
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-innocent P-centers in nonbenzenoid polycyclic aromatic molecules with tunable structures and properties†
Can
Li‡
a,
Wei
Zhou‡
a,
Zhaoxin
Liu‡
a,
Rong
Gao
a,
Qixi
Mi
 a,
Zhijun
Ning
*a and
Yi
Ren
*ab
a,
Zhijun
Ning
*a and
Yi
Ren
*ab
aSchool of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China. E-mail: renyi@shanghaitech.edu.cn; ningzhj@shanghaitech.edu.cn
bShanghai Clinical Research and Trial Center, Shanghai, 201210, People's Republic of China
First published on 22nd October 2024
Abstract
Implanting heteroatoms into polycyclic aromatic molecules (PAMs) offers a great opportunity to fine-tune their optoelectronic properties. Herein, we report a new type of nonbenzenoid PAM in which the sp2 C atoms are replaced by S and P in the azulene moiety. The synthesis harnessed modular P-chemistry and cyclization chemistry, which afforded the first example of P-azulene-based PAMs with isomeric PN- and PC-type structures. Photophysical and theoretical studies revealed that the P-environments have strong impacts on the structures and properties of the P-PAMs. Different from the electronic structure of azulene with strong π conjugation, the PC derivatives maintained effective σ*–π* hyperconjugation in the frontier molecular orbitals via the P-centers. In particular, the PC derivative with a P(III)-center showed unexpected room-temperature phosphorescence in solution, which was attributed to the excited-state aromaticity induced structure change at the P-center. Decoration with various aryl groups further modified the photophysical and redox properties in another dimension. Furthermore, bis(triarylamine)-functionalized P-PAMs formed stable radical cations in which the P-environments strongly influenced the mixed-valence state and open-shell characters. As a proof of concept, bis(triarylamine)-functionalized P-PAMs were explored as the hole-transporting layers in perovskite solar cells, and a power conversion efficiency of 14% was achieved. As a new example of nonbenzenoid PAMs with intriguing optoelectronic properties, our P-PAMs are promising building blocks for diverse optoelectronic applications in the future.
Introduction
Polycyclic aromatic molecules (PAMs) have attracted significant attention in the areas of organic catalysis, bio-imaging/sensing, and organic optoelectronic devices.1–6 Compared with classical benzenoid PAMs, nonbenzenoid PAMs have recently maintained a unique position in the applications mentioned above.7–11 Adding or removing sp2 C atoms in benzenoid PAMs is one popular strategy to build nonbenzenoid PAMs. For example, nonbenzenoid PAMs with five-membered rings have been extensively investigated in the field of organic radical materials, as their open-shell characteristics can be readily activated and tuned.11–16 Azulene, with a fused five-membered and seven-membered ring, is another classical example of a nonbenzenoid PAM with unique anti-Kasha emission properties, redox characters, and optoelectronic functionalities.7,17–21Replacing the sp2 C atoms with main-group elements (such as B,12,22–26 N,7,27–30 S,31–34 Si,7,35 P,36–41etc.) is another popular strategy to construct nonbenzenoid PAMs with diverse chemical/electronic properties. Phosphorus (P) is a special element among the main-group elements that exhibits rich functionalities, such as oxidation, borylation, alkylation, and metal coordination.36,37 These functionalities further facilitate efficient σ(σ*)–π(π*) hyperconjugations between the P-centers and π-conjugated backbones, thus greatly enriching its applications in terms of two-photon emission, electron-accepting character, and electrochromic devices. These intriguing functions are not easily accessible to the traditional purely carbon-based PAMs.
Recently, research into P-six-membered PAMs has started to emerge due to the discovery of new synthetic protocols.41–46 For example, Romero and co-workers recently reported phosphaphenalene derivatives via facile cyclization chemistry.41 In another example, Yamaguchi and co-workers reported new P-xanthene derivatives with excellent near-IR fluorescence imaging properties. Our group also developed a new modular protocol to access highly emissive diazaphosphinines with tunable singlet and triplet emission.46
Compared with P-five-/six-membered PAMs, research into P-seven-membered PAMs is still in its infancy, mainly because of their synthetic challenges and/or instability.47–52 Therefore, it is highly desirable to develop the streamlined protocols that can efficiently construct P-seven-membered PAMs with more complex and rich functionalities. Herein, we report new examples of nonbenzenoid PAMs with S-five-P-seven heterocycles (Chart 1). Leveraging the efficient P-chemistry and cyclization chemistry, the modular synthesis afforded P-PAMs with structural diversity. Distinct structures allowed us to fine-tune the electronic structures and photophysical properties in both the ground and excited state. Further decorating P-PAMs with various aryl substituents systematically modified their photophysical properties, redox behaviors, and radical characteristics. As a proof of concept, P-PAMs with triaryl amine groups were successfully applied as hole-transporting layers in perovskite solar cells.
 | ||
| Chart 1 Nonbenzenoid PAMs in previous studies and the new nonbenzenoid P-PAMs (PN and PC derivatives) in this study. | ||
Results and discussion
The synthesis of the new P-PAMs started with tetrabromothiophene (1), which was converted into various 2,5-diaryl thiophene derivatives (2a,b,c and 2d,e,f) via Suzuki coupling reactions (Scheme 1(i)). Subsequent Suzuki coupling reactions between 2a,b,c/2d,e,f and 2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole afforded 3a,b,c and 3d,e,f in moderate yields. Using P–N and P–C bond formation protocols reported in our previous studies,46,50,51 we were able to synthesize 4a (90%), 4b (93%), 4c (85%), 5d (70%), and 5e (81%) with P-heteropine units. | ||
| Scheme 1 Synthesis of new P-PAMs with isomeric structures. (i) Pd(PPh3)4, base, solvent, Δ. (ii) Pd(OAc)2, Ruphos, K3PO4, solvent, Δ. (iii) PhPCl2, DBU, acetonitrile. (iv) PPhCl2 (1.3 eqv.), TMSOTf (2.6 eqv.), CH3CN, −30 °C → 70 °C. (v) PhP(O)H(OH), DMAP, −30 °C → 70 °C. (vi) H2O2 (excess), r.t. in DCM. (vii) DDQ, 365 nm LED, dry toluene, r.t. (viii) CuI, K3PO4, DMEDA, DMF, 140 °C. | ||
Unlike 5d and 5e, 5f could not be obtained under similar reaction conditions. Alternatively, we used a synthetic method reported by Masahiro and coworkers with modified conditions, in which activated phenylphosphinic acid was used as the starting reagent (Scheme 1(v)).53 This method gave 5fO with a yield of 31%. Oxidation of 4a,b,c and 5d,e with excess H2O2 afforded 4aO/bO/cO and 5dO/5eO in high yields (Scheme 1(vi)). 4a was chosen as the representative molecule for the sulfuration of the P-center. After heating with S8 (10 eqv.) at 140 °C for 10 days, we were able to obtain 4aS in a yield of 96%.
To further planarize the backbones, photocyclization was applied to 4a/b/c and 4aO/bO/cO. In previous studies, Scholl reaction (excess oxidants and Lewis acid) and Mallory reaction (hv, excess I2 and propylene oxide) conditions showed limited compatibility with the cyclization of P-PAMs with various P-centers.38,43,54–56 In our case, the photocyclizations were highly efficient when DDQ was used as the oxidant, giving the PN series in good yields (Scheme 1(vii)). The isomeric counterparts PC1/2 and PC1/2/3O were obtained by applying the intramolecular CuI-catalyzed C–N coupling reaction to 5d/e and 5d/e/fO (Scheme 1(viii)). The results showed that the presence of the P(III) and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O centers did not affect the CuI-catalyzed cyclization. Compared with Pd/Ni-catalyzed cyclizations reported previously,57,58 the CuI-catalyzed cyclization of P-PAMs has rarely been reported in the literature.
O centers did not affect the CuI-catalyzed cyclization. Compared with Pd/Ni-catalyzed cyclizations reported previously,57,58 the CuI-catalyzed cyclization of P-PAMs has rarely been reported in the literature.
We also tried to synthesize PC3 with a P(III) center by reducing PC3O with SiHCl3. However, the easy oxidation of PC3 prevented us from obtaining the pure product during the purification. The synthesis of the new P-PAMs demonstrated a highly modular protocol to access new nonbenzenoid PAMs with the great structural tunability. Due to their large aromatic structures, some of the products exhibited low solubilities in typical organic solvents. The detailed synthesis and characterizations are provided in the ESI.†
We were able to obtain suitable single crystals of PN1S, PN2O, PN3O, PC1O, and PC3O for single-crystal X-ray diffraction experiments (Fig. 1 and Table S1†). The single-crystal structure of PN2O contains a disordered solvent, which resulted in a low crystal data quality (Fig. S9†). In the crystal structures of PN1S and PN3O, the fused polycyclic aromatic structures induced planar backbones. However, the P-centers were significantly twisted out of the central heteropine plane (Fig. 1a and b), with distances between the P-center and the heteropine plane of 0.9 Å for PN1S and 0.7 Å for PN3O, respectively. These results differ from those for previous six-membered diazaphosphinines, in which the central six-membered P-ring showed high planarity.46 The P–N bond lengths of PN1S (1.70 Å) and PN3O (1.69 Å) are similar to those of previous diazaphosphepines.46,50,51
 | ||
| Fig. 1 Single crystal structures of (a) PN1S, (b) PN3O, (c) PC1O, and (d) PC3O (thermal ellipsoids are shown at 50% probability level; hydrogen atoms are omitted for clarity). | ||
Compared with the PN derivatives, PC1O and PC3O showed more planarized P-heteropine planes (Fig. 1c and d). The distances between their P-centers and the heteropine planes are significantly shorter (PC1O: 0.2 Å and PC3O: 0.01 Å) compared with those of PN1S and PN3O. The P–C bond lengths of PC1O (1.77 Å) and PC3O (1.77 Å) are at shorter end of the range for P–C bonds (1.77–1.81 Å) reported in previous P-heteropines, which suggested a strong π-delocalization between the P-center and heteropine backbone.47–50 Furthermore, strong intermolecular π–π interactions were observed in these crystal structures. For example, PN1S and PC3O showed dimeric π–π interaction (Fig. S1 and S7†), whereas PN3O and PC1O showed long-range intermolecular π–π interactions (Fig. S3 and S5†).
We conducted UV-vis and fluorescence spectroscopy experiments on new P-PAMs (Fig. 2a, b and S10–S12†). Due to their rigid structures, both the PN and PC series exhibited small Stokes shifts. Consistently, the absorption and emission spectra showed the typical vibronic structures of rigid aromatic molecules. Upon decoration with electron-withdrawing and electron-donating aryl groups, the PN and PC series showed tunable absorption and emission.
 | ||
| Fig. 2 (a and b) Absorption and emission spectra of PN and PC in CH2Cl2. (c and d) Absorption and emission spectra of the PN1 series and PC1 series in CH2Cl2. (e and f) Low-temperature emission spectra of PN1 and PC1 in 2Me-THF at 77 K. | ||
The absorption and emission spectra of the PC series were systematically red-shifted compared to those of the PN series (Fig. 2a and b). It is rationalized that the strong π-conjugation of the PC series is responsible for the smaller LUMO–HOMO gaps, thus resulting in the red-shifted spectra (vide infra). The results highlighted the distinct structure impacts on the photophysical properties of the P-PAMs. Furthermore, PN1, PN1O and PN1S exhibited similar absorption and emission spectra (Fig. 2c and S10†), suggesting that the changes in the P-chemistry scarcely modified the optical bandgaps. This is not true for the PC series. As shown in Fig. 2d, PC1O (λabs = 453 nm and λem = 475 nm) with a PO center exhibited blue-shifted absorption and emission spectra compared with those of P1C (λabs = 463 nm and λem = 511 nm) with a P(III) center. The emission spectrum of PC1 is also broader than that of PC1O. The photophysical characteristics of PC1 were unexpected, since rigid P-PAMs with a PO center have generally exhibited red-shifted absorption and emission spectra compared with those with a P(III) center in previous studies.9,35 It is rationalized that P1C with a flexible P-center underwent a significant structure change in the excited state (vide infra), thus leading to the red-shifted emission. Although heteropines are well-known for their conformational dynamics in both the S0 and S1 states,47–52,59–61 the conformational dynamics of fused heteropine-PAMs have not been addressed in the literature.
The PN series consistently exhibited lower photoluminescence quantum yields (PLQYs; for example, PN1O: 11% and PN2O: 14%) compared with the PC series (PLQYs: 25% for PC1O, 30% for PC2O), except for those with triaryl amine groups (83% for PN3O, 81% for PN3). We further conducted low-temperature fluorescence spectroscopy experiments on the PN and PC series in 2Me-THF solution (Fig. 2e, f, S13 and S14†). In addition to the fluorescence emission signals, we also observed phosphorescence emission signals at 77 K. The phosphorescence signals of PN1, PN1O, PN1S, PN2, and PN2O were more pronounced than the fluorescence signals (Fig. S13 and S14†). Consistently, the phosphorescence lifetimes of PN series are longer than those of PC series (Table 1). These results suggested that the low PLQYs of the PN series are probably due to the strong intersystem crossing.
| Compoundb | λ abs [nm] | λ em [nm] | ϕ PL [%] | τ FL [ns] | τ Ph [s] |
|---|---|---|---|---|---|
| a Measured in CH2Cl2. b Absorption maximum of the lowest absorption band. c Emission maximum. d Measured by the calibrated integrating sphere. e Measured in DCM at room temperature. f Measured in 2Me-THF at 77 K. g Not detectable by the calibrated integrating sphere. | |||||
| PN1 | 399 | 404 | 11 | 1.50 | 0.93 |
| PN1O | 396 | 402 | 11 | 1.46 | 0.98 |
| PN1S | 398 | 403 | 7 | 0.88 | 0.95 |
| PN2 | 420 | 438 | 10 | 1.41 | 0.41 |
| PN2O | 421 | 441 | 14 | 1.27 | 0.40 |
| PN3 | 428 | 453 | 81 | 1.51 | 0.51 |
| PN3O | 430 | 458 | 83 | 1.61 | 0.51 |
| PC1 | 438 | 511 | NAg | 0.61 | 0.03 |
| 463 (sh) | |||||
| PC1O | 453 | 475 | 25 | 0.74 | 0.09 |
| PC2 | 454 | 535 | 1 | 1.16 | 0.04 |
| PC2O | 473 | 501 | 30 | 1.01 | 0.05 |
| PC3O | 492 | 512 | 53 | 0.48 | 0.04 |
Unexpectedly, PC1 exhibited a low-energy red emission in 2Me-THF solution at room temperature (Fig. 2f). The triplet-state nature of the emission was further confirmed by the long lifetime (0.45 ms) (Fig. S38†). Such observations were not made for PN1 (Fig. 2e). There are only a few molecules that show room-temperature phosphorescence in solution.62–64 To the best of our knowledge, there have been no reports of P-heteropines showing room-temperature phosphorescence in solution. Previous studies showed that the nonbonding electron lone pair of heteroatom groups effectively enhanced the spin–orbit coupling (SOC), thus promoting the triplet emission.65–67 The structure change of PC1 likely enhanced the SOC, further leading to the unexpected room-temperature phosphorescence. At 77 K, at which the conformational changes of PC1 were suppressed, an enhanced fluorescence signal was observed (Fig. 2f).
To shed light on the distinct photophysical characteristics of the PNO and PCO series, we conducted theoretical studies using density functional theory (DFT) and time-dependent (TD)-DFT (see the details in the ESI†). Fig. 3a shows the frontier molecular orbitals (FMOs) of the PN and PC series. The presence of electron-withdrawing substituents mainly lowers the LUMOs, while the presence of electron-donating substituents strongly raised the HOMOs. Consequently, the presence of electron-withdrawing and electron-donating substituents resulted in the lower HOMO–LUMO bandgaps (ΔEHOMO–LUMO) and S0–S1 transition energy in both the PNO and PCO series (Table S2†). Furthermore, the PC series exhibited lower ΔEHOMO–LUMO values and S0–S1 transition energy compared with the PN series (Fig. 3a and S2†), which are consistent with the photophysical characteristics described in the previous section.
 | ||
| Fig. 3 (a and b) FMOs of the PNO series, PCO series, azulene, and heteroatom-azulenes. (c) Optimized structures and FMOs of PC1 in S0 and S1 (TD-DFT calculated at the B3LYP/6-311+G(d,p) level). (d) Intrinsic reaction coordinate of PC1 in S1 (B3LYP/6-311G(d,p) level). Insert: NICS values in S0 and T1 (calculated at the (U)B3LYP/6-311+G(d,p) level in a vacuum). | ||
Different from those of the PNO series, the HOMOs of the central S-five-P-seven moiety clearly show π-bond character in the PCO series, and are very similar to HOMOs of azulene (A, Fig. 3b). However, the LUMOs of the central S-five-P-seven moiety in the PCO series are different from those of azulene. We further conducted natural bond orbital (NBO) analysis, which can provide the second-order perturbation energy (E) associated with the stabilization of hyperconjugation in P-PAMs.68,69 For PC1O, the results suggested a negative hyperconjugation (E = 3.09 kcal mol−1) between the σ*-orbital of the P–C/O bonds and the π* orbitals of heterocycle backbone. These results are in line with the lower-energy bandgaps of the PCO derivatives. Similar results are also observed in the parent PO-azulene (C, Fig. 3b).
We conducted excited-state studies to examine the unexpected room-temperature phosphorescence in solution. As shown in Fig. 3c, PC1 underwent a significant structure change in the S1 state, in which the sum of the three C–P–C bond angles (327°) increased in the S1 state (cf. 308° in S0). The results suggested that the P(III)-center adopted a more planar geometry in the excited state. Furthermore, the lone-pair electrons of PC1 displayed a strong contribution to the HOMO of the S1 state. This observation differs from that for P-azulene, in which the lone-pair electrons display a strong contribution to HOMO of the S0 state (B, Fig. 3b). A similar structure change is not observed for PN1 in the excited state (Fig. S40†). We further conducted intrinsic reaction coordinate (IRC) calculations (Fig. 3d) to visualize the excited state evolution of PC1 between the initial S0 structure and the final S1 structure (see the details in the ESI†). The IRC results showed that P-center planarization occurred downhill, which supported the thermodynamically favorable structure of PC1 in S1.
To probe the driving force for the planarization, we conducted nucleus-independent chemical shift (NICS) calculations of the central heteropine in PC1. According to Baird's rule, conjugated rings with 4n π electrons are aromatic in T1, which was further demonstrated to be true for the rings in S1.70 NICS for S1, which requires a multideterminant wavefunction, is more complicated. Due to the limitations of computational approaches at the current stage, we instead performed NICS calculations at the T1 state, which were used to evaluate the excited state aromaticity in previous studies.71 As we expected, the large negative NICS values (in ppm) implied that the aromaticity gain is likely the driving force for the excited-state planarization (Fig. 3d).
Although previous studies showed that N-heteropines72 and As-heteropines59 underwent excited-state Baird's aromaticity-induced planarization at the As and N centers, similar excited-state planarization of the P-center of heterocycles has not been reported in the literature. In our case, we hypothesized that the planarization of the P-center was more energetically favorable than the change of the fused π-conjugated backbone of PC1 in S1.
Cyclic voltammetry (CV) experiments were conducted to reveal the redox characteristics of the new P-PAMs (Fig. 4a and S41†). Due to their limited solubilities, PC1, PC2, and PC2O were not subjected to the CV experiments. Compared with PN1, PN1O exhibited a more positive reduction potential, likely due to the presence of the strong electron-accepting PO center. In the PC series, PC1O exhibited two reduction potentials. Moreover, the first reversible reduction potential is lower than that of PN1O. Based on the theoretical studies, the stronger electron-accepting ability of PC1O is attributed to the σ*–π* hyperconjugation.
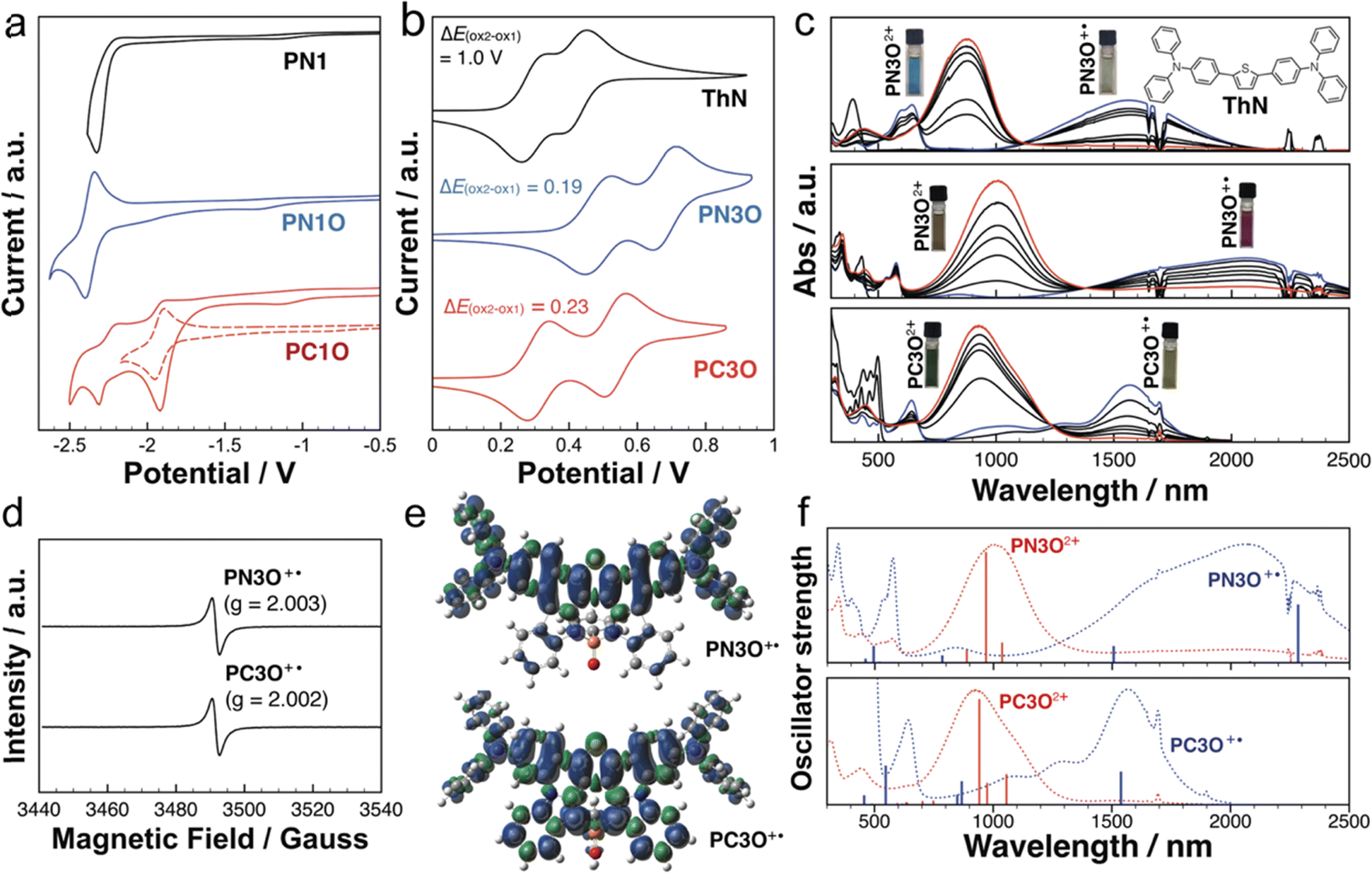 | ||
| Fig. 4 (a and b) Cyclic voltammograms of new P-PAMs. (c) UV-vis-NIR spectra of ThN, PN3O and PC3O (1 × 10−5 M) with different amounts of AgSbF6 in DCM. (d) Solid-state EPR spectra of PN3O and PC3O monocations. (e) Spin density of PN3O and PC3O radical cations calculated at CAM-UB3LYP/6-311+G(d,p) in a vacuum. (f) Theoretical calculated optical transitions of PN3O and PC3O cations calculated at (CAM)-(U)B3LYP/6-311+G(d,p) in a vacuum. | ||
When the strong electron-donating bis(triarylamine) substituents were installed, PN3, PN3O, and PC3O exhibited two reversible oxidation potentials (Fig. 4b). With the electron-accepting PO group, the oxidation potentials of PN3O (Eox1 = 0.49 V, Eox2 = 0.68 V) are slightly higher than those of PN3 (Eox1 = 0.45 V, Eox2 = 0.65 V). PC3O (Eox1 = 0.31 V, Eox2 = 0.54 V) exhibited two lower oxidation potentials than both PN3 and PN3O, suggesting its easier oxidation character. Both PN3 (Eox2–ox1 = 0.20 V) and PN3O (Eox2–ox1 = 0.19 V) exhibited larger peak separations compared with M (Eox2–ox1 = 0.1 V), which implied a stronger electronic communication of the bis(triarylamine) groups in PN3 and PN3O. The peak separation was further enlarged for PC3O (ΔEox2–ox1 = 0.23 V). The results implied that changing the chemical structure of the heteropine ring significantly influenced the electronic communication of the bis(triarylamine) groups, with PC3O maintaining the strongest electronic communication in the current study.
The stable and reversible oxidation character of PN3, PN3O, and PC3O further allowed us to conduct UV-vis-NIR absorption spectroscopy experiments under the different oxidation states (Fig. 4c and d). Upon the addition of AgSbF6 as the oxidation reagent, both PN3O and PC3O showed a two-step oxidation process. In the first process, new peaks at 2000 nm and 1570 nm were observed for PN3O and PC3O, respectively. The low-energy transitions were rationalized to the intervalence charge transfer (IVCT) bands, which are characteristic of mixed-valence states in bis(triarylamine) systems.30,73 Electron paramagnetic resonance (EPR) spectroscopy experiments further revealed a featureless band (Fig. 4d), thus supporting the open-shell character of the PN3O and PC3O monocations. The theoretical studies of the radical cations not only confirmed the lowest transition (2078 nm for PN3O+˙ and 1439 nm for PC3O+˙), but also revealed the different spin densities (Fig. 4e and f). Compared with PN3O+˙, PC3O+˙ showed a broader spin distribution spread over the whole molecular backbone. Particularly, the P-center (spin density: 0.04) of PC3O+˙ showed a higher spin density compared with that of PN3O+˙ (spin density: 0.001). These results suggested that the P-center of PC3O+˙ has a strong impact on the mixed-valence state. Further addition of AgSbF6 converted PN3O and PC3O to the second stage, in which new peaks at 1000 nm for PN3O and 920 nm for PC3O emerged. The results implied that both PN3O and PC3O underwent the transition from the open-shell monocations to the closed-shell quinoid dications, which was supported by the theoretical studies (Fig. 4f).
Previous studies revealed that triaryl amines containing PAMs are promising candidates as hole-transporting layers (HTL) in perovskite solar cells (PSCs). Furthermore, PO functional groups were also demonstrated to be beneficial for the interface coordination and passivation of PSCs.74 Therefore, PN3O and PC3O were explored as new HTLs in inverted PSCs (Fig. 5, and see the details in the ESI†).
 | ||
| Fig. 5 (a) Device structure using PN3O and PC3O as the HTLs. (b) Density–voltage curves and (c) external quantum efficiency curves of the devices without HTL and with HTLs. | ||
We first fabricated the PSC devices in an HTL-free configuration (Fig. 5b and c). From the current density–voltage (J–V) curves of the device, the HTM-free device showed a low power conversion efficiency (PCE) of 7.68%. Then, PN3O was integrated as an HTM into the device, which showed an improved Voc (0.65 V) and FF (73%). The results indicated that PN3O played a better role in hole extraction and hole transport. We further used PC3O as the HTL in the PSCs. The PCE further reached up to 14.0% with a Voc of 0.79 V, FF of 75%, and short-circuit current density (Jsc) of 23.25 mA cm−2. Based on the CV experiments (Fig. 5a), the improved Voc can be attributed to the more suitable HOMO energy level of PC3O (−5.11 eV) compared with that of PN3O (−5.28 eV).
Conclusions
Herein, we have reported a new family of nonbenzenoid PAMs containing S-five-P-seven heterocycles. The selective P-chemistry and cyclization chemistry are modular to afford the target molecules with isomeric PN and PC structures. The distinct P-environments endowed the systems with intriguing photophysical, redox, and radical characteristics. Compared with the PN derivatives, the PC derivatives showed more planar S-five-P-seven heterocycles. Different from the electronic structure of azulene, the S-five-P-seven heterocycles of the PC derivatives maintained the effective σ*–π* hyperconjugation via the P-centers. This was not the case for PN derivatives. Experimental and theoretical results suggested that the PC derivative with a P(III) center underwent a structure planarization in S1, thus resulting in the unexpected room-temperature phosphorescence in solution. After decoration with two triarylamine groups, the P-PAMs readily formed stable radical cations, in which the PC derivative exhibited a stronger mixed-valence state and more delocalized radical density. As a proof of concept, bis(triarylamine) functionalized P-PAMs were successfully used as the HTLs in perovskite solar cells, which showed the promising PCE of 14%. Overall, our work provides a new design strategy for nonbenzenoid PAMs with intriguing optoelectronic properties.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by ShanghaiTech University start-up funding. YR also acknowledges the support from Shanghai Clinical Research and Trial Center, Analytical Instrumentation Center (SPST-AIC10112914), SPST, and the HPC Platform of ShanghaiTech University for providing resources and computing time. The authors cordially thank Shuya Wen for the assistant of the single-crystal X-ray measurements.Notes and references
- J. Corrigan, J. Yeow, P. Judzewitsch, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2019, 58, 5170–5189 CrossRef PubMed
.
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS PubMed
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed
- M. Ball, Y. Zhou, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. X. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267–276 CrossRef CAS PubMed
- H. Xin, B. Hou and X. Gao, Acc. Chem. Res., 2021, 54, 1737–1753 CrossRef CAS PubMed
- S. H. Pun and Q. Miao, Acc. Chem. Res., 2018, 51, 1630–1642 CrossRef CAS PubMed
- M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS PubMed
- C. Chen, J.-Y. Zhang, X.-Y. Wang and J. Pei, Chem. Mater., 2023, 35, 10277–10294 CrossRef CAS
- Chaolumen, I. A. Stepek, K. E. Yamada, H. Ito and K. Itami, Angew. Chem., Int. Ed., 2021, 60, 23508–23532 CrossRef CAS PubMed
- J. Liu, S. Mishra, C. A. Pignedoli, D. Passerone, J. I. Urgel, A. Fabrizio, T. G. Lohr, J. Ma, H. Komber, M. Baumgarten, C. Corminboeuf, R. Berger, P. Ruffieux, K. Müllen, R. Fasel and X. Feng, J. Am. Chem. Soc., 2019, 141, 12011–12020 CrossRef CAS PubMed
- H. Koki, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, A. Konishi and M. Yasuda, J. Am. Chem. Soc., 2022, 144, 3370–3375 CrossRef PubMed
- M. Mamada, M. Hayakawa, J. Ochi and T. Hatakeyama, Chem. Soc. Rev., 2024, 53, 1624–1692 RSC
- C. K. Frederickson, L. N. Zakharov and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 16827–16838 CrossRef CAS PubMed
- B. Prajapati, M. D. Ambhore, D.-K. Dang, P. J. Chmielewski, T. Lis, C. J. Góme-García, P. M. Zimmerman and M. Sępień, Nat. Chem., 2023, 15, 1541–1548 CrossRef CAS PubMed
- A. Shimizu, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Miyata and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 6076–6079 CrossRef CAS PubMed
- P. Cai, X.-Y. Chen, J. Cao, L. Ruppenthal, J. M. Gottfried, K. Müllen and X.-Y. Wang, J. Am. Chem. Soc., 2021, 143, 5314–5318 CrossRef PubMed
- Y. Zhou, G. Baryshnikov, X. Li, M. Zhu, H. Ågren and L. Zhu, Chem. Mater., 2018, 30, 8008–8016 CrossRef CAS
- D. Dunlop, L. Ludvíkova, A. Banerjee, H. Ottosson and T. Slanina, J. Am. Chem. Soc., 2023, 145, 21569–21575 CrossRef CAS PubMed
- Y. Yamaguchi, M. Takubo, K. Ogawa, K. Nakayama, T. Koganezawa and H. Katagiri, J. Am. Chem. Soc., 2016, 138, 11335–11343 CrossRef CAS PubMed
- E. Amir, R. J. Amir, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2011, 133, 10046–10049 CrossRef CAS PubMed
- S. Wang, M. Tang, L. Wu, L. Bian, L. Jiang, J. Liu, Z.-B. Tang, Y. Liang and Z. Liu, Angew. Chem., Int. Ed., 2022, 61, e202205658 CrossRef CAS PubMed
- K. K. Hollister, A. Molino, G. Breiner, J. E. Walley, K. E. Wentz, A. M. Conley, D. A. Dickie, D. J. D. Wilson and R. J. Hilliard Jr, J. Am. Chem. Soc., 2022, 144, 590–598 CrossRef CAS PubMed
- T. Ma, J. Dong and D.-T. Yang, Chem. Commun., 2023, 59, 13679–13689 RSC
- M. Crumbach, J. Bachmann, L. Fritze, A. Helbig, I. Krummenacher, H. Braunschweig and H. Helten, Angew. Chem., Int. Ed., 2021, 60, 9290–9295 CrossRef CAS PubMed
- M. Metzler, M. A. Virovets, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2023, 145, 23824–23831 CrossRef CAS PubMed
- J. Wang, A. Zheng, Y. Xiang and J. Liu, J. Am. Chem. Soc., 2023, 145, 14912–14921 CrossRef CAS PubMed
- S. Ito, Y. Tokimaru and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 7256–7260 CrossRef CAS PubMed
- S. Mishra, M. Krzeszewski, C. A. Pignedoli, P. Ruffieux, R. Fasel and D. T. Gryko, Nat. Commun., 2018, 9, 1714 CrossRef PubMed
- S. Hayakawa, A. Kawasaki, Y. Hong, D. Uraguchi, T. Ooi, D. Kim, T. Akutagawa, N. Fukui and H. Shinokubo, J. Am. Chem. Soc., 2019, 141, 19807–19816 CrossRef CAS PubMed
- G. Tan and X. Wang, Acc. Chem. Res., 2017, 50, 1997–2006 CrossRef CAS PubMed
- T. Jin, L. Kunze, S. Breumaier, M. Bolte, H.-W. Lerner, F. Jäkle, R. F. Winter, M. Braun, J.-M. Mewes and M. Wagner, J. Am. Chem. Soc., 2022, 144, 13704–13716 CrossRef CAS PubMed
- W. Wang, F. Hanindita, Y. Tanaka, Y. Kotaro Ochiai, H. Sato, Y. Li, T. Yasuda and S. Ito, Angew. Chem., Int. Ed., 2023, 62, e202218176 CrossRef CAS PubMed
- S. Tang, L. Zhang, H. Ruan, Y. Zhao and X. Wang, J. Am. Chem. Soc., 2020, 142, 7340–7344 CrossRef CAS PubMed
- S. Takahashi, M. Murai, Y. Hattori, S. Seki, T. Yanai and S. Yamaguchi, J. Am. Chem. Soc., 2024, 146, 22642–22649 CrossRef CAS PubMed
- M. Murai, M. Abe, S. Ogi and S. Yamaguchi, J. Am. Chem. Soc., 2022, 144, 20385–20393 CrossRef CAS PubMed
- R. Deka, M. A. Ansari, S. Chattopadhyay, R. Lomoth, A. Thapper and A. Orthaber, Angew. Chem., Int. Ed., 2024, e202406076 Search PubMed
- N. Asok, J. R. Gaffen and T. Baumgartner, Acc. Chem. Res., 2023, 56, 536–547 CrossRef CAS PubMed
- P.-A. Bouit, A. Escande, R. Szűcd, D. Szieberth, C. Lescop, L. Nyulászi, M. Hissler and R. Réau, J. Am. Chem. Soc., 2012, 134, 6524–6527 CrossRef CAS PubMed
- T. Delouche, E. Caytan, M. Cordier, T. Roisnel, G. Taupier, Y. Molard, N. Vanthuyne, B. L. Guennic, M. Hissler, D. Jacquemin and P.-A. Bouit, Angew. Chem., Int. Ed., 2022, 61, e202205548 CrossRef CAS PubMed
- N. Hashimoto, R. Y. Umano, S. Nakamura, S. Mori, H. Ohta, Y. Watanabe and M. Hayashi, J. Am. Chem. Soc., 2018, 140, 2046–2049 CrossRef CAS PubMed
- C. Romero-Nieto, A. López-Andarias, C. Egler-Lucas, F. Gebert, J.-R. Neus and O. Pilgram, Angew. Chem., Int. Ed., 2015, 54, 15872–15875 CrossRef CAS PubMed
- P. Hindenberg, M. Bush, A. Paul, M. Bernhardt, P. Gemessy, F. Rominger and C. Romero-Nieto, Angew. Chem., Int. Ed., 2018, 57, 15157–15161 CrossRef CAS PubMed
- J. D. R. Asherl, C. Neiβ, A. Vogel, J. Graf, F. Rominger, T. Oeser, F. Hampel, A. Görling and M. Kivala, Chem.–Eur. J., 2020, 26, 13157–13162 CrossRef PubMed
- L. Xu, D. Wu, W. Lv, Y. Xiang, Y. Liu, Y. Tao, J. Yin, M. Qian, P. Li, L. Zhang, S. Chen, O. F. Mohammed, O. M. Bakr, Z. Duan, R. Chen and W. Huang, Adv. Mater., 2022, 34, 2107111 CrossRef CAS PubMed
- M. Grzybowski, M. Taki, K. Senda, Y. Sato, T. Ariyoshi, Y. Okada, R. Kawakami, T. Imamura and S. Yamaguchi, Angew. Chem., Int. Ed., 2018, 57, 10137–10141 CrossRef CAS PubMed
- Z. Yang, C. Li, C. Liu, X. Li, N. Yu and Y. Ren, Angew. Chem., Int. Ed., 2022, 61, e202212844 CrossRef CAS PubMed
- H. Jansen, J. C. Slootweg and K. Lammertsma, Beilstein J. Org. Chem., 2011, 7, 1713–1721 CrossRef CAS PubMed
- K. Padberg, J. D. R. Ascherl, F. Hampel and M. Kivala, Chem.–Eur. J., 2020, 26, 3474–3478 CrossRef CAS PubMed
- T. Delouche, A. Mocanu, T. Roisnel, R. Szűcs, E. Jacques, Z. Benkő, L. Nyulászi, P.-A. Bouit and M. Hissler, Org. Lett., 2019, 21, 802–806 CrossRef CAS PubMed
- Z. Yang, X. Li, K. Yang, Z. Zhang, Y. Wang, N. Yu, T. Baumgartner and Y. Ren, Org. Lett., 2022, 24, 2045–2049 CrossRef CAS PubMed
- Y. Ren, M. Sezen, F. Gao, F. Jäkle and Y.-L. Loo, Chem. Sci., 2016, 7, 4211–4219 RSC
- K. Andoh, M. Murai, P.-A. Bouit, M. Hissler and S. Yamaguchi, Angew. Chem., Int. Ed., 2024, e202410204 CAS
- K. Nishimura, K. Hirano and M. Miura, Org. Lett., 2020, 22, 3185–3189 CrossRef CAS PubMed
- M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900–9930 CrossRef CAS PubMed
- K. B. Jørgensen, Molecules, 2010, 15, 4334–4358 CrossRef PubMed
- O. Fadhel, D. Szieberth, V. Deborde, C. Lescop, L. Nyulászi, M. Hissler and R. Réau, Chem.–Eur. J., 2009, 15, 4914–4924 CrossRef CAS PubMed
- T. Delouche, R. Mokrai, T. Roisnel, D. Tondelier, B. Geffroy, L. Nyulászi, Z. Benkö, M. Hissler and P.-A. Bouit, Chem.–Eur. J., 2020, 26, 1856–1863 CrossRef CAS PubMed
- Y. Kuninobu, T. Yoshida and K. Takai, J. Org. Chem., 2011, 76, 7370–7376 CrossRef CAS PubMed
- I. Kawashima, H. Imoto, M. Ishida, H. Furuta, S. Yamamto, M. Mitsuishi, S. Ianaka, T. Fujii and K. Naka, Angew. Chem., Int. Ed., 2019, 131, 11812–11816 CrossRef
- D. Shukla and P. Wan, J. Am. Chem. Soc., 1993, 115, 2990–2991 CrossRef CAS
- R. Kotani, L. Liu, P. Kumar, H. Kuramochi, T. Tahara, P. Liu, A. Osuka, P. B. Karadakov and S. Saito, J. Am. Chem. Soc., 2020, 142, 14985–14992 CrossRef CAS PubMed
- J. Xu, A. Takai, Y. Kobayashi and M. Takeuchi, Chem. Commun., 2013, 49, 8447–8449 RSC
- H. Shu, J. Rao, J. Chen, X. Wang, X. Wu, H. Tian, H. Tong and L. Wang, J. Mater. Chem. C, 2020, 8, 14360–14364 RSC
- A. Lv, W. Ye, X. Jiang, N. Gan, H. Shi, W. Yao, H. Ma, Z. An and W. Huang, J. Phys. Chem. Lett., 2019, 10, 1037–1042 CrossRef CAS PubMed
-
B. Valeur and M. N. Serberan-Santos, Molecular Fluorescence: Principles and Applications, Wiley-VCH, Weinheim, Germany, 2nd edn, 2012 Search PubMed
- Y. Tao, R. Chen, H. Li, J. Yuan, Y. Wan, H. Jiang, C. Chen, Y. Si, C. Zheng, B. Yang, G. Xing and W. Huang, Adv. Mater., 2018, 30, 1803856 CrossRef PubMed
- Z. An, C. Zhen, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed
- A. d. Cózar and C. Romero-Nieto, Inorg. Chem., 2023, 62(10), 4097–4105 CrossRef PubMed
- O. Larrañaga, C. Romero-Nieto and A. d. Cózar, Chem.–Eur. J., 2019, 25, 9035–9044 CrossRef PubMed
- N. C. Baird, J. Am. Chem. Soc., 1972, 94, 4941–4948 CrossRef CAS
- X. Jin, S. Li, L. Guo, J. Hua, D.-H. Qu, J. Su, Z. Zhang and H. Tian, J. Am. Chem. Soc., 2022, 144, 4883–4896 CrossRef CAS PubMed
- Y. Chen, K.-H. Chang, F.-Y. Meng, S. M. Tseng and P.-T. Chou, Angew. Chem., Int. Ed., 2016, 60, 7205–7212 CrossRef PubMed
- M. Krug, N. Fröhlich, D. Fehn, A. Vogel, F. Rominger, K. Meyer, T. Clark, M. Kivala and D. M. Guldi, Angew. Chem., Int. Ed., 2021, 60, 6771–6777 CrossRef CAS PubMed
- X. Chen, J. Huang, F. Gao and B. Xu, Chem, 2023, 9, 562–575 CAS
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2379333–2379336. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05857g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |