DOI:
10.1039/D4SC05740F
(Edge Article)
Chem. Sci., 2024,
15, 18387-18394
Uncovering diverse reactivity of NHCs with diazoalkane: C–H activation, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) C bond formation, and access to N-heterocyclic methylenehydrazine†
C bond formation, and access to N-heterocyclic methylenehydrazine†
Received
26th August 2024
, Accepted 5th October 2024
First published on 8th October 2024
Abstract
N-heterocyclic carbenes (NHCs) have attracted significant attention due to their strong σ-donating capabilities, as well as their transition-metal-like reactivity towards small molecules. However, their interaction with diazoalkanes remains understudied. In this manuscript, we explore the reactivity of a series of stable carbenes, encompassing a wide range of electronic properties, with Me3SiCHN2. 5-SIPr activates the C–H bond of Me3SiCHN2, resulting in the formation of a novel diazo derivative (1), while carbenes such as 5-IPr, 6-SIPr, and diamido carbene yield N-heterocyclic methylenehydrazine derivatives (3, 4, and 8). The reaction of Me3SiCHN2 with 5-ItBu unexpectedly leads to the formation of a triazole ring linked with the imidazole moiety via a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond (6) alongside the azine product (7). Substituting the diazoalkane with diazoester consistently yields azine derivatives (9–12 and 14). Only in the case of 5-ItBu, an imidazolium salt with tetrazenide anion (13) was obtained as a side product. The reaction of 4 with HCl resulted in the desilylprotonation to form a salt, 5a, which undergoes deprotonation upon using bases such as Et3N and KHMDS to form N-heterocyclic methylene hydrazine, 5. Theoretical calculations have been conducted to elucidate the diverse mechanisms underlying product formation.
C double bond (6) alongside the azine product (7). Substituting the diazoalkane with diazoester consistently yields azine derivatives (9–12 and 14). Only in the case of 5-ItBu, an imidazolium salt with tetrazenide anion (13) was obtained as a side product. The reaction of 4 with HCl resulted in the desilylprotonation to form a salt, 5a, which undergoes deprotonation upon using bases such as Et3N and KHMDS to form N-heterocyclic methylene hydrazine, 5. Theoretical calculations have been conducted to elucidate the diverse mechanisms underlying product formation.
Introduction
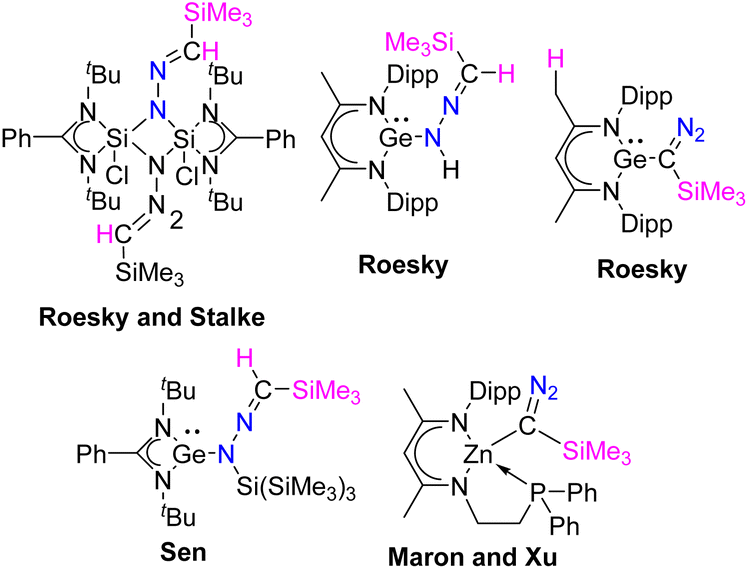
Diazoalkanes have been recognized for over a century and have demonstrated their versatility as significant building blocks in organic synthesis. Over the past two decades, there has been a substantial increase in research focused on the reactions between diazoalkanes and transition metal complexes. By examining these structures, it is conceivable that diazoalkanes engage in reactions with transition metal complexes, by serving as 1,3-dipoles, N-nucleophiles, or C-nucleophiles.1 In contrast, to this well-explored chemistry, the reactions of diazoalkanes with compounds having low-valent main-group elements are poorly studied (Scheme 1). Roesky and coworkers reported the reaction of silicon(II) species, LSiCl [L = PhC(NtBu)2], with Me3SiCHN2.2 The same group documented the reactions between β-diketiminate germylene hydride, L1GeH [L1 = HC{(CMe)(NAr)}2, Ar = 2,6-iPr2C6H3] and Me3SiCHN2, which resulted in the end-on insertion of the diazoalkane moiety into the germanium hydride.3 Our group made a similar observation when we studied a reaction involving PhC(NtBu)2GeSi(SiMe3)3 and Me3SiCHN2, leading to the insertion of the diazoalkane motif into the Ge–Si bond.4 Very recently, the groups of Maron and Xu reported the synthesis of the first example of a well-defined zinc α-diazoalkyl complex by reacting a zinc hydride with Me3SiCHN2.5
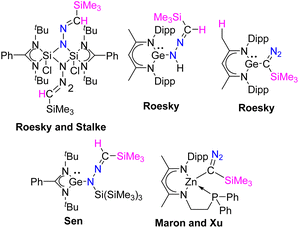 |
| | Scheme 1 Examples of the reactivity of diazoalkane with main-group species. | |
It is explicit from the aforementioned discussion that while the reactions of diazoalkane with Si(II), Ge(II) are reported, there is only one paper reported describing the interaction of diazoalkane with stable N-heterocyclic carbene (NHC) resulting in azine formation.6 In addition, there is another report on the reaction of 2,2,2-trifluorodiazoethane with the in situ generated N-heterocyclic carbenes leading to azine formation.7 As a consequence, systematic investigation of the reaction of diazoalkane with NHCs is important. Here, we report the reactions of Me3SiCHN2 with a diverse array of NHCs, which resulted in divergent reactivities: (a) 5-IPr, 6-SIPr, and diamido carbene led to N-heterocyclic methylenehydrazine derivatives; (b) 5-SIPr inserts into the C–H bond of Me3SiCHN2 reminiscent to Xu's dehydrocoupling reaction of Me3SiCHN2 with zinc hydride; (c) 5-ItBu led to a 1,2,4-triazole ring formation connected to the imidazole unit with a CC bond; (d) the reaction of 5-IPr and 5-ItBu with Me3SiCHN2 and water led to the formation of novel N-heterocyclic hydrazines, which were structurally unknown so far.
Results and discussion
Case I: reaction with 5-SIPr
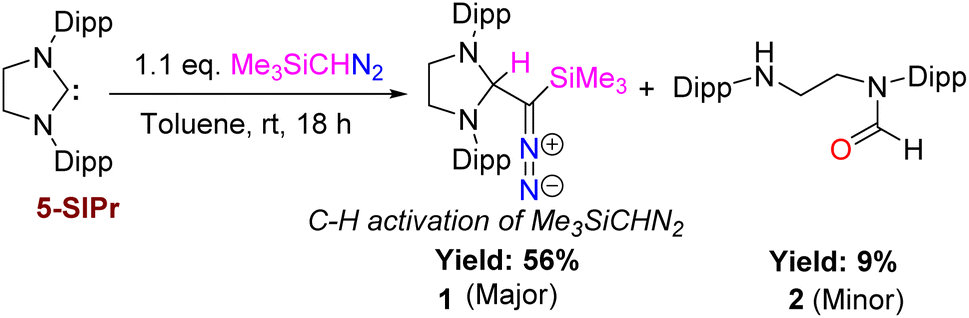
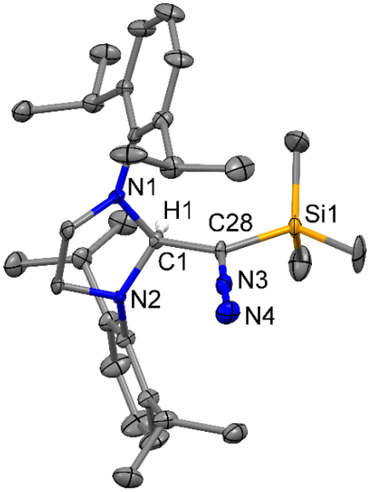
Because of the report on the reactivity of 5-IMes with diazoalkanes,6 we have initiated our investigation with the more nucleophilic and saturated 5-SIPr to determine whether the reaction proceeds similarly or diverges in reactivity. Interestingly, treatment of 5-SIPr with Me3SiCHN2 activated the C–H bond of diazoalkane yielding diazoalkyl compound 1, accompanied by the formation of compound 2via hydrolysis due to the presence of adventitious water (Scheme 2).8 NHCs have shown a remarkable ability to mimic transition metals in activating robust C–H bonds.9 Following the seminal work on the C–H activation using 5-SIMes by Arduengo's group in 1999,10 the groups of Whittlesey,11 Bielawski,12 Turner13 and Bertrand14 explored various NHCs, including ring-expanded, diamido, and cyclic alkyl amino carbenes for C–H bond activation. However, the insertion of NHC into the C–H bond of diazoalkane is unprecedented, as the C–H bond of diazoalkanes is not highly acidic. To the best of our knowledge, there is only one report published by Roesky and coworkers, where the C–H activation of diazoalkane takes place at the exocyclic double bond of nacnac N-heterocyclic germylene (Scheme 1).15 Crystallization of the crude reaction mixture in n-hexane at room temperature yielded block-shaped yellow crystals. The 1H NMR spectrum shows a peak at −0.38 ppm for the SiMe3 groups, and the corresponding peak appears at −2.1 ppm in the 29Si NMR spectrum. The molecular structure of compound 1 reveals a linear geometry of C28–N3–N4 (178.8(3)°) (Fig. 1).
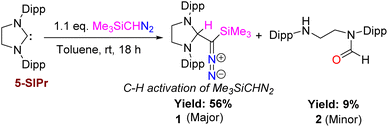 |
| | Scheme 2 Insertion of NHC into the C–H bond of diazoalkane in the reaction of 5-SIPr with Me3SiCHN2. | |
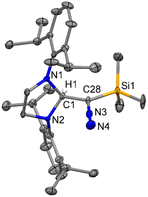 |
| | Fig. 1 The molecular structure of 1 with anisotropic displacement parameters are depicted at the 50% probability level respectively. The hydrogen atoms are not shown for clarity. Selected distances (Å) and angles (deg): C1–C28 1.530(4), N3–N4 1.143(4), N3–C28 1.307(4), Si1–C28 1.861(3), N1–C1–N2 102.4(2), N4–N3–C28 178.8(3), N3–C28–C1 117.4(3), N3–C28–Si1 117.8(2). | |
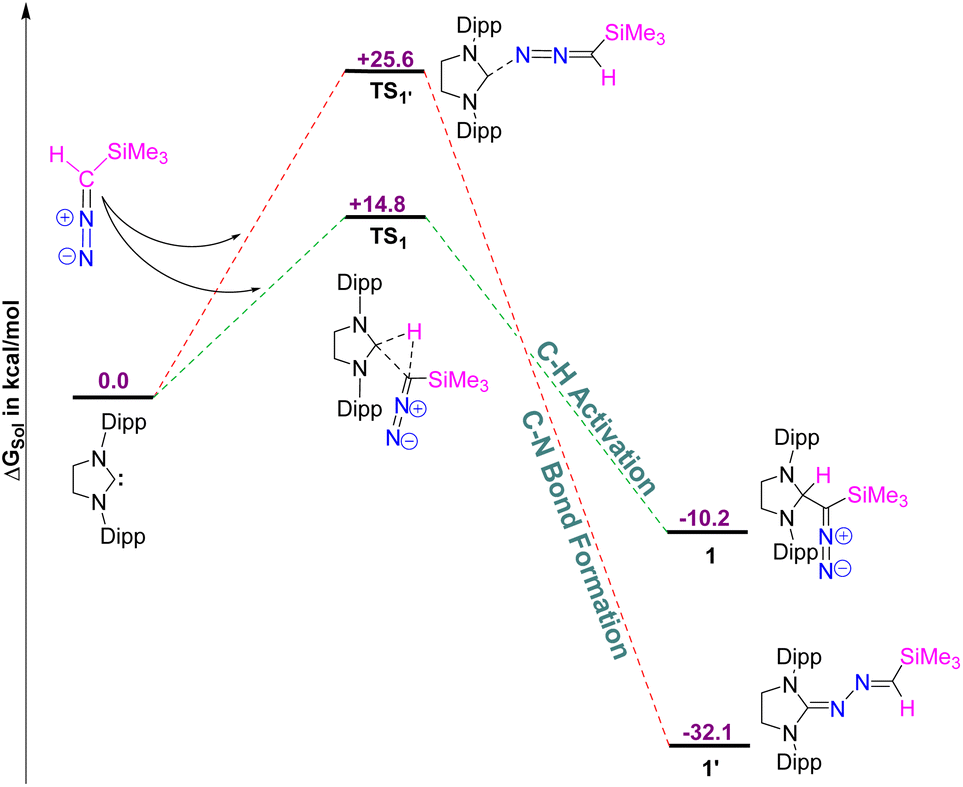
Using density functional theory (DFT) calculations at the PBE0-D3/def2TZVP//PBE-D3/defTZVP level of theory, incorporating the COSMO solvation model for toluene (∈ = 2.38), we systematically investigated the formation of compound 1 (see the ESI† for detailed computational methodology). Additionally, we explored the reasons why the putative azine product (1′) was not obtained (Fig. 2).
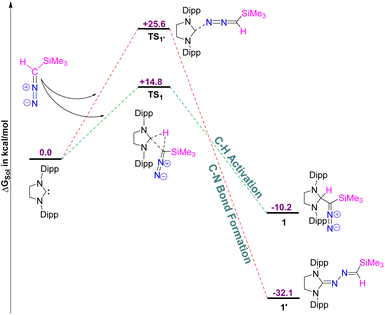 |
| | Fig. 2 The free energy profile for the formation of 1 and 1′. The energy values are in kcal mol−1. Level of theory: PBE0-D3/def2TZVP//PBE-D3/defTZVP with solvent toluene (∈ = 2.38). | |
The DFT results reveal that the formation of 1 is exergonic, with a free energy (ΔGsol) release of 10.2 kcal mol−1, attributed to C–H bond activation of the diazoalkane. Conversely, the formation of 1′ is significantly more thermodynamically favorable, releasing 32.1 kcal mol−1 of free energy, indicating that 1′ is 21.9 kcal mol−1 more stable than 1. However, kinetic analysis shows that the free energy barrier (ΔG‡sol) for the formation of 1′ is 10.8 kcal mol−1 higher than that for 1. Specifically, the free energy barrier for 1 is calculated to be 14.8 kcal mol−1, whereas for 1′, it is 25.6 kcal mol−1. Therefore, despite the greater thermodynamic stability of 1′ the reaction favors the formation of 1, as it proceeds via a lower free energy pathway, making 1 the kinetically preferred product.
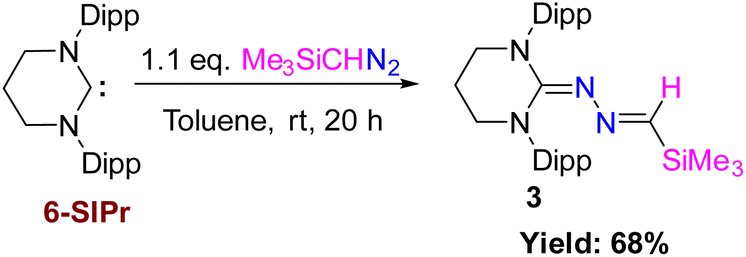
Case II: reaction with 6-SIPr
Very recently, we employed a six-membered saturated NHC, 6-SIPr, to isolate parent oxoborane,16 cationic bori-17 and boronic acid,18 and investigated their ring-expansion chemistry in the presence of borane19 and alane.20 The unexpected results obtained from the reaction involving 5-SIPr prompted us to investigate the reaction with 6-SIPr. In contrast to the previously mentioned results, the reaction of 6-SIPr with Me3SiCHN2 in toluene for 20 hours yielded a yellow-colored solution. Subsequent workup and characterization confirmed the formation of an unprecedented trimethylsilyl-substituted N-heterocyclic methylene hydrazine (3) (Scheme 3). The characteristic peak of Me3SiCHN2 appeared as a singlet at 7.40 ppm for C–H and −0.21 ppm for Me3Si group in the 1H NMR spectrum, and a resonance at −11.2 ppm in the 29Si NMR spectrum confirmed the presence of the trimethylsilyl group. Compound 3 crystallized in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, and its molecular structure is depicted in Fig. 3. The CN and N–N bond lengths in 3 are 1.308(3) and 1.389(3) Å, respectively, consistent with the bond lengths observed in azines.
space group, and its molecular structure is depicted in Fig. 3. The CN and N–N bond lengths in 3 are 1.308(3) and 1.389(3) Å, respectively, consistent with the bond lengths observed in azines.
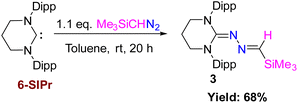 |
| | Scheme 3 Reaction of 6-SIPr with Me3SiCHN2 and the formation of trimethylsilyl protected N-heterocyclic methylene hydrazine (3). | |
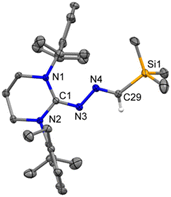 |
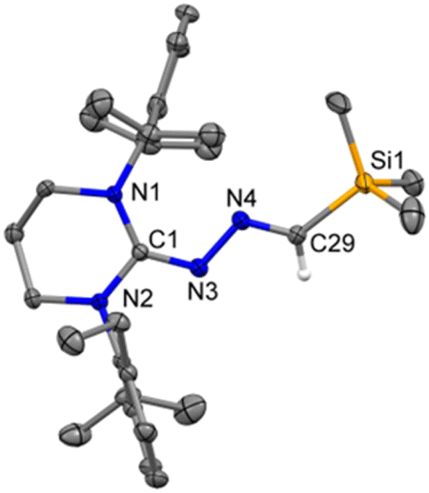
| | Fig. 3 The molecular structure of 3 with anisotropic displacement parameters are depicted at the 50% probability level respectively. Hydrogen atoms and vibrational atoms are not shown for clarity. Selected distances (Å) and angles (deg): N3–C1 1.308(3), N3–N4 1.389(3), N4–C29 1.288(4), Si1–C29 1.861(3), N1–C1–N2 119.2(2), C1–N3–N4 115.7(2), C29–N4–N3 112.8(2), N4–C29–Si1 122.8(2). | |
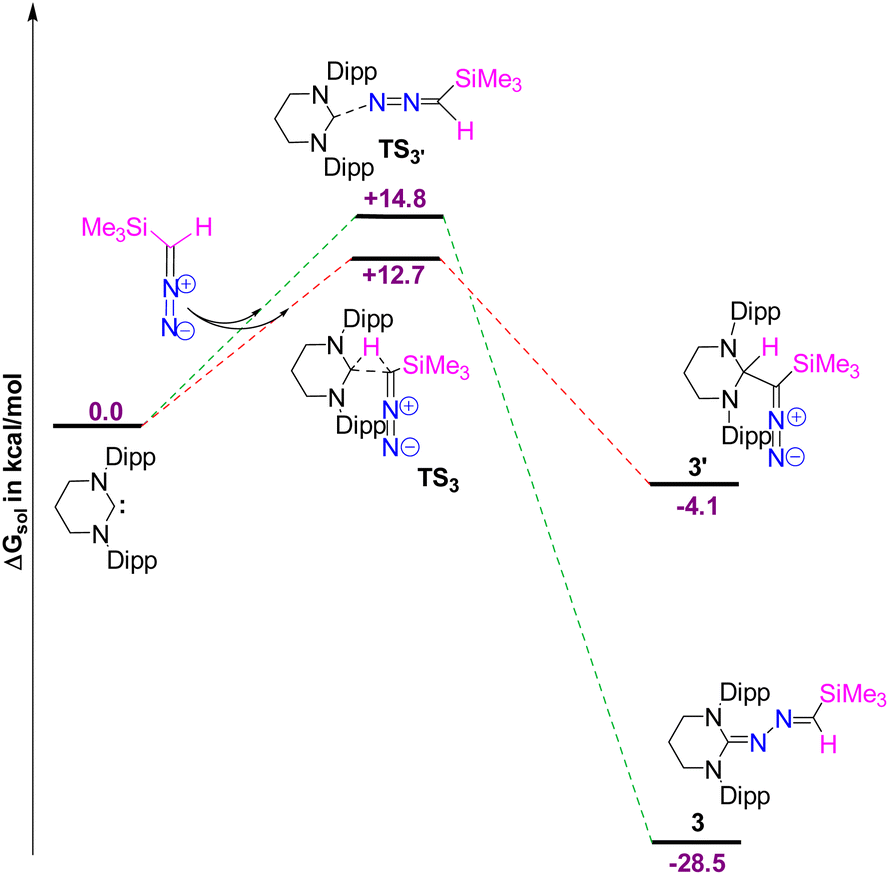
The DFT calculations reveal that the formation of 3 is thermodynamically favored over 3′ by 24.4 kcal mol−1 (Fig. 4). Although the free energy barrier for forming 3′ is 12.7 kcal mol−1, only 2.1 kcal mol−1 lower than that of 3, the weakly exergonic C–H bond activation in 3′ (releasing 4.1 kcal mol−1 of free energy) may allow the system to overcome the backward barrier of 16.8 kcal mol−1, potentially reverting to the initial reactants at room temperature. In contrast, the formation of 3 is significantly more exergonic, releasing 28.5 kcal mol−1 of free energy (ΔGsol). Once formed, 3 is unlikely to revert to the initial reactants due to the high backward energy barrier of 43.3 kcal mol−1, which is not surmountable at room temperature. Therefore, 3 is identified as the thermodynamically controlled product.
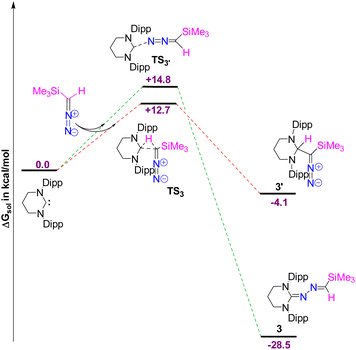 |
| | Fig. 4 The free energy profile for the formation of 3 and 3′. The energy values are in kcal mol−1. Level of theory: PBE0-D3/def2TZVP//PBE-D3/defTZVP with solvent toluene (∈ = 2.38). | |
Case III: reaction with 5-IPr
With these two divergent reactivities in hand, we wondered how Me3SiCHN2 would react with unsaturated carbenes. The C–H activation was not expected because Bertrand's group noted no reaction of terminal alkyne with 5-IPr.14 The reaction of 5-IPr with Me3SiCHN2 for 24 h in toluene at room temperature resulted in a deep red-colored solution. The NMR spectrum of the crude mixture reveals the formation of a product (4) (Scheme 4), akin to 3. The 1H NMR shows a peak at −0.18 ppm for the Me3Si protons. The corresponding resonance in the 29Si NMR appears at −10.9 ppm. Attempts to hydrolyze the Me3Si group in compound 4 using methanol and water were unsuccessful. However, treatment with HCl led to the unexpected formation of 5avia desilylprotonation. Our investigation revealed that the reaction consistently yields product 5a regardless of the equivalence of HCl used (0.5–1.1 equiv.) with a maximum yield of 91% obtained upon using 1.1 equivalents of HCl in toluene. Interestingly, the dehydrohalogenation of 5a using bases such as Et3N and KHMDS yielded 5 (Scheme 4). Likewise, the treatment of 5 with HCl gives back 5a. We have also noted that the reaction of 5-IPr with Me3SiCHN2 in the presence of a stoichiometric amount of water straightforwardly afforded compound 5 exclusively, presumably via in situ generation of CH2N2. Attempts to prepare analogous compound with 6-SIPr led to an intractable mixture of products.
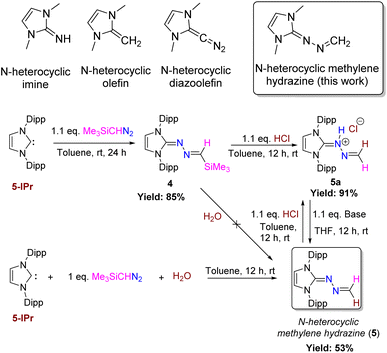 |
| | Scheme 4 Reaction of 5-IPr with Me3SiCHN2 (base = Et3N, KHMDS). The yield of 5a was determined from 4. The yield of compound 5 was determined from 5a. | |
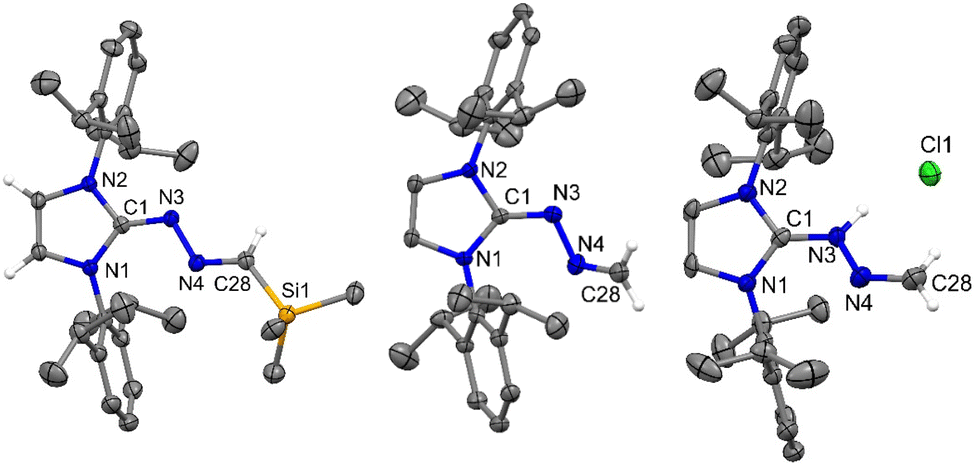
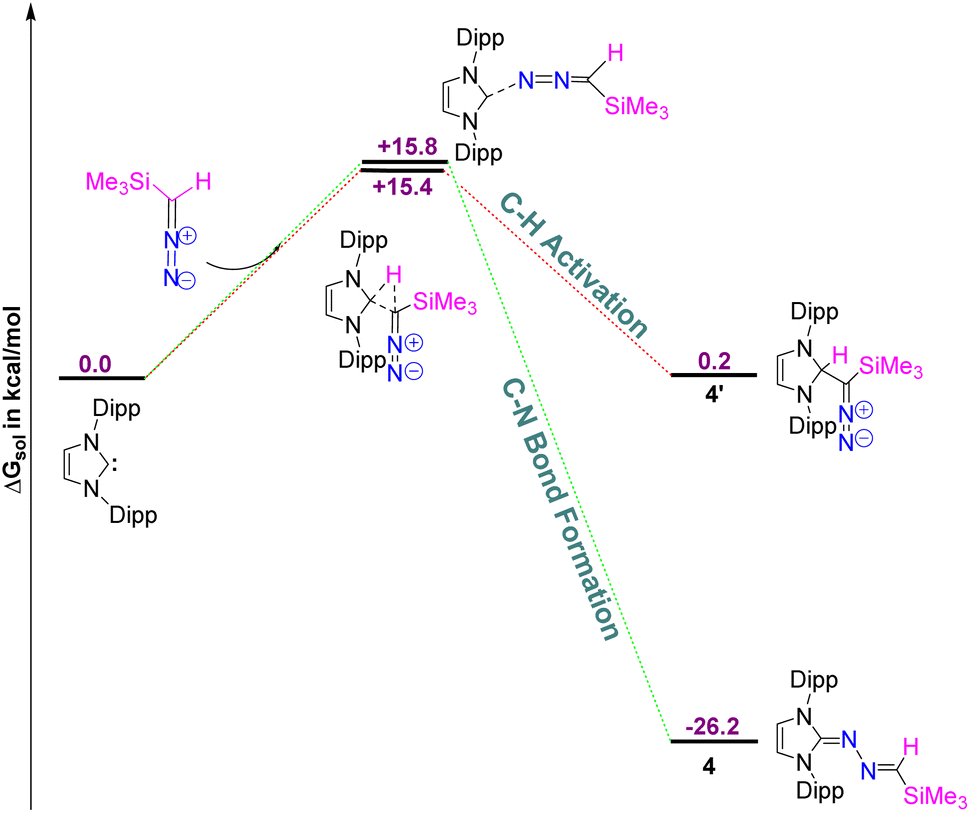
While there are a huge number of azines reported in the literature,21 an azine with a terminal methylene group (5) is unprecedented, and a new addition to the tapestry of NHC based organic functionalities such as N-heterocyclic imine,22 N-heterocyclic olefin,23 N-heterocyclic diazoalkene24etc. The molecular structures of 4, 5, and 5a are shown in Fig. 5. Similar to 6-SIPr, the formation of 4 is favored over the alternative C–H activation product, 4′, with a free energy (ΔGsol) of −26.2 kcal mol−1 and a free energy barrier (ΔG‡sol) of +15.8 kcal mol−1, making it the thermodynamically controlled product (Fig. 6).
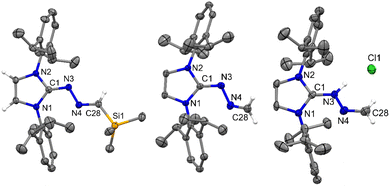 |
| | Fig. 5 The molecular structures of 4 (left), 5 (middle), and 5a (right) with anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms (except on terminal C) are not shown for clarity. Selected distances (Å) and angles (deg) for 4 (Vibrational atoms are omitted for clarity): N3–C1 1.309(2), N3–N4 1.394(2), N4–C28 1.286(2), Si1–C28 1.871(2), N2–C1–N1 109.85(14), C1–N3–N4 112.62(15), C28–N4–N3 114.07(15), N4–C28–Si1 119.82(14). 5: N3–C1 1.308(3), N3–N4 1.397(3), N4–C28 1.275(3), N1–C1–N2 105.61(17), C1–N3–N4 112.95(17), C28–N4–N3 112.9(2); 5a (H2O molecule is not shown for clarity): N3–C1 1.334(4), N3–N4 1.375(3), N4–C28 1.271(4), N2–C1–N1 107.7(3), C1–N3–N4 116.0(3), C28–N4–N3 116.1(3). | |
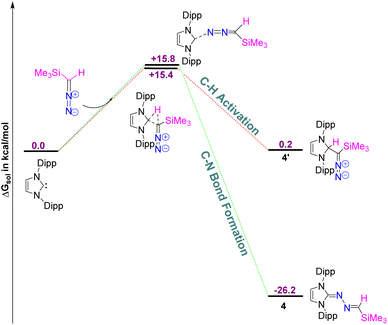 |
| | Fig. 6 The free energy profile for the formation of 4 and 4′. The energy values are in kcal mol−1. Level of theory: PBE0-D3/def2TZVP//PBE-D3/defTZVP with solvent toluene (∈ = 2.38). | |
To gain insight into the composition of the reaction mixture, we performed the reaction at a low temperature for 1.5 hours to trap the kinetically controlled product. However, the 1H NMR spectrum primarily displayed resonances corresponding to the starting material, with a slight formation of compound 4. Furthermore, we monitored the reaction progress by recording 1H NMR spectra at various time intervals, which confirmed the formation of a single product (ESI, Fig. MS1–MS3).†
Case IV: reaction with 5-ItBu
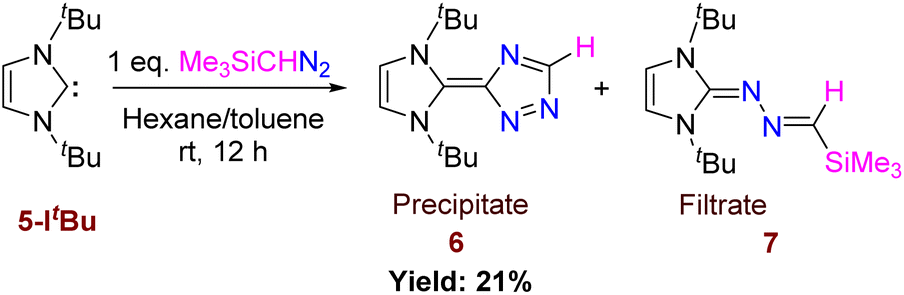
Differing reactivities of Me3SiCHN2 with different nucleophilic carbenes prompted us to perform the reaction with the more basic ItBu.25 The reactivity of 5-ItBu towards Me3SiCHN2 was not selective and resulted in the formation of a mixture of products, 6 and 7 (Scheme 5), which were confirmed by single crystal XRD studies (Fig. 7 and 8). The most interesting feature of 6 is the triazole ring formation from diazoalkane. The 1H NMR of 6 shows three distinct peaks at 1.48, 7.37, and 8.27 ppm corresponding to methyl protons of tBu group, carbene backbone, and triazole ring protons, respectively. While the generation of 6 is quite unprecedented, and can be considered as a new type of N-heterocyclic olefin, the formation of 7 was anticipated from the aforementioned reactions with other NHCs. The separation of compounds 6 and 7 is simple. Compound 6 gets precipitated in hexane or toluene, while the filtrate contains a mixture of both 6 and 7. Hence, we could not determine the yield of 7 as well as it’s complete spectroscopic characterization was not possible. The solid-state structure of 6 reveals that the C–C bond length between the triazole and imidazole ring is significantly longer than a CC bond with a bond distance of 1.4651(15) Å. The tau value for 6 is 0.6 (Tables S1 and S2†). The increased bond length allows to circumvent the cross-conjugation and large charge separation between the two rings. We have tried to understand the mechanism pertaining to the formation of compound 6 and explored several potential pathways for its formation, but none of them presented a barrier(s) that is acceptable for a reaction occurring at room temperature.
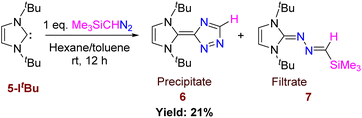 |
| | Scheme 5 Reaction of 5-ItBu with Me3SiCHN2 and formation of CC bound product. | |
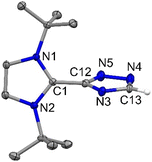 |
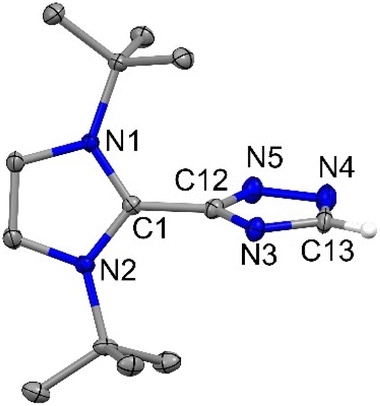
| | Fig. 7 The molecular structure of 6 with anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms (except C13) are not shown for clarity. Selected distances (Å) and angles (deg): C1–C12 1.4651(15), N5–C12 1.3362(15), N3–C12 1.3499(14), N3–C13 1.3467(15), N4–C13 1.3233(17), N4–N5 1.3751(15), N1–C1–N2 107.94(9), N1–C1–C12 125.79(10), N2–C1–C12 126.22(10), N5–C12–N3 115.29(10), N5–C12–C1 121.52(10), C12–N5–N4 104.37(10), N4–C13–N3 115.40(11), C13–N3–C12 99.52(10). All atoms are sp2 hybridized and the tau value is 0.6 (Tables S1 and S2†). | |
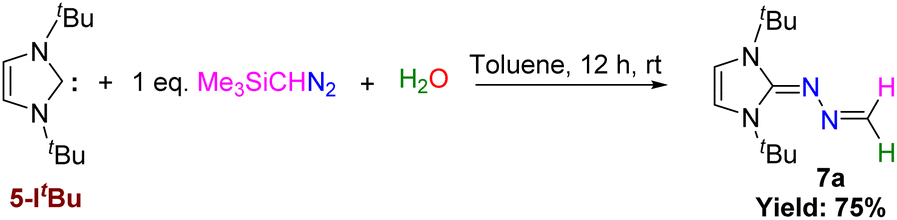
As we could not isolate 7 in a sufficient quantity, we could not study its further reaction with HCl like we did in case of 5-IPr. However, the reaction between 5-ItBu and Me3SiCHN2 in the presence of stoichiometric amount of water yielded the novel N-heterocyclic hydrazine, 7a (Scheme 6). The molecular structure of 7a is depicted in Fig. 8 (right).
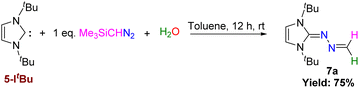 |
| | Scheme 6 Reaction of 5-ItBu with Me3SiCHN2 and water. | |
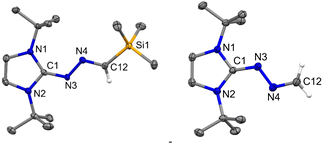 |
| | Fig. 8 The molecular structure of 7 (left) and 7a (right) with anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are not shown for clarity. Selected distances (Å) and angles (deg): N3–C1 1.321(7), N3–N4 1.378(6), N4–C12 1.293(7), Si1–C12 1.875(6), N2–C1–N1 106.2(4), C1–N3–N4 118.3(4), C12–N4–N3 113.9(4), N4–C12–Si1 121.4(4). 7a: N3–C1 1.3864(10), N3–N4 1.3706(9), N4–C12 1.2808(11), N1–C1–N2 106.18(6), C1–N3–N4 117.70(7), C12–N4–N3 113.19(7). | |
Case V: reaction with DAC
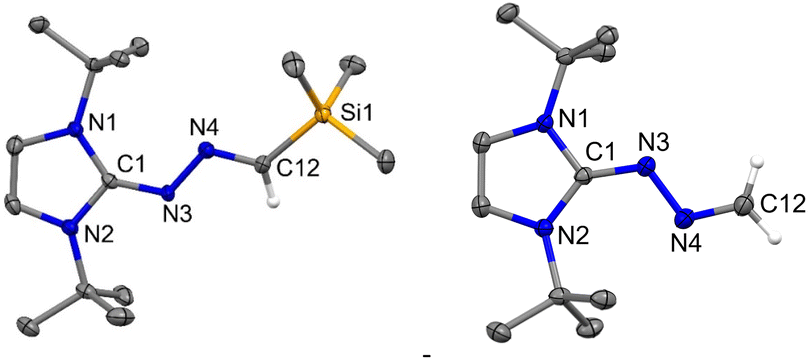
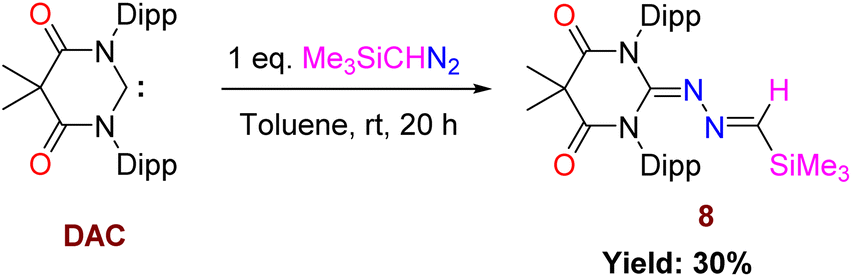
Now, the question remains what will happen with an electrophilic carbene such as diamido carbene (DAC).26 The reaction of DAC with Me3SiCHN2 in toluene at room temperature for 20 h resulted in a yellow-colored solution and formation of 8 (Scheme 7). The 1H NMR shows a sharp singlet at −0.24 ppm for the Me3Si group and a resonance at −7.63 ppm in the 29Si NMR spectrum. The crystallization in CH2Cl2 and hexane mixture afforded yellow-colored crystals of 8. The molecular structure of 8 is shown in Fig. 9. The reaction of the DAC with Me3SiCHN2 and a stoichiometric amount of water to form N-heterocyclic hydrazines similar to 5 and 7a was unsuccessful. It can be due to the fact that DAC reacts with water before Me3SiCHN2 leading to inhibit the possible generation of diazomethane (CH2N2).
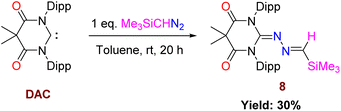 |
| | Scheme 7 Reaction of Me3SiCHN2 with DAC. | |
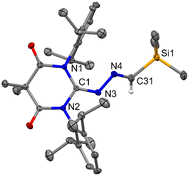 |
| | Fig. 9 The molecular structure of 8 with anisotropic displacement parameters are depicted at the 50% probability level. Hydrogen atoms are not shown for clarity. Selected distances (Å) and angles (deg): N3–C1 1.272(3), N3–N4 1.404(2), N4–C31 1.275(3), C1–N1–C4 123.67(16), C1–N3–N4 119.95(17), C31–N4–N3 113.57(18). | |
Case VI: reactions of NHCs with diazoester
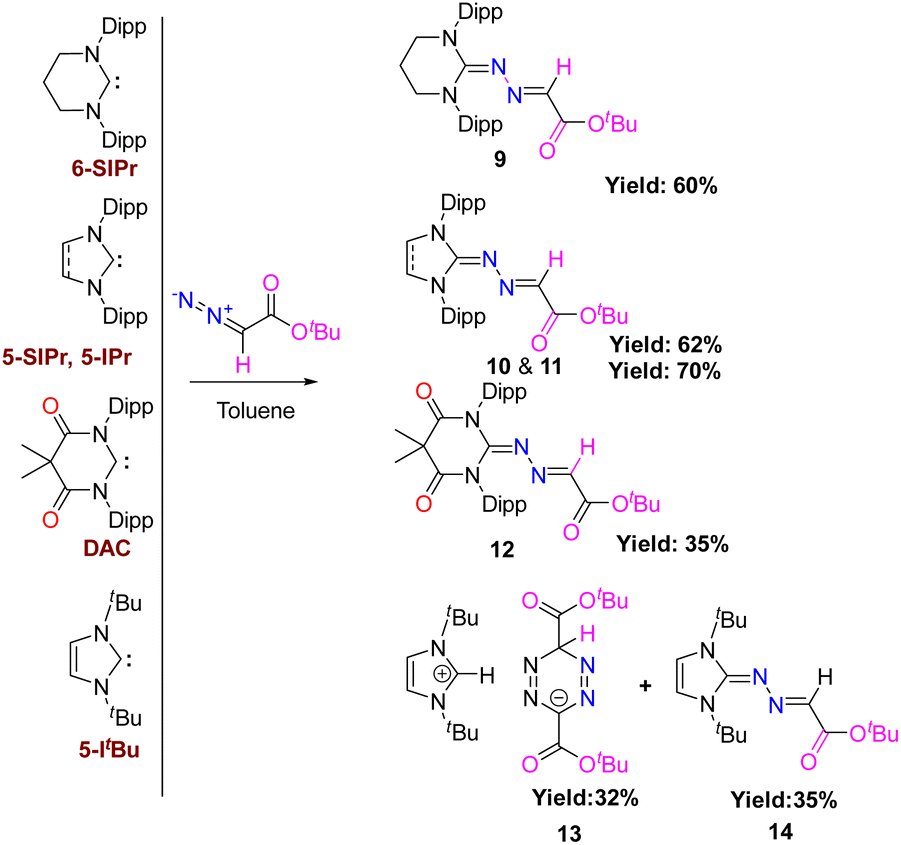
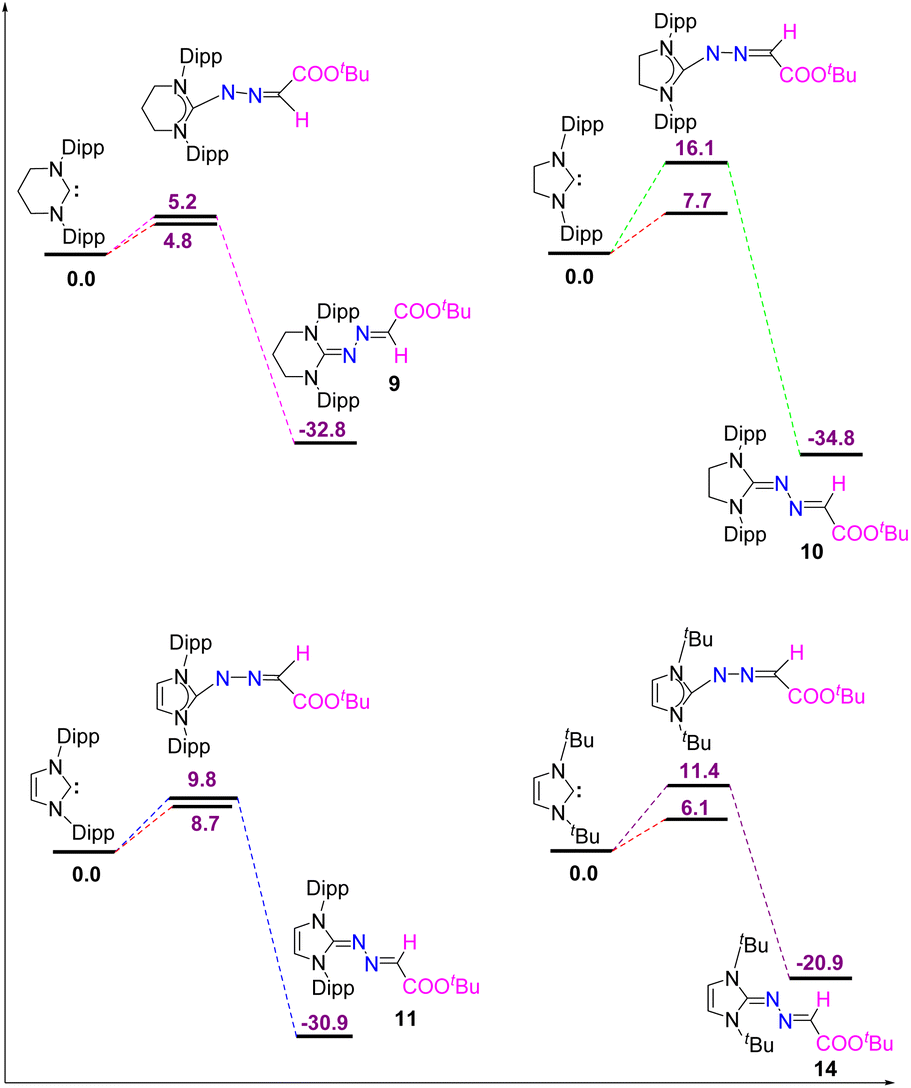
To examine whether varying the substituent on diazo alkane influences the reactivity of carbenes, we have also performed the reaction of different types of carbenes with electron-withdrawing diazoester, COOtBuCHN2, which resulted in the formation of the same kind of products (9–12 and 14) except 5-ItBu as shown in Scheme 8. Their moelcular structures are shown in Fig. 10.
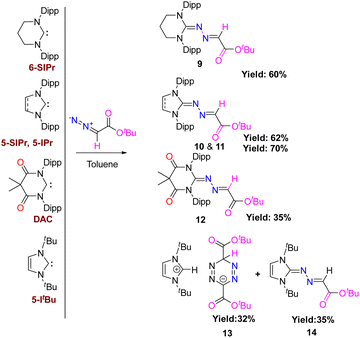 |
| | Scheme 8 Reaction of different carbenes with COOtBuCHN2. | |
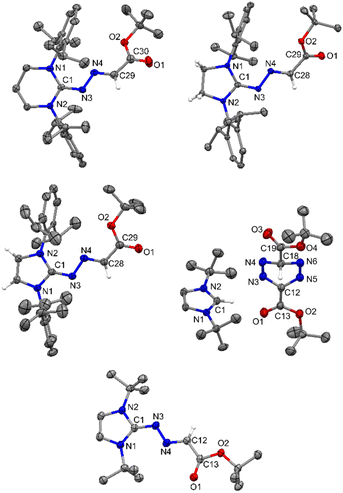 |
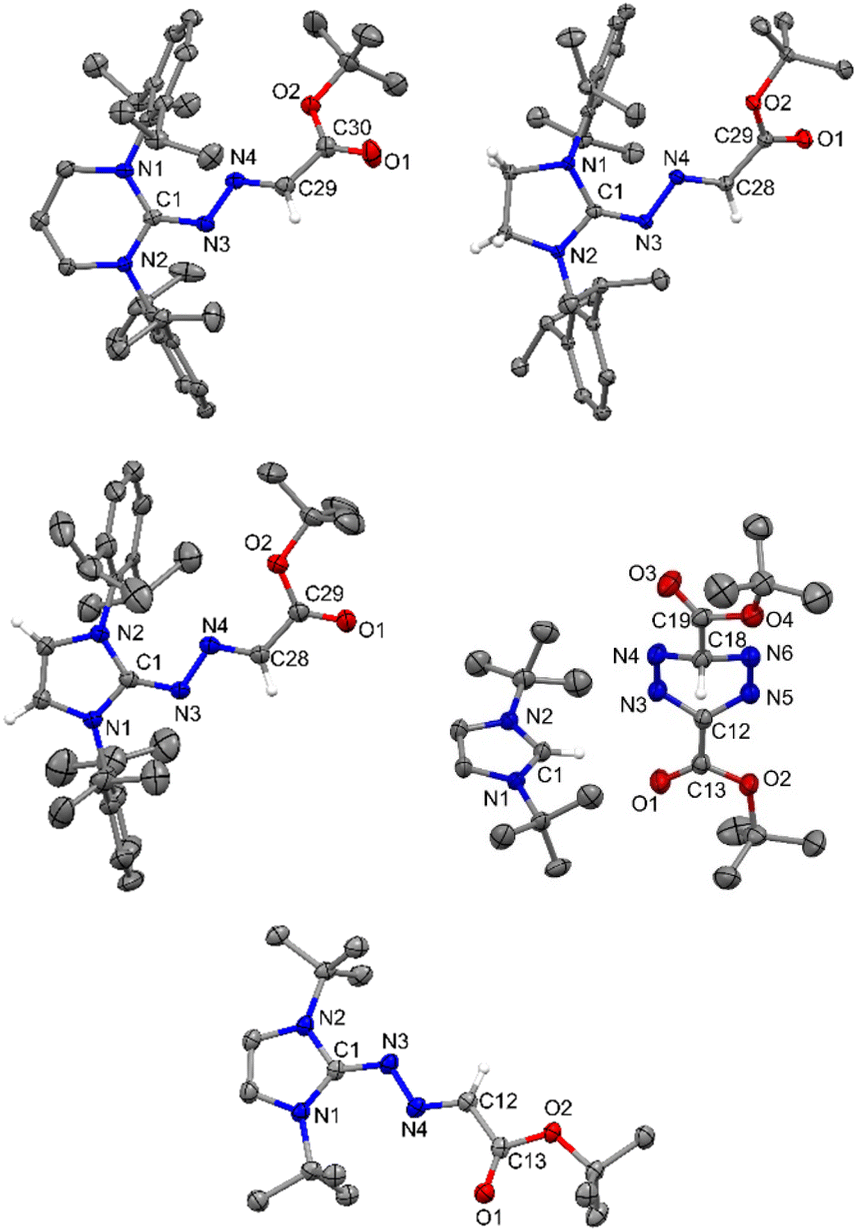
| | Fig. 10 The molecular structures of 9, 10, 11, 13, and 14 with anisotropic displacement parameters are depicted at the 50% probability level and 13 at the 30% probability level. Hydrogen atoms are not shown for clarity. Selected distances (Å) and angles (deg) 9: N3–C1 1.327(2), N3–N4 1.366(2), N4–C29 1.287(3), C29–C30 1.462(3), O1–C30 1.216(3), O2–C30 1.333(2), N2–C1–N1 117.53(17), C1–N3–N4 116.22(15), C29–N4–N3 111.92(16), O1–C30–O2 124.66(19), O1–C30–C29 120.08(19). 10: N3–C1 1.313(2), N3–N4 1.3745(18), N4–C28 1.283(2), C28–C29 1.474(2), O1–C29 1.213(2), O2–C29 1.3402(19), C1–N1–C2 109.54(13), C1–N3–N4 111.97(13), C28–N4–N3 113.59(14), N4–C28–C29 121.63(15), O1–C29–C28 120.95(15), O2–C29–C28 113.27(14). 11: (Vibrational atoms are omitted for clarity): N3–C1 1.328(2), N3–N4 1.365(2), N4–C28 1.288(2), C28–C29 1.461(3), O1–C29 1.216(2), O2–C29 1.336(2), C1–N1–C2 109.99(14), C1–N3–N4 112.05(15), C28–N4–N3 113.42(15), N4–C28–C29 122.29(17), O1–C29–C28 121.50(18), O2–C29–C28 114.02(16). 13: (Vibrational atoms are omitted for clarity): N1–C1 1.326(3), N2–C1 1.328(3), N3–N4 1.289(3), N5–N6 1.287(3), N3–C12 1.366(3), N5–C12 1.369(3), C12–C13 1.460(3), C18–C19 1.507(4), N1–C1–N2 109.8(2), N4–C18–N6 109.4(2), N3–C12–N5 117.7(2), N6–N5–C12 117.2(2), N4–N3–C12 117.2(2), N3–N4–C18 110.55(19), N5–N6–C18 110.31(19). 14: C1–N3 1.342(2), N3–N4 1.342(2), N4–C12 1.292(3), C12–C13 1.462(3), O1–C13 1.216(2), O2–C13 1.355(2), N1–C1–N2 106.78(16), C1–N3–N4 114.86(17), N3–N4–C12 114.94(17), O1–C12–C13 126.27(19), O2–C12–C13 109.21(17). | |
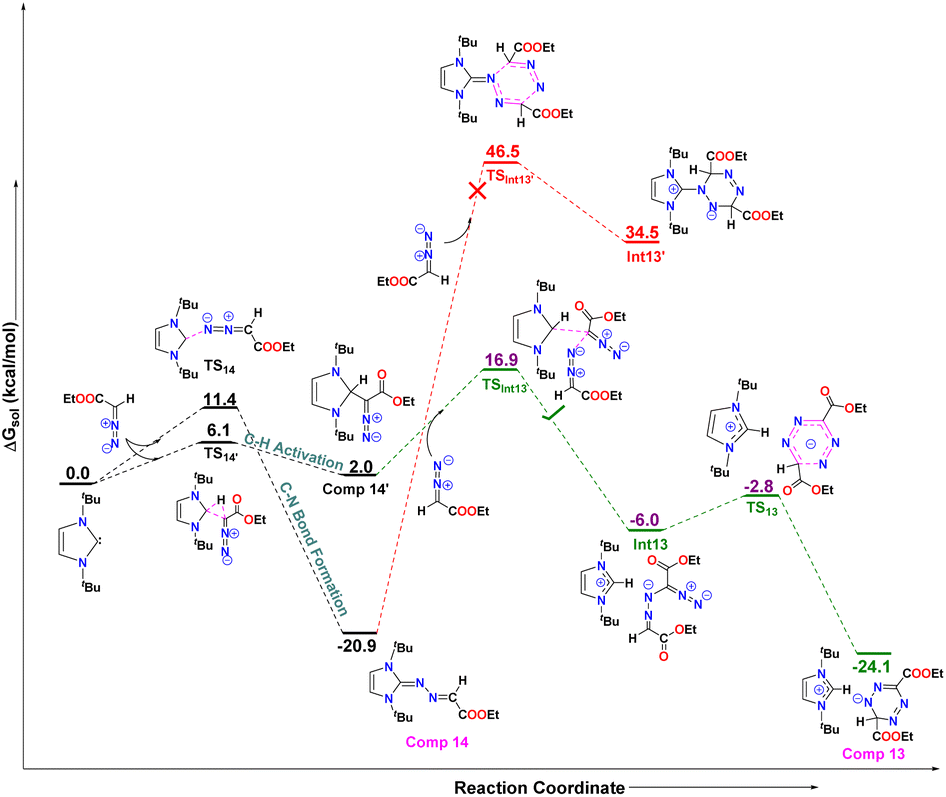
The reaction of 5-ItBu with diazoester also resulted in the formation of the corresponding imidazolium salt with tetrazenide counter anion (13)27 alongside 14. Even though X-ray diffraction of a single crystal provided clear proof into the connectivity of compound 12 (see Fig. S61, ESI†), we abstain from discussing bonding parameters due to the poor quality of the data. The DFT calculations for the reactions of various NHCs with diazoester are presented in Fig. 11. The energy profile diagram clearly indicates that for diazoester, all products (9–11 and 14) are formed through thermodynamically controlled pathways. This is evidenced by the higher free energy barriers for all direct C–N bond formation reactions compared to their competing C–H bond activation pathways.
Finally, in order to understand the formation of the new imidazolium salt with a tetrazenide counterion (13) from the reaction of a diazoester with 5-ItBu, comprehensive DFT calculations were performed (Fig. 11). As with other NHCs, 5-ItBu reacts with the diazoester in two possible ways, leading to either compound 14, via direct C–N bond formation, or compound 14′, through C–H bond activation. The free energy difference between these pathways is 5.3 kcal mol−1, with the C–N bond formation requiring 11.4 kcal mol−1, and C–H activation requiring 6.1 kcal mol−1. This suggests that both pathways are kinetically accessible. Despite the kinetic feasibility of both routes, compound 14 is thermodynamically more stable than compound 14′ by 18.9 kcal mol−1 (see Fig. 12).
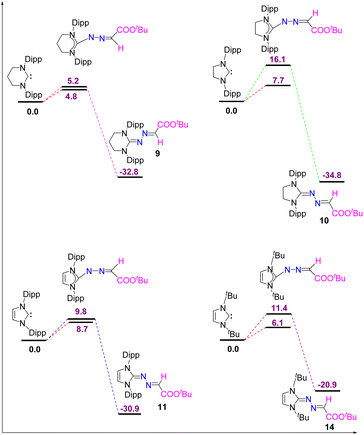 |
| | Fig. 11 The free energy profiles for the reaction of diazoester with different NHCs have been shown here. All values are in kcal mol−1. Level of theory: PBE0-D3/def2TZVP//PBE-D3/defTZVP with solvent toluene. | |
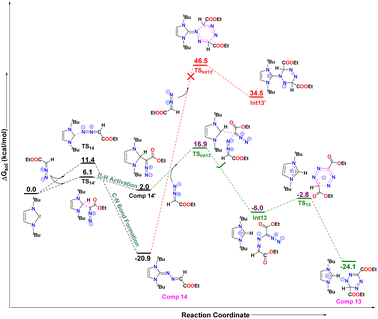 |
| | Fig. 12 The free energy profiles for the formation of compound 13 and compound 14 have been shown here. All values are in kcal mol−1. Level of theory: PBE0-D3/def2TZVP//PBE-D3/defTZVP with solvent toluene. | |
Moreover, the formation of compound 13 involves the reaction of an additional diazoester unit with either compound 14 or 14′, so both pathways were explored as depicted in Fig. 12 below. The DFT calculations revealed that the reaction of diazoester with compound 14 leads to a six-membered cyclized product, Int1′, which is endergonic, requiring 55.4 kcal mol−1 of free energy. The free energy barrier (TSInt13′) for this [3 + 3] cyclization is 67.4 kcal mol−1, which is too high to occur at room temperature. This suggests that this pathway is unlikely.
In contrast, when the diazoester reacts with compound 14′, it forms the exergonic intermediate Int13 (Fig. 12), passing through the transition state TSInt13 with a barrier of 16.9 kcal mol−1, which is 29.6 kcal mol−1 lower than the corresponding barrier (TSInt13′) for compound 14. Finally, Int13 undergoes a six-membered cyclization to yield the final product, compound 13, releasing 18.1 kcal mol−1 of free energy. The activation energy for this step is only 3.2 kcal mol−1, making it highly feasible.
Conclusions
From this comparative study, it can be concluded that the reactivity of one particular NHC is not representative for the whole range of stable carbenes that are available. 5-IPr, 6-SIPr, and diamido carbene afforded N-heterocyclic methylene hydrazine derivatives upon reaction with Me3SiCHN2. 5-IPr and 5-ItBu led to the formation of N-heterocylic methylene hydrazines with terminal CH2 formed via dehydrohalogenation of 5a using base or reacting 5-IPr and 5-ItBu with Me3SiCHN2 in presence of stoichiometric amount of water. However, 5-SIPr activates the C–H bond of Me3SiCHN2. Another surprising results were obtained from the reaction 5-ItBu, which undergoes triazole ring formation with an exocyclic CC double bond. The same set of reactions were performed with COOtBuCHN2 instead of Me3SiCHN2, and in every case azine derivatives were obtained. These results should encourage the organometallic community to broaden the range of carbenes they use for small molecule activation or as ligands for transition metals.
Data availability
The data supporting this article have been included as part of the ESI.† CCDC 2347293 (1), 2347305 (3), 2363380 (4), 2347306 (5), 2347322 (5a), 2347308 (6), 2347310 (7), 2368081 (7a), 2347311 (8), 2347313 (9), 2347315 (10), 2347321 (11), 2375811 (13) and 2363384 (14) contain the supplementary crystallographic data for this paper.
Author contributions
K. B. and S. S. S. conceived the idea. K. B. has carried out the synthesis and characterization of the compounds. H. S. and K. V. executed the theoretical calculations. K. B. and R. G. G. performed the single-crystal X-ray structures of the molecules.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
S. S. S. and K. V. are grateful to the Science and Engineering Science Board, India, for financial assistance (CRG/2023/000780). K. B. and H. S. thanks CSIR and UGC, India, for their research fellowships. The authors thank S. Gayathridevi for doing the preliminary DFT calculations for the systems.
Notes and references
-
(a) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911–936 CrossRef CAS PubMed;
(b) W. Kirmse, Angew. Chem., Int. Ed., 2003, 42, 1088–1093 CrossRef CAS PubMed;
(c) D. Marcoux and A. B. Charette, Angew. Chem., Int. Ed., 2008, 47, 10155–10158 CrossRef CAS PubMed;
(d) R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 3748–3759 CrossRef CAS PubMed;
(e) R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760–3765 CrossRef CAS PubMed.
- S. S. Sen, J. Hey, D. Kratzert, H. W. Roesky and D. Stalke, Organometallics, 2012, 31, 435–439 CrossRef CAS.
- A. Jana, S. S. Sen, H. W. Roesky, C. Schulzke, S. Dutta and S. K. Pati, Angew. Chem., Int. Ed., 2009, 48, 4246–4248 CrossRef CAS PubMed.
- V. S. Ajithkumar, P. B. Ghanwat, K. V. Raj, K. Vanka, R. G. Gonnade and S. S. Sen, Organometallics, 2023, 42, 2983–2990 CrossRef CAS.
- S. Jiang, Y. Cai, T. Rajeshkumar, I. Rosal, L. Maron and X. Xu, Angew. Chem., Int. Ed., 2023, 62(1–6), e202307244 CAS.
- J. M. Hopkins, M. Bowdridge, K. N. Robertson, T. S. Cameron, H. A. Jenkins and J. A. C. Clyburne, J. Org. Chem., 2001, 66, 5713–5716 CrossRef CAS PubMed.
- R. Guo, Y. Zheng and J.-A. Ma, Org. Lett., 2016, 18, 4170–4173 CrossRef PubMed.
- M. E. Gunay, N. Ozdemir, M. Ulusoy, M. Ucak, M. Dincer and B. Cetinkaya, J. Organomet. Chem., 2009, 694, 2179–2184 CrossRef.
-
(a) D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC;
(b) P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed;
(c) T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- A. J. Arduengo III, J. C. Calabrese, F. Davidson, H. V. Rasika Dias, J. R. Goerlich, R. Krafczyk, W. J. Marshall, M. Tamm and R. Schmutzler, Helv. Chim. Acta, 1999, 82, 2348–2364 CrossRef.
- R. S. Holdroyd, M. J. Page, M. R. Warren and M. K. Whittlesey, Tetrahedron Lett., 2010, 51, 557–559 CrossRef CAS.
-
(a) T. W. Hudnall and C. W. Bielawski, J. Am. Chem. Soc., 2009, 131, 16039–16041 CrossRef CAS PubMed;
(b) J. P. Moerdyk and C. W. Bielawski, Chem.–Eur. J., 2013, 19, 14773–14776 CrossRef CAS PubMed;
(c) J. P. Moerdyk and C. W. Bielawski, Chem. Commun., 2014, 50, 4551–4553 RSC.
- Z. R. Turner, Chem.–Eur. J., 2016, 22, 11461–11468 CrossRef CAS PubMed.
-
(a) S. Sole, H. Gornitzka, W. W. Schoeller, D. Bourissou and G. Bertrand, Science, 2001, 292, 1901–1903 CrossRef CAS PubMed;
(b) J. Vignolle, M. Asay, K. Miqueu, D. Bourissou and G. Bertrand, Org. Lett., 2008, 10, 4299–4302 CrossRef CAS PubMed;
(c) F. Vermersch, R. Jazzar, V. T. Wang and G. Bertrand, Chem. Sci., 2024, 15, 3707–3710 RSC;
(d) D. Martin, Y. Canac, V. Lavallo and G. Bertrand, J. Am. Chem. Soc., 2014, 136, 5023–5030 CrossRef CAS PubMed.
- A. Jana, I. Objartel, H. W. Roesky and D. Stalke, Inorg. Chem., 2009, 48, 7645–7649 CrossRef CAS PubMed.
- G. Kundu, P. R. Amrutha, K. V. Raj, S. Tothadi, K. Vanka and S. S. Sen, Chem. Sci., 2023, 14, 5894–5898 RSC.
- G. Kundu, K. Balayan, S. Tothadi and S. S. Sen, Inorg. Chem., 2022, 61, 12991–12997 CrossRef CAS PubMed.
- G. Kundu, V. S. Ajithkumar, K. V. Raj, K. Vanka, S. Tothadi and S. S. Sen, Chem. Commun., 2022, 58, 3783–3786 RSC.
- G. Kundu, R. Dixit, S. Tothadi, K. Vanka and S. S. Sen, Dalton Trans., 2022, 51, 14452–14457 RSC.
- K. Balayan, H. Sharma, K. Vanka, S. Ravindranathan, R. G. Gonnade and S. S. Sen, Chem. Commun., 2023, 59, 8540–8543 RSC.
-
(a) J. Safari and S. Gandomi-Ravandia, RSC Adv., 2014, 4, 46224–46249 RSC;
(b) S. S. Chourasiya, D. Kathuria, A. A. Wani and P. V. Bharatam, Org. Biomol. Chem., 2019, 17, 8486–8521 RSC.
- T. Ochiai, D. Franz and S. Inoue, Chem. Soc. Rev., 2016, 45, 6327–6344 RSC.
-
(a) M. M. D. Roy and E. Rivard, Acc. Chem. Res., 2017, 50, 2017–2025 CrossRef CAS PubMed;
(b) G. Mahantesh, D. Sharma, R. Dandela and V. Dhayalan, Chem.–Eur. J., 2023, 29, e202302106 CrossRef PubMed.
-
(a) P. Varava, Z. Dong, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Nat. Chem., 2021, 13, 1055–1060 CrossRef CAS PubMed;
(b) P. W. Antoni, C. Golz, J. J. Holstein, D. A. Pantazis and M. M. Hansmann, Nat. Chem., 2021, 13, 587–593 CrossRef CAS PubMed;
(c) T. Eisner, A. Kostenko, F. J. Kiefer and S. Inoue, Chem. Commun., 2024, 60, 558–561 RSC;
(d) M. M. Hansmann, Angew. Chem., Int. Ed., 2023, 62, e202304574 CrossRef CAS PubMed.
- S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, J. Am. Chem. Soc., 2010, 132, 1123–1126 CrossRef CAS PubMed.
- J. P. Moerdyk, D. Schilter and C. W. Bielawski, Acc. Chem. Res., 2016, 49, 1458–1468 CrossRef CAS PubMed.
- M. Schwach, H.-D. Hausen and W. Kaim, Inorg. Chem., 1999, 38, 2242–2243 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2347293 (1), 2347305 (3), 2363380 (4), 2347306 (5), 2347322 (5a), 2347308 (6), 2347310 (7), 2386081 (7a), 2347311 (8), 2347313 (9), 2347315 (10), 2347321 (11), 2375811 (13) and 2363384 (14). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05740f |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Himanshu
Sharma
ab,
Himanshu
Sharma