounds was tested upon the repeated photodimerization and self-activated heat release processes, revealing no degradation as confirmed by NMR analysis.
Remarkably, the released energy (ΔGstorage) is comparable to or even exceeds the activation energy (ΔG‡) for thermal cycloreversion, which allows for the observation of self-activated energy release in the solid MOST compounds. Locally triggered energy release can induce further cycloreversion from neighboring areas of unreacted dianthracenes, resulting in a rapid cascade of reactions and heat propagation throughout the solid materials within 15–20 seconds (Fig. 6d). The use of an IR laser allows for short and localized thermal triggering as well as the estimation of the thermal activation efficiency; approximately 7.5% of energy input triggered the complete energy release from dianthracenes upon the self-activated cascade process. Such cycloreversion of dianthracenes is also facilitated by molecular cooperativity in the solid state, which lowers the activation barrier as the reaction progresses, displaying the characteristic kinetics of auto-catalysis.109
Recap
| MOST systems | Melting temperatures of monomers | Charging wavelength(s) | Half-lives of dimers | Cycloreversion temperatures | Energy storage densities |
|---|---|---|---|---|---|
| Styrylpyrylium/cyclobutane dimerization44 | 150–250 °C | 470 or 530 nm | 4 days to 32 years | 71–165 °C | 6–51 J g−1 |
| Styryldipyrylium/cyclobutane polymerization85 | — | 470 nm | — | 263 °C | 13 J g−1 |
| Benzoxazole/diazetidine dimerization94 | 101–149 °C | 300 nm | 135 days to 23 years | 60–71 °C | 224–318 J g−1 |
| Anthracene/dianthracene dimerization45 | 176–207 °C | 365 or 405 nm | 1 day to 190 years | 59–140 °C | 195–221 J g−1 |
Conclusions and outlook
The rational molecular designs and precise molecular arrangements in crystals have enabled quantitative topochemical cycloaddition reactions, which are critical for effective photon energy storage in solid-state MOST systems. The reversible and repeatable cycloaddition reactions, occurring in the absence of solvents, catalysts, or side reactions, allow for greener, safer, and more efficient MOST energy storage and release. Because of the solvent-free operation, the cycloaddition systems can be easily integrated into various materials including garments, windows, surfaces of solid-state consumer products, and so on for various self-charging and heating applications. The solid-state materials can be processed into diverse forms such as films, pellets, fibers, and powder, which is difficult to achieve for conventional MOST systems that operate in solutions. Most critically, the overall energy densities of cycloaddition MOST systems (i.e., gravimetric and volumetric energy densities) are substantially higher than those of solution-state systems, in which the large excess solvent contributes to diluting the energy densities of molecules in a large volume and mass of solution. These desirable features of solid-state cycloaddition chemistry drive further development of topochemical MOST systems, also supported by the rich literature in the fields of crystal engineering and topochemical reactions. Translating the design principles gained from traditional solid-state photochemical reactions to MOST applications will be an important step towards achieving practical and novel molecular solids that harness solar energy.However, this burgeoning topic of chemical research has many remaining challenges and unanswered questions. For example, how can we control the impact of lattice energy change upon cycloaddition to favorably contribute to the overall energy storage? The prominent intermolecular interactions in close-packed crystals will affect the relative stability of reactants and cycloadducts, in turn changing the energy storage densities. Are there ways to predict such an effect, for example, by the computation of molecular crystals? The emerging machine-learning strategies could possibly accelerate the discovery of novel materials.110
The mechanistic and kinetic analysis of solid-state reactions, both photochemical and thermal transformations, will allow for a deeper understanding of energy storage and release processes. The advantages and disadvantages of solid-state designs for achieving rapid photo-charging, long-term energy storage, and facile heat release will need to be further scrutinized by fundamental studies. The cooperativity of molecules for reactions in a given crystalline lattice will either accelerate or hinder the desired processes.
Lastly, new method development for catalyzing the energy release will be critical. Unlike solution-state heat release for which photochemical, chemical, and electrochemical triggering has been rigorously developed, solid-state systems present unique difficulties such as the increased light scattering and limited diffusion of triggers. Fortunately, some solid-state compounds, such as dianthracenes, have shown auto-catalyzed thermal cycloreversion, which efficiently releases the stored energy. Further studies on controlling the self-activated energy release process and discovering alternative triggers will be important for elevating and expanding the emerging topic.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
C. R. created the original draft and figures. H. P. Q. N. revised the figures and contributed to writing. G. G. D. H. edited the text and figures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the NSF CAREER award (DMR-2142887), the Air Force Office of Scientific Research YIP award (FA9550-22-1-0254), the Alfred P. Sloan Foundation (FG-2022-18328), and the Camille and Henry Dreyfus Foundation (TC-23-028).References
- T. J. Kucharski, Y. Tian, S. Akbulatov and R. Boulatov, Energy Environ. Sci., 2011, 4, 4449–4472 RSC.
- C. L. Sun, C. Wang and R. Boulatov, ChemPhotoChem, 2019, 3, 268–283 CrossRef CAS.
- Z. Wang, P. Erhart, T. Li, Z.-Y. Zhang, D. Sampedro, Z. Hu, H. A. Wegner, O. Brummel, J. Libuda, M. B. Nielsen and K. Moth-Poulsen, Joule, 2021, 5, 3116–3136 CrossRef CAS.
- A. Giménez-Gómez, L. Magson, C. Merino-Robledillo, S. Hernáez-Troya, N. Sanosa, D. Sampedro and I. Funes-Ardoiz, React. Chem. Eng., 2024, 9, 1629–1640 RSC.
- A. Gimenez-Gomez, L. Magson, B. Peñin, N. Sanosa, J. Soilán, R. Losantos and D. Sampedro, Photochem, 2022, 2, 694–716 CrossRef CAS.
- J. Usuba and G. G. D. Han, Trends Chem., 2023, 5, 577–580 CrossRef.
- Z. Wang, H. Hölzel and K. Moth-Poulsen, Chem. Soc. Rev., 2022, 51, 7313–7326 RSC.
- A. K. Jaiswal, P. Saha, J. Jiang, K. Suzuki, A. Jasny, B. M. Schmidt, S. Maeda, S. Hecht and C.-Y. D. Huang, J. Am. Chem. Soc., 2024, 146, 21367–21376 CrossRef PubMed.
- A. Lennartson, A. Roffey and K. Moth-Poulsen, Tetrahedron Lett., 2015, 56, 1457–1465 CrossRef CAS.
- R. Schulte, S. Afflerbach, T. Paululat and H. Ihmels, Angew. Chem., Int. Ed., 2023, 62, e202309544 CrossRef CAS PubMed.
- J. Orrego-Hernández, A. Dreos and K. Moth-Poulsen, Acc. Chem. Res., 2020, 53, 1478–1487 CrossRef PubMed.
- L. Dong, Y. Feng, L. Wang and W. Feng, Chem. Soc. Rev., 2018, 47, 7339–7368 RSC.
- M. A. Gerkman, R. S. L. Gibson, J. Calbo, Y. Shi, M. J. Fuchter and G. G. D. Han, J. Am. Chem. Soc., 2020, 142, 8688–8695 CrossRef PubMed.
- A. Kunz, A. H. Heindl, A. Dreos, Z. Wang, K. Moth-Poulsen, J. Becker and H. A. Wegner, ChemPlusChem, 2019, 84, 1145–1148 CrossRef CAS PubMed.
- S. Sun, S. Liang, W.-C. Xu, M. Wang, J. Gao, Q. Zhang and S. Wu, Soft Matter, 2022, 18, 8840–8849 RSC.
- Q. Qiu, S. Yang, M. A. Gerkman, H. Fu, I. Aprahamian and G. G. D. Han, J. Am. Chem. Soc., 2022, 144, 12627–12631 CrossRef CAS PubMed.
- Z. Wang, J. Udmark, K. Börjesson, R. Rodrigues, A. Roffey, M. Abrahamsson, M. B. Nielsen and K. Moth-Poulsen, ChemSusChem, 2017, 10, 3049–3055 CrossRef CAS PubMed.
- M. Brøndsted Nielsen, N. Ree, K. V. Mikkelsen and M. Cacciarini, Russ. Chem. Rev., 2020, 89, 573–586 CrossRef.
- Y. Kanai, V. Srinivasan, S. K. Meier, K. P. C. Vollhardt and J. C. Grossman, Angew. Chem., Int. Ed., 2010, 49, 8926–8929 CrossRef CAS PubMed.
- A. Lennartson, A. Lundin, K. Börjesson, V. Gray and K. Moth-Poulsen, Dalton Trans., 2016, 45, 8740–8744 RSC.
- M. Le and G. G. D. Han, Acc. Mater. Res., 2022, 3, 634–643 CrossRef CAS.
- X. Xu and G. Wang, Small, 2022, 18, 2107473 CrossRef CAS PubMed.
- Y. Yang, S. Huang, Y. Ma, J. Yi, Y. Jiang, X. Chang and Q. Li, ACS Appl. Mater. Interfaces, 2022, 14, 35623–35634 CrossRef CAS PubMed.
- S. Wu, T. Li, Z.-Y. Zhang, T. Li and R. Wang, Matter, 2021, 4, 3385–3399 CrossRef CAS.
- Z. Wang, H. Moïse, M. Cacciarini, M. B. Nielsen, M.-a. Morikawa, N. Kimizuka and K. Moth-Poulsen, Adv. Sci., 2021, 8, 2103060 CrossRef CAS PubMed.
- Q. Qiu, Y. Shi and G. G. D. Han, J. Mater. Chem. C, 2021, 9, 11444–11463 RSC.
- W.-C. Xu, S. Sun and S. Wu, Angew. Chem., Int. Ed., 2019, 58, 9712–9740 CrossRef CAS PubMed.
- A. Gonzalez, E. S. Kengmana, M. V. Fonseca and G. G. D. Han, Mater. Today Adv., 2020, 6, 100058 CrossRef.
- X. Li, S. Cho, J. Wan and G. G. D. Han, Chem, 2023, 9, 2378–2389 CAS.
- A. B. Grommet, L. M. Lee and R. Klajn, Acc. Chem. Res., 2020, 53, 2600–2610 CrossRef CAS PubMed.
- G. C. Thaggard, J. Haimerl, K. C. Park, J. Lim, R. A. Fischer, B. K. P. Maldeni Kankanamalage, B. J. Yarbrough, G. R. Wilson and N. B. Shustova, J. Am. Chem. Soc., 2022, 144, 23249–23263 CrossRef CAS PubMed.
- D. Zhitomirsky, E. Cho and J. C. Grossman, Adv. Energy Mater., 2016, 6, 1502006 CrossRef.
- X. Li, S. Cho and G. G. D. Han, ACS Mater. Au, 2023, 3, 37–42 CrossRef CAS PubMed.
- K. Griffiths, N. R. Halcovitch and J. M. Griffin, Chem. Mater., 2020, 32, 9925–9936 CrossRef CAS.
- K. Griffiths, N. R. Halcovitch and J. M. Griffin, Inorg. Chem., 2021, 60, 12950–12960 CrossRef CAS.
- K. Griffiths, N. R. Halcovitch and J. M. Griffin, Chem. Sci., 2022, 13, 3014–3019 RSC.
- D. Gupta, A. K. Gaur, H. Kumar, S. Singh and S. Venkataramani, ChemPhotoChem, 2023, 7, e202300068 CrossRef CAS.
- O. S. Bushuyev and C. J. Barrett, in Photomechanical Materials, Composites, and Systems, ed. T. J. White, John Wiley & Sons, UK, 2017, pp. 37–77 Search PubMed.
- B. Zhang, Y. Feng and W. Feng, Nano-Micro Lett., 2022, 14, 138 CrossRef CAS PubMed.
- X. Xu, J. Feng, W.-Y. Li, G. Wang, W. Feng and H. Yu, Prog. Polym. Sci., 2024, 149, 101782 CrossRef CAS.
- S. Wu and H.-J. Butt, Macromol. Rapid Commun., 2020, 41, 1900413 CrossRef CAS PubMed.
- W. Luo, Y. Feng, C. Cao, M. Li, E. Liu, S. Li, C. Qin, W. Hu and W. Feng, J. Mater. Chem. A, 2015, 3, 11787–11795 RSC.
- Y. Feng, H. Liu, W. Luo, E. Liu, N. Zhao, K. Yoshino and W. Feng, Sci. Rep., 2013, 3, 3260 CrossRef PubMed.
- S. Cho, J. Usuba, S. Chakraborty, X. Li and G. G. D. Han, Chem, 2023, 9, 3159–3171 CAS.
- S. Chakraborty, H. P. Q. Nguyen, J. Usuba, J. Y. Choi, Z. Sun, C. Raju, G. Sigelmann, Q. Qiu, S. Cho, S. M. Tenney, K. E. Shulenberger, K. Schmidt-Rohr, J. Park and G. G. D. Han, Chem, 2024, 10, 1–14 Search PubMed.
- X. Wang, B. Xu and W. Tian, Acc. Mater. Res., 2023, 4, 311–322 CrossRef CAS.
- F. Sun and D. Wang, J. Mater. Chem. C, 2022, 10, 13700–13716 RSC.
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, 1905966 CrossRef CAS PubMed.
- S. Kobatake and T. Nakahama, in Advances in Organic Crystal Chemistry, eds. M. Sakamoto and H. Uekusa, Springer Singapore, Singapore, 2020, pp. 299–323 Search PubMed.
- J. W. Lauher, F. W. Fowler and N. S. Goroff, Acc. Chem. Res., 2008, 41, 1215–1229 CrossRef CAS PubMed.
- K. Biradha and R. Santra, Chem. Soc. Rev., 2013, 42, 950–967 RSC.
- K. Hema, A. Ravi, C. Raju, J. R. Pathan, R. Rai and K. M. Sureshan, Chem. Soc. Rev., 2021, 50, 4062–4099 RSC.
- R. Medishetty, I.-H. Park, S. S. Lee and J. J. Vittal, Chem. Commun., 2016, 52, 3989–4001 RSC.
- M. N. Tahir, A. Nyayachavadi, J.-F. Morin and S. Rondeau-Gagné, Polym. Chem., 2018, 9, 3019–3028 RSC.
- K. Hema and K. M. Sureshan, Acc. Chem. Res., 2019, 52, 3149–3163 CrossRef CAS PubMed.
- J.-G. Yu, M.-M. Gan, S. Bai and Y.-F. Han, CrystEngComm, 2019, 21, 4673–4683 RSC.
- M. D. Cohen, G. M. J. Schmidt and F. I. Sonntag, J. Chem. Soc., 1964, 2000–2013 RSC.
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CrossRef CAS.
- V. Ramamurthy and J. Sivaguru, Chem. Rev., 2016, 116, 9914–9993 CrossRef CAS PubMed.
- T. Friščić and L. R. MacGillivray, Z. Kristallogr. Cryst. Mater., 2005, 220, 351–363 CrossRef.
- L. R. MacGillivray, G. S. Papaefstathiou, T. Friščić, T. D. Hamilton, D.-K. Bučar, Q. Chu, D. B. Varshney and I. G. Georgiev, Acc. Chem. Res., 2008, 41, 280–291 CrossRef CAS PubMed.
- K. M. Hutchins, R. Soc. Open Sci., 2018, 5, 180564 CrossRef PubMed.
- G. Campillo-Alvarado, C. Li and L. R. MacGillivray, in Reactivity in Confined Spaces, eds. G. Lloyd and R. S. Forgan, The Royal Society of Chemistry, 2021, pp. 322-339 Search PubMed.
- D. S. Reddy, D. C. Craig and G. R. Desiraju, J. Am. Chem. Soc., 1996, 118, 4090–4093 CrossRef CAS.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS.
- G. Resnati, E. Boldyreva, P. Bombicz and M. Kawano, IUCrJ, 2015, 2, 675–690 CrossRef CAS PubMed.
- M. K. Corpinot and D.-K. Bučar, Cryst. Growth Des., 2019, 19, 1426–1453 CrossRef CAS.
- C. A. Gunawardana and C. B. Aakeröy, Chem. Commun., 2018, 54, 14047–14060 RSC.
- E. Schaumann and R. Ketcham, Angew Chem. Int. Ed. Engl., 1982, 21, 225–247 CrossRef.
- A. Chanthapally, H. Yang, H. S. Quah, R. D. Webster, M. K. Schreyer, M. W. Wong and J. J. Vittal, Chem. - Eur. J., 2014, 20, 15702–15708 CrossRef CAS PubMed.
- H. Hopf, H. Greiving, P. G. Jones and P. Bubenitschek, Angew Chem. Int. Ed. Engl., 1995, 34, 685–687 CrossRef CAS.
- P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774–778 CrossRef CAS PubMed.
- M. J. Kory, M. Wörle, T. Weber, P. Payamyar, S. W. van de Poll, J. Dshemuchadse, N. Trapp and A. D. Schlüter, Nat. Chem., 2014, 6, 779–784 CrossRef CAS PubMed.
- G. C. George III and K. M. Hutchins, Chem. - Eur. J., 2023, 29, e202302482 CrossRef PubMed.
- R. Medishetty, Z. Bai, H. Yang, M. W. Wong and J. J. Vittal, Cryst. Growth Des., 2015, 15, 4055–4061 CrossRef CAS.
- T. Jadhav, Y. Fang, C.-H. Liu, A. Dadvand, E. Hamzehpoor, W. Patterson, A. Jonderian, R. S. Stein and D. F. Perepichka, J. Am. Chem. Soc., 2020, 142, 8862–8870 CrossRef CAS PubMed.
- G. K. Kole, T. Kojima, M. Kawano and J. J. Vittal, Angew. Chem., Int. Ed., 2014, 53, 2143–2146 CrossRef CAS PubMed.
- F.-L. Hu, H.-F. Wang, D. Guo, H. Zhang, J.-P. Lang and J. E. Beves, Chem. Commun., 2016, 52, 7990–7993 RSC.
- B. B. Rath and J. J. Vittal, Acc. Chem. Res., 2022, 55, 1445–1455 CrossRef CAS PubMed.
- K. Novak, V. Enkelmann, G. Wegner and K. B. Wagener, Angew Chem. Int. Ed. Engl., 1993, 32, 1614–1616 CrossRef.
- J. W. Chung, Y. You, H. S. Huh, B.-K. An, S.-J. Yoon, S. H. Kim, S. W. Lee and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 8163–8172 CrossRef CAS PubMed.
- T. Nishiuchi, K. Kisaka and T. Kubo, Angew. Chem., Int. Ed., 2021, 60, 5400–5406 CrossRef CAS PubMed.
- K. Hesse and S. Hünig, Liebigs Ann. Chem., 2006, 1985, 715–739 CrossRef.
- K. Novak, V. Enkelmann, G. Wegner and K. B. Wagener, Angew Chem. Int. Ed. Engl., 2003, 32, 1614–1616 CrossRef.
- J. Usuba, Z. Sun, H. P. Q. Nguyen, C. Raju, K. Schmidt-Rohr and G. G. D. Han, Commun. Mater., 2024, 5, 98 CrossRef CAS.
- V. Buchholz and V. Enkelmann, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 2001, 356, 315–325 CrossRef CAS.
- N. Kaur, Elsevier, in 4-Membered Heterocycle Synthesis, ed. N. Kaur, Elsevier, 2023, pp. 1–42 Search PubMed.
- M. D'Auria, A. Guarnaccio, R. Racioppi, S. Stoia and L. Emanuele, in Photochemistry of Heterocycles, eds. M. D'Auria, A. Guarnaccio, R. Racioppi, S. Stoia and L. Emanuele, Elsevier, 2023, pp. 219–296 Search PubMed.
- R. O. Kan and R. L. Furey, J. Am. Chem. Soc., 1968, 90, 1666–1667 CrossRef CAS.
- J. Roussilhe, B. Despax, A. Lopez and N. Paillous, J. Chem. Soc. Chem. Commun., 1982, 380–381 RSC.
- J. Roussilhe, E. Fargin, A. Lopez, B. Despax and N. Paillous, J. Org. Chem., 1983, 48, 3736–3741 CrossRef CAS.
- S. Fery-Forgues and N. Paillous, J. Org. Chem., 1986, 51, 672–677 CrossRef CAS.
- N. Paillous, S. F. Forgues, J. Jaud and J. Devillers, J. Chem. Soc. Chem. Commun., 1987, 578–579 RSC.
- H. P. Q. Nguyen, A. Mukherjee, J. Usuba, J. Wan, and G. G. D. Han, Under Review.
- C. Carayon and S. Fery-Forgues, Photochem. Photobiol. Sci., 2017, 16, 1020–1035 CrossRef CAS.
- A. Ghodbane, N. Saffon, S. Blanc and S. Fery-Forgues, Dyes Pigm., 2015, 113, 219–226 CrossRef CAS.
- D. Lawrenz, S. Mohr and B. Wendländer, J. Chem. Soc. Chem. Commun., 1984, 863–865 RSC.
- L. Zhu, R. O. Al-Kaysi and C. J. Bardeen, J. Am. Chem. Soc., 2011, 133, 12569–12575 CrossRef CAS PubMed.
- C. Sun, J. J. Oppenheim, G. Skorupskii, L. Yang and M. Dincă, Chem, 2022, 8, 3215–3224 CAS.
- H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2001, 30, 248–263 RSC.
- S. Kataoka, D. Kitagawa, H. Sotome, S. Ito, H. Miyasaka, C. J. Bardeen and S. Kobatake, Chem. Sci., 2024, 15, 13421–13428 RSC.
- J. Brancart, J. Van Damme, F. Du Prez and G. Van Assche, Phys. Chem. Chem. Phys., 2021, 23, 2252–2263 RSC.
- G. Ganguly, M. Sultana and A. Paul, J. Phys. Chem. Lett., 2018, 9, 328–334 CrossRef CAS PubMed.
- W. R. Bergmark, G. Jones, II, T. E. Reinhardt and A. M. Halpern, J. Am. Chem. Soc., 1978, 100, 6665–6673 CrossRef CAS.
- S. Grimme, S. D. Peyerimhoff, H. Bouas-Laurent, J.-P. Desvergne, H.-D. Becker, S. M. Sarge and H. Dreeskamp, Phys. Chem. Chem. Phys., 1999, 1, 2457–2462 RSC.
- D. Donati, G. G. T. Guarini and P. Sarti-Fantoni, J. Therm. Anal., 1991, 37, 1917–1922 CrossRef CAS.
- T. Nishiuchi, S.-y. Uno, Y. Hirao and T. Kubo, J. Org. Chem., 2016, 81, 2106–2112 CrossRef CAS PubMed.
- D. Donati, G. Guarini and P. Sarti-fantoni, Mol. Cryst. Liq. Cryst., 1972, 17, 187–195 CrossRef CAS.
- C. Raju, Z. Sun, R. Koibuchi, J. Y. Choi, S. Chakraborty, J. Park, H. Houjou, K. Schmidt-Rohr and G. G. D. Han, J. Mater. Chem. A, 2024 10.1039/D4TA05282J.
- K. Wang, H. Yu, J. Gao, Y. Feng and W. Feng, J. Mater. Chem. C, 2024, 12, 3811–3837 RSC.
| This journal is © The Royal Society of Chemistry 2024 |